

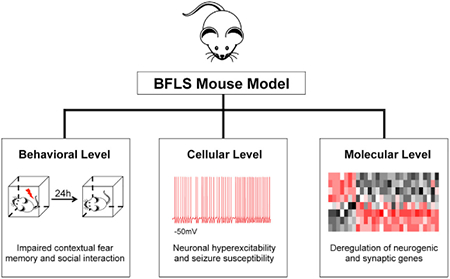
SUMMARY
Mutations of the transcriptional regulator PHF6 cause the X-linked intellectual disability disorder Börjeson-Forssman-Lehmann syndrome (BFLS), but the pathogenesis of BFLS remains poorly understood. Here, we report a mouse model of BFLS, generated using a CRISPR-Cas9 approach, in which cysteine 99 within the PHD domain of PHF6 is replaced with phenylalanine (C99F). Mice harboring the patient-specific C99F mutation display deficits in cognitive functions, emotionality, and social behavior, as well as reduced threshold to seizures. Electrophysiological studies reveal that the intrinsic excitability of entorhinal cortical stellate neurons is increased in PHF6 C99F mice. Transcriptomic analysis of the cerebral cortex in C99F knockin mice and PHF6 knockout mice show that PHF6 promotes the expression of neurogenic genes and represses synaptic genes. PHF6-regulated genes are also overrepresented in gene signatures and modules that are deregulated in neurodevelopmental disorders of cognition. Our findings advance our understanding of the mechanisms underlying BFLS pathogenesis.
Graphical Abstract
In Brief
Cheng et al. generated a mouse model of Börjeson-Forssman-Lehmann syndrome containing a patient-specific mutation of PHF6. PHF6 knockin mice display cognitive impairments, neuronal hyperexcitability, and seizure susceptibility. PHF6 promotes neurogenic and repressed synaptic genes in the cortex. This study advances understanding of the cellular and molecular underpinnings of BFLS.
INTRODUCTION
Intellectual disability (ID) is a prevalent developmental disorder that affects 1% to 3% of the general population (Bhasin et al., 2006). With no effective treatments, there is an urgent need to better understand the pathogenesis of ID. Human genetics studies have uncovered a large number of gene mutations associated with ID. Although a few genetic causes such as fragile X and Rett syndromes have been the subject of intense scrutiny (Schubert et al., 2015), the functions of most ID proteins and the mechanisms by which these proteins regulate pathologically relevant biological processes remain poorly understood.
Börjeson-Forssman-Lehmann syndrome (BFLS) was identified >5 decades ago as an X-linked syndrome of moderate to severe impaired cognitive function associated with developmental delay, truncal obesity, and seizures (Börjeson et al., 1962; Lower et al., 2002). Forty years later, mutations in the gene encoding the plant homeodomain (PHD) and zinc-finger domain-containing protein PHF6 were identified as the genetic cause of BFLS (Lower et al., 2002). Since then, multiple mutations in distinct regions of PHF6, including within its two PHDs, have been identified in BFLS patients (Jahani-Asl et al., 2016). However, how PHF6 mutations trigger the pathogenesis of BFLS remains to be elucidated. Modeling of cognitive disorders such as fragile X and Rett syndromes in mice has helped to advance our understanding of these disorders (Schubert et al., 2015). However, a mouse model of BFLS using patient-specific PHF6 mutations has yet to be established.
Knockdown of PHF6 in the mouse cerebral cortex impairs the migration of cortical neurons, leading to white matter heterotopias (Zhang et al., 2013); this suggests a role for PHF6 in the regulation of cortical neuron positioning. PHF6 protein is localized in the nucleus and interacts with the polymerase II associated factor (PAF1) transcription elongation factor complex, nucleosome remodeling deacetylase (NuRD) chromatin remodeling complex, and upstream binding factor (UBF1) (Liu et al., 2014; Todd et al., 2015; Zhang et al., 2013). Accordingly, PHF6 is thought to regulate transcription in neurons (Zhang et al., 2013). How PHF6 regulation of gene expression, particularly at a genome-wide level, affects BFLS pathogenesis remains incompletely understood.
In this study, we report a patient-specific PHF6 mutant mouse model of BFLS. Using a CRISPR-Cas9 approach, we have generated mice in which cysteine 99 within the PHD domain of PHF6 is replaced with phenylalanine (C99F). Mice with the patient-specific PHF6 C99F mutation display deficits in cognitive functions, emotionality, and social behaviors, and are susceptible to seizures. In electrophysiological studies, the intrinsic excitability of entorhinal cortical layer II stellate neurons is increased in PHF6 C99F mice, providing a cellular basis for the susceptibility of BFLS mice to seizures. Genome-wide RNA sequencing (RNA-seq) analyses of the cerebral cortex in PHF6 C99F and PHF6 knockout (KO) mice show that PHF6 promotes the expression of neurogenic genes and concomitantly suppresses the expression of synaptic genes. PHF6-regulated genes are also overrepresented in gene signatures and modules that are deregulated in neurodevelopmental disorders of cognition. Our findings broaden our understanding of the cellular and molecular underpinnings of BFLS pathogenesis.
RESULTS
Generation of a Patient-Specific PHF6 Mutant Mouse Model of BFLS
We characterized the expression of different patient-specific mutations of PHF6 in cells. Among patient-specific mutations of PHF6, three mutations led to a substantial reduction in PHF6 protein levels (Figure 1A), suggesting that BFLS may result from hypomorphic PHF6 function. Among the three mutations, substitution of cysteine 99 with phenylalanine (C99F), located in the first PHD of PHF6, dramatically reduced the levels of PHF6 protein (Figure 1A).
Figure 1. Generation of Patient-Specific PHF6 Mutant Mouse Model of BFLS.
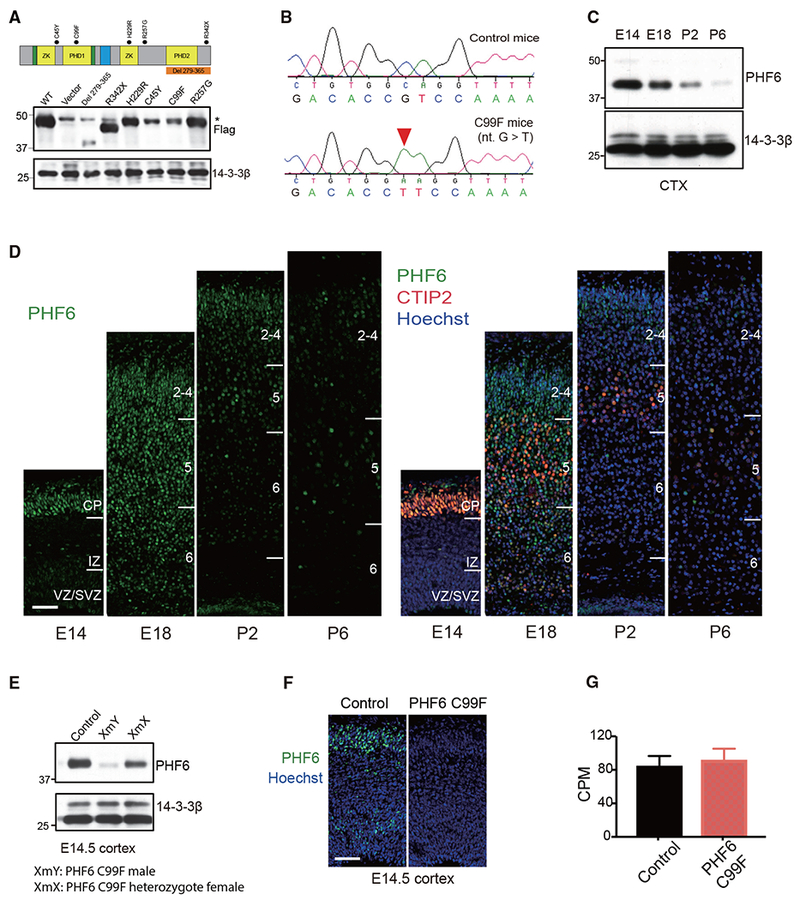
(A) Top: schematic of PHF6 protein structure. Dots: patient-specific mutations in BFLS; nucleus localization sequence (green);nucleolar localization sequence (blue). Bottom: immunoblot of lysates of 293T cells transfected with expression plasmids encoding FLAG-tagged PHF6, patient-specific mutations of PHF6, or control vector using an antibody to FLAG or 14-3-3β, the latter serving as loading control. Non-specific immunoreactive band (*).
(B) Sequencing traces of wild-type and C99F PHF6 alleles.
(C) Immunoblot of PHF6 in lysates of the cerebral cortex in mice at embryonic day 14 (E14), E18, postnatal day 2 (P2), and P6.
(D) Immunohistochemical analyses of PHF6 (left) and CTIP2 (right) in sections of the cerebral cortex from E14, E18, P2, and P6 mice. DNA dye bisbenzimide (Hoechst) was also used (right). CP, cortical plate; IZ, intermediate zone; VZ/SVZ, ventricular zone/subventricular zone. Scale bar, 50 μm.
(E) Immunoblot of PHF6 in lysates of the cerebral cortex from control male, C99F male, and C99F heterozygote female mice.
(F) Immunohistochemical analyses of PHF6 in sections of the cerebral cortex from E14.5 control and C99F mice. Hoechst was also used. Scale bar, 50 μm.
(G) The levels of PHF6 mRNA in the cerebral cortex of control and C99F mice in RNA-seq analysis (n = 5). CPM, counts per million.
Data are presented as means ± SEMs. See also Figure S1 and Table S1.
We next generated C99F knockin mice. Using a CRISPR-Cas9 method, the single nucleotide mutation nt.296G > T was successfully introduced in the mouse PHF6 gene, which was confirmed with Sanger sequencing (Figures 1B and S1A). PHF6 C99F mice were backcrossed to mice with C57BL/6 background for >10 generations. Screening of potential CRISPR-Cas9 off-target sites in the mouse genome predicted by COSMID (Cradick et al., 2014) and CRISPOR (Haeussler et al., 2016), as well as 1-kb regions flanking the C99F point mutation, revealed no other mutations (Figures S1A and S1B; Table S1). PHF6 C99F knockin mice were born with the Mendelian ratio; had slightly reduced body weight, body length, and body surface area; and survived to adulthood (Figures S1D-S1F). Notably, additional morphological analyses of PHF6 C99F and control littermate mice showed little or no differences in BMI, ear length, facial length, interocular distance, and testis weight when normalized to body weight (Figures S1G-S1K). These data suggest that PHF6 C99F mice do not phenocopy syndromic features of BFLS, such as obesity, facial dysmorphisms, small genitalia, and large ears.
Having generated the PHF6 C99F knockin mouse, we characterized the expression of PHF6 in the cerebral cortex in these mice. As expected, immunoblotting analyses in control mice revealed that PHF6 was expressed in the mouse cerebral cortex at early embryonic ages and was substantially downregulated postnatally (Figure 1C) (Zhang et al., 2013). PHF6 co-localized with the layer V marker Ctip2 in the cortical plate of control mice at embryonic day 14 (E14), and was expressed in all of the layers of the cortical plate at E18 (Figure 1D). The levels of PHF6 protein were substantially reduced, although not eliminated, in the cerebral cortex in PHF6 C99F mice at E14.5 and postnatal day 0 (P0) (Figures 1E, 1F, and S1C). However, the levels of PHF6 mRNA in the cerebral cortex in the C99F mice were not reduced (Figure 1G), suggesting that the stability of PHF6 protein in C99F mice may be reduced.
PHF6 C99F Knockin Mice Display Impairments in Cognition, Social Interaction, and Emotionality
We conducted behavioral analyses on PHF6 C99F mice in a battery of tests. In analyses of contextual fear conditioning, PHF6 C99F mice exhibited significantly reduced freezing levels compared with control littermate animals on day 2 (Figure 2A), suggesting that contextual memory is impaired in PHF6 C99F mice. No differences were observed between groups of mice during either the baseline period or tone-shock training on day 1 (Figure S2A), and no differences in shock sensitivity were evident between groups (data not shown). Also, the groups did not differ during the altered context baseline on day 3, although moderate performance deficits were observed in the PHF6 C99F mice during the auditory cue test on day 3 (Figures S2A and S3A). Defects in contextual fear memory were observed in another independent cohort of mice (Figure S3A). In other analyses, PHF6 C99F mice did not exhibit performance deficits during the cued, place (Figure S2B), or probe (not shown) conditions when tested in the Morris water maze (MWM), suggesting that PHF6 C99F mice may not be impaired in spatial learning acquisition or retention. In a novel object location test to evaluate recognition memory, control mice but not PHF6 C99F mice spent more time investigating the moved object than the stationary object (Figure 2B). Thus, our data suggest that PHF6 C99F mice have deficits in contextual fear conditioning and in some forms of spatial learning.
Figure 2. PHF6 C99F Mice Exhibit Deficits in Cognition, Social Interaction, and Emotionality.
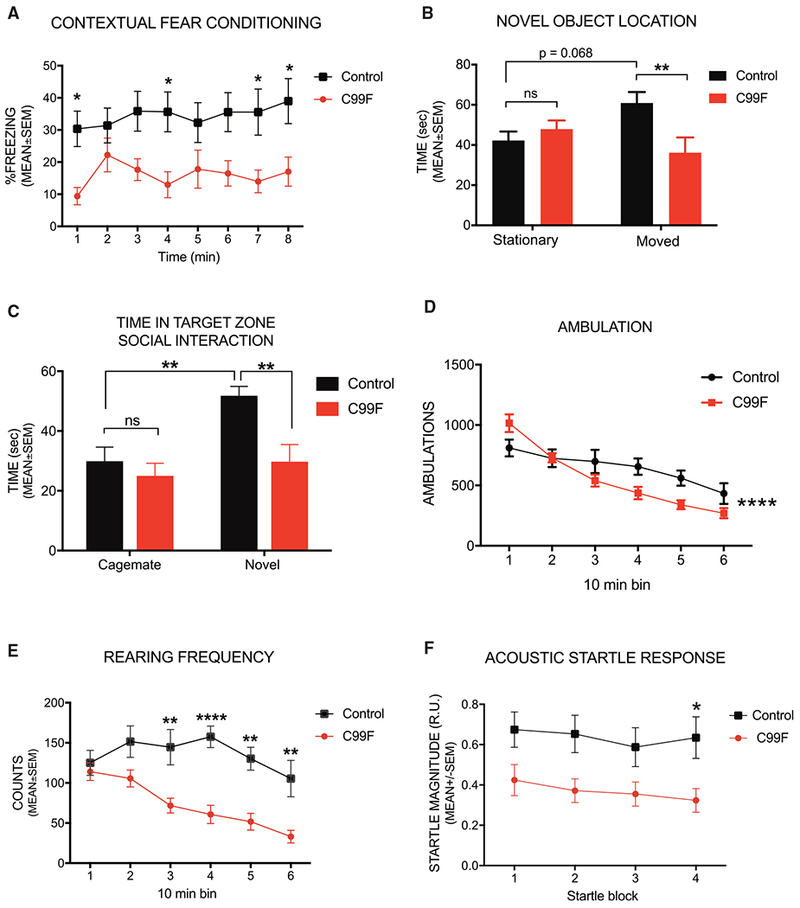
(A) Contextual fear conditioning test.
(B) Novel object-location test.
(C) Three-chambered social approach test.
(D) Ambulations.
(E)Vertical rearing frequency.
(F) Acoustic startle.
*p < 0.05, **p < 0.01, ****p < 0.0001 for genotype × time (10 min bins) interaction, ns, non-significant; two-way repeated-measures ANOVA, followed by pairwise comparisons that were Bonferroni adjusted. Data are presented as mean ± SEM. See also Figures S2 and S3.
To characterize sociability and preference for social novelty, mice were subjected to a three-chamber social approach task. Both control and PHF6 C99F mice exhibited a preference for investigating a novel stimulus mouse versus an empty withholding cage (data not shown). However, control littermate mice but not PHF6 C99F mice showed a preference for a novel mouse compared to a familiar (cagemate) mouse (Figures 2C and S3B). A similar preference for the novel mouse in control but not PHF6 C99F mice was observed in an independent cohort (Figures S3C and S3D). These data suggest that PHF6 C99F mice display normal levels of sociability, but they may be impaired in social recognition memory.
Ambulatory activity and exploratory behavior during a control 1-hr locomotor activity test before the conditioned fear procedure revealed reduced activity in PHF6 C99F mice, but only in the latter part of the test session (Figure 2D). Vertical rearing frequency was also significantly reduced in PHF6 C99F mice (Figure 2E). Similar effects of the PHF6 C99F mutation on ambulatory activity and vertical rearing were observed in an independent cohort (Figures S3E and S3F). These results suggest that PHF6 C99F mice may show reduced levels of attention or interest toward novel environments. In two independent cohorts, PHF6 C99F mice also exhibited significantly reduced acoustic startle response throughout the test session (Figures 2F and S3G), although no significant effects were observed on pre-pulse inhibition (PPI) (not shown). These data suggest that sensorimotor reactivity but not gating was attenuated in PHF6 C99F mice. Our data suggest that PHF6 plays critical roles in normal cognitive development as it relates to certain forms of learning and memory, emotionality, and social behaviors.
PHF6 C99F Mice Exhibit Increased Neuronal Excitability and Seizure Susceptibility
Besides impaired cognitive functions, seizures represent an important aspect of the clinical presentation of BFLS (Jahani-Asl et al., 2016). To characterize susceptibility to seizures, we injected PHF6 C99F and control littermate mice intraperitoneally with the GABA antagonist pentylenetetrazol (PTZ). PHF6 C99F mice showed reduced time to reach generalized seizures (Figure 3A). Because PTZ-induced seizures progress rapidly (Zhang and Wong, 2012), we used the glutamate agonist kainic acid, allowing for scoring of slowly progressing seizure stages, according to the Racine scale (Racine et al., 1972). PHF6 C99F animals showed more rapid seizure progression and greater number of seizure events at higher Racine scores (Figure 3B). These results suggest that PHF6 C99F mice phenocopy the predisposition of BFLS patients to seizures.
Figure 3. PHF6 C99F Mice Exhibit Seizure Susceptibility and Increased Neuronal Excitability.
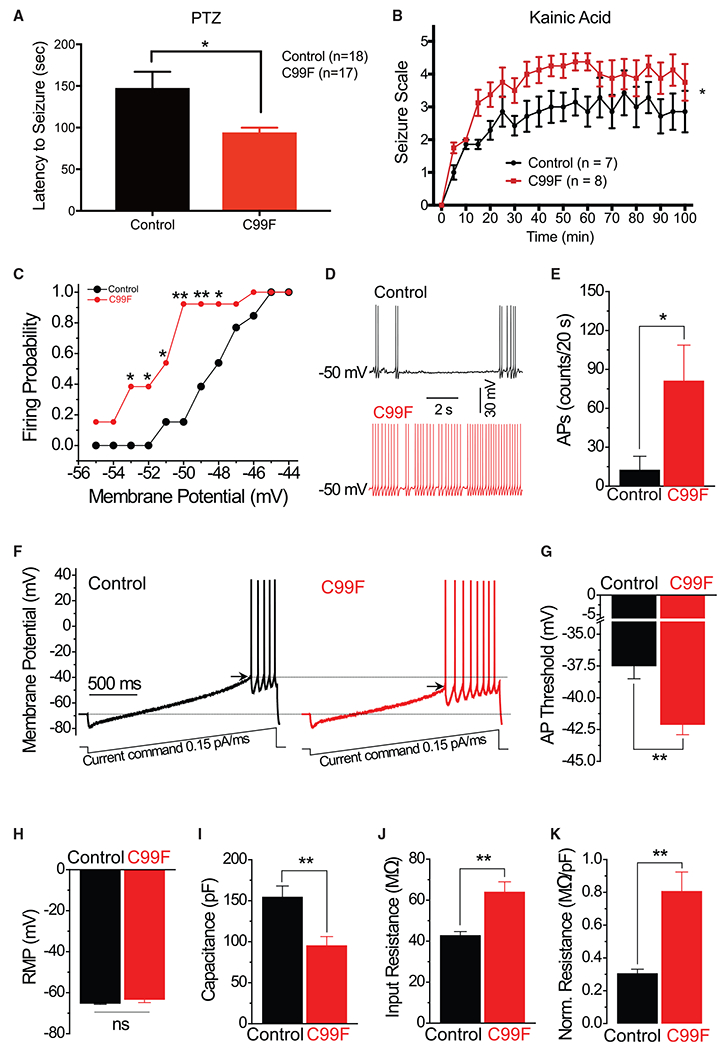
(A) Time to generalized seizure upon PTZ induction. Unpaired t test, *p < 0.05.
(B) Racine scale scoring of animal behavior upon kainic acid seizure induction. Two-way repeated-measures ANOVA. *p < 0.05.
(C) Current-clamped layer II stellate cells in acute slices of the entorhinal cortex were depolarized to potentials from −55 to −44 mV with a step of 1 mV via automatic current injection to induce action potential (AP) firing. *p < 0.05, **p < 0.01; chi-square test within corresponding potentials.
(D) Sample traces of spontaneous AP firing recorded at −50 mV in control and C99F neurons.
(E) AP firing rate was measured as number of AP fired during a 20-s period. *p < 0.05, t test.
(F) APs were evoked by a ramp current injection (0.15 pA/ms, lower traces). Representative AP traces are shown (upper traces). Only the first AP was used to estimate the AP threshold (arrows).
(G) Summary of the data in (F) revealed that AP threshold is decreased in C99F mice. **p < 0.01, t test.
(H) Average rest membrane potential (RMP).
(I) Membrane capacitance.
(J) Input resistance.
(K) Capacitance-normalized input resistance.
**p < 0.01, t test, unless specified. Data are presented as means ± SEMs.
Alterations of neuronal excitability in diverse neuronal populations may be epileptogenic (Scharfman, 2007). We focused our analyses on layer II stellate neurons in the entorhinal cortex, whose excessive activation triggers seizures (Vismer et al., 2015). Current-clamped stellate neurons in acute brain slices from C99F and control littermate mice were depolarized to a range of potentials from −55 to −44 mV with a 1-mV step via automatic current injection to induce action potential firing. Nearly 40% of all of the neurons in the brain slices of PHF6 C99F mice fired action potentials at −53 mV, compared to none from control mice, and >90% of all neurons from C99F mice fired action potentials at −50 mV, in contrast to only 15% of control neurons (Figure 3C). In addition, the number of action potentials fired at −50 mV during a 20-s period was markedly increased in C99F mice (Figures 3D and 3E). In a standard ramp protocol (Deng and Klyachko, 2016), the action potential threshold of stellate neurons in C99F mice was significantly decreased (Figures 3F and 3G). These results suggest that entorhinal cortical stellate neurons have increased excitability in PHF6 C99F mice.
We next determined the physiological basis for increased neuronal excitability in PHF6 C99F mice. No detectable changes in the resting membrane potential were observed in stellate neurons of C99F mice (Figure 3H). However, membrane capacitance was significantly decreased and input resistance was significantly increased in PHF6 C99F mice (Figures 3I and 3J). Input resistance normalized to cell capacitance in each recorded cell was also significantly increased in C99F mice (Figure 3K), suggesting that alterations in input resistance occur independently of changes in capacitance. Our data suggest that stellate neurons in C99F mice are hyperexcitable due at least in part to diminution of the action potential firing threshold associated with increased input resistance and decreased cell capacitance.
PHF6 Regulates Neurogenic and Synaptic Genes during Brain Development
Having identified PHF6 C99F knockin mice as a model of BFLS, we next characterized the mechanism by which PHF6 deficiency may contribute to BFLS pathogenesis. Because PHF6 regulates transcription (Jahani-Asl et al., 2016), we subjected the cerebral cortex from P0 PHF6 C99F mice and control littermates to RNA-seq analyses. The expression of only 48 genes was altered in PHF6 C99F mice with a false discovery rate (FDR) <0.05 in edgeR analyses. Because the C99F mutation leads to substantial but incomplete downregulation of the PHF6 protein (Figure 1E), residual PHF6 protein in these mice may not allow detection of robust transcriptional changes. It was fortuitous that, in generating the C99F allele, a nucleotide insertion adjacent to the Cas9 cleavage site was also obtained, leading to a frameshift mutation and consequent KO of PHF6 in mice (Figures S4A and S4B). Unlike the C99F knock-in mice, PHF6 KO animals died perinatally. Consistent with the prediction that PHF6 KO mice may provide a more robust means for the identification of PHF6-regulated genes, the expression of 193 genes was significantly altered (FDR <0.05 in edgeR) in the cerebral cortex of PHF6 KO mice compared to control littermate animals (Figure 4A; Table S2). Transcriptomic analysis using DEseq2 showed that 87.5% of differentially expressed genes upon PHF6 KO overlapped with those obtained by edgeR analyses. Notably, alterations in gene expression in the cerebral cortex upon PHF6 KO with FDR <0.05 correlated significantly with changes in their expression in PHF6 C99F mice (Figure 4B), supporting the interpretation that the C99F mutation may act as a hypomorphic allele in the regulation of gene expression. In control permutation analyses, the correlation of differentially regulated genes upon PHF6 KO with their changes in PHF6 C99F mice was highly significant compared to 10,000 random samplings of 193 gene pairs in PHF6 KO and C99F mice (Figure 4C). In addition, significantly downregulated and upregulated genes upon PHF6 KO (FDR <0.05) were also downregulated and upregulated, respectively, in PHF6 C99F mice (Figure 4D). We therefore focused subsequent analyses on deregulated genes in the cerebral cortex of PHF6 KO mice.
Figure 4. Transcriptional Alterations in the Cerebral Cortex in PHF6-Deficient Mice.
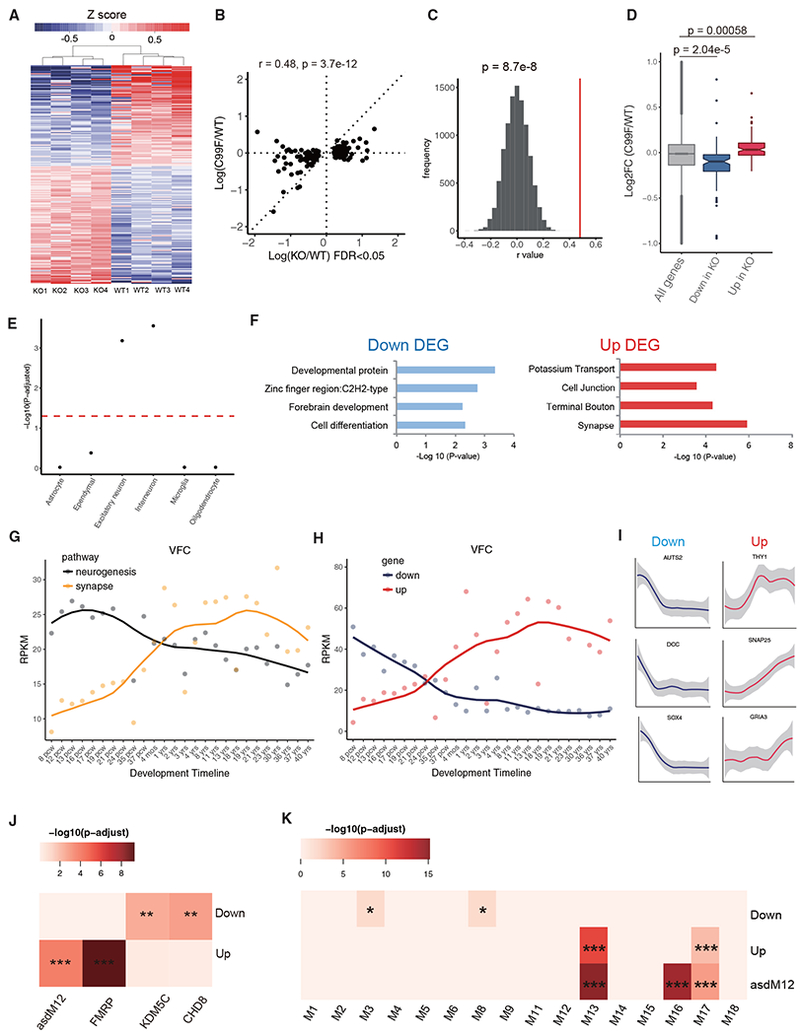
(A) Heatmap of 193 differentially expressed genes upon PHF6 KO by RNA-seq (edgeR, FDR <0.05).
(B) Correlation analysis of alteration of gene expression in the cerebral cortex of PHF6 C99F mice that were deregulated upon PHF6 KO (193 genes; FDR <0.05).
(C) Spearman correlation rho distribution by comparing gene expression changes in PHF6 KO and C99F mice, calculated by random sampling of 193 genes 10,000 times. Red line: rho = 0.48 (see B). Z score statistics; p = 8.7e-8.
(D) Boxplot of log fold change of upregulated and downregulated genes upon PHF6 KO in C99F. Wilcoxon rank sum test; p = 5.8e-4 (upregulated), p = 2.04e-5 (downregulated).
(E) Gene enrichment analysis of major brain cell types. Dotted red line: p-adjusted = 0.05.
(F) Gene Ontology analysis by DAVID for downregulated and upregulated genes upon PHF6 KO. DEG, differentially expressed gene.
(G) Average expression for neurogenic (GO: 0050769) and synaptic (GO: 0007268) genes across human brain developmental time in the VFC based on BrainSpan datasets.
(H) Average expression of downregulated and upregulated genes upon PHF6 KO across developmental time in the VFC based on BrainSpan datasets.
(I) Example of individual gene expression across development time.
(J) Enrichment analysis of upregulated and downregulated genes upon PHF6 KO with gene targets of FMRP, KDM5C, CHD8, and the autism module asdM12, containing misregulated genes in the frontal and temporal cerebral cortex in the brains of patients with autism spectrum disorder.
(K) Enrichment analysis of misregulated genes upon PHF6 KO and the asdM12 gene module with previously described developmental gene co-expression network modules.
Enrichment analysis was tested by hypergeometric test, followed by Benjamini-Hochberg multiple testing corrections. *p-adjusted < 0.05, **p-adjusted < 0.01, ***p-adjusted < 0.001. See also Figure S4.
Using the dataset that contains genes enriched in major brain cell types (Zeisel et al., 2015), we found that PHF6-regulated genes were enriched in interneurons and excitatory neurons (Figure 4C) but not in glia cells. In DAVID Gene Ontology (GO) analyses, PHF6-repressed genes included those encoding potassium transport, terminal bouton, and synapse proteins, whereas PHF6-activated genes included those encoding developmental, zinc finger, and cell differentiation proteins (Figure 4F). These analyses suggest that PHF6 promotes the expression of differentiation genes and concomitantly suppresses the expression of synaptic genes in the cerebral cortex.
We next characterized the developmental expression pattern of PHF6-regulated genes in the human brain using a BrainSpan dataset (BrainSpan, 2013). PHF6 mRNA was significantly downregulated beginning at post-conceptual week 8 (PCW8) during human brain development in the ventral frontal cortex (VFC) (Figure S4C), mimicking its expression pattern in mice. In contrast, doublecortin (DCX) was upregulated from PCW8 to PCW24 and subsequently downregulated (Figure S4C). The expression of genes upregulated upon PHF6 loss steadily increased throughout human development from PCW8 to 40 years of age in the VFC, similar to the expression pattern of genes for chemical synaptic transmission (Figures 4G-4I). Concomitantly, genes downregulated upon PHF6 loss had high levels of expression at early time points during human brain development, but steadily declined starting at PCW8, displaying a similar pattern as genes encoding neurogenesis (Figures 4G-4I). These developmental expression patterns were also observed for upregulated and downregulated genes upon PHF6 loss identified using DEseq2 analyses (Figure S4D). Similar results were observed in other brain regions, including the dorsal frontal cortex (DFC), medial frontal cortex (MFC), and primary somatosensory cortex (S1C) (Figure S4F). Our data suggest that PHF6 loss may lead to premature downregulation of neurogenic genes and activation of synaptic genes. Notably, the density of dendritic spines in layer II stellate neurons in the entorhinal cortex did not appear to increase prematurely in PHF6 C99F mice (Figures S4I and S4J), although premature maturation of other aspects of synaptic development upon PHF6 loss in the brain remains to be explored.
PHF6-Regulated Genes Are Implicated in Neurodevelopmental Disorders of Cognition
Because PHF6 mutations cause ID, we reasoned that differentially expressed genes in the PHF6-deficient cortex also may be regulated by proteins implicated in other neurodevelopmental disorders of cognition. Upregulated genes upon PHF6 loss overlapped significantly with targets of fragile X mental retardation protein (FMRP) (Figure 4J; Darnell et al., 2011), suggesting that PHF6 and FMRP regulate a common group of genes. In other analyses, downregulated genes upon PHF6 loss overlapped significantly with differentially expressed genes in the cerebral cortex of mice in which the histone demethylase protein KDM5C was knocked out and of mice with haploinsufficiency of the chromatin remodeling protein CHD8 (Figure 4J; Gompers et al., 2017; Iwase et al., 2016). In other analyses, upregulated genes upon PHF6 loss also overlapped significantly with the gene module termed asdM12, which is misregulated in the cerebral cortex in patients with autism spectrum disorders (Figure 4J; Voineagu et al., 2011). These observations suggest that PHF6-regulated genes overlap with genes implicated in other neurodevelopmental disorders of cognition beyond BFLS.
We also mapped PHF6-regulated genes onto available human developmental modules containing gene networks co-regulated across developmental stages, generated by weighted gene co-expression network analysis (WGCNA) using BrainSpan whole-genome transcriptomic data (Parikshak et al., 2013). Downregulated genes upon PHF6 loss were overrepresented in module 3 (M3), enriched for genes encoding chromatin organization, and M8, enriched for genes encoding neuron differentiation. In addition, upregulated genes upon PHF6 loss were overrepresented in M13 and M17 (Figure 4K), enriched for synaptic genes. The GO terms and temporal expression of overrepresented gene modules were similar to misregulated genes upon PHF6 loss (Figures 4F, S4G, and S4H). Notably, upregulated genes upon PHF6 loss and the autism-associated asdM12 gene module converged onto the developmental modules M13 and M17, representing synaptic terms (Figures 4K, S4E, and S4H). These results suggest that PHF6-regulated genes coalesce in distinct developmental gene modules in the human brain.
DISCUSSION
In this study, we report a mouse model of Börjeson-Forssman-Lehmann syndrome (BFLS). Mice harboring the patient-specific PHF6 C99F mutation display deficits in cognitive functions, emotionality, and social behaviors, and are susceptible to seizures, phenocopying key clinical features of BFLS. Electrophysiological analyses reveal increased excitability of entorhinal cortical stellate neurons secondary to reduced threshold to action potential firing in PHF6 C99F mice, providing a cellular basis for seizure predisposition of the brain upon PHF6 deficiency. Gene profiling analyses in PHF6-deficient mice show that PHF6 promotes the expression of neurogenesis genes and concomitantly represses synaptic genes. PHF6-regulated genes are overrepresented in gene signatures and modules that are deregulated in the brains of patients with neurodevelopmental disorders of cognition. Taken together, our findings provide insights into the cellular and molecular underpinnings of BFLS.
The identification of a novel mouse model of BFLS provides an excellent tool for studies of the pathogenesis of BFLS. In addition to their mild to severe intellectual disability, BFLS patients display disturbances in adaptive behaviors, including aggressive behavior, poor spatial awareness, and short attention span (Jahani-Asl et al., 2016). Consistent with the diverse picture of cognitive impairment in BFLS patients, behavior analyses reveal that PHF6 C99F mice have deficits in multiple domains, including some forms of learning and memory, social interactions, and emotionality.
The identification of increased seizure susceptibility in the C99F mouse sheds light on the cell types and neural circuits that may contribute to epilepsy in BFLS patients. The finding of increased excitability associated with reduced action potential and increased input resistance in entorhinal cortical layer II stellate neurons provides a cellular basis for seizures in BFLS. Because increased excitation from stellate cells to the hippocampal dentate gyrus is associated with temporal lobe epilepsy (Janz et al., 2017), it will be interesting to determine whether BFLS patients have focal seizures of temporal lobe origin. Notably, layer II stellate neurons project to the hippocampus via the perforant pathway, which plays a critical role in contextual fear memory and object location recognition (Kitamura et al., 2015; Tennant et al., 2018). Thus, electrophysiological abnormalities of stellate neurons may also contribute to defects in spatial recognition and contextual fear memory in PHF6 C99F knockin mice. Beyond neurons in the entorhinal cortex, it will be important in future studies to explore the role of other populations of neurons and their networks in the pathogenesis of epilepsy in BFLS.
Our study also provides insights into the role of PHF6 in brain development. The findings that PHF6 promotes the expression of neurogenesis genes and downregulates synaptic genes suggest that PHF6 may control the timing of neuronal differentiation, possibly preventing premature connectivity. Thus, deregulation of temporal control of neuronal maturation may contribute to the pathogenesis of neurodevelopmental disorders of cognition.
Analyses of PHF6-regulated genes also suggest molecular convergence with gene signatures and modules implicated in other neurodevelopmental disorders, including, potentially, autism spectrums disorders. Notably, clinical descriptions of BFLS are focused on intellectual disability and syndromic features of the disorder (Jahani-Asl et al., 2016). However, in a recent series of patients with neurodevelopmental disorders undergoing exome sequencing, a patient with autism spectrum disorder was found to harbor a pathogenic nonsense mutation of PHF6 at C305 (Baldridge et al., 2017). In future studies, it will be interesting to explore whether autistic features and behaviors are present in other BFLS patients and PHF6 C99F knockin mice, respectively.
In summary, our study advances understanding of the cellular and molecular underpinnings of BFLS and lays the foundation for potential treatments for neurodevelopmental disorders of cognition.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti PHF6 | Novus Biologicals | NB100-68262; RRID: AB_1109819 |
| Rat anti CTIP2 | Abcam | Ab18465; RRID: AB_2064130 |
| Mouse anti 14-3-3β | Santa Cruz | Sc-1657; RRID: AB_626618 |
| Rabbit anti FLAG | Sigma | F7425; RRID: AB_439687 |
| Alexa-Fluro 488-conjugated Avidin | Thermo Fisher Scientific | A21370 |
| Alexa-Fluro 488-conjugated goat anti rabbit | Abcam | Ab150077; RRID: AB_2630356 |
| Cy3-conjugated goat anti rat | Invitrogen | A10522 |
| Goat anti-mouse HRP | Calbiochem | 401253; RRID: AB_437779 |
| Goat anti-rabbit HRP | VWR | 80056-796 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kainic Acid | Sigma | K2389 |
| Pentylenetetrazole | Sigma | P6500 |
| Biocytin | Sigma | B4261 |
| Hoechst 33258 Solution | Sigma | 94403 |
| T7 Endonuclease I | NEB | M0302S |
| BbsI | NEB | R0539S |
| Critical Commercial Assays | ||
| Q5® Site-Directed Mutagenesis Kit | NEB | E0554S |
| Deposited Data | ||
| Raw RNaseq data | This paper | GSE119973 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: PHF6 C99F knock-in:B6.PHF6(nt.296G > T) | Laboratory of Dr. Azad Bonni | N/A |
| Mouse: PHF6 knockout:B6 | Laboratory of Dr. Azad Bonni | N/A |
| Oligonucleotides | ||
| sgRNAs (5′-GTGATGTGAAAACCTGCCAC-3′) | IDT | N/A |
| Off-target primers: 1F (5′- TTGGGAGCTTGTGGGACCTG-3′) | IDT | N/A |
| Off-target primers: 1R (5′- CTTGGCTCCTGATGACGGCA-3′) | IDT | N/A |
| Off-target primers: 2F (5′- TTTGGAAACTTTCCGGTTGGGGG –3′) | IDT | N/A |
| Off-target primers:2R (5′-GGCACTTGCTGTCCATAATGCTG-3′) | IDT | N/A |
| Off-target primers: 3F (5′- GGAAGTGGTCCATGCCACTG-3′) | IDT | N/A |
| Off-target primers: 3R (5′-GCCAAAGCTCGTTCTGAAGTGATG-3′) | IDT | N/A |
| Off-target primers: 4F (5′- CTTTAACCAGGTACTAAGTGTGACAGTGTG –3′) | IDT | N/A |
| Off-target primers: 4R (5′- GGAATGGACGTGGCCTTTAAAGC –3′) | IDT | N/A |
| Off-target primers: 5F (5′- GTTGGCAACCCAGAGAGACA –3′) | IDT | N/A |
| Off-target primers: 5R (5′-ACCACCAGTGTTGTGTCCAG-3′) | IDT | N/A |
| Off-target primers: 6F (5′-ACCTGAGTAACCAAGTCCACAC-3′) | IDT | N/A |
| Off-target primers: 6R (5′-TTACAGCAGACCTCTAGGGATT-3′) | IDT | N/A |
| Off-target primers: 7F (5′- CCACTGTGGTTAGACTCCCTC –3′) | IDT | N/A |
| Off-target primers: 7R (5′- AGACATGCCTGTAGCAGACT –3′) | IDT | N/A |
| Upstream F1 (5′- GAACAAGGCGTTTGGCAACCAGG-3′) | IDT | N/A |
| Upstream R1 (5′- GGAGCTGAGCTAGATACAACTG –3′) | IDT | N/A |
| Middle F2 (5′-GTATCCAGCAAGTTCTGGATTGC-3′) | IDT | N/A |
| Middle R2 (5′-GGAAAAACTTAGTAGAGGACTAC-3′) | IDT | N/A |
| Downstream F3 (5′-CTGCACATAACTCGGAAGGTATG-3′) | IDT | N/A |
| Downstream R3 (5′-GGTACACAGACATACCCGCAGG-3′) | IDT | N/A |
| Recombinant DNA | ||
| pHAGE-NTAP-del279-365-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-R342X-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-H229R-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-C45Y-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-C99F-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-C305X-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-R257G-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| pHAGE-NTAP-PHF6 | Lab: Prof. Dr. Azad Bonni | N/A |
| PX330 | addgene | Addgene #42230 |
| Software and Algorithms | ||
| R | Open Source, Software | R Project for Statistical Computing, RRID:SCR_001905 |
| GraphPad Prism 6.0 | GraphPad Software |
https://www.graphpad.com/ RRID: SCR_002798 |
| EdgeR | Open source | RRID:SCR_012802 |
| DEseq2 | Open source | RRID:SCR_015687 |
| BrainSpan | Allen Institute for Brain Science | RRID:SCR_008083 |
| HUGO multi-symbol checker | Open source | https://www.genenames.org/cgi-bin/symbol_checker |
| SPSS (v.2.5)/Systat | SPSS software | RRID:SCR_002865 |
| Adobe Photoshop | Adobe | RRID:SCR_014199 |
| COSMID | Cradick et al., 2014 | https://crispr.bme.gatech.edu/ |
| CRISPOR/v.4.4 | Haeussler et al., 2016 | http://crispor.tefor.net/ |
| Fiji | ImageJ software | https://imagej.nih.gov/ij/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Azad Bonni (bonni@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were approved by the institutional Animal Care and Use Committees (IACUC) at Washington University in St. Louis. Mice were maintained under regular housing conditions with standard access to food and drink in a pathogene-free facility. The immunohistochemistry experiments were carried out using mice from embryonic day 14 (E14) to postnatal day 6 (P6). Western Blot experiments were carried out on E14.5-P6 mice. Behavior experiments were carried out using 4-6 month old male mice. Electrophysiology recordings were carried out on male mice from P18-23. The RNA-seq experiments were performed at P0. Biocytin injection was performed in male mice from postnatal week 1 (P7-9), postnatal week 2 (P14-16) and postnatal week 3 (P21-P25). Genotypes were determined using PCR from tail or ear punches.
PHF6 C99F knock-in and knockout mice
Target specific sgRNAs (5′-GTGATGTGAAAACCTGCCAC-3′) were cloned into BbsI digested plasmid pX330 (addgene # 42230) and sgRNA cleavage activity was validated in vitro by transfecting NIH 3T3 cells, followed by T7E1 assay (NEB). T7 sgRNA and Cas9 templates for in vitro transcription were PCR amplified, gel purified, and in vitro transcribed with the MEGAshortscript T7 kit (sgRNA, Life Technologies) or the T7 mMessage mMachine Ultra kit (Cas9, Life technologies). After transcription, both RNAs were purified with the Megaclear kit (Life Technologies). A 200nt ssODN donor DNA with the mutation(s) centered within the oligo was synthesized by IDT as an ultramer oligo. Injection concentrations were: 50ng/ul Cas9, 25ng/ul gRNA, 100ng/ul ssODN. All animal experiments were approved by institutional IACUC protocols. B6/CBA F1 mice at 3-4 weeks of age (JAX Laboratories, Bar Harbor ME, USA) were superovulated by intraperitoneal injection of 5 IU pregnant mare serum gonadotropin, followed 48h later by intraperitoneal injection of 5 IU human chorionic gonadotropin (PMS from SIGMA, HGC from Millipore USA). Mouse zygotes were obtained by mating B6/CBA stud males with superovulated B6/CBA females at a 1:1 ratio. One-cell fertilized embryos were injected into the pronucleus and cytoplasm of each zygote. Microinjections and mouse transgenesis experiments were performed as described (Behringer et al., 2014). Our founder animals that were mosaic for the nt.296G > T (p.C99F) missense mutation and nt. 298insG frameshift mutation using a pyrosequencing. Two mutation alleles were segregated in F1 generation, and their genotypes were confirmed with Sanger sequencing. Based on detailed molecular characterization, we confirmed nt. 298insG line is a null. To conduct the behavior and cellular experiments, PHF6 C99F knock-in mice were backcrossed to C57BL6 line for more than ten generations.
METHOD DETAILS
Immunohistochemistry
Immunohistochemical analyses were performed as described (Huynh et al., 2011; Yamada et al., 2014). The brain from mice at distinct ages were fixed with 2% PFA and 4% sucrose and subjected to cryosectioning. Sections were blocked with blocking buffer (10% goat serum, 3% BSA, and 0.4% Triton-X in PBS), and subjected to immunofluorescence analyses with rabbit anti-PHF6 and/or rat anti-CTIP2. The DNA dye bisbenzimide (Hoechst 33258) was used to label cell nuclei. Confocal images were acquired with Olympus FV1200 with identical scanning configurations for all samples in the same experiment.
Plasmids
PHF6 patient mutations del 279-365, R342X, H229R, C45Y, C99F, C305X and R257G were generated using Q5 site-directed mutagenesis and subcloned into the pHAGE-NTAP vector.
Behavioral tests
Three separate cohorts of 4-6 month old male PHF6 mutant and control littermate mice were used for phenotypical analysis. The initial cohort (n = 12 per group) was assessed first on a 1-h locomotor activity test, then on a battery of sensorimotor measures, followed by Morris water maze (MWM), acoustic startle/prepulse inhibition (PPI), social approach, and conditioned fear paradigms. In a second cohort (n = 11 per group), mice were evaluated on 1-h locomotor activity, novel object and novel location recognition memory, followed by the conditioned fear test. In the third cohort (n = 12 for C99F and n = 13 for control), ambulatory activity/exploration and anxiety-related behaviors were assessed for 1 h in an open field and in an elevated plus maze. Acoustic startle/PPI and social approach tests were also conducted in this cohort. The detailed behavioral tests were listed as follows:
One-hour locomotor activity and open-field
Mice in cohorts 1 and 2 were evaluated over a 1-h period in transparent enclosures as described (Dougherty et al., 2013). Each cage was surrounded by a frame containing a 7 × 15 matrix of photocell pairs, the output of which was fed to an on-line computer (Kinder Scientific, LLC, Poway, CA). The system software was used to define a 33 × 11 cm central zone and a peripheral or surrounding zone that was 5.5 cm wide with the sides of the cage being the outermost boundary. In cohort 3, ambulatory activity and exploratory behaviors of the C99F and control mice were quantified over a 1-h period in an open-field (41 × 41 × 38.5 cm high) constructed of Plexiglas, which utilized computerized photobeam instrumentation that included a frame containing a 16 × 16 matrix of photocell pairs surrounding the apparatus. Total ambulations (whole body movements) and vertical rearing frequency were computed using system software (Kinder Scientific, LLC) and were analyzed for both tests.
Conditioned fear
Mice were trained and tested using two clear-plastic conditioning chambers (26 × 18 × 18 cm high) (Med-Associates, St. Albans, VT) easily distinguished by different olfactory, visual, and tactile cues present in each chamber as described (Wozniak et al., 2007). On day 1, each mouse was placed into the conditioning chamber for 5 min and freezing behavior was quantified using FreezeFrame image analysis software during a 2 min baseline period. After baseline measurements, a conditioned stimulus (CS) consisting of an 80 dB tone (white noise) was presented for 20 s followed by an unconditioned stimulus (US) consisting of a 1 s, 1.0 mA continuous foot shock. This tone-shock (T/S) pairing was repeated each minute over the next 2 min, and freezing was quantified after each of the three tone-shock pairings. Twenty-four hours after training, each mouse was placed back into the original conditioning chamber to test for fear conditioning to the contextual cues in the chamber. Twenty-four hours later, each mouse was placed into the other chamber containing distinct cues. Freezing was quantified during a 2 minutes “altered context” baseline period as well as over a subsequent 8 min period during which the auditory cue (CS) was presented. Shock sensitivity was evaluated following completion of the conditioned fear test, which involved determining shock levels that elicited flinching, escape-related behaviors and vocalization.
Novel object and novel location recognition
Using a published protocol (Brown et al., 2010), each mouse was habituated to the test chamber over two days and on the following day given two sample trials and two test trials. For novel object recognition, a sample trial consisted of placing a mouse in the familiar arena that contained two copies of the same object and allowing it to explore for 10 min, and then returning it to its home cage. A 10-min test trial was conducted following a 50-min delay, when a mouse was placed back into the test arena, where a novel object was presented along with the copy of the familiar object used during the sample trial. Fifty minutes later, another 10-min sample trial was administered in which two new novel objects were placed in opposite corners of the arena. Mice were again returned to their home cage for a 50 min delay, then placed back into the test arena where one of the objects had been moved to a novel location. The amount of time each animal spent actively investigating the objects was manually scored and the investigation times for the novel versus the familiar object (or locations) were analyzed as a measure of recognition memory.
Social Approach
Sociability and preference for novel social interactions were evaluated in a rectangular apparatus divided into three chambers each measuring 19.5cm × 39cm × 22 cm according to published methods (Dougherty et al., 2013). Two small stainless steel withholding cages (Galaxy Pencil/Utility Cup, Spectrum Diversified Designs, Streetsboro, OH) were used to prevent contact between test and stimulus mice with one being placed in each outer chamber. Test sessions were recorded using a digital camera connected to a PC and scored using the ANY-maze (Stoelting, Co, Wood Dale, IL) software program. Time spent, distance traveled, entries into, and latency to enter each chamber and the investigation zones around the holding cages were analyzed. Each test session contained five consecutive, 10-min trials, with the first two trials being dedicated to habituation to the apparatus without stimulus animals present followed by three test trials. After habituation, sociability exhibited toward a novel stimulus mouse versus a familiar, empty withholding cage was assessed in the first test trial by placing an unfamiliar, gender-matched stimulus mouse in one withholding cage while the other was left empty, and the test mouse was allowed to freely explore the apparatus and investigate the novel mouse in the withholding cage. For the second test trial, a gender-matched cagemate was placed in one withholding cage while the other remained empty, and the test mouse could freely explore the apparatus and investigate the cagemate in the withholding cage. During the third test trial, the familiar cagemate remained in the withholding cage, although a new, unfamiliar gender-matched stimulus mouse was now placed in the other withholding cage, and the test mouse was allowed to explore the apparatus and investigate the two mice contained in the withholding cages. Entries, latency to enter, and time spent in each chamber and investigation zone were recorded for each mouse.
Acoustic startle response/prepulse inhibition
Sensorimotor reactivity and gating were evaluated in the mice by quantifying the magnitude of their acoustic startle (ASR) and PPI responses (Hamilton Kinder, LLC) as described (Yuede et al., 2010). In the first cohort, a startle response to a 120 dB auditory stimulus pulse (40 ms broadband burst) and PPI (response to a prepulse plus the startle pulse) were measured concurrently in mice. Beginning at stimulus onset, 1 ms force readings were averaged to obtain an animal’s startle amplitude. A total of 20 startle trials were presented over a 20 min test period during which the first 5 min served as an acclimation period when no stimuli above the 70 dB white noise background were presented. The session began and ended by presenting 5 consecutive startle (120 db pulse alone) trials unaccompanied by other trial types. The middle 10 startle trials were interspersed with PPI trials (consisting of an additional 30 presentations of 120 dB startle stimuli preceded by pre-pulse stimuli of either 4, 12, or 20 dB above background (10 trials for each PPI trial type). A percent PPI score for each trial was calculated using the following equation: %PPI = 100*(ASRstartle pulse alone - ASRprepulse+startle pulse)/ASRstartle pulse alone Another cohort was tested using an upgraded instrumentation system (Kinder Scientific, LLC), and a slightly modified protocol. This involved evaluating startle responses to increasing sound pressure levels (80 to 110 dB), as well as the startle response to 120 dB collected over 20 trials as described above. Pre-pulse levels tested in this cohort were 4, 8, and 16 dB above background, and PPI was calculated using the same equation.
Morris Water Maze Test
Reference memory was evaluated in the mice using the Morris water maze navigation test similar to published procedures (Wozniak et al., 2004). The protocol included cued (visible platform), place (submerged, not visible platform), and probe trials (platform removed). Testing took place in a round pool (118 cm diameter) containing water made opaque with non-toxic white tempura paint. All trials were monitored through a live video feed by computer software (Any-maze, Stoelting Co., Wood Dale, IL) which calculated swim speed, escape path length, escape latency, and time and distance spent in each of the four quadrants of the pool. The maximum score for all water maze trials was 60 s. Cued trials were conducted first to determine if nonassociative deficits (e.g., visual or sensorimotor disturbances, or alterations in motivation) were likely to affect swimming performance and confound interpretation of the acquisition data collected during the place trials. Mice received 4 cued trials per day for 2 consecutive days. Mice were placed in the quadrant directly opposite a submerged platform marked with a visible cue. The platform was moved to a different location for each trial within a day and there were very few distal cues available during this time, both of which limited spatial learning. Escape path length and latency and swimming speeds were used to evaluate performance during the cued and place conditions. Place trials were performed following the cued trials. Stationary distal cues to facilitate learning were placed around the room and mice were evaluated on their ability to learn the location of a hidden, unmarked platform. Four place trials per day were administered for 5 consecutive days during which the platform remained in the same location for all trials. The mice were released from 4 different locations each day. The daily protocol involved administering 2 sets of 2 trials each, with sets being separated by approximately 1 hr. A single probe trial was administered approximately 1 hr after completion of place trials on the fifth day. During the probe trial, the platform was removed from the pool and the animals placed in the quadrant directly opposite the former platform location. Mice were allowed to explore for 60 s during which time various aspects of their search behaviors for the platform were quantified. The number of times a mouse passed directly over the platform location (platform crossings), the time spent in the target quadrant that had contained the platform, and spatial bias (preference for the target quadrant compared to other quadrants) were used to evaluate retention performance during the probe trial.
Seizure induction
Pentylenetetrazole (Sigma) dissolved in 0.9% saline was injected intraperitoneally (75mg/kg) into 5-7 month old mice as described (Zhang and Wong, 2012). Mice then were placed individually into cages for observation. A trained blinded observer performed the experiment. Generalized seizure was characterized by sudden loss of upright posture with diffuse tonic posturing followed by clonic shaking. The time to the generalized seizure was recorded for each mouse.
Kainic acid (Sigma) dissolved in the 0.9% saline was injected intraperitoneally (25mg/kg) into 5-7 month old mice. Mice then were placed individually in cages for observation and only two mice were simultaneously observed. Mouse genotypes were blinded from the experimenter. The Racine scale was used to score seizure progression in mice as described (Racine et al., 1972). A score of 1 was assigned when mice displayed absence-like immobility and oro-alimentary movements. Mice received a score of 2 for hunching with facial or manual automatisms and head bobbing. Mice scored 3 rearing with facial or manual automatisms and forelimb clonus, and achieved a score of 4 with repeated rearing with continuous forelimb clonus and falling. Score of 5 was assigned if mice displayed generalized tonic-clonic seizure with lateral recumbence or jumping. Death was also scored as 5. Mice were scored every five minutes for 100 minutes.
Electrophysiology
Animals and slice preparation
Electrophysiological recordings were conducted blind to genotype. Slices were prepared as described (Deng and Klyachko, 2016). In brief, 18-23-day-old male mice were used. After deep anesthesia with CO2, mice were decapitated and the brain was dissected out in ice-cold saline containing (in mM): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 5.0 MgCl2, and 10 glucose, pH 7.4 (saturated with 95% O2 and 5% CO2). Horizontal brain slices (350 mm) including the entorhinal cortices were cut using a vibrating microtome (Leica VT1100S). Slices were initially incubated in the above solution at 35°C for 1 h for recovery and then kept at room temperature (~23°C) until use. All animal procedures were in compliance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals, and conformed to Washington University Animal Studies Committee guidelines.
Action potential recording and threshold determination
Whole-cell patch-clamp recordings using a Multiclamp 700B amplifier (Molecular Devices) in current-clamp mode were made from stellate cells of EC superficial layers visually identified with differential interference contrast optics (Olympus BX51WI). Current-clamp recordings were made with pipette capacitance compensation and bridge-balance compensation. Stellate cells were identified according to morphology and electrophysiological properties as described (Deng et al., 2007). All recordings were conducted at near-physiological temperature (33–34°C). The recording electrodes were filled with (in mM): 130 K-gluconate, 10 KCl, 0.1 EGTA, 2 MgCl2, 2 ATPNa2, 0.4 GTPNa, and 10 HEPES, pH 7.3. The extracellular solution contained (in mM): 125 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose, pH 7.4 (saturated with 95% O2 and 5% CO2).
For neuronal excitability measurements, cell membrane potential was set to given potentials (−55 to −44 mV with 1-mV step) through automatic current injection. Action potential (AP) firing probability and the number of APs within 20 s was averaged from 4-5 trials for each cell. For AP threshold determination, APs were evoked by a ramp current injection (0.15 pA/ms) (Deng and Klyachko, 2016) with a hyperpolarizing onset to ensure maximal Na+ channel availability before the 1st AP. The threshold was defined as the voltage at the voltage trace turning point, corresponding to the first peak of 3rd order derivative of AP trace (Deng and Klyachko, 2016). Ramp current-evoked AP threshold was then defined as the mean threshold of the first AP in the train averaged over 5-8 trials.
Determination of resting membrane potential, capacitance and input resistance
Resting membrane potential (RMP) was measured immediately after whole-cell formation. Cell capacitance was determined by the amplifier’s auto whole-cell compensation function with slight manual adjustment to optimize the measurement if needed. The obtained values then were used for input resistance normalization. In current-clamp mode, a negative current (−50 pA for 500 ms) was injected every 5 s to assess the input resistance.
RNA-seq
The RNaseq analyses were conducted as described (Yamada et al., 2014; Yang et al., 2016). For control and PHF6 knockout (KO) experiment, four pairs of animals were harvested from two litters. For control and PHF6 C99F knock-in experiment, five pairs were harvested from three litters. Animals were harvested at postnatal day 0 (P0). The cerebral cortex was dissected and subjected to RNA extraction using Trizol (Invitrogen). RNA was sequenced on Illumina HiSeq 2500. EdgeR and DEseq2 were used to analyze the differentially expressed genes. The Benjamini-Hochberg false discovery rate (FDR) was calculated for all genes.
Permutation testing
Permutation testing was performed to assess the correlation between gene expression alteration in the cerebral cortex of PHF6 knockout and C99F knock-in mice by comparing the observed correlation value with expected correlation value. Observed valued was calculated by Spearman correlation (rho) between log fold change (logFC) KO/control and C99F/control of 193 differentially expressed genes upon PHF6 KO, whereas the expected correlation distribution was generated via iterative sampling of random sets of 193 genes and calculating the logFC correlation between KO and C99F. Random sampling was repeated 10,000 times, allowing us to estimate the mean and standard deviation of the expected rho distribution. Empirical z-scores and p value were calculated for the observed rho value based on the expected rho distribution. A custom R script was used for the analysis.
Gene enrichment analyses
For cell type enrichment, we used cell marker gene set previously described ((Zeisel et al., 2015). We used the expressed genes in the postnatal day 0 (P0) mouse cortex averaging the RPKM (mean > 0.1) across control samples, and overlapping with all the expressed genes in the single cell RNA-seq dataset (Zeisel et al., 2015) as our background list.
For gene enrichment analysis with developmental modules and other ID proteins, all gene symbols were converted into human orthologs using web based HUGO multi-symbol checker. Expressed protein-coding genes in the P0 mouse cortex (RPKM > 0.1) were used as background for KDM5C and CHD8 enrichment analysis. Protein-coding genes that had probes called in Voineagu et al. were used as background for asdM12 enrichment analysis. For FMRP enrichment analysis, all protein-coding genes detected in HITS-CLIP (Darnell et al., 2011) were used as the background list, and FMRP enriched genes (765) were used as FMRP-target. Expressed genes in the P0 mouse cortex (RPKM > 0.1) that had human orthologs were used as background for developmental module enrichment analysis.
All gene enrichment analyses were performed using one-sided hypergeometric test calculated according to the R function phyper(). Each p value was adjusted by Benjamini-Hochberg correction.
BrainSpan developmental analyses
Normalized RPKM values of RNaseq data were obtained from Allen Brain Atlas BrainSpan dataset (http://www.brainspan.org/). Datasets from four brain regions, including the ventral frontal cortex (VFC), medial frontal cortex (MFC), dorsal frontal cortex (DFC) and primary somatosensory cortex (S1C) were used for developmental expression analysis. Genes from GO:0050769 and GO: 0007268, representing neurogenesis and synaptic genes respectively were first extracted from the Brainspan dataset and the average RPKM value in VFC was calculated and plotted across developmental time. Differentially expressed genes (DEGs) upon PHF6 KO were divided into upregulated and downregulated genes. These genes were extracted from BrainSpan data for each brain region, and the average RPKM was calculated and analyzed across the developmental time. A custom R script was used for analyses.
Biocytin injection and post hoc staining
Biocytin labeling was performed as described (Huang et al., 2010). Intracellular recording solution containing 1% biocytin was loaded into layer II stellate neurons in the entorhinal cortex from P7-9 (C99F: n = 8; control: n = 7), P14-16 (C99F: n = 6; control: n = 7) and P21-25 (C99F: n = 7; control: n = 7) animals through recording electrode. Slices were fixed in 4% PFA for 2 hours. Slices were subjected to histochemical analyses with Alexa Fluor 488-conjugated Avidin (1:1000) for 4°C overnight.
QUANTIFICATION AND STATISTICAL ANALYSIS
For electrophysiology, data are presented as mean ± SEM. Student’s unpaired t test was used for statistical analysis as appropriate. A Chi-square test was used in analysis of AP firing probability (n = 13 neurons from 2 mice) at different membrane potentials. Unpaired t test was used in rest of the analysis (n = 14 neurons from 2 mice). For seizure induction, unpaired t tests for PTZ and two-way repeated-measure ANOVA for Kainic acid were used. Significance was set as p < 0.05.
For behavioral tests, statistical analyses were performed using GraphPad Prism 6.0, and SPSS (v2.5)/Systat statistical software packages. Independent t tests, one-sample t tests, factorial ANOVAs, including repeated-measures (rm) ANOVAs were used where appropriate. The Huynh-Feldt correction was applied to all within-subjects effects containing more than two levels to protect against violations of sphericity/compound symmetry assumptions underlying rmANOVA models. Specific planned comparisons were conducted to provide further clarification of significant interactions between main factors. Simple main effects were calculated for significant main effects of factors, and Bonferroni corrections were used for multiple comparisons. Probability value for statistical significance for all analyses was p < 0.05.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE119973.
Supplementary Material
Highlights.
Generation of patient-specific PHF6 mutant mice using CRISPR-Cas9
PHF6 C99F mice display impairments in cognition and social interaction
PHF6 C99F mice exhibit neuronal hyperexcitability and seizure susceptibility
PHF6 promotes neurogenic genes and suppresses synaptic genes in the cortex
ACKNOWLEDGMENTS
The authors thank Chi Zhang for providing plasmids for the study, Anna Oldenberg for initial maintenance of the mouse line, and members of the Bonni laboratory for helpful discussions. This work was supported by NIH grants RO1NS088378 (to A.B.), RO1NS081972 (to V.A.K.), RO11HG007175 (to T.W.), and RF1MH117070-01 (to J.D.D.); Washington University Intellectual and Developmental Disabilities Research Center (IDDRC) NIH U54 HD087011; the Mouse Genetics core; the Hope Center Transgenic Vectors core; the Washington University Center for Cellular Imaging (WUCCI); and the Genome Technology Access Center (GTAC) at the Washington University School of Medicine.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.043.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baldridge D, Heeley J, Vineyard M, Manwaring L, Toler TL, Fassi E, Fiala E, Brown S, Goss CW, Willing M, et al. (2017). The exome clinic and the role of medical genetics expertise in interpretation of exome sequencing results. Genet. Med 19, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer R, Gertsenstein M, Nagy K, and Nagy A (2014). Manipulating the Mouse Embryo: A Laboratory Manual (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Bhasin T, Brocksen S, Avchen R, and Van Naarden Braun K (2006). Prevalence of four developmental disabilities among children aged 8 years – Metropolitan Atlanta Developmental Disabilities Surveillance Program, 1996 and 2000. MMWR Morb. Mortal. Wkly. Rep 55 (SS01), 1–9. [PubMed] [Google Scholar]
- Börjeson M, Forssman H, and Lehmann O (1962). An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med. Scand 171, 13–21. [DOI] [PubMed] [Google Scholar]
- BrainSpan (2013).http://www.brainspan.org/.
- Brown JA, Emnett RJ, White CR, Yuede CM, Conyers SB, O’Malley KL, Wozniak DF, and Gutmann. (2010). Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet 19, 4515–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Qiu P, Lee CM, Fine EJ, and Bao G (2014). COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 3, e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, and Klyachko VA (2016). Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of Fmr1 knockout mice. Cell Rep 16, 3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P-Y, Poudel SKS, Rojanathammanee L, Porter JE, and Lei S (2007). Serotonin inhibits neuronal excitability by activating two-pore domain k+ channels in the entorhinal cortex. Mol. Pharmacol 72, 208–218. [DOI] [PubMed] [Google Scholar]
- Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, Mahieu NG, Zhang J, Cai J, Patti GJ, et al. (2013). The disruption of Celf6, a gene identified by translational profiling of serotonergic neurons, results in autism-related behaviors. J. Neurosci 33, 2732–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, Stradleigh TW, Pride MC, Schaffler MD, Wade AA, Catta-Preta R, et al. (2017). Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang W, Qiao W, Hu A, and Wang Z (2010). Functional connectivity and selective odor responses of excitatory local interneurons in Drosophila antennal lobe. Neuron 67, 1021–1033. [DOI] [PubMed] [Google Scholar]
- Huynh MA, Ikeuchi Y, Netherton S, de la Torre-Ubieta L, Kanadia R, Stegmüller J, Cepko C, Bonni S, and Bonni A (2011). An isoform-specific SnoN1-FOXO1 repressor complex controls neuronal morphogenesis and positioning in the mammalian brain. Neuron 69, 930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, Tomassy GS, Kasza T, Lin G, Thompson A, et al. (2016). A mouse model of X-linked intellectual disability associated with impaired removal of histone methylation. Cell Rep 14, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A, Cheng C, Zhang C, and Bonni A (2016). Pathogenesis of Börjeson-Forssman-Lehmann syndrome: insights from PHF6 function. Neurobiol. Dis 96, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz P, Savanthrapadian S, Häussler U, Kilias A, Nestel S, Kretz O, Kirsch M, Bartos M, Egert U, and Haas CA. (2017). Synaptic remodeling of entorhinal input contributes to an aberrant hippocampal network in temporal lobe epilepsy. Cereb. Cortex 27, 2348–2364. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Sun C, Martin J, Kitch LJ, Schnitzer MJ, and Tonegawa S (2015). Entorhinal cortical ocean cells encode specific contexts and drive context-specific fear memory. Neuron 87, 1317–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li F, Ruan K, Zhang J, Mei Y, Wu J, and Shi Y (2014). Structural and functional insights into the human Börjeson-Forssman-Lehmann syndrome-associated protein PHF6. J. Biol. Chem 289, 10069–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, Schelley S, Hoyme HE, White SM, Delatycki MB, et al. (2002). Mutations in PHF6 are associated with Börjeson-Forssman-Lehmann syndrome. Nat. Genet 32, 661–665. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, Okujava V, and Chipashvili S (1972). Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Scharfman HE (2007). The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep 7, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Martens GJM, and Kolk SM (2015). Molecular underpinnings of prefrontal cortex development in rodents provide insights into the etiology of neurodevelopmental disorders. Mol. Psychiatry 20, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant SA, Fischer L, Garden DLF, Gerlei KZ, Martinez-Gonzalez C, McClure C, Wood ER, and Nolan MF (2018). Stellate cells in the medial entorhinal cortex are required for spatial learning. Cell Rep 22, 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd MAM, Ivanochko D, and Picketts DJ (2015). Phf6 degrees of separation: the multifaceted roles of a chromatin adaptor protein. Genes (Basel) 6, 325–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vismer MS, Forcelli PA, Skopin MD, Gale K, and Koubeissi MZ (2015). The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, and Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis 17, 403–414. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Xiao M, Xu L, Yamada KA, and Ornitz DM (2007). Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol. Dis 26, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K, and Bonni A (2014). Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 83, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. (2016). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede CM, Wozniak DF, Creeley CE, Taylor GT, Olney JW, and Farber NB (2010). Behavioral consequences of NMDA antagonist-induced neuroapoptosis in the infant mouse brain. PLoS One 5, e11374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, Manno G, La Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang B, and Wong M (2012). Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia 53, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, Jahani-Asl A, Gallardo G, Ikeuchi Y, Yamada T, et al. (2013). The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron 78, 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.