

Pathological drug withdrawal syndrome is linked to accumulation of JAK2 phosphorylation in V617F myelofibrosis.
Abstract
Treatment of patients with myelofibrosis with the type I JAK (Janus kinase) inhibitor ruxolitinib paradoxically induces JAK2 activation loop phosphorylation and is associated with a life-threatening cytokine-rebound syndrome if rapidly withdrawn. We developed a time-dependent assay to mimic ruxolitinib withdrawal in primary JAK2V617F and CALR mutant myelofibrosis patient samples and observed notable activation of spontaneous STAT signaling in JAK2V617F samples after drug washout. Accumulation of ruxolitinib-induced JAK2 phosphorylation was dose dependent and correlated with rebound signaling and the presence of a JAK2V617F mutation. Ruxolitinib prevented dephosphorylation of a cryptic site involving Tyr1007/1008 in JAK2 blocking ubiquitination and degradation. In contrast, a type II JAK inhibitor, CHZ868, did not induce JAK2 phosphorylation, was not associated with withdrawal signaling, and was superior in the eradication of flow-purified JAK2V617F mutant CD34+ progenitors after drug washout. Type I inhibitor–induced loop phosphorylation may act as a pathogenic signaling node released upon drug withdrawal, especially in JAK2V617F patients.
INTRODUCTION
JAK (Janus kinase) family kinases are nonreceptor tyrosine kinases that are crucial for signal transduction of many cytokines and growth factors and comprise four members: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2) (1). Increased activity of these kinases due to mutation or overexpression leads to hematopoietic malignancies, whereas genetic inactivation in mouse models mostly results in a lack of definitive erythropoiesis (2, 3). JAK family kinases are preassociated with the cytoplasmic portion of their cognate receptors, and ligand-induced receptor dimerization facilitates JAK transactivation and tyrosine phosphorylation in the activation loop of the kinase (4). JAKs contain a C-terminal tyrosine kinase domain termed JAK homology (JH) 1 domain and an adjacent pseudokinase domain termed JH2, as well as a 4.1/Ezrin/Radixin/Moesin (FERM) domain required for receptor binding. The JH2 domain has no definitive kinase activity, as it lacks an Asp residue in the His/Arg/Asp motif of its catalytic loop. However, the JH2 domain is required for cytokine receptor activation of JAK2 and is mutated at high frequency in polycythemia vera, myelofibrosis, and essential thrombocythemia, exchanging valine at position 617 for phenylalanine, JAK2V617F.
Historically, pan-JAK inhibitors such as AG-490 were first described, which mechanistically were substrate competitive for tyrosine residues and either noncompetitive or mixed competitive for adenosine triphosphate (ATP) (5). More recently, ATP-competitive inhibitors such as ruxolitinib have emerged, which bind and stabilize the kinase-active conformation, termed type I inhibitors, and show clinical activity in patients with JAK2V617F myeloproliferative neoplasms (MPNs). Paradoxically, type I inhibitors can induce the accumulation of JAK activation loop phosphorylation for unknown reasons, despite blockade of kinase function and inhibition of signal transducer and activator of transcription (STAT) phosphorylation. Whether this phenomenon contributes to clinically relevant pathological signaling is not clear. In recent studies, type I inhibitor–induced JAK2 activation loop phosphorylation has been shown to be (i) staurosporine sensitive and ATP dependent; (ii) require cytokine receptor interaction and intact JH1, FERM, and JH2 domains; and (iii) can occur in the absence of JAK1 and TYK2, among other kinases (6). Type II inhibitors, on the other hand, are ATP-competitive small molecules that stabilize the inactive kinase conformation. Their clinical utility and any molecular advantages they offer in the treatment of JAK2V617F myeloid malignancies are now being investigated.
Heterodimerization of different JAK molecules is a postulated mechanism for clinically observed resistance to JAK inhibitors (7). Ruxolitinib, a type I inhibitor of both JAK1 and JAK2, is approved for the treatment of MPNs. Patients with myelofibrosis who are treated with ruxolitinib derive symptomatic benefit through improvement of cytokine-mediated symptoms and show cytoreductive responses in disease manifestations, such as splenomegaly or leukocytosis. However, ruxolitinib treatment does not consistently reduce JAK2 mutant allele burden nor eradicate the disease (8). Second, when ruxolitinib is withdrawn, there is a rapid recrudescence of cytokine-mediated symptoms within days of treatment stopping, and in rare cases, this has led to a life-threatening “ruxolitinib discontinuation syndrome” characterized by an acute relapse of disease symptoms, splenomegaly, worsening cytopenia, and a cytokine storm akin to septic shock (9).
In this paper, we measured the kinetics of type I inhibitor withdrawal signaling in a series of patients with myelofibrosis and observed notable activation of spontaneous STAT signaling in JAK2V617F samples. Inhibitor withdrawal–induced signaling was not observed with a type II JAK2–biased inhibitor. The mechanism of ruxolitinib withdrawal signaling is linked to the accumulation of activation loop phosphorylation induced by ruxolitinib that prevents dephosphorylation and ubiquitination of JAK2. High levels of phosphorylated JAK2 may then lead to ruxolitinib withdrawal signaling when drug is removed. Our findings have important clinical implications for future drug development and for the clinical management of patients carrying JAK2 mutations.
RESULTS
Abrupt withdrawal of type I JAK inhibitor triggers STAT activation in samples with JAK2V617F myelofibrosis
Early clinical trials with ruxolitinib observed a number of cases of ruxolitinib discontinuation syndrome after abrupt or rapid tapering of drug (9), but the molecular mechanisms have not been investigated. We designed a time-course assay (fig. S1) using samples of primary JAK2V617F mutant, Ficoll gradient–separated, mononuclear cells from patients with myelofibrosis (table S1) to mimic ruxolitinib exposure, followed by rapid withdrawal. Patient cells were cultured in vehicle or 280 nM ruxolitinib for 12 hours and then rapidly washed out of drug and transferred to fresh media in the absence of any extrinsic cytokine or fetal calf serum (FCS). Cell lysates were then immunoblotted for phosphorylation signaling events at the indicated time points (Fig. 1, A and B, and fig. S2, A and B). The dose of ruxolitinib used is the lowest dose that consistently induced JAK2 phosphorylation in SET-2 cells (Fig. 1C) and is equivalent to 100 × the in vitro JAK2 median inhibitory concentration (IC50) of 2.8 nM in a recombinant kinase assay. Consistent with recent publications (6, 7), treatment of SET-2 cells with ruxolitinib-induced phosphorylation of Tyr1007/1008 was present in the JAK2 activation loop and was not observed with vehicle treatment. During ruxolitinib exposure, no detectable STAT3 or STAT5 phosphorylation and minimal STAT1 phosphorylation were observed [Fig. 1, A and B (lane 7)]. However, following drug washout, strong activation of STAT1, STAT3, STAT5, and extracellular signal–regulated kinase (ERK) was demonstrated, which was still sustained at 30 and 60 min after ruxolitinib withdrawal [Fig. 1, A and B (lanes 8 to 12), and fig. S2A]. Unexpectedly, the duration and amplitude of downstream signaling induced by drug withdrawal were greater than that observed in vehicle control [Fig. 1, A and B (lane 1)]. Myelofibrosis cells from a patient with a calreticulin (CALR) mutation and wild-type JAK2 did not show accumulation of phosphorylated JAK2 in the presence of ruxolitinib and also showed less sustained STAT activation following drug withdrawal (fig. S2B). The lack of accumulated phosphorylation of JAK2 in the presence of ruxolitinib is consistent with previous reports investigating CALR mutations in mouse models (10).
Fig. 1. Ruxolitinib washout triggers intracellular signaling in patients with myelofibrosis.
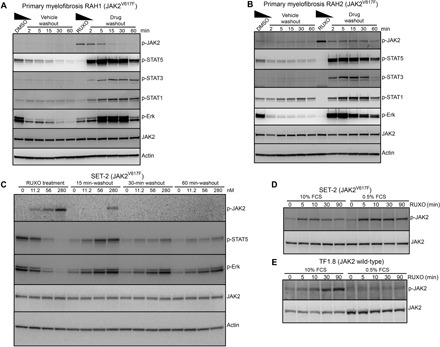
(A and B) Mononuclear cells from patients with JAK2V617F myelofibrosis RAH1 and RAH2 were cultured for 12 hours in media containing 10% FCS and erythropoietin (EPO) and interleukin-3 (IL-3) (1 ng/ml each) with 280 nM ruxolitinib or dimethyl sulfoxide (DMSO) as vehicle control. Cells were washed in cold media and transferred into prewarmed and CO2-equilibrated media without additives for the indicated periods of time, and cell lysates were prepared and immunoblotted for the indicated proteins. (C) JAK2V617F positive SET-2 cells were cultured with ruxolitinib (0, 11.2, 56, or 280 nM) for 12 hours, followed by washout of drug. Whole-cell lysates were prepared at 0, 15, 30, and 60 min and analyzed by immunoblotting with p-JAK2, p-STAT5, and p-ERK antibodies. SET-2 (D) or TF1.8 (E) cells were incubated for 6 hours in media containing either 10 or 0.5% FCS before the addition of 280 nM ruxolitinib for 0, 5, 10, 30, and 90 min. Whole-cell lysates were prepared and immunoblotted with p-JAK2 and total JAK2 antibodies.
Cytokine signaling inputs regulate type I inhibitor–induced JAK2 phosphorylation
Time-dependent accumulation of type I inhibitor–induced JAK2 phosphorylation was first described in SET-2 cells, a JAK2V617F-mutated cell line derived from a patient with essential thrombocythemia (11). We used SET-2 cells to investigate dose dependency of ruxolitinib treatment in the activation of withdrawal signaling. The dose of ruxolitinib used during drug exposure correlated with the amplitude of withdrawal signaling (Fig. 1C). SET-2 cells incubated with low dose of ruxolitinib (4 × in vitro IC50) for 12 hours elicited less downstream STAT activation upon drug withdrawal compared with 56 nM (20 × IC50) or 280 nM (100 × IC50). The degree of JAK2 activation loop phosphorylation during ruxolitinib exposure was strongly associated with the magnitude of spontaneous signal activation upon drug withdrawal.
To investigate the signaling inputs that regulate this phenomenon, we incubated SET-2 cells with 280 nM ruxolitinib at varying time points in the presence of low (0.5%) or high (10%) FCS. We observed rapid accumulation of phosphorylated JAK2 after 5 min in both high and low FCS (Fig. 1D), although notably higher levels of phosphorylated JAK2 were seen in low FCS.
In contrast, when we performed the same experiment using wild-type JAK2 TF1.8 cells cultured with 280 nM of ruxolitinib, we noted a clear FCS dependence (Fig. 1E). Cells cultured in 10% FCS displayed a strong JAK2 phosphorylation signal accumulation over 90 min of ruxolitinib exposure; however, there was no accumulation of phosphorylated JAK2 when cells were cultured in 0.5% FCS. These data suggest that, in wild-type JAK2 cells, ruxolitinib-mediated phosphorylation of JAK2 is partially dependent on extrinsic inputs provided by FCS. Wild-type JAK2 cells cultured in high FCS also show ruxolitinib withdrawal signaling similar to primary cells (figs. S1B and S2C), with an increase in spontaneous STAT5 and ERK phosphorylation after washout. Wild-type JAK2 cells showed minimal withdrawal signaling in low FCS, compared to SET-2 cells that showed prominent STAT activation after ruxolitinib withdrawal in low FCS (fig. S2D). This suggests that the JAK2V617F mutation is linked to the amplitude of spontaneous STAT signaling observed after ruxolitinib withdrawal.
Ruxolitinib prevents dephosphorylation and ubiquitination of JAK
The delayed accumulation of JAK2 activation loop phosphorylation observed in TF1.8 cells implies that ruxolitinib does not directly induce Tyr1007/1008 phosphorylation upon binding JAK2, but may prevent other down-regulating signaling events. In this regard, while investigating regulation of ruxolitinib-induced JAK2 phosphorylation following cytokine stimulation, we noted differences in the Tyr1007/1008 phosphorylation profile of JAK2, depending on the method used before immunoblotting. Immunoprecipitation of lysates from TF1.8 cells using an anti-phosphotyrosine antibody, followed by immunoblot with anti-JAK1 or anti-JAK2 antibodies, resulted in clear bands from vehicle-treated cells at 5 and 15 min after cytokine stimulation but not in ruxolitinib-treated cells (Fig. 2, A and C). Similarly, immunoprecipitation with a p-JAK2 antibody, followed by immunoblot with an anti-JAK2 antibody, resulted in a clear band from vehicle-treated cells but not in ruxolitinib-treated cells (Fig. 2B and fig. S3A). This is in contrast to the results obtained with direct immunoblot analysis of whole-cell lysates where Tyr1007/1008-phosphorylated JAK2 is readily observed from vehicle- and ruxolitinib-treated cells [Fig. 2, A to C, and fig. S3A (lower panels)]. We noted the same discrepancy in SET-2 cells where, following immunoprecipitation with anti-phosphotyrosine antibody, a significantly reduced phosphorylated JAK2 signal was observed, despite strong phosphorylated JAK2 signals in whole-cell lysates (Fig. 2D). This is consistent with a recent report in which a C-terminal–directed antibody against total JAK2 protein failed to precipitate JAK2 in the presence of type I inhibitor (6). These data suggest that ruxolitinib binding to JAK2 hides phosphorylated residues in the activation loop from the surface of the protein. Because of presumed structural flexibility, high-resolution crystallographic structural data are not available for the activation loop during type I inhibitor binding.
Fig. 2. Type I JAK2 inhibitor protects JAK2 from degradation and down-regulation.
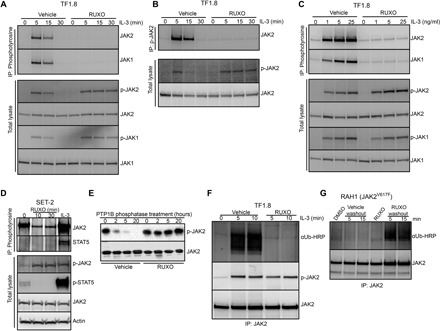
(A and B) TF1.8 cells were starved overnight in the presence of 0.5% FCS, preincubated with either vehicle or 280 nM ruxolitinib for 10 min, and stimulated with IL-3 (50 ng/ml) for different times. Cells were lysed and subjected to immunoprecipitation with (A) anti-phosphotyrosine (4G10) or (B) anti-phosphorylated JAK2 (Y1007/1008) antibodies, followed by immunoblotting with JAK2 and JAK1 antibodies. As a control, total lysates from the same experiment were immunoblotted with p-JAK1, p-JAK2, JAK1, and JAK2 antibodies as indicated. (C) TF1.8 cells were starved overnight in the presence of 0.5% FCS, preincubated with either vehicle or 280 nM ruxolitinib for 10 min, and stimulated with different doses of IL-3 for 5 min. Cells were lysed and subjected to immunoprecipitation (IP) with anti-phosphotyrosine (4G10) antibody, followed by immunoblotting with JAK1 and JAK2 antibodies. As a control, total lysates from the same experiment were immunoblotted with p-JAK1, p-JAK2, JAK1, and JAK2 antibodies. (D) SET-2 cells were incubated in 0.5% FCS for 6 hours before the addition of 280 nM ruxolitinib for 0, 10, or 30 min or stimulation with IL-3 (50 ng/ml) and EPO for 5 min. Cells were lysed and subjected to immunoprecipitation with anti-phosphotyrosine (4G10) antibody, followed by immunoblotting with JAK2 and STAT5 antibodies. As a control, total lysates from the same experiment were immunoblotted with p-JAK2, p-STAT5, JAK2, and actin antibodies. (E) Recombinant JAK2 kinase domain was mixed with recombinant tyrosine phosphatase PTP1B and ruxolitinib or no inhibitor in phosphatase assay buffer. Phosphatase reactions were incubated at room temperature for 0, 2, 5, and 20 hours, fractionated by SDS–polyacrylamide gel electrophoresis (PAGE), and immunoblotted with p-JAK2 and JAK2 antibody. (F) TF1.8 cells were starved overnight in the presence of 0.5% FCS, preincubated for 10 min with MG132 plus either vehicle or 280 nM ruxolitinib, and stimulated with IL-3 (50 ng/ml) for 0, 5, or 10 min. Cells were lysed and subjected to immunoprecipitation with JAK2 antibody, followed by immunoblotting with ubiquitin antibody conjugated to horseradish peroxidase (αUb-HRP) or p-JAK2 antibody. (G) Mononuclear cells from patient with myelofibrosis RAH1 were cultured in 10% FCS with EPO and IL-3 (1 ng/ml each) and 280 nM ruxolitinib or DMSO for 12 hours. Cells were then washed in cold RPMI and cultured in MG132 without additives for 5 or 15 min. Cells were lysed and subjected to immunoprecipitation with JAK2 antibody, followed by immunoblotting with ubiquitin-HRP or JAK2 antibody.
To further test this hypothesis, we preincubated recombinant phosphorylated kinase domain of JAK2 with ruxolitinib and performed in vitro phosphatase assays (Fig. 2E). Ruxolitinib blocked the dephosphorylation of JAK2 kinase domain by protein tyrosine phosphatase nonreceptor type 1 (PTP1B) for up to 20 hours, whereas vehicle-treated samples displayed a significant reduction of Tyr1007/1008 phosphorylation within 2 hours. Similar results were obtained with another type I JAK inhibitor, CMP6 (fig. S3B). In contrast, no difference in dephosphorylation was observed for recombinant phosphorylated gp130 in the presence of ruxolitinib or vehicle, indicating that ruxolitinib specifically prevents dephosphorylation of JAK2 (fig. S3C).
Phosphorylated JAKs are also targets for ubiquitination and degradation. We noted that, upon ruxolitinib withdrawal in primary samples, the activation loop–phosphorylated pool of JAK2 rapidly declines (lanes 10 to 12, Fig. 1A). Accordingly, any change induced by ruxolitinib that prevents exposure of the JAK2 phosphorylation loop and limits phosphatase accessibility may also prevent ubiquitination and degradation of phosphorylated JAK2. We analyzed the ubiquitination status of phosphorylated JAK2 in TF1.8 cells using an anti-ubiquitin antibody while blocking proteosomal degradation with MG132. Stimulation by IL-3 resulted in significant JAK2 ubiquitination after 5 and 10 min; however, the presence of ruxolitinib prevented detectable ubiquitination of JAK2 (Fig. 2F). Similar results were obtained using a ubiquitinated protein pull-down [tandem ubiquitin-binding entity (TUBE)], followed by immunoblot with anti-JAK2 antibodies (fig. S3D). If the presence of ruxolitinib protects phosphorylated JAK2 from degradation, then proteasome inhibition should have a similar effect and promote prolonged JAK2 phosphorylation after IL-3 treatment. We observed that treatment of TF1.8 cells with MG132, before IL-3 stimulation, mimicked the effect of ruxolitinib treatment and prolonged JAK2 phosphorylation but without the inhibition of STAT5 phosphorylation associated with ruxolitinib treatment (fig. S3E). Conversely, removal of ruxolitinib should permit ubiquitination and degradation of accumulated phosphorylated JAK2. In a complementary experiment, we observed that primary JAK2V617F myelofibrosis cells showed evidence of delayed JAK2 ubiquitination after ruxolitinib washout (Fig. 2G). No JAK2 ubiquitination was observed in the presence of vehicle or vehicle washout or during ruxolitinib treatment, but only after ruxolitinib withdrawal (Fig. 2G and fig. S3F). These results are consistent with a model where, upon ruxolitinib withdrawal, the accumulated pool of phosphorylated JAK2 is not only able to promote signaling but also subsequently becomes susceptible to ubiquitination and degradation, in agreement with the dephosphorylation of JAK2 observed in patient samples (lanes 10 to 12, Fig. 1A).
Ruxolitinib withdrawal induces cytokine receptor phosphorylation and stabilizes JAK1
Factor-independent oncogenic signaling by JAK2V617F in Ba/F3 cells requires the presence of a cytokine receptor (12). Thus, STAT phosphorylation induced by ruxolitinib withdrawal presumably occurs in the context of an activated cytokine receptor complex. We confirmed that SET-2 cells express the β common (βc) subunit of the IL-3 and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor (fig. S4A), display STAT5 phosphorylation after IL-3 treatment (fig. S4B), and proliferate in response to IL-3 and GM-CSF (13). Similar to STAT proteins, immunoprecipitation with anti-βc antibody showed strong βc phosphorylation within 5 min of ruxolitinib washout (Fig. 3A), indicating that type I inhibitor withdrawal can induce cytokine receptor phosphorylation in addition to STAT phosphorylation, and these phosphorylated cytokine receptors may serve as a connecting point for activated JAK molecules and STAT proteins.
Fig. 3. Effect of ruxolitinib on cytokine receptor phosphorylation and JAK1 down-regulation.
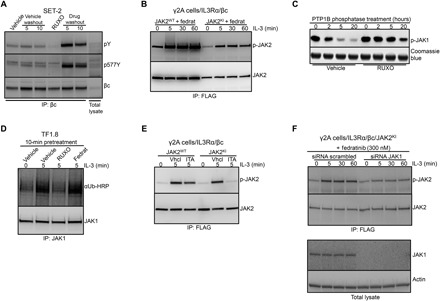
(A) SET-2 cells were treated with ruxolitinib or DMSO for 12 hours, followed by washout as described in Fig. 1A. Whole-cell lysates were prepared and subjected to immunoprecipitation with human βc receptor antibody and immunoblotted with phosphotyrosine, p577Y βc, and βc antibodies. (B) γ2A cells that are JAK2 deficient were stably transfected with βc and IL3Rα receptors (γ2A/IL3Rα/βc) and then additionally stably transfected with either JAK2WT-FLAG or JAK2KI-FLAG. After overnight starvation, cells were pretreated with 150 nM of the type I inhibitor fedratinib and stimulated for 0, 5, 30, or 60 min with IL-3 (25 ng/ml). Cells were lysed and subjected to immunoprecipitation with anti-FLAG antibody, followed by immunoblotting with p-JAK2 and JAK2 antibodies. (C) Recombinant JAK1 kinase domain was mixed with recombinant tyrosine phosphatase PTP1B and ruxolitinib or no inhibitor in phosphatase assay buffer. Phosphatase reactions were incubated at room temperature for 0, 2, 5, and 20 hours, fractionated by SDS-PAGE, and immunoblotted with p-JAK1 antibody. Coomassie blue staining was used as a loading control. (D) TF1.8 cells were starved overnight in 0.5% FCS and then treated with DMSO, 280 nM ruxolitinib, or 150 nM fedratinib for 10 min before stimulation with IL-3 (50 ng/ml) for 0 or 5 min. Whole-cell lysates were prepared and subjected to immunoprecipitation with anti-JAK1 antibody, followed by immunoblotting with ubiquitin-HRP or JAK1 antibody. (E) γ2A/IL3Rα/βc cells were stably transfected with either JAK2WT-FLAG or JAK2KI-FLAG. After overnight starvation, cells were pretreated with DMSO or 11 nM itacitinib and stimulated for 5 min with IL-3 (50 ng/ml). Cells were lysed and subjected to immunoprecipitation with anti-FLAG antibody, followed by immunoblotting with p-JAK2 and JAK2 antibodies. (F) γ2A/IL3Rα/βc cells expressing JAK2KI-FLAG were transfected with control small interfering RNA (siRNA) or JAK1 siRNA. After overnight starvation, cells were pretreated with fedratinib for 10 min and stimulated with IL-3 (50 ng/ml). Cells were lysed and subjected to immunoprecipitation with anti-FLAG antibody, followed by immunoblotting with p-JAK2 and JAK2 antibodies. As a control, total lysates from the same experiment were immunoblotted with JAK1 and actin antibodies.
To test whether kinases beyond JAK2 are specifically involved in activation loop phosphorylation in the setting of a type I kinase inhibitor, we expressed wild-type or mutant forms of JAK2 and IL-3 receptor components into gamma2A (γ2A) cells derived from human mesenchymal cells, which lack endogenous JAK2 while retaining endogenous JAK1 expression (14). We created γ2A/IL3Rα/βc cells stably expressing the βc and IL3Rα chains, which together can bind IL-3 with high affinity as well as FLAG-tagged wild-type or kinase-inactive JAK2 (JAK2KI). Treatment of γ2A/IL3Rα/βc cells with the JAK2-selective type I inhibitor, fedratinib, resulted in accumulation of phosphorylated JAK2 after IL-3 stimulation in cells expressing wild-type or kinase-inactive JAK2, indicating that other kinases (including but not limited to JAK1) can promote type I inhibitor–induced JAK2 phosphorylation (Fig. 3B). Ruxolitinib is a potent nanomolar inhibitor of both JAK1 and JAK2. We found that ruxolitinib also blocked dephosphorylation of phosphorylated JAK1 by PTB1B in vitro for up to 20 hours (Fig. 3C) and ubiquitination of JAK1 in TF1.8 cells (Fig. 3D). Fedratinib is a type I JAK2 inhibitor, which has 35-fold lower potency toward JAK1. When this inhibitor was applied to TF1.8 cells at 150 nM (50 × JAK2 IC50, 1.4 × JAK1 IC50), it was not able to prevent JAK1 ubiquitination (Fig. 3D). JAK2 phosphorylation induced by IL-3 in the setting of the kinase-inactive JAK2 mutant was abrogated by the JAK1-selective type I inhibitor, itacitinib (Fig. 3E; experimentally determined IC50 value for JAK1 and JAK2 in fig. S4C). In addition, siRNA knockdown of JAK1 resulted in a significant reduction of kinase-inactive JAK2 mutant phosphorylation in γ2A/IL3Rα/βc cells treated with fedratinib (Fig. 3F). Together, these results show that βc phosphorylation can be triggered by ruxolitinib withdrawal and that JAK1 is sufficient to activate physiological JAK2 phosphorylation in response to βc signaling through IL-3. Furthermore, ruxolitinib can also protect JAK1 from degradation and dephosphorylation. These results indicate that cytokine receptors and JAK1 may participate in ruxolitinib withdrawal signaling.
Type II JAK2 inhibitors do not cause JAK2 phosphorylation accumulation or pathological signaling upon withdrawal
Recently, type II JAK2 inhibitors have been developed that bind to the inactive conformation of JAK2 and reportedly do not lead to an accumulation of phosphorylated JAK2 in SET-2 cells (6). CHZ868 is a potent, JAK2-biased type II inhibitor that has efficacy in preclinical models of MPN at high nanomolar concentrations (250 to 500 nM) (15), but its effects on IL-3 signaling or drug withdrawal signaling in primary patient samples have not been explored. In TF1.8 cells stimulated with IL-3, CHZ868 treatment inhibited STAT5 and ERK phosphorylation from 500 nM CHZ868 (Fig. 4A), while TF1.8 proliferation induced by IL-3 was completely blocked at 500 nM CHZ868 (fig. S5A). In contrast to type I inhibitors, CHZ868 did not prevent JAK2 activation loop dephosphorylation, as shown by in vitro phosphatase assay (Fig. 4B). CHZ868 and ruxolitinib were equally effective at inhibiting the proliferation of JAK2V617F mutant cells, while wild-type JAK2 cells were significantly more sensitive to ruxolitinib treatment (Fig. 4C and fig. S5, B and C). To exclude off-target effects, we performed proliferation assays with γ2A cells using doses of ruxolitinib or CHZ868 ranging from 300 to 33,000 nM. γ2A cells, which are not dependent on JAK signaling and are deficient in JAK2, were completely resistant to both of these inhibitors up to 33,000 nM (fig. S5D). Similarly, in survival assays, wild-type JAK2 cells were significantly more sensitive to ruxolitinib treatment compared to CHZ868 treatment than JAK2V617F mutant cells (Fig. 4, D and E). A similar observation was noted for EPO signaling comparing cells expressing wild-type JAK2 or JAK2V617F after treatment with ruxolitinib or CHZ868 (15).
Fig. 4. Type II JAK2 inhibitor CHZ868 blocks proliferation of cells expressing wild-type JAK2 and JAK2V617F.
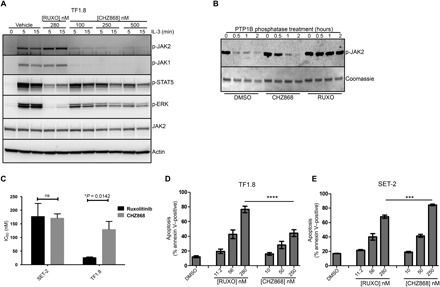
(A) TF1.8 cells were starved overnight in the presence of 0.5% FCS and stimulated for 5 or 15 min with IL-3 (25 ng/ml) in the presence of 280 nM ruxolitinib or different concentrations of CHZ868. Cells were lysed and immunoblotted with p-JAK2, p-JAK1, p-ERK, JAK2, and actin antibodies. (B) Recombinant JAK2 kinase domain was mixed with recombinant tyrosine phosphatase PTP1B and either DMSO, CHZ868, or ruxolitinib in phosphatase assay buffer. Phosphatase reactions were incubated at room temperature for 0, 0.5, 1, and 2 hours, fractionated by SDS-PAGE, and immunoblotted with p-JAK2 antibody. Coomassie blue staining was used as a loading control. (C) Starved TF1.8 cells and SET-2 cells were incubated in IL-3 (50 ng/ml) and 10% FCS with titrations of ruxolitinib or CHZ868 in triplicate. After 48 hours, cell proliferation was assessed using CellTiter 96 reagent. The ruxolitinib and CHZ868 IC50 values required to inhibit proliferation of TF1.8 and SET-2 cells, as shown in fig. S5 (B and C), were determined using GraphPad Prism. Data are presented as the means ± SEM of IC50 values determined from three independent biological replicates. Statistical significance was determined using an unpaired two-tailed t test (*P < 0.05). ns, not significant. (D and E) FCS-starved TF1.8 or SET-2 cells were treated with increasing concentrations of ruxolitinib or CHZ868 in triplicate for 48 hours. Apoptosis was determined by annexin V staining. Bars show means ± SEM of three independent biological replicates, ***P < 0.01 and ****P < 0.001 determined by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons post-test.
CHZ868 was tested for withdrawal signaling after drug washout in hematopoietic cell lines and primary JAK2V617F patient samples using the same assay format as in Fig. 1. Unlike ruxolitinib, CHZ868 did not induce accumulation of JAK2 phosphorylation and did not show any evidence of residual JAK2 activation in low FCS, drug-free conditions. CHZ868 treatment did not produce rebound JAK2 signaling upon drug washout (Fig. 5, A and B, and fig. S6, A and B).
Fig. 5. Type II inhibitor withdrawal does not trigger intracellular STAT signaling and has superior activity compared to type I inhibitor.
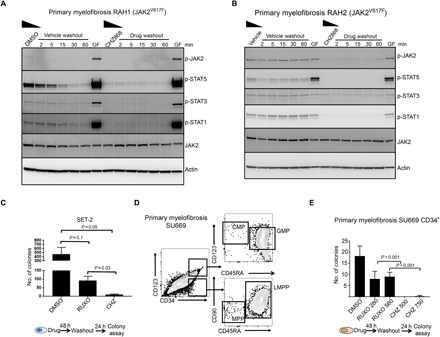
Mononuclear cells from patients with myelofibrosis RAH1 (A) or RAH2 (B) were cultured for 12 hours in 10% FCS and EPO and IL-3 (1 ng/ml) with either 250 nM CHZ868 or DMSO. In addition, cells were transiently stimulated 60 min after washout with IL-3 and EPO (25 ng/ml each) for 5 min (GF, growth factors) as a positive control to show that cells are still competent to signal through JAK/STAT. Cells were then washed extensively in cold media and transferred into prewarmed and CO2-equilibrated media without additives for the indicated periods of time. Cell lysates were prepared and immunoblotted for the indicated proteins. (C) SET-2 cells were treated for 48 hours in either 280 nM ruxolitinib or 500 nM CHZ868 and then cultured in 0.5% FCS to mimic drug withdrawal for 24 hours before plating in a colony assay. Blast-forming unit colonies were scored 10 days after plating. (D) Flow cytometry of peripheral blood mononuclear cells purified from a patient with primary myelofibrosis (SU669) and flow sorted for CD34+ stem progenitor cells. Most of CD34+ cells resembled CD123hiCD45RA+ granulocyte macrophage progenitor (GMPs) or lymphoid-restricted progenitor (LMPPs). Few common myeloid progenitor (CMPs) and multipotent progenitor (MPPs) were observed. (E) CD34+ stem cells were treated with either 280 nM ruxolitinib, 560 nM ruxolitinib, 500 nM CHZ868, or 750 nM CHZ868 for 48 hours then washed and plated in Iscove’s modified Dulbecco’s medium (IMDM) 0.5% FCS with thrombopoietin (TPO), FLT3 ligand (FLT3L), stem cell factor (SCF), and IL-6 (0.1 ng/ml each) to mimic drug withdrawal, followed by plating in methylcellulose in triplicate. Colonies were scored 14 days after plating, and the total number of colonies is shown. Bars show means ± SEM of three independent experiments. Student’s t test was used to compare differences.
A type II JAK2 inhibitor is superior to type I inhibitors after drug withdrawal in JAK2V617F and CALR mutant myelofibrosis cells
To understand the clinical significance of type I inhibitor withdrawal signaling, we performed clonogenic colony-forming assays with JAK2V617F mutant cells, 24 hours after ruxolitinib or CHZ868 washout. We noted more colony-forming blast colonies derived from SET-2 cells treated with ruxolitinib before washout compared to cells similarly treated with CHZ868 (Fig. 5C). Similar assays were performed with primary, flow cytometry–purified, CD34+ stem/progenitor cells collected from a patient with JAK2V617F myelofibrosis and plated in methylcellulose after CHZ868 or ruxolitinib treatment and a 24-hour washout. Immunophenotypically, CD34+ progenitor cells from these patients most closely resembled CD34+CD38+CD45RA+CD123hi granulocyte-myeloid progenitors and lymphoid-restricted multipotential progenitors (Fig. 5D) (16). JAK2V617Fpatients exhibited higher levels of the CD34+CD38− subpopulation in peripheral blood compared with patients with CALR mutant myelofibrosis (fig. S5, E and F). Compared to ruxolitinib-treated cells, we observed significantly reduced colony numbers after 24 hours of drug washout in CHZ868-treated cells, at all doses tested (Fig. 5E). All residual colonies from ruxolitinib-treated cells were confirmed to be JAK2V617F positive by digital droplet polymerase chain reaction (ddPCR). We performed similar experiments in an extended panel of three heterozygous JAK2V617F mutant myelofibrosis samples, two homozygous JAK2V617F mutant samples (clinical details in Table 1), and four CALR mutant samples (Fig. 6). JAK2V617F was confirmed to be present in more than 95% of colonies from ruxolitinib-treated JAK2V617F positive samples by ddPCR. Residual colonies after treatment with CHZ868 were also confirmed to be JAK2V617F positive in these cases. Our data suggest that the lack of phosphorylated JAK2 accumulation and the decreased rebound signaling after cessation of type II inhibitor treatment are associated with superior eradication of disease-associated progenitor cells in both JAK2 mutant and CALR mutant myelofibrosis.
Fig. 6. Type II JAK inhibitor has activity in primary CALR mutant samples and homozygous JAK2 mutant samples.
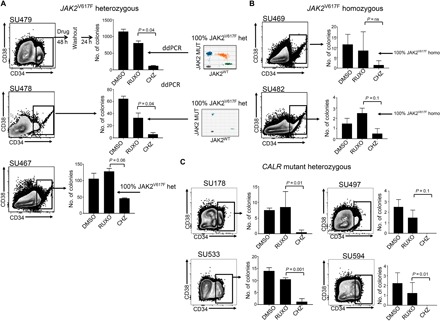
Mononuclear cells obtained from the peripheral blood of patients with myelofibrosis with (A) heterozygous JAK2V617F, (B) homozygous JAK2V617F mutations, or (C) confirmed CALR mutations were flow sorted for CD34+ stem progenitor cells and treated with either 560 nM ruxolitinib or 750 nM CHZ868 for 48 hours and then washed into media for 24 hours in IMDM 0.5% FCS with TPO, FLT3L, SCF, and IL-6 (0.1 ng/ml each) to mimic drug withdrawal. This was followed by plating in methylcellulose in triplicate at a density of ~300 CD34+ input cells per plate. Colonies were scored 14 days after plating. Bars show average colony numbers ± SD. An unpaired Student’s t test was used to compare the differences between drug washouts. Inset panels in (A) show a representative fluorescent droplet distribution of a genotyped colony from ruxolitinib-treated cells from two samples. Twenty colonies were genotyped per treatment when numbers were sufficient. Dots represent droplets containing at least one copy of mutant or wild-type JAK2 alleles as analyzed by ddPCR. The variant allele frequency (VAF) is determined by the fraction of single-allele droplets containing the variant allele.
DISCUSSION
A number of cases of ruxolitinib withdrawal syndrome have been described, including three patients who developed acute respiratory distress syndrome in the original phase 1/2 trial of ruxolitinib (17). In the phase 3 Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment I (COMFORT-I) study, one patient was reported to develop acute respiratory distress, pyrexia, and splenic infarction following ruxolitinib discontinuation (18). Other reports include a patient who developed tumor lysis–like syndrome (19), a case of acute respiratory failure (20), and a case of acute respiratory distress syndrome that resolved after ruxolitinib reintroduction (21). Ruxolitinib discontinuation syndrome is a diagnosis of exclusion based on a temporal relationship between drug withdrawal and onset of clinical manifestations that can appear within 24 hours and up to 3 weeks after discontinuation (table S2).
Our data provide a mechanistic explanation for some clinical observations regarding withdrawal effects seen during the treatment of patients with MPNs with type I JAK inhibitors. Using primary human samples and cell lines, we show that (i) abrupt ruxolitinib withdrawal induces phosphorylation of STAT, ERK, and cytokine receptor βc phosphorylation in a dose-dependent fashion; (ii) type I inhibitor–induced accumulation of JAK phosphorylation is sensitive to extrinsic signals, including exposure to FCS and IL-3; (iii) type I JAK inhibitors induce a change in the accessibility of a cryptic site in the activation loop of both JAK1 and JAK2 that protects them from dephosphorylation and ubiquitination; (iv) JAK1 can phosphorylate kinase-inactive JAK2 in the presence of a specific JAK2 type I inhibitor in response to IL-3 stimulation and leads to the accumulation of p-JAK2; and (iv) treatment of JAK2V617F cells with a type II JAK inhibitor did not elicit inhibitor withdrawal signaling or the accumulation of JAK2 with activation loop phosphorylation. Our results indicate that ruxolitinib withdrawal syndrome is linked to the latent signaling activity contained within the accumulated pool of phosphorylated JAK. More severe withdrawal effects have occurred when ruxolitinib is discontinued during an acute illness when inflammatory cytokines may be elevated, and this observation concurs with our observation that extrinsic signaling may exacerbate the accumulation of phosphorylated JAK2 in both wild-type and JAK2 mutant cells. Our data strongly suggest a need to assess withdrawal signaling in clinical drug development programs using type I kinase inhibitors, in addition to conventional on-target effect assessments. The inhibition of phosphorylated JAK2 degradation, leading to aberrant accumulation and withdrawal signaling, may be an inherent limitation of type I JAK inhibitors and could potentially be relevant for inhibitors of other tyrosine kinases. Flow cytometric measurement of activation loop phosphorylation could be developed as a biomarker to assess the risk of ruxolitinib withdrawal syndrome.
We note that cells from one patient with MPN with a CALR mutant did not exhibit the same degree of spontaneous withdrawal signaling as seen with JAK2V617F samples (fig. S1B). Similarly, Tyr1007/1008 phosphorylation of JAK2 was not detectable in CALR mutant myelofibrosis cells in the presence of ruxolitinib. This is consistent with a number of emerging reports using CALR-mutated models of myelofibrosis but needs to be confirmed with additional patient samples (10, 22, 23). These results suggest that there are fundamental differences in the nature of JAK activation in CALR-mutated cells compared to JAK2-mutated cells. We performed an extensive search of medical literature and adverse drug reaction databases and were unable to find any reports of withdrawal syndrome in CALR-mutated patients (9, 17, 19–21, 24). Analysis of 10 published cases of severe ruxolitinib withdrawal syndrome (table S2) showed a high frequency of JAK2 mutant cases (100%) compared to expected mutation frequencies (χ2 2 × 2 contingency table, P = 0.0203). In most of the cases, symptoms began within 72 hours of drug withdrawal, suggesting a rapid rather than a delayed phenomenon. Although intriguing, this retrospective analysis is limited by potential publication bias and availability of JAK2 mutation testing versus CALR mutation testing at pathology laboratories.
Our data indicate that the JAK2 activation loop undergoes a change in surface accessibility in the presence of ruxolitinib and other type I inhibitors. This presumptive conformational change prevents immunoprecipitation with anti-phosphotyrosine antibodies and confers resistance to the action of tyrosine phosphatases. Thus, type I inhibitors cause accumulation of phosphorylated JAK2 by preventing dephosphorylation, ubiquitination, and subsequent degradation by the proteasome. In contrast, these phenomena are not seen with a type II inhibitor, CHZ868. While CHZ868 is not currently being developed for clinical use, our data provide a strong rationale to develop selective type II JAK inhibitors for use in patients with MPN. This strategy may lead to more effective targeting of clonal cells in MPN without the risk of severe drug withdrawal phenomena.
METHODS
Cell lines and cytokines
TF1.8 cells were cultured in RPMI with 10% (v/v) FCS supplemented with 10 mM Hepes and GM-CSF (2 ng/ml). SET-2 cells were cultured in RPMI with 10% (v/v) FCS. γ2A cells were cultured in Dulbecco’s modified Eagle’s medium with 10% (v/v) FCS.
Expression constructs
Human JAK2 complementary DNA (cDNA) containing a C-terminal Flag epitope tag was purchased from Sino Biological Inc., and the V617F and K882E kinase inactivation mutation were generated by PCR. JAK2-FLAG cDNA fragments were cloned into the pRufBlast retroviral expression vector to produce the pRufBlast:JAK2-Flag expression plasmids that were transfected into γ2A cells using Lipofectamine 2000 (Invitrogen) and then selected with (5 μg/ml) blasticidin to create stable cell lines.
Wild-type IL3Rα cDNA was cloned into the retroviral expression vector pRufHygro to produce pRufHygro:IL3Rα. Wild-type βc cDNA was cloned into the pRufPuro retroviral expression vector to produce pRufPuro:βc. The pRufHygro:IL3Rα and pRufPuro:βc plasmids were cotransfected into γ2A cells using Lipofectamine 2000 (Invitrogen) and then selected with hygromycin and puromycin to create stable cell lines. After selection, pools of hygromycin- and puromycin-resistant cells were sorted for IL3Rα and βc expression by flow cytometry. ON-TARGETplus Human JAK1 siRNA from Dharmacon was transfected using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen).
Cell proliferation
TF1.8 or SET-2 cells were resuspended in RPMI supplemented with 10% (v/v) FCS, plated at 2 × 104 cells per well in 96-well plates, and incubated with titrations of cytokine or inhibitors for 2 days. Cell proliferation was assessed using CellTiter 96 AQueous (#G3581, Promega) following the manufacturer’s protocol. Data are expressed as means ± SEM from triplicates, and n = 3 independent experiments.
Cell viability assay
TF1.8 or SET-2 cells were starved for 3 hours in RPMI containing 0.5% (v/v) FCS and then plated at 1.5 × 105 cells/ml with the addition of human IL-3 (2 ng/ml) in the presence of either 0.1% (v/v) DMSO or a titration of ruxolitinib or CHZ868. Cells were analyzed for apoptosis after 48 hours by staining with 1:100 annexin V–allophycocyanin (APC) in 1× annexin V binding buffer (BD Pharmingen) for 20 min at 4°C. The percentage of cells that were annexin V positive was determined by flow cytometry using an LSRFortessa Special Order Research Flow Cytometer with FACSDiva Software version 8.0 (BD Biosciences, San Diego, CA, USA). Data are expressed as means ± SEM from triplicates, and n = 3 independent experiments.
Colony assays
SET-2 cells were plated in MethoCult H4435 (STEMCELL Technologies) after 48 hours of drug treatment, followed by washout in low FCS media and low-dose cytokines (24 hours) and scored for colony-forming unit blast at 10 days. Primary CD34+ stem/progenitor cells from peripheral blood samples from patients with JAK2V617F mutant positive myelofibrosis were purified by flow cytometry using anti–CD34-APC (clone 8G12), anti-human CD45RA Brilliant Violet 605, anti-CD90 fluorescein isothiocyanate, anti-human CD123-phycoerythrin (PE), and anti-human CD38 PE-Cy7 and plated in MethoCult H4435 at 1000 CD34+ per plate after ruxolitinib treatment and washout.
Digital droplet PCR
Colonies were picked in up to 10 μl of media and added to 20 μl of QuickExtract Solution (Lucigen, USA) to extract DNA. DNA was diluted 1:10 before PCR. JAK2V617F (SNP ID: rs77375493) mutant and wild-type specific fluorescent probes were obtained from Thermo Fisher Scientific (#4351379). For droplet generation and analysis, we used the QX200 Droplet Digital PCR Systems consisting of two instruments: the droplet generator and the droplet reader. The droplet generator divided the sample by creating 20,000 partitions (droplets). The droplets were then transferred into PCR plates and, at the end of the amplification cycles, placed into the droplet reader, where each droplet is read as mutated or wild-type by issuing specific fluorescence signals (FAM for the mutation and Hex for the wild type). These signals, after being counted, were redistributed according to the Poisson’s law. All reagents were purchased from Bio-Rad. Quantitative PCR was performed with annealing/extension temperatures of 59°C × 40 cycles. The VAF was determined from the fraction of the single-allele droplets containing the variant allele.
Protein lysate preparation, immunoprecipitation, and immunoblot analysis
For immunoblotting of total lysates, cells were first lysed in NP-40 buffer containing 150 mM NaCl, 50 mM tris (pH 7.6), and 1% (v/v) NP-40, supplemented with protease inhibitors (cOmplete, Roche) and phosphatase inhibitor cocktails. Lysates were boiled for 5 min after the addition of SDS sample buffer [60 mM tris-HCl (pH 6.8), 5% (v/v) glycerol, 1% (w/v) SDS, 2% (v/v) β-mercaptoethanol, and 0.02% (w/v) bromophenol blue] before SDS gel electrophoresis and immunoblotting. Primary antibodies against p-JAK2 (#3771), p-JAK1 (#3331), p-AKT S473 (#9271), p-ERK (#9101), p-STAT5 (#9359), Akt (#4685), and STAT5 (#9363) were purchased from Cell Signaling Technology. Primary antibodies against p-STAT1 (#612132), p-STAT3 (#612356), STAT3 (#612257), JAK1 (#610232), and phosphotyrosine-HRP conjugate (#610012) were obtained from BD Biosciences. A primary antibody against actin (#MAB1501) and anti-phosphotyrosine clone 4G10 (#05-777) were obtained from EMD Millipore. A primary antibody against TYK2 (#SC-169) and ubiquitin-HRP conjugate (SC-8017) were purchased from Santa Cruz Biotechnology. Primary antibodies against IL3Rα and βc were previously developed and characterized in our laboratory (25). Secondary antibodies were obtained from Thermo Fisher Scientific. The immunoprecipitation protocol has been described elsewhere (26). For immunoprecipitation, cells were lysed in NP-40 buffer containing 150 mM NaCl, 50 mM tris (pH 7.6), 1% (v/v) NP-40, and 50 mM iodoacetamide supplemented with protease inhibitors (cOmplete, Roche) and phosphatase inhibitor cocktails. For TUBE assays, cells were pretreated with MG132 (Sigma-Aldrich) to prevent degradation of ubiquitinated proteins for 10 min and stimulated with IL-3 in the presence or absence of ruxolitinib. Cells were lysed according to the manufacturer’s recommendations, and Agarose-TUBE (LifeSensors) was added for 2 hours. After two washes with lysis buffer, SDS sample buffer was used to elute bound ubiquitinated proteins, which were subsequently analyzed by immunoblotting with different JAK antibodies. Immunoblot analysis is representative of three independent experiments, with the exception of patient samples, where n = 4.
PTP1B dephosphorylation assay
Recombinant JAK2 kinase domain (270 nM) was mixed with 80 nM recombinant PTP1B and no inhibitor, ruxolitinib, CHZ868, or itacitinib. The mixture was incubated at room temperature, and at various time points, 15 μl of reaction mix was added to 5 μl of SDS-PAGE reducing buffer [50 mM tris-HCl (pH 7.4), 200 mM β-mercaptoethanol, 10% (v/v) glycerol, 4% (w/v) SDS, and 0.2% (w/v) bromophenol blue]. Samples were blotted using primary antibody specific for the phosphorylated JAK activation loop (sc-10176, Santa Cruz Biotechnology) and an infrared fluorescent secondary antibody (925-32211, LI-COR) and imaged using an Odyssey infrared imager (LI-COR). Coomassie staining was used to monitor JAK1 levels due to unavailability of an appropriate antibody.
In vitro kinase assay
JAK domain (3 nM) was mixed with 1 mM STAT5b substrate peptide, 0.5 mM ATP, 1 μCi [γ-32P] ATP, 30 mM tris-HCl (pH 8.0), 100 mM NaCl, bovine serum albumin (0.2 mg/ml), 1 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and various concentrations of JAK inhibitor. After incubation at room temperature for 20 to 60 min, 3.5 μl of reaction mix was spotted onto P81 phosphocellulose paper (Whatman, GE Healthcare), which was washed four times for 15 min with 100 ml of 5% (v/v) H3PO4. The paper was dried and exposed to a phosphorimager plate overnight, which was then scanned using a Typhoon FLA 7000 PhosphorImager (GE Life Sciences).
Reagents and JAK inhibitors
Fedratinib, itacitinib, and ruxolitinib were obtained from Sellecta. CHZ868 was from APExBIO, and CMP6 was from Sigma-Aldrich. All inhibitors were diluted in DMSO as 1 mM stock solutions and used at the indicated concentrations.
Statistical data analysis
Unless otherwise stated, P values comparing two means were calculated using the two-tailed unpaired Student’s t test in Prism version 6 (GraphPad Software Inc., La Jolla, CA). A P value of less than 0.05 was considered statistically significant. IC50 values were determined using the dose-response (inhibition) function in Prism version 6.0. The data were normalized and fitted using a variable Hill slope model.
Patient samples and human ethics approval
Primary peripheral blood de novo myelofibrosis samples were obtained before treatment with informed consent according to institutional guidelines. Stanford University Institutional Review Board no. 6453 or South Australian Cancer Research Biobank, Royal Adelaide Hospital Human Research Ethics Committee; project title: South Australian Cancer Research Biobank (protocol nos. 110304, 110304b, and 110304c). Patient details are provided in table S1.
Supplementary Material
Acknowledgments
Funding: Funding for this project was provided through a National Health and Medical Research Council of Australia (NHMRC) Program grant to A.F.L., M.W.P., and T.P.H. (APP 1071897). We acknowledge funding from the Victorian Government Operational Infrastructure Support Scheme to St. Vincent’s Institute. This work was also supported by CSL Limited, Australia. T.P.H. is an NHMRC Practitioner Fellow and M.W.P. is an NHMRC Research Fellow. D. Thomas was funded by NIH/NCI Pathway-to-Independence (K99) grant number 5K99CA207731-02. Author contributions: D. Tvorogov designed and executed experiments, analyzed data, and wrote the manuscript. D. Thomas designed and performed experiments, analyzed data, and wrote the manuscript. N.P.D.L. and J.J.B. designed and performed experiments and supplied critical reagents. M.D., E.F.B., W.L.K., and F.S. performed experiments, analyzed data, and reviewed the manuscript. M.L. performed database searches and data analysis. T.R.H. performed DNA manipulations and reviewed the manuscript. D.M.R. provided clinical material and reviewed the manuscript. M.W.P. discussed the structural biology aspects and reviewed the manuscript. T.P.H., V.T., and R.M. provided advice and reviewed the manuscript. A.F.L. and D. Tvorogov conceived the study and wrote the manuscript. Competing interests: D.M.R. receives honorarium and research funding from Novartis. T.P.H. receives honorarium and research funding from Novartis, BMS, and Ariad. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaat3834/DC1
Table S1. Primary myelofibrosis patient characteristics.
Table S2. Clinical characteristics and genotype of reported cases of ruxolitinib withdrawal syndrome.
Fig. S1. Graphical representation of experiments used in JAK inhibitor withdrawal studies in patient samples and TF1.8 cells.
Fig. S2. Ruxolitinib washout triggers intracellular signaling.
Fig. S3. Type I JAK2 inhibitor protects JAK2 from degradation and down-regulation.
Fig. S4. Selective targeting of JAK1 kinase by itacitinib.
Fig. S5. Type II JAK2 inhibitor CHZ868 blocks proliferation of cells expressing wild-type JAK2 and JAK2V617F.
Fig. S6. Type II inhibitor withdrawal does not trigger intracellular signaling.
REFERENCES AND NOTES
- 1.Stark G. R., Darnell J. E. Jr., The JAK-STAT pathway at twenty. Immunity 36, 503–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen E., Staudt L. M., Green A. R., Janus kinase deregulation in leukemia and lymphoma. Immunity 36, 529–541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parganas E., Wang D., Stravopodis D., Topham D. J., Marine J. C., Teglund S., Vanin E. F., Bodner S., Colamonici O. R., van Deursen J. M., Grosveld G., Ihle J. N., Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93, 385–395 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Brooks A. J., Dai W., O’Mara M. L., Abankwa D., Chhabra Y., Pelekanos R. A., Gardon O., Tunny K. A., Blucher K. M., Morton C. J., Parker M. W., Sierecki E., Gambin Y., Gomez G. A., Alexandrov K., Wilson I. A., Doxastakis M., Mark A. E., Waters M. J., Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Wang L. H., Kirken R. A., Erwin R. A., Yu C.-R., Farrar W. L., JAK3, STAT, and MAPK signaling pathways as novel molecular targets for the tyrphostin AG-490 regulation of IL-2-mediated T cell response. J. Immunol. 162, 3897–3904 (1999). [PubMed] [Google Scholar]
- 6.Andraos R., Qian Z., Bonenfant D., Rubert J., Vangrevelinghe E., Scheufler C., Marque F., Régnier C. H., De Pover A., Ryckelynck H., Bhagwat N., Koppikar P., Goel A., Wyder L., Tavares G., Baffert F., Pissot-Soldermann C., Manley P. W., Gaul C., Voshol H., Levine R. L., Sellers W. R., Hofmann F., Radimerski T., Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2, 512–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koppikar P., Bhagwat N., Kilpivaara O., Manshouri T., Adli M., Hricik T., Liu F., Saunders L. M., Mullally A., Abdel-Wahab O., Leung L., Weinstein A., Marubayashi S., Goel A., Gönen M., Estrov Z., Ebert B. L., Chiosis G., Nimer S. D., Bernstein B. E., Verstovsek S., Levine R. L., Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer S. C., Levine R. L., Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 20, 2051–2059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coltro G., Mannelli F., Guglielmelli P., Pacilli A., Bosi A., Vannucchi A. M., A life-threatening ruxolitinib discontinuation syndrome. Am. J. Hematol. 92, 833–838 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Marty C., Pecquet C., Nivarthi H., El-Khoury M., Chachoua I., Tulliez M., Villeval J.-L., Raslova H., Kralovics R., Constantinescu S. N., Plo I., Vainchenker W., Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127, 1317–1324 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Quentmeier H., MacLeod R. A. F., Zaborski M., Drexler H. G., JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia 20, 471–476 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Lu X., Huang L. J.-S., Lodish H. F., Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 283, 5258–5266 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Uozumi K., Otsuka M., Ohno N., Moriyama T., Suzuki S., Shimotakahara S., Matsumura I., Hanada S., Arima T., Establishment and characterization of a new human megakaryoblastic cell line (SET-2) that spontaneously matures to megakaryocytes and produces platelet-like particles. Leukemia 14, 142–152 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Kohlhuber F., Rogers N. C., Watling D., Feng J., Guschin D., Briscoe J., Witthuhn B. A., Kotenko S. V., Pestka S., Stark G. R., Ihle J. N., Kerr I. M., A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol. Cell. Biol. 17, 695–706 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer S. C., Keller M. D., Chiu S., Koppikar P., Guryanova O. A., Rapaport F., Xu K., Manova K., Pankov D., O’Reilly R. J., Kleppe M., McKenney A. S., Shih A. H., Shank K., Ahn J., Papalexi E., Spitzer B., Socci N., Viale A., Mandon E., Ebel N., Andraos R., Rubert J., Dammassa E., Romanet V., Dölemeyer A., Zender M., Heinlein M., Rampal R., Weinberg R. S., Hoffman R., Sellers W. R., Hofmann F., Murakami M., Baffert F., Gaul C., Radimerski T., Levine R. L., CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell 28, 15–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majeti R., Park C. Y., Weissman I. L., Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 1, 635–645 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tefferi A., Pardanani A., Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin. Proc. 86, 1188–1191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verstovsek S., Mesa R. A., Gotlib J., Levy R. S., Gupta V., DiPersio J. F., Catalano J. V., Deininger M., Miller C., Silver R. T., Talpaz M., Winton E. F., Harvey J. H. Jr., Arcasoy M. O., Hexner E., Lyons R. M., Paquette R., Raza A., Vaddi K., Erickson-Viitanen S., Koumenis I. L., Sun W., Sandor V., Kantarjian H. M., A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai T., Friedman E. W., Barta S. K., Ruxolitinib withdrawal syndrome leading to tumor lysis. J. Clin. Oncol. 31, e430–e432 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Beauverd Y., Samii K., Acute respiratory distress syndrome in a patient with primary myelofibrosis after ruxolitinib treatment discontinuation. Int. J. Hematol. 100, 498–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman D. D., Kempe C. B., Thomson C. C., McCallister J. W., Recurrent hypoxemic respiratory failure. Beyond the usual suspects. Ann. Am. Thorac. Soc. 11, 1145–1148 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Balligand T., Achouri Y., Pecquet C., Chachoua I., Nivarthi H., Marty C., Vainchenker W., Plo I., Kralovics R., Constantinescu S. N., Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia 30, 1775–1778 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Nivarthi H., Chen D., Cleary C., Kubesova B., Jäger R., Bogner E., Marty C., Pecquet C., Vainchenker W., Constantinescu S. N., Kralovics R., Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 30, 1759–1763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatt V. R., Bociek R. G., Yuan J., Fu K., Greiner T. C., Dave B. J., Rajan S. K., Armitage J. O., Leukemic diffuse large B-cell lymphoma in a patient with myeloproliferative disorder. J. Natl. Compr. Canc. Netw. 13, 281–287 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Sun Q., Jones K., McClure B., Cambareri B., Zacharakis B., Iversen P. O., Stomski F., Woodcock J. M., Bagley C. J., D’Andrea R., Lopez A. F., Simultaneous antagonism of interleukin-5, granulocyte-macrophage colony-stimulating factor, and interleukin-3 stimulation of human eosinophils by targetting the common cytokine binding site of their receptors. Blood 94, 1943–1951 (1999). [PubMed] [Google Scholar]
- 26.Tvorogov D., Anisimov A., Zheng W., Leppänen V. M., Tammela T., Laurinavicius S., Holnthoner W., Heloterä H., Holopainen T., Jeltsch M., Kalkkinen N., Lankinen H., Ojala P. M., Alitalo K., Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. Cancer Cell 18, 630–640 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaat3834/DC1
Table S1. Primary myelofibrosis patient characteristics.
Table S2. Clinical characteristics and genotype of reported cases of ruxolitinib withdrawal syndrome.
Fig. S1. Graphical representation of experiments used in JAK inhibitor withdrawal studies in patient samples and TF1.8 cells.
Fig. S2. Ruxolitinib washout triggers intracellular signaling.
Fig. S3. Type I JAK2 inhibitor protects JAK2 from degradation and down-regulation.
Fig. S4. Selective targeting of JAK1 kinase by itacitinib.
Fig. S5. Type II JAK2 inhibitor CHZ868 blocks proliferation of cells expressing wild-type JAK2 and JAK2V617F.
Fig. S6. Type II inhibitor withdrawal does not trigger intracellular signaling.