

The advent of JAK inhibitors has provided a powerful new set of immunomodulatory agents to treat rheumatoid arthritis (RA). JAK inhibitors bind to and interfere with the function of Janus kinases (JAKs), which mediate signaling downstream of multiple cytokine and growth factor receptors(1). Given the wide range of cytokines that JAK inhibition can potentially influence, it is important to establish the dominant immunologic effects that these molecules exert in patients. In this issue of Arthritis and Rheumatology, Tanaka and colleagues describe the changes in circulating lymphocyte populations induced by baricitinib using data from 3 Phase 3 randomized controlled trials(2). By examining alterations in lymphocyte populations in over 2000 patients, with some patients followed for 2 years, the authors provide a robust set of observations that can be compared to experience with other JAK inhibitors and can yield insights into the effects of baricitinib on immune cell homeostasis.
Randomized, placebo-controlled trials provide a powerful infrastructure upon which to build correlative immunologic analyses. Blood samples can be obtained before and after initiation of therapy and can be collected longitudinally throughout the trial to determine changes induced by therapy over time (Figure 1). While measurements of cytokines in serum have been frequently employed in such studies, detailed assessments of the circulating immune cells themselves may provide a higher resolution view of the most active immune cell populations and pathways in individual patients(3). Immune cell phenotyping by flow cytometry can quantify many leukocyte populations with diverse functions and has the potential to identify cell populations that are strongly influenced by drug therapy(4). These cell populations then serve as candidates that can be evaluated as potentially important targets in the mechanism of action of the drug. In addition, immunophenotyping data can be mined for cellular biomarkers that predict response to therapy or adverse events.
Figure 1:
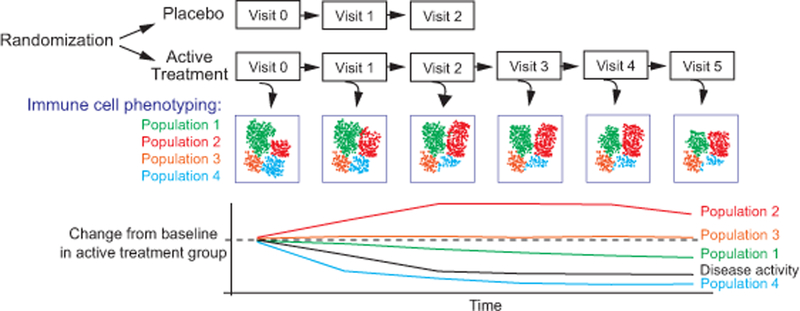
Schematic of immune cell phenotyping within a clinical trial design. Quantitative immunophenotyping can track the frequency of different leukocyte populations longitudinally. In addition to comparisons with the placebo control arm, changes in leukocyte frequencies in the active treatment arm can be tracked over time, allowing for correlation with clinical parameters.
Correlative assessments of immune cell alterations may be particularly valuable in evaluating therapies that target complex pathways such as the JAK-STAT pathway. There are 4 JAKs (JAK1, JAK2, JAK3, and TYK2), which pair in defined combinations to transduce signals downstream of cytokine receptors. JAKs typically act by phosphorylating specific Signal Transducers and Activators of Transcription (STAT) proteins, which then translocate to the nucleus and regulate gene expression. JAK inhibitors bind individual JAKs with varying affinities; thus, each JAK inhibitor may affect different cytokine pathways to varying degrees(1). Tofacitinib, the first FDA-approved JAK inhibitor for treatment of moderate to severe RA, preferentially binds JAK1 and JAK3. Baricitinib preferentially binds JAK1 and JAK2 and has recently received FDA approval at the 2 mg dose for treatment of moderate to severe RA with an inadequate response to anti-TNF therapy. Both tofacitinib and baricitinib potently interfere with interferon and IL-6 signaling, 2 important pathways in RA pathology. However, because of differences in JAK binding, the 2 drugs may also exert distinct immunologic effects. Other selective JAK inhibitors are also being evaluated in RA, with filgotinib and upadacitinib preferentially targeting JAK1 and decernotinib preferentially targeting JAK3. The selectivity of different inhibitors for the 4 JAKs is relative and not absolute; nonetheless, comparing the overlap and differences in their effects on immunologic parameters, clinical endpoints, and adverse events may prove informative in identifying the major immune functions affected by each JAK inhibitor.
One strategy to gain insights into the critical effects of these drugs in clinical disease is to evaluate the cellular and molecular changes in the target tissues of RA patients exposed to the drug. Using synovial tissue biopsies obtained from RA patients before and 1 month after treatment with tofacitinib, Boyle and colleagues demonstrated a significant decrease in the expression of matrix metalloproteinases and the interferon-inducible chemokine CXCL10 following treatment with tofacitinib(5). These same studies demonstrated that decreases in the phosphorylation of STAT1 and STAT3, which are substrates of JAKs, were associated with a good clinical response to tofacitinib(5). These observations suggest that tofacitinib may inhibit phosphorylation and activation of STAT1 and STAT3 to dampen actions of interferons and IL-6 in RA synovium.
Studies of the effects of JAK inhibitors on circulating immune cells may also provide insights into immunologic mechanisms associated with clinical outcomes. While immune cells in the blood may not fully reflect the pathologic mechanisms in the target tissue, blood samples can be routinely collected from large cohorts of patients at regular intervals, bolstering the power of these analyses. In this issue of Arthritis and Rheumatology, Tanaka and colleagues evaluate lymphocyte counts and composition in 3 Phase 3 randomized controlled trials that compared baricitinib to placebo in active RA patients (RA-BEAM, RA-BEACON, and RA-BUILD), as well as in a long-term extension study (RA-BEYOND). Blood samples were collected longitudinally throughout the trials from 2,186 study participants and shipped to a commercial laboratory with 8 harmonized analysis locations. The laboratory sites used validated methods to generate robust flow cytometric quantification of lymphocytes from the blood samples. By combining data collected in a uniform way from 3 large clinical trials at defined intervals, the authors amass a substantial cytometry dataset with which to assess the effects of baricitinib exposure on lymphocyte populations over time.
The authors find that initiation of baricitinib induces a transient increase in lymphocyte counts within the first 4 weeks of treatment, with increases observed across CD4+ T cells, CD8+ T cells, NK cells, and B cells. CD4+ and CD8+ T cell counts returned to baseline by 12 weeks. NK cell counts dipped below baseline by 12 weeks and then stabilized and returned to baseline by 1 year. B cell counts remained stably elevated through 24–32 weeks and then returned to baseline by 1 year. The transient increase in T cell counts and the more prolonged increase in B cell counts appear highly reproducible, observed with 2 different doses of baricitinib across the 3 Phase 3 trials. Notably, a very similar pattern of a transient increase in T cells and a more persistent increase in B cells has been observed with tofacitinib treatment, suggesting likely shared mechanisms for these effects(6, 7).
The reasons for transient increases in lymphocytes induced by baricitinib are unclear. It is possible that baricitinib alters the migration of lymphocytes into and out of secondary lymphoid organs, which can be strongly influenced by environmental cues such as chemokines and sphingosine 1-phosphate(8, 9). Alterations in lymphocyte maturation or survival could also influence total circulating lymphocyte counts. Nonetheless, the return of T cell counts to baseline by 24–32 weeks, despite continued drug treatment, suggests that this modest change may be of little clinical significance. The authors also demonstrate that despite causing increased B cell counts, baricitinib treatment reduces rheumatoid factor and anti-citrullinated peptide antibody levels, suggesting that the increase in total B cell counts is unlikely to reflect a major expansion of RA antigen-specific B cells.
Tanaka and colleagues utilize the long-term extension study in RA-BEYOND to follow lymphocyte counts in patients exposed to baricitinib for up to 2 years, and at these later time points some notable trends emerge. When followed out to 2 years, the CD3+ T cell count in baricitinib treated patients showed a steady downward trend, although levels remained above the lower limit of normal. Both CD4+ T cells and CD8+ T cells showed a significant decrease from baseline levels, with more than a 15% decrease in CD4+ T cells and over 20% decrease in CD8+ T cells. Notably, a similar downward trend in CD3+ T cell counts was observed with 1 year of tofacitinib use(6). Longer term observations of tofacitinib use have indicated that total lymphocyte counts continue to decrease for up to 4 years, with a 28% reduction from baseline in CD4+ T cells and a 27% reduction in CD8+ T cells after a median of 5 years of treatment(10, 11). Evaluation of longer periods of baricitinib use will be important to determine whether the T cell counts plateau or whether they continue to decrease with extended use. In addition, comparison with other selective JAK inhibitors (e.g. JAK1- or JAK3-selective inhibitors) as the data become available will be very interesting to try to decipher which JAKs exert the relevant effects.
Detailed phenotyping of the lymphocyte populations may suggest possible mechanisms and implications of decreased lymphocyte counts over time. Using 4–7 parameter flow cytometry panels, Tanaka and colleagues tracked Th1, Th17, and T regulatory (Treg) cell subsets, the latter requiring a more complicated protocol to detect intracellular expression of the Treg master regulator FoxP3. The authors demonstrate that Th1 (CD4+ CXCR3+ CCR6-) T cell counts appear to trend down over 1 year of treatment, while Treg (CD4+ CD25+ CD127low FoxP3+) counts remain stable over the course of 2 years. The preferential loss of Th1 cells, compared to Th17 cells, might suggest that Th1 cells rely on a cytokine that is strongly inhibited by baricitinib. The preserved Treg counts with baricitinib treatment might also imply effects on specific cytokines. Inhibition of IL-6, an effect of JAK inhibitors, can increase the frequency of Tregs in the circulation, as has been observed with tocilizumab (anti-IL-6 receptor)(12). The ability of tocilizumab to increase Treg populations in RA patients is consistent with a role for IL-6 in blunting Treg differentiation. In addition, it has been suggested that the limited effect of baricinib on JAK3 (compared to tofacitinib) may allow for relatively unimpeded IL-2 signaling through STAT5 to preserve Treg survival(13). Thus, an intriguing possibility is that baricitinib may act to preserve Treg counts while other effector T cell populations contract. Further evaluation of the frequency and function of Tregs after treatment with different JAK inhibitors will be of substantial interest.
Beyond acquiring potential mechanistic insights, a major motivation to pursue these phenotyping analyses is to develop cellular biomarkers to predict clinical outcomes. The authors begin to evaluate lymphocyte population counts as potential biomarkers of treatment-emergent infection, with a particular focus on NK cell changes. While suggestive, these analyses have not yet produced actionable biomarkers, perhaps in part because the characterization of lymphocyte populations still remains somewhat superficial. As lymphocyte biology becomes increasingly sophisticated, the desire to subclassify cell populations in finer detail grows. One wonders about the effects of baricitinib on lymphocyte subpopulations not captured in the 4–7 parameter flow cytometry panels used. For example, are there selective differences in CD56dim CD16+ versus CD56bright NK cell populations? What are the effects on lymphocyte populations expressing markers of activation, proliferation, exhaustion, or senescence? Is there a skewing of T cells across differentiation stages, such as recent thymic emigrants, T memory stem cells, or late-stage CD45RA+ T effector memory cells? Are there effects on innate-like T cell populations, such as MAIT cells, gamma delta T cells, or natural killer T cells?
Rapid advances in high-dimensional single cell analyses, coupled with development of novel data analysis methods, are enabling increasingly powerful, high-resolution maps of cell populations within patient samples(4). Mass cytometry, which can measure 40 or more parameters simultaneously on single cells, is being implemented in large-scale immunophenotyping studies of RA patient synovial and blood samples in the Accelerating Medicines Partnership RA/SLE network(14). Single cell RNA-seq technologies make even genome-wide gene expression measurements of single cells feasible. If such technologies can be applied in a cost-effective manner to samples collected from patients within the defined structure of large clinical trials, this would provide a remarkable resource to interrogate for biomarkers of treatment response and adverse events. The work from Tanaka and colleagues illustrates a satisfying example of the foundations that might support large-scale, high-dimensional biomarker discovery efforts in rheumatic diseases.
ACKNOWLEDGMENTS
D.A.R. is supported by the Rheumatology Research Foundation Tobe and Stephen E. Malawista, MD Endowment in Academic Rheumatology and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K08 AR072791–01.
REFERENCES
- 1.Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. Journal of leukocyte biology 2018. doi: 10.1002/JLB.5RI0218-084R. PubMed PMID: 29999544. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka Y Characterization and changes of lymphocyte subsets in baricitinib-treated patients with rheumatoid arthritis: an integrated analysis. Arthritis and Rheumatology 2018:this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ermann J, Rao DA, Teslovich NC, Brenner MB, Raychaudhuri S. Immune cell profiling to guide therapeutic decisions in rheumatic diseases. Nature reviews Rheumatology 2015. doi: 10.1038/nrrheum.2015.71. PubMed PMID: 26034835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fonseka CY, Rao DA, Raychaudhuri S. Leveraging blood and tissue CD4+ T cell heterogeneity at the single cell level to identify mechanisms of disease in rheumatoid arthritis. Current opinion in immunology 2017;49:27–36. doi: 10.1016/j.coi.2017.08.005. PubMed PMID: 28888129; PubMed Central PMCID: PMC5705469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Annals of the rheumatic diseases 2015;74(6):1311–6. doi: 10.1136/annrheumdis-2014-206028. PubMed PMID: 25398374; PubMed Central PMCID: PMC4431345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clinical and experimental rheumatology 2016;34(2):318–28. PubMed PMID: 26966791. [PubMed] [Google Scholar]
- 7.Strober B, Buonanno M, Clark JD, Kawabata T, Tan H, Wolk R, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. The British journal of dermatology 2013;169(5):992–9. doi: 10.1111/bjd.12517. PubMed PMID: 23855761. [DOI] [PubMed] [Google Scholar]
- 8.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annual review of immunology 2005;23:127–59. doi: 10.1146/annurev.immunol.23.021704.115628. PubMed PMID: 15771568. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, et al. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nature immunology 2006;7(12):1299–308. doi: 10.1038/ni1406. PubMed PMID: 17086187. [DOI] [PubMed] [Google Scholar]
- 10.Schulze-Koops H, Strand V, Nduaka C, DeMasi R, Wallenstein G, Kwok K, et al. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology 2017;56(1):46–57. doi: 10.1093/rheumatology/kew329. PubMed PMID: 28028154. [DOI] [PubMed] [Google Scholar]
- 11.Xeljanz, INN-tofacitinib citrate. Annex I Summary of Product Characteristics European Medicines Agency European Public Assessment Report.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004214/WC500224911.pdf.
- 12.Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis and rheumatism 2012;64(8):2499–503. doi: 10.1002/art.34477. PubMed PMID: 22488116. [DOI] [PubMed] [Google Scholar]
- 13.Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia 2018. doi: 10.1038/s41375-018-0123-z. PubMed PMID: 29691471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donlin LT, Rao DA, Wei K, Slowikowski K, McGeachy MJ, Turner JD, et al. Methods for high-dimensonal analysis of cells dissociated from cyropreserved synovial tissue. Arthritis research & therapy 2018;20(1):139. doi: 10.1186/s13075-018-1631-y. PubMed PMID: 29996944; PubMed Central PMCID: PMC6042350. [DOI] [PMC free article] [PubMed] [Google Scholar]