

Abstract
In females with X-linked genetic disorders, wild-type and mutant cells coexist within brain tissue because of X-chromosome inactivation, posing challenges for interpreting the effects of X-linked mutant alleles on gene expression. We present a single-nucleus RNA sequencing approach that resolves mosaicism by using SNPs in genes expressed in cis with the X-linked mutation to determine which nuclei express the mutant allele even when the mutant gene is not detected. This approach enables gene expression comparisons between mutant and wild-type cells within the same individual, eliminating variability introduced by comparisons to controls with different genetic backgrounds. We apply this approach to mosaic female mouse models and humans with Rett syndrome, an X-linked neurodevelopmental disorder caused by mutations in the methyl-DNA-binding protein MECP2 and observe that cell-type-specific DNA methylation predicts the degree of gene up-regulation in MECP2-mutant neurons. This approach can be broadly applied to study gene expression in mosaic X-linked disorders.
INTRODUCTION
The diversity of cell types in the brain has largely precluded the characterization of cell-type-specific features of neurodevelopmental diseases. For X-linked neurodevelopmental disorders, this cellular heterogeneity poses an additional challenge in females where random X-chromosome inactivation (XCI) results in a mixture of wild-type and mutant cells within the brain of the same individual1.
These challenges are exemplified by Rett syndrome, an X-linked neurodevelopmental disorder predominantly affecting girls and characterized by speech delay, repetitive hand movements, seizures, and autism-like behavior2. Rett syndrome is caused by mutations in the MECP2 gene on the X chromosome, and disease severity is thought to be correlated with the fraction of brain cells expressing the mutant allele after X-inactivation1,3. In individuals with Rett syndrome, neural circuits will thus consist of wild-type and mutant cells, raising the possibility that both cell-autonomous and non-cell-autonomous effects contribute to the pathophysiology of Rett syndrome at the cellular and circuit levels. Better understanding of these effects of the MECP2 mutation will be critical for developing targeted therapeutics, but it has been difficult to distinguish gene expression in MECP2-mutant neurons from that of normal neurons within the same brain.
MECP2 encodes a nuclear protein that is enriched in neurons, binds to methylated cytosines broadly across the genome and has been suggested to act as a transcriptional repressor by recruiting co-repressor complexes (e.g. NCOR) to sites of methylated DNA2,4–7. Consistent with this finding, we have found in male mice where all cells express a single allele of Mecp2, that when MeCP2 function is disrupted, genes with the highest level of gene body DNA methylation and MeCP2 binding in wild-type neurons exhibit the largest degree of up-regulation in gene expression in Mecp2-mutant neurons8–10. However, numerous reports have proposed additional functions of MeCP2 at specific loci, including the regulation of mRNA splicing, transcriptional activation, and chromatin structure2,11–14. At present, it is not clear whether these effects are due to direct or indirect actions of MeCP2. Notably, since these previous studies of MeCP2 function have mostly focused on male hemizygous animals in which all cells lack functional MeCP2, the extent to which the effects observed in male mice accurately reflect the effects of MeCP2 loss in the mosaic brains of female heterozygous mice or humans with Rett syndrome remains unclear.
The recent development of high-throughput single-cell RNA sequencing (scRNA-seq) technologies has revolutionized gene expression analysis of complex tissues and enabled the characterization of cell-type-specific transcriptional programs in various brain regions in mice and humans15–17. While these advances have permitted the identification and characterization of unique cell types within complex tissues, until now it has not been possible, even with scRNA-seq, to reliably distinguish between cells that express the wild-type or mutant allele in mosaic females with X-linked disorders because the sequencing reads generated from single cells rarely include the disease-causing mutations. Here, we describe an approach, single-cell SNP-seq, that reliably determines whether individual cells derived from mosaic murine and post-mortem human brain express the wild-type or mutant X-chromosome allele, enabling gene expression profiles of wild-type and mutant cells from the same individual to be distinguished from each other. Using this approach, we find that in the brains of female heterozygous mouse models and humans with Rett syndrome, MECP2 selectively and cell-autonomously represses the expression of highly methylated genes in a cell-type-specific manner in wild-type but not MECP2-mutant neurons. The methods and analyses outlined here for Rett syndrome can be broadly applied to the characterization of gene expression patterns in additional mosaic X-linked disorders such as Fragile X syndrome, CDKL5 deficiency disorder, X-linked intellectual disability, and multiple X-linked genetic causes of autism.
RESULTS
Single-cell SNP sequencing in mouse models of Rett syndrome
Droplet-based high-throughput scRNA-seq methods employ poly-A transcript selection in which the majority of sequence information is restricted to the distal 3’ end of genes, a region that often does not include the disease-causing mutations under investigation15,16. Moreover, these methods typically sample a fraction of the total transcripts per cell, which further limits the ability to reliably detect the expressed mutant allele even when the variant of interest lies within the 3’ sequenced region. For the same reason, a failure to detect expression of a given gene in mutant cells is not a reliable way to discriminate between mutant and wild-type cells. However, we reasoned that single nucleotide polymorphisms (SNPs) that differ between the two X chromosomes and are within genes expressed in cis with the mutant allele might provide a reliable way to determine whether a given cell expresses the mutant or wild-type allele, hereafter defined as the cell’s transcriptotype.
To determine the utility of this approach, we first attempted to distinguish between cells expressing wild-type or mutant alleles in female Mecp2+/− mice. These mice were generated by deleting the majority of the Mecp2 gene (exons 3 and 4) and recapitulate key features of Rett syndrome18. The absence of Mecp2 expression is not a reliable indicator of a mutant cell, however, both because expression of the Mecp2 3’ UTR is still detectable at low levels in mutant cells and because scRNA-seq only captures a fraction of genes per cell. Thus, we searched expressed genes for SNPs that were maintained in cis with the mutant Mecp2 allele during the process of backcrossing the 129/OlaHsd strain of mice in which the Mecp2-mutant mice were generated. Despite extensive backcrossing (>38 generations at Jackson Labs) of the Mecp2-mutant mice with the C57BL/6J strain, we identified four 129P2/OlaHsd-specific SNPs in cis with the Mecp2-mutant allele that were present in the expressed 3’ UTR regions of two genes that are closely linked to Mecp2 and well sampled in the scRNA-seq datasets (Supplementary Fig. 1).
We performed scRNA-seq on visual cortex from five adult (12-to-20-week-old) female Mecp2+/− mice and obtained 12,451 cells that passed initial quality-control tests. Consistent with data from wild-type cortex19, cells from Mecp2+/− cortex were clustered into eight major cell types using the Seurat single-cell analysis pipeline20 (Supplementary Fig. 2A). We focused on excitatory neurons because they have previously been directly implicated in Rett syndrome pathophysiology21,22 and are the most abundant cell type in our dataset (Fig. 1A). Sequencing reads encompassing the identified strain-specific SNPs allowed 1,289 out of 5,761 excitatory neurons to be identified as expressing either the wild-type or mutant Mecp2 allele (Fig. 1B, Supplementary Fig. 2B). In support of the SNP-based transcriptotype classification, the resulting Mecp2-mutant population of cells exhibited significantly reduced levels of the Mecp2 transcript relative to wild-type cells, or groups of excitatory neurons with randomly assigned transcriptotypes (Fig. 1C). Gene expression analysis of the transcriptotyped mutant versus wild-type cells identified 734 differentially expressed genes (366 that were up-regulated, 368 that were down-regulated, false-discovery rate (FDR) < 0.1, Supplementary Table 1). By contrast, only four significantly misregulated genes were identified when cell populations with randomly assigned transcriptotypes were compared (Fig. 1D). These data indicate that we can successfully study gene expression in wild-type and mutant cells by single-cell SNP-seq, making it possible to address whether MeCP2 function in mosaic females is accurately modeled in male hemizygous mice in which all cells express the mutant form of the protein.
Figure 1.
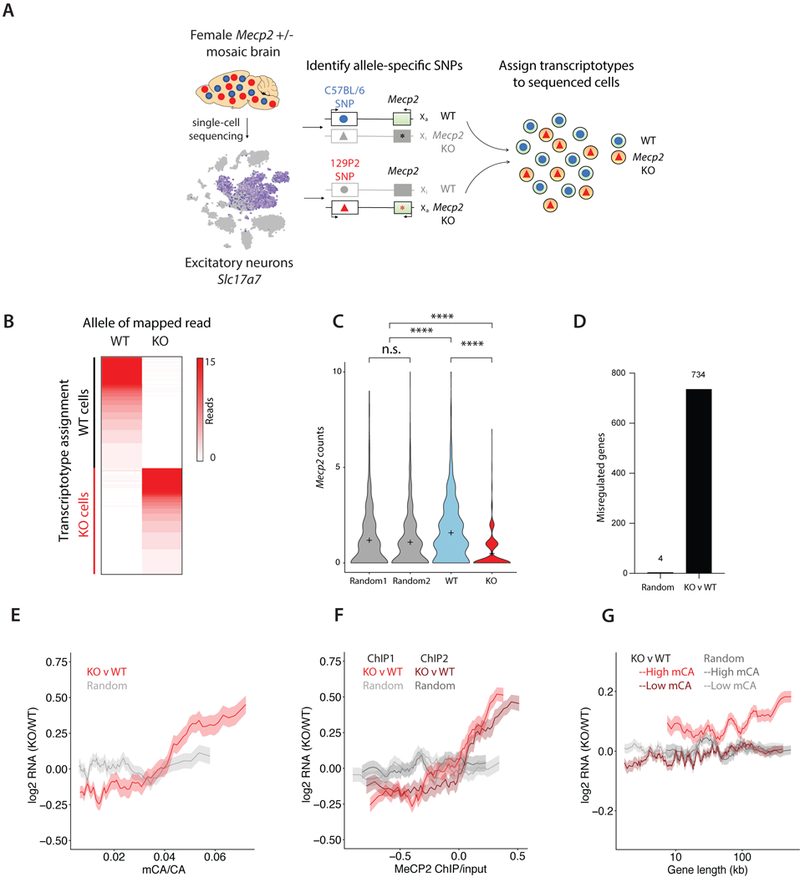
Single-cell SNP sequencing in a female mouse model of Rett syndrome. A) Flow chart of single-cell SNP sequencing pipeline. Single-cell RNA sequencing was performed on visual cortex from five female Mecp2+/− mice followed by graph clustering to identify the group of excitatory neurons (Slc17a7 +). Allele-specific SNPs in genes expressed in cis with the Mecp2 mutation were identified by variant calling and then used to assign the corresponding transcriptotype to the individually sequenced cells. B) Heatmap of reads per analyzed cell (rows of the heatmap) that map to wild-type (WT)- or knockout (KO)-specific SNPs (columns of the heatmap). C) Violin plots of Mecp2 mRNA counts in cells that were grouped based on their SNP-identified transcriptotype (WT, Mecp2+/− wild-type excitatory neurons, KO, Mecp2+/− mutant excitatory neuron, tails represent min and max of data) or by randomly assigned transcriptotypes (Random 1, Random 2). Mecp2 expression was significantly higher in the WT cells (sampled n = 593) compared to KO cells (n = 593) (Kruskal-Wallis test, H = 210, ****P < 0.0001, + indicates mean) and the populations with randomly assigned transcriptotypes (Random 1, n = 593, Random 2, n = 593; ****P < 0.0001). The groups with randomly assigned transcriptotypes had similar levels of Mecp2 expression (P > 0.9999). For the transcriptotyped excitatory neurons, we obtained an average of 7,634 transcripts per cell representing 3,879 distinct genes. D) The number of significantly misregulated genes (FDR < 0.1, monocle2) when comparing gene expression differences between groups of mutant and wild-type excitatory neurons (KO v WT, 734 genes) or two groups of randomly assigned transcriptotypes (Random, 4 genes). E) The mean fold-changes of the misregulated genes described in D (KO v WT, Random) are displayed as a function of excitatory neuron gene body DNA methylation (mCA/CA) (KO v WT, Pearson’s r = 0.38, Random, Pearson’s r = 0.04). The correlation between MeCP2-dependent gene expression and mCA/CA was significantly greater in KO v WT than Random (permutation test, P < 0.001). F) The fold-change of genes in D (KO v WT, Random) binned by gene body MeCP2 ChIP enrichment over input. The correlations between MeCP2-dependent gene expression and two MeCP2 ChIP replicates from purified cortical excitatory neurons (ChIP1, Pearson’s r = 0.41, ChIP2, Pearson’s r = 0.31) are significantly greater than the correlations observed in the Random controls (Random ChIP 1, Pearson’s r = 0.06, Random ChIP 2, Pearson’s r = 0.04) (permutation test, P < 0.001). G) Mean fold-change in gene expression of mutant excitatory neurons (KO) compared to wild-type excitatory neurons (WT) from Mecp2+/− mice, with genes separated into groups of highly methylated genes (normalized expression > 0.1, high mCA, top 25%) or lowly methylated genes (normalized expression > 0.1, low mCA, bottom 66%) and binned by their gene length. MeCP2-dependent gene expression and gene length were significantly more correlated in KO v WT than Random for high mCA genes (KO v WT, Pearson’s r = 0.10, Random, Pearson’s r =0.00, permutation test P < 0.001). The correlations between MeCP2-dependent gene expression and gene length were not statistically different between KO v WT and Random for low mCA genes (KO v WT, Pearson’s r = 0.04, Random, Pearson’s r = 0.02, permutation test P = 0.23). In E-G, the lines represent mean fold-change in expression for genes binned according to gene length (250 gene bins, 25 gene step), methylation (100 gene bins, 10 gene step), or MeCP2 enrichment (100 gene bins, 10 gene step); the ribbon displays s.e.m. of each bin.
Previous reports in male mice indicate that gene bodies of MeCP2-repressed genes are highly methylated, have increased levels of MeCP2 binding, and tend to be long compared to genes that are not repressed by MeCP28,10,23–25. These previous observations of MeCP2 dysfunction provided a molecular signature for assessing the ability of single-cell RNA sequencing data to detect relevant gene expression changes in mosaic tissue. Consistent with previous observations, we found that in mosaic female mice the degree of gene up-regulation in Mecp2-mutant compared to wild-type excitatory neurons directly correlates with gene body DNA methylation (Pearson’s r = 0.38) as well as the length of highly-methylated genes (Pearson’s r = 0.10) (Fig. 1E,G). In the brains of mosaic female Mecp2+/− mice, we also observed that the degree of gene up-regulation in mutant-expressing excitatory neurons directly correlates with increasing levels of gene body MeCP2 binding in excitatory neurons (ChIP1, Pearson’s r = 0.41; ChIP2, Pearson’s r = 0.31) (Fig. 1F). MeCP2 binding was characterized by chromatin immunoprecipitation of MeCP2 in CaMKIIα-positive excitatory neurons isolated from wild-type male mice using INTACT, a method in which genetically tagged nuclei can be immune-purified26. These findings suggest that the up-regulation of highly methylated genes is a cell-autonomous signature of MeCP2 dysfunction, consistent with the observation that Rett syndrome severity correlates with the number of MeCP2-mutant cells1. Notably, the differentially expressed genes between mutant and wild-type excitatory neurons within mosaic female heterozygous mice significantly overlap with the misregulated genes we identified when comparing excitatory neurons from male Mecp2-mutant mice and their wild-type controls (hypergeometric test, P = 7.2 × 10−14, Supplementary Fig. 3, Supplementary Tables 2-5). Thus, by resolving mosaicism with single-cell SNP-seq in a female mouse model of Rett syndrome and comparing the patterns of cell-type-specific gene misregulation to those of male mouse models (Supplementary Fig. 4), we have identified a reproducible set of cell-autonomous MeCP2-dependent genes in excitatory neurons.
While our data indicate that the relationships between MeCP2-dependent gene expression and gene body DNA methylation, MeCP2 occupancy, and gene length are cell-autonomous, it has been difficult to characterize if there are also non-cell-autonomous effects of Mecp2-mutant cells on wild-type cells within the same tissue. Previous attempts to identify such effects have relied on tagged forms of MeCP2 that were not expressed at normal levels27. We overcame these challenges by using scRNA-seq to compare wild-type excitatory neurons (671 cells) from five female Mecp2+/− mice with wild-type excitatory neurons from four female Mecp2+/+ control mice (671 sampled cells). We observed 233 differentially expressed genes (FDR < 0.1) between these conditions, many of which involve key neuronal processes such as neuronal activity-dependent gene expression and neurotrophin signaling (Supplementary Table 6). Importantly, these differentially expressed genes between wild-type cells from Mecp2+/− and Mecp2+/+ mice do not appear to be directly repressed by MeCP2 (e.g. their degree of gene misregulation does not correlate with the level of gene body DNA methylation (permutation test, P = 0.55) or gene length (permutation test, P = 0.73) (Supplementary Fig. 5). These data suggest that gene expression abnormalities are present in wild-type cells from Mecp2+/− mice and are likely due to indirect effects of neighboring Mecp2-mutant cells. This non-cell-autonomous misregulation of gene expression in wild-type neurons of mosaic individuals with Rett syndrome could in principle contribute to disease pathophysiology.
Single-nucleus SNP sequencing of human Rett brain tissue
Given the successful implementation of single-cell SNP-seq in rodent models of Rett syndrome, we reasoned that this method could also be used to characterize MECP2-dependent gene expression changes in post-mortem human Rett brain tissue. This approach is potentially powerful because mutant and wild-type cells of the same age and genetic background can be compared directly in a single experiment, largely eliminating the transcriptional consequences of genetic variation that are introduced when comparing donor samples to unrelated age-matched controls (an especially important advantage in the study of Rett syndrome where the differences in gene expression are expected to be small in magnitude2).
We performed single-nucleus RNA sequencing on occipital cortex from three post-mortem females with Rett syndrome, each harboring the second most common nonsense mutation (c.763C>T) in a single MECP2 allele that generates the R255X truncated gene product lacking the MECP2 transcriptional repressor domain (Supplementary Fig. 6). We isolated nuclei for these experiments because nuclei are more reliably extracted than entire cells from post-mortem tissue samples and can provide sufficient gene expression information for cell type classification and analysis28. We successfully sequenced a total of 43,558 nuclei, with 30,293 nuclei passing the minimum required threshold of 500 uniquely expressed genes. In line with previous single-cell/single-nucleus RNA-seq experiments15,16,19, the nuclei analyzed had an average of 2,800 transcripts per nucleus from 1,671 unique genes. Using Seurat20 and known excitatory neuron and interneuron marker genes19, the nuclei cluster into a large excitatory population (18,545 nuclei) and multiple distinct interneuron populations (5,952 nuclei total) (Fig. 2A). The heterogeneity of cells in the interneuron cluster prompted us to further subdivide this population into their known functional classes by the expression of specific marker genes (e.g. VIP, PVALB, SST, or CCK) (Fig. 2A, Supplementary Fig. 7).
Figure 2.
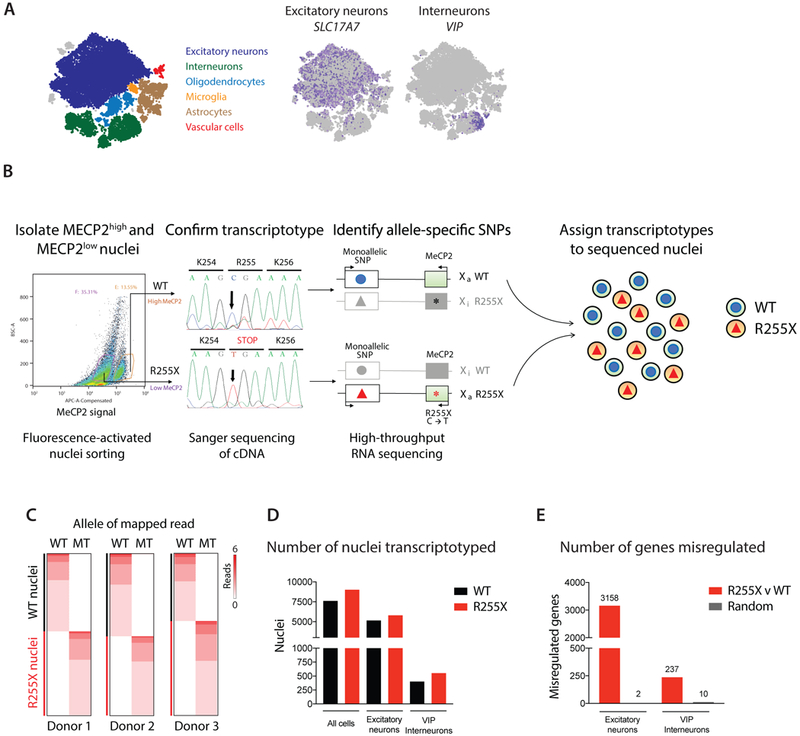
Single-nucleus SNP sequencing of human Rett brain tissue. A) Single-nucleus RNA sequencing of occipital cortex from three females with Rett syndrome. Graph clustering nuclei from the three individuals together according to their respective brain cell types. B) Flowchart for the identification and assignment of allele-specific SNPs for each Rett donor. Single nuclei suspensions from each Rett donor were sorted based on their level of immunoreactivity to a C-terminal MeCP2 antibody (MECP2high and MECP2low). The weak staining observed in MECP2low nuclei represents background immunofluorescence. cDNA from the MECP2high and MECP2low nuclei was Sanger sequenced to confirm that the sorted populations expressed the expected MECP2 allele. Deep high-throughput RNA sequencing of these populations followed by variant calling identified the allele-specific SNPs that were used to assign transcriptotypes to each nucleus from the single-nucleus RNA sequencing dataset shown in A. C) Heatmap of reads per cell (rows of the heatmap) that map to WT- or MECP2 mutant (MT)-specific SNPs (columns of the heatmap) for each of the three donors. D) The number of total nuclei, excitatory neuronal nuclei, and VIP interneuronal nuclei that could be transcriptotyped from the single-nucleus RNA sequencing dataset of Rett donors. E) The number of significantly misregulated genes (FDR < 0.01, monocle2, R255X v WT, 3158 genes in excitatory neurons, 237 genes in VIP interneurons) identified when comparing gene expression differences between groups of mutant and wild-type neurons, or two groups of neurons with randomly assigned transcriptotypes (Random, 2 genes in excitatory neurons, 10 genes in VIP interneurons). The difference in number of misregulated genes between excitatory and inhibitory neurons is largely explained by the number of cells analyzed (Supplementary Fig. 8B). The number of excitatory neuronal nuclei and VIP interneuronal nuclei used for differential expression analysis is shown in D.
Once each nucleus was assigned to its respective cell type cluster, we next turned to identifying its transcriptotype. Because there were no sequencing reads that included the R255 position of MECP2, we reasoned that the large number of SNPs that differ between an individual’s two X chromosomes might allow us to identify allele-specific SNPs that are in cis with the mutant MECP2 locus and therefore expressed only in MECP2-mutant neurons. To identify the transcriptotype-specific SNPs in each Rett donor, we took advantage of an MECP2-specific antibody that was raised against a region of the C-terminus that is truncated by the R255X mutation. We used this antibody to separate high-staining (MECP2high) and low-staining (MECP2low) nuclei by fluorescence-activated sorting (Fig. 2B). Sanger sequencing of isolated cDNA from the two populations confirmed that the MECP2high population expressed wild-type MECP2 and that the MECP2low population expressed the R255X mutant MECP2.
Having isolated the two populations from each donor, we next performed total RNA sequencing on both populations and identified between 69–75 allele-specific SNPs that were uniquely expressed in MECP2high nuclei (Supplementary Fig. 8, see methods). Expression of these allele- and transcriptotype-specific SNPs was then queried in the corresponding single-nucleus RNA-seq dataset from the same donor sample and used to assign the corresponding wild-type or R255X MECP2 transcriptotypes (Fig. 2C). Using the allele-specific SNPs identified from each Rett donor, we could assign transcriptotypes to 16,627 nuclei, or 55% of the nuclei assayed (Fig. 2D); the remaining 45% of nuclei were excluded from further analysis. The ratio of wild-type to mutant nuclei was approximately even across the three donor samples (donor 1 = 49% WT, 51% R255X; donor 2 = 51% WT, 49% R255X; donor 3 = 42% WT, 58% R255X), which suggests that there was not significant skewing of XCI and that Rett syndrome in the three donors is likely due to the loss of MECP2 function in approximately 50% of brain cells.
For subsequent analyses, we focused on the excitatory neuron population (SLC17A7-expressing, 18,545 cells) and on the most abundant subtype of interneurons in our datasets (VIP-expressing, 1,839 cells) (Fig. 2A). Importantly, the neuronal subtype clusters were similar between wild-type and mutant cells (Supplementary Fig. 9A), enabling the direct comparison of gene expression between wild-type and mutant cells of the same neuronal subtype. To maximize the number of nuclei and statistical power for cell-type-specific gene expression comparisons, we combined nuclei of the same neuronal subtype and transcriptotype from the three Rett donors. We identified significant gene expression differences between mutant and wild-type excitatory neurons (3,158 genes, Supplementary Table 7) and VIP interneurons (237 genes, Supplementary Table 8) (Fig. 2E). Importantly, these findings were dependent on proper transcriptotype assignment, as gene expression analysis between populations of cells that were randomly assigned transcriptotypes consistently recovered ≤ 10 differentially expressed genes (Fig. 2E). It should be noted that the difference in numbers of significantly misregulated genes between excitatory neurons and VIP interneurons is largely attributable to the greater number of excitatory nuclei sampled with higher transcript coverage because the number of misregulated genes are similar in excitatory and VIP interneurons if equal numbers of nuclei and transcripts are sampled for both cell types (Supplementary Fig. 9B).
Cell-type-specific DNA methylation patterns predict gene misregulation in Rett syndrome
These new human datasets provided the opportunity to determine whether features described in mouse models regarding MECP2-dependent gene expression are also observed in neurons from human individuals with Rett syndrome. It is not known, for example, if in fact MECP2 in human neurons represses highly-methylated long genes in a neuronal subtype-specific manner, as has been observed in mice8,10,25. In mice, DNA methylation in both the CG and CA dinucleotide contexts recruits MeCP2 binding and contributes to MeCP2-dependent gene repression8,29. While both CG and non-CG methylation (mCH, comprised of mCA, mCT, and mCC) display cell-type-specific patterns, mCH is significantly more divergent across neuronal cell types26,30 and, in mice, contributes to cell-type-specific MeCP2-dependent gene repression31. To determine whether cell-type-specific patterns of mCH predict the degree of MECP2-dependent gene repression in human females with Rett syndrome, we compared the set of genes that are differentially expressed in human female MECP2-mutant-expressing and wild-type-expressing nuclei with recently published human single-cell methylation data from cerebral cortex32. We found that in humans, the degree of gene misregulation in MECP2-mutant compared to wild-type excitatory neurons and VIP interneurons is directly correlated with the level of gene body mCH in neurons of the respective subtype (excitatory neurons, Pearson’s r = 0.22, VIP interneurons, Pearson’s r = 0.18, Fig. 3A, E). These correlations are dependent on the correct assignment of transcriptotype, as gene expression differences between groups of randomly assigned transcriptotypes do not correlate with gene body mCH for either excitatory neurons (Pearson’s r = −0.01) or VIP interneurons (Pearson’s r = −0.05). The relationship between neuronal subtype-specific mCH and MECP2-dependent gene expression is highly reproducible and can be observed in each of the three donor samples by directly comparing MECP2-mutant and wild-type neurons from the same individual (Supplementary Fig. 10). The direct correlation between MECP2-dependent gene repression and gene body mCH for each neuronal subtype depends on its subtype-specific DNA methylation patterns, as MECP2-dependent gene repression in excitatory neurons does not correlate with the extent of gene body mCH from VIP interneurons (Pearson’s r = −0.01, Fig. 3B) and MECP2-dependent gene repression in VIP interneurons poorly correlates with mCH from excitatory neurons (Pearson’s r = 0.05, Fig. 3D). Of note, the direct correlation between MECP2-dependent gene repression and DNA methylation was also observed in the CG dinucleotide context in both excitatory neurons and VIP interneurons (Supplementary Fig. 11).
Figure 3.
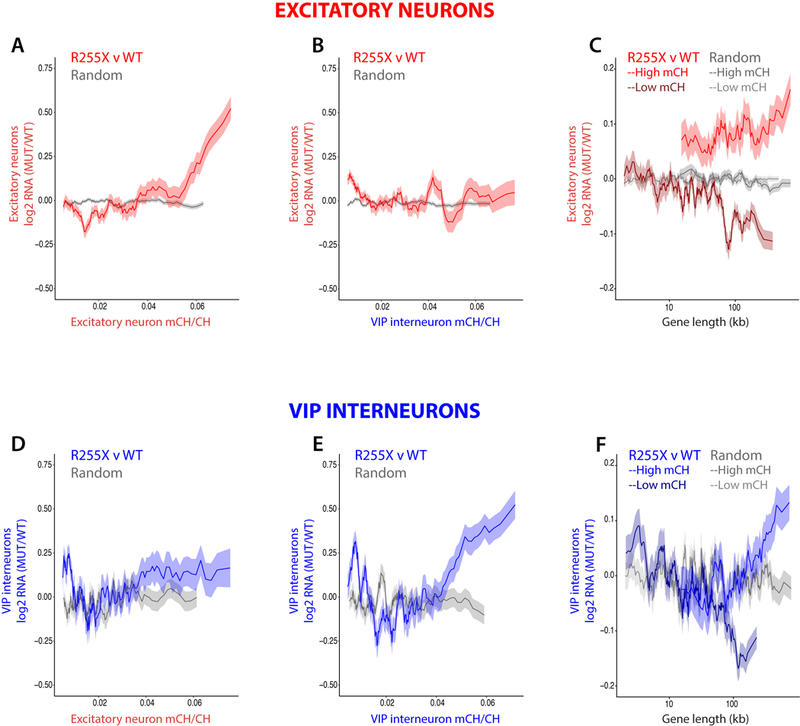
Cell-type-specific DNA methylation patterns predict gene misregulation in Rett syndrome. For each graph in A-B and D-E, mean fold-change in gene expression of R255X MECP2 nuclei compared to WT nuclei (R255X v WT) or of two groups of the respective cell type that were randomly assigned transcriptotypes (Random) is binned according to the fraction of gene body DNA methylation (mCH/CH). Gene expression changes (FDR < 0.01, monocle2) from R255X v WT or Random excitatory neurons are compared to patterns of DNA methylation from (A) excitatory neurons (Pearson’s r = 0.22) or from (B) VIP interneurons (Pearson’s r = −0.01) (250 gene bins, 25 gene step). R255X v WT is significantly more correlated with excitatory neuron mCH/CH than Random (A) (permutation test, P < 0.001) and significantly more correlated with excitatory mCH/CH patterns than mCH/CH patterns from VIP interneurons (B, R255X v WT (A) correlation compared to R255X v WT (B), permutation test, P < 0.001). Gene expression changes (FDR < 0.25) from R255X v WT or Random VIP interneurons are compared to DNA methylation patterns from (D) excitatory neurons (Pearson’s r = 0.05, R255X v WT; Pearson’s r = 0.03 Random) or from (E) VIP interneurons (Pearson’s r = 0.18, R255X v WT; Pearson’s r = −0.05 Random) (50 gene bins, 5 gene step). R255X v WT is significantly more correlated with mCH/CH than Random in (E) (permutation test, P < 0.001) but not in (D) (permutation test, P = 0.71). In VIP interneurons, the correlation of R255X v WT with mCH/CH is significantly greater for mCH/CH patterns from VIP interneurons (E) than mCH/CH patterns from excitatory neurons (D) (permutation test, P = 0.008). C,F) Mean fold-change in gene expression of R255X v WT excitatory neuronal nuclei (C) or VIP interneuronal nuclei (F) for expressed genes (> 0.1 normalized counts) with high mCH (top 25% mCH/CH) or low mCH (bottom 66% mCH/CH) binned according to gene length (250 gene bins, 25 gene step). MECP2-dependent gene expression and gene length were significantly more correlated in R255X v WT than Random for high mCH/CH genes (R255X v WT: C, Pearson’s r = 0.07; F, Pearson’s r = 0.08; Random: C, Pearson’s r = 0.02; F, Pearson’s r = 0.00, C, permutation test, P = 0.007, F, permutation test, P < 0.001) and significantly more anti-correlated for low mCH/CH genes (R255X v WT: C, Pearson’s r = −0.07; F, Pearson’s r = −0.09; Random: C, Pearson’s r = 0.00; F, Pearson’s r = 0.00, C, F, permutation test, P < 0.001). The lines represent mean fold-change in expression for genes binned as described; the ribbon is s.e.m. of each bin.
As described above, for highly methylated genes, gene length predicts the degree of gene up-regulation in Mecp2-mutant mice compared with their wild-type counterparts10. Consistent with this observation, we find that in humans with Rett syndrome the level of gene body methylation together with gene length predicts the degree of gene up-regulation in both MECP2-mutant excitatory and VIP interneuronal nuclei (Fig. 3C,F). We further find that in human females, as in mice, gene length does not positively correlate with MECP2-dependent gene repression for lowly methylated genes, underscoring the importance of accounting for DNA methylation in the analysis of MECP2-dependent gene regulation in humans8,10. These findings in human females with Rett syndrome are consistent with our findings in male and female Mecp2-mutant mouse models and indicate that MeCP2 acts through an evolutionarily conserved, cell-autonomous mechanism to preferentially repress the expression of highly methylated long genes.
The large number of excitatory neuronal nuclei sequenced from each individual provided sufficient power to study gene expression differences between mutant and wild-type nuclei of this neuronal subtype within the same individual’s brain (Supplementary Tables 9-14), thus eliminating much of the genetic and environmental heterogeneity that is inherent to previous studies of MECP2-dependent gene expression33,34. We were thus able to identify genes that are consistently misregulated in mutant excitatory neurons across all three Rett syndrome donors. This analysis demonstrated a highly significant overlap in affected genes across the three Rett donor samples, identifying 537 genes that are consistently up-regulated in mutant-MECP2 excitatory neurons compared to wild-type neurons and 395 genes that are reproducibly down-regulated (Fig. 4A, Supplementary Fig. 12, Supplementary Table 15). As might be predicted, the up-regulated genes had significantly higher levels of gene body methylation than the down-regulated genes (Fig. 4B). Genes that control metabolism or regulate neuronal processes such as ion transport or nervous system development were significantly enriched in the set of up-regulated or down-regulated genes (Fig. 4C,D), and misregulation of these genes may contribute to the metabolic and neuronal deficits observed in Rett syndrome35. The ability of single-nucleus SNP-seq to reliably transcriptotype and reproducibly identify gene expression changes between mutant and wild-type cells within the same individual largely overcomes the previous reliance on age-matched controls for molecular characterization of mosaic X-linked disorders, and will significantly improve our ability to distinguish gene expression differences that are due directly to the mutation under investigation rather than to unrelated genomic variation between cases and controls.
Figure 4.
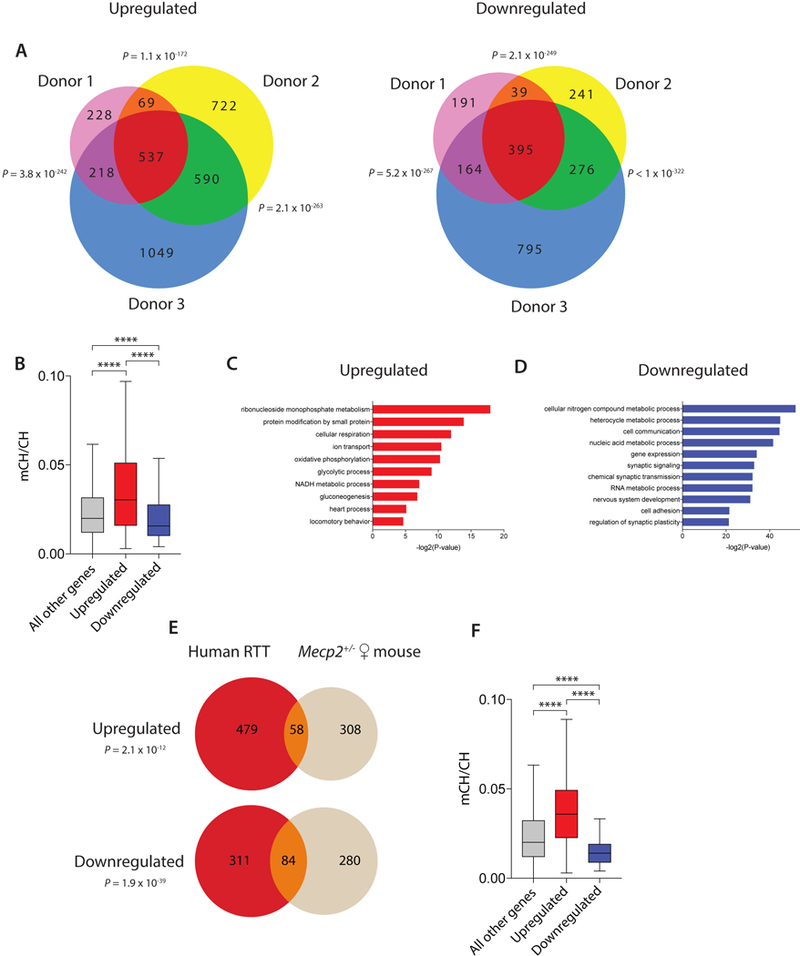
Characterization of MECP2-regulated genes in human and mouse A) Venn diagram of the number of overlapping significantly up-regulated (left) or down-regulated (right) genes (FDR < 50.1, monocle2) between R255X MECP2 mutant and wild-type nuclei in excitatory neurons of each donor. P-values describing the significance of overlap between pairs of up- or down-regulated gene lists were calculated by hypergeometric testing. B) Boxplot of the gene body DNA methylation level (mCH/CH) of the 537 overlapping up-regulated genes and 395 overlapping down-regulated genes in the 3 donors, as well as all other expressed genes (****P < 0.0001 (Dunn’s), Kruskal-Wallis test H(2) = 146.6) C-D) Lists of the most highly significant gene ontology terms (Fisher’s Exact test with FDR) enriched in the 537 overlapping genes that are up-regulated (C) or the 395 overlapping genes that are down-regulated (D) between R255X MECP2 mutant and WT excitatory neurons. E) Venn diagram of the genes that are commonly up-regulated (top, P = 2.1 × 10−12, hypergeometric test) or down-regulated (bottom, P = 1.9 × 10−39, hypergeometric test) in mutant MECP2 compared to wild-type excitatory neurons in human and female heterozygous mice. F) Boxplot of the fraction of gene body DNA methylation (mCH/CH) of the 58 overlapping up-regulated genes and 84 overlapping down-regulated genes between human and mouse (****P < 0.0001 (Dunn’s), Kruskal-Wallis test H(2) = 52.35). Boxplots show the median (line), 25th to 75th percentiles (box), and 1.5X the interquartile range (whiskers).
We next sought to identify genes that are controlled by MECP2 in both humans and mice, reasoning that despite the significant species differences, the evolutionarily conserved MECP2 targets might provide an opportunity to investigate MECP2 function in mouse models that might be relevant to human pathophysiology. To this end, we identified the genes that are up-regulated or down-regulated in excitatory neurons across all three Rett syndrome donor samples (537 and 395 genes, respectively) and asked which of these are also significantly misregulated in female Mecp2+/− excitatory neurons from mice. We identified 58 evolutionarily conserved genes that are up-regulated and 84 genes that are down-regulated in MECP2-mutant compared to wild-type excitatory neurons in both mouse and human (Fig. 4E, Supplementary Fig. 13, Supplementary Table 16-17). These evolutionarily conserved MECP2-regulated genes represent high-confidence MECP2 targets in excitatory neurons because of their reproducibility across multiple datasets. However, we stress that deeper sequencing would provide greater statistical power and the ability to identify many additional evolutionarily conserved MECP2 targets. We note that the high-confidence evolutionarily conserved genes identified here that are up-regulated in MECP2-mutant excitatory neurons have significantly higher levels of gene body DNA methylation than the set of genes that are down-regulated in MECP2-mutant neurons (Fig. 4F), suggesting that the up-regulated gene set may be enriched for direct MECP2 targets. However, it seems likely that the misregulation of both MECP2-repressed and MECP2-activated genes contribute to Rett syndrome pathophysiology as 25% of the MECP2-repressed genes (enrichment P = 1.0 × 10−6, hypergeometric test) and 13% of the MECP2-activated genes (enrichment P = 0.02, hypergeometric test) have been previously shown to be mutated in intellectual disability or autism (see methods). Many of the MECP2-repressed genes (e.g. AUTS2, RBFOX1) are transcriptional regulators and are known to control neuronal gene expression36–38. The MECP2-repressed genes that encode neuronal ion channels such as GABRA1 and SCN1B are known to cause epilepsy when mutated39,40 and thus could contribute to this comorbidity in individuals with Rett syndrome. The evolutionarily conserved genes that are down-regulated in MECP2-mutant neurons include the neurotrophin BDNF and the presynaptic adhesion molecule NRXN2, both of which have also been shown to contribute to neurological disorders when mutated41,42. Given that the selective disruption of Mecp2 in excitatory neurons is sufficient to cause Rett-like phenotypes in mice22, further investigation of evolutionarily-conserved MECP2-regulated genes in this cell type could both yield new mechanistic insight into MECP2 function and help characterize the role of these genes in specific aspects of Rett syndrome pathophysiology.
DISCUSSION
Here we present a new experimental approach that leverages the power of single-cell or single-nucleus RNA sequencing and individual genetic variation to simultaneously characterize cell-type-specific gene expression and allele-specific X-chromosome activation status in individual cells within mosaic mouse and human brains. This approach has broad applicability for studying gene expression abnormalities in X-linked neurodevelopmental disorders such as Rett syndrome, Fragile X syndrome, CDKL5 disorder, X-linked intellectual disability, and multiple X-linked genetic causes of autism (e.g. NLGN3, NLGN4, SLC6A8, PLXNA3, DDX3X, WDR45, CASK) in females where mosaicism between wild-type and mutant cells has hindered previous analyses. This method can be easily adapted (see Methods) to female mouse models of X-linked disorders that were generated in mixed genetic backgrounds by using strain-specific SNPs to identify the cells expressing the mutant allele. Moreover, this approach is particularly useful for studying mosaic disorders in human samples because the wealth of natural genetic variation across individuals provides many opportunities to identify allele-specific SNPs that are expressed from the same X-chromosome as the mutant allele under investigation43,44. Indeed, SNPs have been recently used in conjunction with scRNA-seq data to determine the sample identity of individual cells within a pool of human samples44 and to study genes that escape X-chromosome inactivation45.
In addition to validating the single-cell SNP-seq approach, our study provides further insight into important aspects of Rett syndrome pathophysiology and the consequences of MECP2 dysfunction. The inherent X-linked mosaicism in females with Rett syndrome has hampered prior efforts to determine if genes that are differentially expressed in Rett and age-matched controls are due to the MECP2 mutation itself or a consequence of genetic and environmental variation between individuals. Our study overcame these limitations and directly assessed the MECP2-dependent gene expression changes in the same cell type and genetic background. We found that cell-type-specific patterns of DNA methylation largely predict the degree of gene up-regulation within each subtype of mutant MECP2 (R255X)-expressing neuron from humans with Rett syndrome. Importantly, our approach confirmed that the preferential up-regulation of highly methylated long genes is a cell-autonomous molecular signature of MECP2 dysfunction that is conserved between mutant MeCP2 mouse models and humans with Rett syndrome.
The relative contribution of gene length and DNA methylation to MECP2-dependent gene regulation is complex because long genes tend to have a higher level of gene body methylation compared to shorter genes10 (Supplementary Fig. 14). Partial correlation analysis was previously used to parse the relative contribution of gene length and DNA methylation to MeCP2-dependent gene regulation in mouse cortical tissue and found that the total number of gene body methyl-cytosine binding sites within a given gene, rather than gene length alone, best predicts MECP2-dependent gene repression8. While our scRNA-seq data suggest a role for MECP2 in regulating cell-type-specific gene expression in a DNA methylation-dependent manner, MeCP2-dependent gene expression also correlates with DNA methylation patterns in whole cortical tissue10,24. This finding is likely explained both by an averaging effect due to the most abundant cell type driving the observed DNA methylation and gene expression patterns, as well as the presence of commonly methylated regions that would be expected to result in similar MECP2-dependent gene expression across cell types.
The power of single-cell and single-nucleus RNA sequencing to identify MECP2-regulated genes in a given individual with Rett syndrome and in specific cell types enabled the identification of MECP2-repressed genes and MECP2-activated genes that are evolutionarily conserved in both mouse and human excitatory neurons. While deeper single-cell sequencing will provide the statistical power necessary to identify many additional conserved MECP2-regulated genes, the set of genes described here has the potential to provide some new insight into Rett syndrome pathophysiology and provides an opportunity to link mechanistic studies of MECP2 function in mouse models to Rett syndrome in humans. Notably, the conserved MECP2-repressed genes have significantly higher levels of gene body DNA methylation than the set of conserved MECP2-activated genes. The high levels of DNA methylation within the transcribed region of these MECP2-repressed genes, taken together with abundant evidence that MECP2 binds preferentially to methyl cytosines10,29, suggest that the conserved MECP2-repressed genes are direct targets of MECP2. However, it remains to be determined whether the conserved genes that are down-regulated in the absence of MECP2 are down-regulated due to a secondary change in neurons that occurs as a consequence of the disrupted expression of highly methylated genes or if these genes are activated directly by MECP2 via a distinct mechanism. It should be noted that a previous report suggested that MeCP2 may regulate long gene expression through a post-transcriptional mechanism, but in this study gene body DNA methylation was not considered27. Reanalysis of the data in this study with respect to DNA methylation supports the conclusion that MeCP2 represses gene expression at the level of transcription (Supplementary Fig. 15). Additional studies into the regulation of nuclear/nascent RNA by MeCP2 will likely reveal valuable new insights into MeCP2’s function.
It remains challenging to reconcile the small magnitude of misregulation that occurs for an individual gene when MECP2 is mutated with the dramatic neurological sequelae of Rett syndrome. It is possible that the deleterious effect of mutating MECP2 may summate across hundreds to thousands of genes to cause Rett syndrome10 or that only a small subset of the misregulated genes are responsible for the neurological phenotypes. It is also possible that the kinetics of gene transcription (e.g. elongation rates) are altered in the absence of MECP2, which could result in abnormal timing of transcriptional programs in addition to subtle changes in steady-state gene expression9. Further study of the proximal mechanisms by which MECP2 regulates gene expression is needed to identify therapeutic approaches for normalizing the diverse gene expression abnormalities that occur across cell types in Rett syndrome.
Taken together, we have shown that single-cell and single-nucleus SNP sequencing enables the cell-type-specific characterization of gene expression in mosaic mouse models and post-mortem tissue of human brain donors. In the present study, we have leveraged this approach to glean new insights into Rett syndrome pathophysiology, and in the future, we envision its broad application to the study of additional X-linked disorders in both the brain and other tissues.
METHODS:
Mice
All animal experiments were approved by the National Institutes of Health and the Harvard Medical School Institutional Animal Care and Use Committee and were conducted in compliance with the relevant ethical regulations. Male and female Mecp2 knockout mice and their wild-type controls were obtained from Jackson Labs (Stock No. 003890). This line was originally generated by Adrian Bird 18. Mice were housed under a standard 12 hr light cycle before being placed in constant darkness for 7 days prior to sacrificing. Mecp2 mutant mice all demonstrated decreased locomotor activity at time of analysis; male mice were 8 weeks old and female mice were 12–20 weeks old. Mice of the respective genotype, age, and sex were randomly selected for inclusion in the study.
Brain tissue samples from donors with Rett syndrome
Post-mortem cortical tissue (visual cortex, BA17) was obtained from the National Institutes of Health NeuroBioBank and Harvard Brain Bank with approval from the coordinating foundation Rettsyndrome.org. The study was conducted in compliance with relevant consent and ethical considerations. Work was approved by Harvard Medical School and is compliant with all ethical regulations. Rett donor samples were genotyped by the NeuroBioBank/Harvard Brain Bank and were confirmed by Sanger sequencing.
Single-cell isolation from male and female mouse cortex
Single-cell suspensions from adult male and female visual cortex were prepared as described in 19. Briefly, mice were euthanized with isofluorane and perfused with an ice-cold choline solution. Visual cortices were dissected, chopped into 300-μm fragments, and dissociated with papain (Worthington). Cells were then triturated into a single-cell suspension and collected by gradient centrifugation.
Single-nuclei isolation from human post-mortem cortex
Single nuclei suspensions from post-mortem human occipital cortex were collected as described previously 26 with minor modifications. Cortical tissue was removed from dry ice and placed directly into a Dounce with homogenization buffer (0.25 M sucrose, 25 mM KCl, 5mM MgCl2, 20 mM Tricine-KOH, pH 7.8, 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, protease inhibitors, 5 μg/mL actinomycin, 0.04% BSA). After 10 strokes with the tight pestle, a 5% IGEPAL (Sigma) solution was added to a final concentration of 0.32% and 5 additional strokes with the tight pestle were performed. The tissue homogenate was then passed through a 40-μm filter, and diluted 1:1 with OptiPrep and layered onto an OptiPrep gradient as described previously 26. After ultracentrifugation, nuclei were collected between the 30% and 40% Optiprep layers, confirmed to be single nuclei, and diluted to 80,000 nuclei/mL for inDrops. All buffers and gradient solutions for nuclei extraction contained RNAsin (Promega) and 0.04% BSA.
Nuclei sorting and RNA sequencing
Cortical tissue from each Rett donor was dounce homogenized in Buffer HB (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH pH 7.8, 1 mM DTT, 0.15 mM spermine, 0.5mM spermidine, protease inhibitors). A 5% IGEPAL solution was added to a final concentration of 0.16% followed by five additional dounce strokes, then the lysate was filtered through a 40-μm strainer. Nuclei were pelleted by centrifuging at 500 g for 5 min at 4°C and washed once with PBS with 1% BSA. To stain nuclei for sorting, nuclei were incubated with a C-terminal MeCP2 antibody46 at 1:500 for 1 hour at 4°C, washed once with Wash buffer (PBS with 1% BSA and 0.16% IGEPAL), incubated with a goat anti-rabbit 647 secondary antibody (Life Technologies, cat# A21244) at 1:500 for 30 min at 4°C, then washed once with Wash buffer. All washes were performed by centrifuging at 500g for 5 min at 4°C. Nuclei were then resuspended in PBS with 1% BSA and sorted on a Sony SH800Z Cell Sorter (100 μm nozzle, default laser settings). Nuclei were sorted into TRIzol LS (Invitrogen), and total RNA was chloroform extracted and purified with the Qiagen RNeasy Micro Kit with on-column DNase treatment. For Sanger sequencing of the MECP2 R255X mutation, cDNA was generated with the SuperScript III First-strand Synthesis System (Invitrogen). The MECP2 R255X region was amplified with Q5 Hot Start High-Fidelity Master Mix (NEB) with the following primers: MECP2 R255X F: AAGATGCCTTTTCAAACTTCG and MECP2 R255X R: CCCAGGGCTCTTACAGGTCT, and Sanger sequencing was performed with the MECP2 R255X R primer at the DF/HCC DNA Sequencing Facility. To identify monoallelic SNPs in the two populations of nuclei, total RNA-seq libraries were generated with the NEBNext Ultra Directional Library Prep Kit with rRNA depletion. Libraries were sequenced on an Illumina Nextseq 500 with 85 bp single-end reads. Reads were mapped to the hg38 genome with Tophat2.
Single-cell/single-nucleus RNA sequencing (inDrops)
Single-cell or single-nuclei suspensions were encapsulated into droplets, lysed, and the RNA within each droplet was reverse-transcribed using unique nucleotide barcode as described previously15. Cell or nuclei encapsulation was performed in a blinded fashion. Approximately 3000 cells were processed per library and sequenced on an Illumina Nextseq 500 to achieve at least 5 reads on average per unique molecular index (typically about 500 million reads per 30,000 droplets collected by inDrops). Transcripts were processed and mapped using a previously described pipeline15. Briefly, a custom transcriptome was built from Ensembl GRCh38 (GRCm38.85 annotation) and GRCm38 (GRCm38.84 annotation) with the referenced pipeline.
Quality control for cell or nuclei inclusion
Cells or nuclei with greater than 500 unique genes detected per cell were included for further consideration. Cells or nuclei with greater than 15,000 unique molecular identifiers detected were omitted to minimize inclusion of data that represented the common barcoding of two or more cells.
Cell type identification by dimensionality reduction
We used the R-package Seurat20 to cluster cells based on similar gene expression profiles. The raw counts obtained from the mapping pipeline described above were log normalized and scaled to 10,000 transcripts per cell. Variable genes were identified by the MeanVariablePlot() function with the following parameters: x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5. Principle component analysis was then performed, and the top 30 principle components were used for the FindClusters() function (kNN clustering) and RunTSNE function (for t-distributed stochastic neighbor embedding). Clusters with fewer than 100 cells were omitted from further analysis. Classification of cell types were determined by visualizing known marker gene expression within each identified cluster. Excitatory neurons were marked by the expression of vesicular glutamate transporter 1 (Slc17a7) and Calcium/Calmodulin Dependent Protein Kinase II Alpha (Camk2a). Interneurons were marked by glutamate decarboxylase 1 (Gad1), and were further separated into three major subtypes by the expression of parvalbumin (Pvalb), vasoactive intestinal peptide (Vip), or somatostatin (Sst). Astrocytes were marked by the expression of aldolase dehydrogenase (Aldoc), oligodendrocytes by the expression of Olig1, microglia by the expression of Cx3cr1, and endothelial cells by the expression of Cldn5. Cells expressing significant levels of two or more of the above marker genes were considered doublets and discarded from further analysis.
General approach to single-cell/nucleus SNP sequencing
There are four general strategies to identify SNPs that are in genes expressed in cis with the mutant or wild-type form of a gene: 1) Identify cells that have transcripts covering the mutated genomic region of interest. Because of low per cell sequencing coverage, it is rare that an individual cell will have coverage of this precise genomic region to directly determine its transcriptotype. Therefore, the few definitively mutant and wild-type cells can be used to search for genomic variation in the expressed X-chromosome genes between mutant and wild-type cells. This provides a set of allele-specific SNPs that can be used in addition to the gene of interest itself to increase the likelihood that a given cell can be transcriptotyped; 2) Long-read DNA sequencing to directly confirm which SNPs are in cis with the wild-type and mutant gene of interest. This approach would start by identifying SNPs in the single-cell RNA sequencing dataset (e.g. half of the reads mapping to the reference nucleotide and the other half mapping to an alternate nucleotide) and perform long-read DNA sequencing (e.g. Pacific Biosciences, Oxford Nanopore) to directly confirm which neighboring SNPs are in cis. Once the allele containing a SNP is confirmed to be expressed in cis with either the wild-type or mutant allele of interest, this SNP can be used in turn to identify additional allele-specific SNPs as described in approach 1; 3) Identify SNPs that are in cis with the mutant allele by sequencing members of the donor’s family. For example, if the mutation is inherited, DNA sequencing of the X-chromosome of each parent can provide the set of allele-specific SNPs that are unique to the wild-type or mutant alleles. This approach has been employed to catalogue XCI status of human cells 45 and is the approach we used for the analysis of the Mecp2 mutant mouse; and 4) Separate wild-type and mutant cells from an individual sample and perform deep RNA sequencing to identify the set of expressed SNPs that are unique to the wild-type or mutant population of cells. This is the approach we used to transcriptotype cells from the human Rett syndrome brain donors.
An additional consideration when implementing single-cell SNP sequencing to study X-linked disorders is that some X-linked disease-causing genes escape X-chromosome inactivation (e.g. IQSEC2). In these cases, both the mutant and wild-type allele will be expressed in each cell. Therefore, it is important to assess the X-inactivation status of the gene under investigation to ensure it is not biallelically expressed in the cell types of interest. While there is a report that a small percentage of neuroprogenitor cells express Mecp2 biallelically47, this event is exceedingly rare in post-natal mouse brain tissue and has not been observed in humans despite an in-depth genome-wide search for X-inactivation escape genes45.
After transcriptotypes are assigned, it should be noted that while the wild-type and mutant cells have the same genetic background, there are also allele-specific X-chromosome SNPs expressed in cis with the mutant or wild-type gene. Thus, it is important to have multiple donors with the same mutation to confirm the gene expression differences observed are not secondary to differences in X-chromosome SNPs between mutant and wild-type cells. In our data, these X-chromosome SNPs do not contribute substantially to the gene expression changes observed between mutant and wild-type cells because the three individuals have similar patterns of gene misregulation despite having unique sets of X-chromosome SNPs.
Single-cell SNP sequencing in mosaic female brain tissue
To transcriptotype cells from mosaic female Mecp2+/− mutant mice, we first identified SNPs that were consistently inherited with the mutant Mecp2 allele. Because this line has been inbred (backcrossed > 38 generations), sequencing offspring from previous litters was equivalent to sequencing the parents directly. For the same reason, however, we also expected that the only retained SNPs from the 129/OlaHsd strain in which the mutant Mecp2 allele was made would be closely linked to the Mecp2 locus itself. Indeed, variant calling (Freebayes using default settings, discussed further below) on single-cell RNA seq data from either Mecp2 WT or KO male hemizygous mice identified four SNPs within 2 MB of the Mecp2 locus that were confirmed by manually browsing the RNA sequencing tracks (Integrative Genomics Viewer – Broad Institute, see Supplementary Fig 1). All male Mecp2 knockout mice across two separate generations (WT1–3 and WT4–6 were from separate generations) contained the same SNPs, indicating that these SNPs can be used as a reliable marker of the mutant allele. Given the small number of SNPs that were identified, we attempted to maximize their detection by modifying the standard inDrops single-cell library preparation. Specifically, half of the amplified RNA was processed according to the published protocol using random hexamers and universal primers for PCR amplification, and the other half was reverse-transcribed and then PCR-amplified with gene-specific primers for each allele-specific SNP (see primer sequences below).
To identify the set of expressed SNPs that are unique to wild-type or mutant nuclei from post-mortem human Rett syndrome brain donor samples, we first separated wild-type and mutant cells by FACS (described above) and performed deep total RNA-seq on the separate populations. Unlike the highly backcrossed Mecp2-mutant mice, there was a wealth of genomic variability that could be used to transcriptotype cells once the variants were confirmed to be expressed in cis selectively with the mutant or wild-type MECP2 allele. After performing RNA-seq on sorted wild-type (MECP2high) and mutant (MECP2low) populations of cells, we performed X-chromosome variant calling on these datasets using Freebayes version 1.1.0–448 with default parameters. The genomic location of SNPs with a Freebayes score > 10 were used to extract reads (Samtools version 1.2) from the mapped RNA-seq data of wild-type and mutant Mecp2 populations. Based on the reference genome, each SNP sequence was assigned to be either the reference sequence, the alternate sequence or other. For both the sorted wild-type and mutant samples, the fraction of “reference”, “alternate”, or “other” reads that cover each SNP was calculated. If a gene were expressed from both alleles, approximately 50% of the reads sequenced would be expected to map to each allele. Because X-inactivation typically results in monoallelic expression, for a given cell, most of the sequencing reads (allowing for some sequencing error and/or sorting error) would be expected to map to a single allele. We thus considered the expression of a SNP to be allele-specific if ≥ 85% of wild-type (MECP2high) reads and < 85% of the mutant (MECP2low) reads encompassing this region contain the same sequence variant (e.g. “reference” or “alternate”). We used the MECP2high population as the primary filter for monoallelic SNP expression because the MECP2low population, while mainly defined by the low background immunofluorescence signal from MECP2-mutant cells (Sanger sequencing confirmed the cell population expresses the mutant allele), could also contain small numbers of wild-type cells that have background levels of fluorescence because they express low levels of MECP2 (e.g. non-neuronal cells). The identification of allele-specific SNPs with these parameters is supported by the observation that for a given SNP, an average of 98% of the reads from wild-type cells (MECP2high) map to the same SNP (e.g. “reference”) and an average of 76% of the reads from the mutant (MECP2low) cells map to the alternative SNP (e.g. “alternate”). The allele-specific SNPs identified from each donor sample (Donor 1 = 69 SNPs, Donor 2 = 69 SNPs, Donor 3 = 75 SNPs) were then used to mark the X chromosome alleles that are expressed in cis with either the wild-type or mutant allele of MECP2. Custom R-scripts were written to process BAM output files from the inDrops mapping pipeline or total RNAseq mapping pipeline for the identification of allele-specific SNPs.
Assignment of transcriptotype to individual cells
After the identification of the allele-specific SNPs that are expressed in cis with either the wild-type or mutant allele, we next used this information to assign transcriptotypes to the individually sequenced cells. To do this, we used Samtools to identify the sequencing reads within the single-cell or single-nucleus RNA sequencing datasets that contained both the allele-specific SNPs identified above and a unique cell barcode. We then grouped the reads from each cell or nucleus and assigned the MECP2 transcriptotype corresponding to the profile of allele-specific SNPs expressed. Specifically, a transcriptotype was assigned if ≥ 85% of the reads covering an individual allele-specific SNP mapped to the same allele (e.g. ≥ 85% of the reads were “reference”) and ≥ 80% of the total SNPs covered in each cell were concordant with the same transcriptotype (e.g. ≥ 80% of the SNPs covered in a cell were expressed in cis with the R255X allele of MECP2). Some cells or nuclei only had one or two reads mapping to an allele-specific SNP, which increases the chance of an incorrect transcriptotype call. After estimating the mean error rate for transcriptotype assignments was only 0.5% in female Mecp2+/− mice and 4.6% in human Rett donors, we chose to include these cells in the differential gene expression analysis to maximize the number of cells and resulting statistical power. The estimated transcriptotype error rate for a given cell with only one or two reads encompassing allele-specific SNPs was determined as the percent of genotype discordant reads in cells with at least three reads (defined as cells with confident transcriptotypes). The mean estimated transcriptotype error was then calculated by averaging the error rates for each cell in the dataset. The lower estimated error rate in mouse was accomplished by deeper sequencing of the allele-specific SNPs using gene-specific library preparations, an approach that can be used to further improve the confidence of transcriptotype calls in any dataset. Custom R-scripts were written to process BAM output files from the inDrops mapping pipeline for the assignment of transcriptotypes to specific cells based on allele-specific SNPs.
Gene-specific primers for enriching SNP coverage in Mecp2+/− mouse single-cell libraries
Reverse transcription:
rs13468851: TGTATGTCGGACTTGATGTACT
rs13468852: TTTACAGTATTCTTTCTACATGGA
rs31144974: GATTAACTGTAACAACGATCACAAC
rs29035084: GGTTTCAAAGTACCCAGCATAAAT
PCR:
rs13468851: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNCTTGCTCTGTCAAGCTCTTTGC
rs13468852: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNGATTACATCCGACACGTCTGC
rs31144974: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNGCATGTTGGATTAGATTGTC
rs29035084: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNCAGCAGAGGTGGCTGAACTT
Differential gene expression analysis
We used Monocle 2 to identify differentially expressed genes between wild-type and mutant cells49. The single-cell and single-nucleus RNA-seq data was modeled by a negative binomial distribution, consistent with the expression profiles of our data. Differential expression analysis was conducted independently for each cell type by aggregating the gene counts from the population of cells or nuclei within a given cell type (median counts per gene in human excitatory neuron cluster = 2,029), which provided sufficient coverage of expressed genes for differential expression analysis between mutant and wild-type cells. Certain analyses, where described, required combining cells or nuclei of the same cell-type from multiple mice or human donor samples (e.g. Fig. 3 because of limited inhibitory neuron populations). Otherwise, differential expression was performed between mutant and wild-type cells from each human donor sample individually (e.g. Fig. 4 excitatory neurons). A gene was included for differential expression analysis if its minimum expression was ≥ 0.1 and it was detected in at least 100 cells or nuclei. Significantly misregulated genes were identified by the FDR cutoff described for the specific analysis and number of cells studied. Differential expression analysis of randomly transcriptotyped cell populations resulted in few, if any, significantly misregulated genes. Thus, the uncorrected p-values were ranked from smallest to largest and the number of genes selected for analysis was determined by the corresponding number of significantly misregulated genes identified in the respective mutant to wild-type comparison. To generate randomly transcriptotyped groups of cells, the sample function in R was used to randomly select the same number of cells from each individual (without respect to transcriptotype) as was used for the SNP-seq-based transcriptotype analyses. Differential expression analysis was performed on two groups of randomly transcriptotyped cells (the same cell could not exist in both randomly generated lists). If analyses combined cells that were transcriptotypted from multiple individual donor samples (e.g. Figs. 2–3), the corresponding number of cells was first randomly sampled on a per-individual basis and then combined to form the control group. To ensure the randomly sampled groups in each figure were representative of the entire dataset, differential expression was performed on 3 independent pairs of randomly sampled cells.
Correlations of MeCP2-dependent gene expression with DNA methylation, MeCP2 ChIP, and gene length
To generate the smooth-line correlation plots, genes were sorted by their gene length, DNA methylation, or MeCP2 ChIP signal, and a sliding window was defined by the indicated bin and step sizes for each analysis. The bin and step sizes were adjusted to the size of the gene list. The log2 fold-change for each bin was averaged and plotted with the standard error for each bin. The gene length for a gene was obtained from RefSeq annotation (gene end – gene start). Cell-type-specific mouse DNA methylation data were obtained from26. Gene body level cell-type-specific human DNA methylation data were obtained from32 and averaged across all cells within the indicated cell type. Excitatory neuron-specific MeCP2-ChIP-seq data was generated as described below.
Gene ontology analysis and cell-type-specific enrichment analysis
Gene ontology analysis was performed at geneontology.org using the PANTHER overrepresentation test (Fisher’s Exact with FDR). All expressed genes (normalized expression > 0.1 in both mutant and wild-type cells of the corresponding cell type) for the respective comparison were used as the background lists. Gene ontology biological processes were reported with redundant/overlapping pathways only displayed once. Single-cell mRNA sequencing data from >160,000 cells and 39 distinct cell types were obtained from www.mousebrain.org50. For each gene, the normalized gene expression counts were averaged across all cells of the same cell type. The mean gene expression level within each cell type was then row-normalized using the Morpheus heatmap tool (https://software.broadinstitute.org/morpheus). Enrichment statistics were calculated by the following formula: the number of times out of 1000 iterations the mean expression of randomly sampled cells was greater than or equal to the cell type of interest / 1000 iterations.
INTACT nuclei isolation and MeCP2 chromatin immunoprecipitation sequencing (ChIP-seq):
CamkIIa-cre mice were crossed with mice that express the SUN1-sfGFP-MYC protein in the nuclear membrane in a CRE-dependent manner, and SUN1-GFP-expressing nuclei were isolated from the forebrain of 8-week-old male Sun1-GFP; CamkIIa-cre mice as previously described 26. Nuclei were immunoprecipitated with a GFP antibody (Fisher G10362) and Protein G Dynabeads (Invitrogen). Nuclei were cross-linked in 1% formaldehyde in PBS for 10 min at room temperature, quenched with 125 mM glycine for 5 min, and washed twice with PBS. Nuclei were then resuspended in LB3 buffer (10 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-Lauroylsarcosine, protease inhibitors), and sonicated in a Diagenode Bioruptor. Insoluble material and beads were removed by spinning at 16,000 g for 10 min at 4°C, and Triton X-100 was added to soluble chromatin at a final concentration of 1%. Chromatin was pre-cleared for two hours with Protein A Dynabeads, then incubated with Protein A Dynabeads conjugated to an MeCP2 antibody 46 overnight at 4°C. Beads were washed twice with Low Salt Buffer (20 mM Tris pH 8, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice with High Salt Buffer (20 mM Tris pH 8, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice with LiCl Wash Buffer (10 mM Tris pH 8, 1 mM EDTA, 1% NP-40, 250 mM LiCl, 1% sodium deoxycholate) and once with TE Buffer (50 mM Tris pH 8, 10 mM EDTA) at 4°C. Chromatin was eluted off the beads by incubating in TE Buffer with 1% SDS at 65°C for one hour, and crosslinks were reversed by incubating overnight at 65°C. Chromatin was treated with RNase A for 30 min at 37°C and Proteinase K for 2 hours at 55°C. DNA was phenol-chloroform extracted and purified with the Qiagen PCR purification kit. Libraries were generated using the NuGEN Ovation Ultralow System V2 following manufacturer instructions. Libraries were sequenced on an Illumina Nextseq 500 with 85 bp single-end reads. Reads were mapped to the mm10 genome with Bowtie2, and PCR duplicates were removed using SAMtools rmdup. Mapped reads from MeCP2 ChIP and input were randomly down-sampled to the same number of reads. Bedtools map was used to count ChIP and input reads mapped to gene bodies for comparison to gene expression.
Re-analysis of published RNA sequencing data:
Gene read counts tables for male 6-week-old WT and R106W excitatory neuron nuclear RNA-seq and female 18-week-old R106WWT and R106WMUT excitatory neuron nuclear RNA-seq were downloaded from GEO (GSE83474). Differential expression analysis was performed with the R package edgeR. A FDR < 0.1 was used to identify differentially expressed genes. For comparison to DNA methylation, excitatory neuron mCA26 was mapped to the gene body locations in the Johnson et al. counts tables using bedtools map.
Overlap with autism and intellectual disability genes
Rett syndrome gene lists were compared to the autism genes list at SFARI (gene.sfari.org) and to the intellectual disability gene lists at the University of Colorado: (gfuncpathdb.ucdenver.edu/iddrc/iddrc/home.php). Enrichment statistics were calculated using the hypergeometric test in R 3.3.2.
Statistical analysis
Enrichment statistics of pairwise comparisons between two gene lists was calculated using the hypergeometric test as calculated in R 3.3.2. Pearson correlations between gene expression and DNA methylation, gene length, or MeCP2 ChIP were compared by permutation. P-values for these comparisons were estimated by calculating: (# of events where | corr1permutation – corr2permutation | > | corr1observed – corr2observed |) / 1000 permutations. Kruskal-Wallis tests and Mann-Whitney tests were performed using Prism v7. No statistical methods were used to pre-determine sample size but our samples sizes are similar or larger to those reported in previous publications19,28.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the Rett Syndrome Research Trust for support of this work, along with NIH K08NS101064 (WR), F32NS101739 (LDB), and R01NS048276 (MEG). We are also grateful to Rettsyndrome.org and the Harvard Brain Bank for providing tissue from brain donors with Rett syndrome. D. Harmin assisted with data processing and scripting. H. Gabel provided thoughtful comments on the manuscript, and A. Ratner provided technical assistance. The single-cell methylation data was graciously provided by C. Luo and J. Ecker.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests
ACCESSION CODES:
Life Sciences Reporting Summary
Additional information about the experimental design is available in the Life Sciences Reporting Summary associated with this manuscript.
Code availability
Custom R scripts are available upon request.
Data availability
All sequencing data reported in this study have been deposited in the NCBI Gene Expression Omnibus under accession GSE113673.
REFERENCES
- 1.Amir RE et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol 47, 670–679 (2000). [PubMed] [Google Scholar]
- 2.Lyst MJ & Bird A Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16, 261–275, doi:10.1038/nrg3897 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics 23, 185–188, doi:10.1038/13810 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Ebert DH et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345, doi:10.1038/nature12348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis JD et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Lyst MJ et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature neuroscience 16, 898–902, doi:10.1038/nn.3434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skene PJ et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Molecular cell 37, 457–468, doi:10.1016/j.molcel.2010.01.030 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinde B, Wu DY, Greenberg ME & Gabel HW DNA methylation in the gene body influences MeCP2-mediated gene repression. Proceedings of the National Academy of Sciences of the United States of America 113, 15114–15119, doi:10.1073/pnas.1618737114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinde B, Gabel HW, Gilbert CS, Griffith EC & Greenberg ME Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proceedings of the National Academy of Sciences of the United States of America 112, 6800–6806, doi:10.1073/pnas.1411269112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabel HW et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93, doi:10.1038/nature14319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brero A et al. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol 169, 733–743, doi:10.1083/jcb.200502062 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chahrour M et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229, doi:10.1126/science.1153252 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng TL et al. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell 28, 547–560, doi:10.1016/j.devcel.2014.01.032 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Young JI et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proceedings of the National Academy of Sciences of the United States of America 102, 17551–17558, doi:10.1073/pnas.0507856102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201, doi:10.1016/j.cell.2015.04.044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214, doi:10.1016/j.cell.2015.05.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142, doi:10.1126/science.aaa1934 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Guy J, Hendrich B, Holmes M, Martin JE & Bird A A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature genetics 27, 322–326, doi:10.1038/85899 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Hrvatin S et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nature neuroscience 21, 120–129, doi:10.1038/s41593-017-0029-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502, doi:10.1038/nbt.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson ED, Kavalali ET & Monteggia LM MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol 16, 710–716, doi:10.1016/j.cub.2006.02.062 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Peterson M, Beyer B, Frankel WN & Zhang ZW Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J Neurosci 34, 2754–2763, doi:10.1523/JNEUROSCI.4900-12.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugino K et al. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci 34, 12877–12883, doi:10.1523/JNEUROSCI.2674-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo JU et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nature neuroscience 17, 215–222, doi:10.1038/nn.3607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L et al. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proceedings of the National Academy of Sciences of the United States of America 112, 5509–5514, doi:10.1073/pnas.1505909112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mo A et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384, doi:10.1016/j.neuron.2015.05.018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson BS et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med 23, 1203–1214, doi:10.1038/nm.4406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lake BB et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590, doi:10.1126/science.aaf1204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagger S et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet 13, e1006793, doi:10.1371/journal.pgen.1006793 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lister R et al. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905, doi:10.1126/science.1237905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stroud H et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164 e1116, doi:10.1016/j.cell.2017.09.047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo C et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604, doi:10.1126/science.aan3351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng V et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet 16, 640–650, doi:10.1093/hmg/ddm007 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Lin P et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q Genes in Rett syndrome. BMC Genomics 17, 427, doi:10.1186/s12864-016-2746-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kyle SM, Vashi N & Justice MJ Rett syndrome: a neurological disorder with metabolic components. Open Biol 8, doi:10.1098/rsob.170216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sultana R et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 80, 129–134 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Fogel BL et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet 21, 4171–4186, doi:10.1093/hmg/dds240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao Z et al. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354, doi:10.1038/nature13921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cossette P et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nature genetics 31, 184–189, doi:10.1038/ng885 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Wallace RH et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nature genetics 19, 366–370, doi:10.1038/1252 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Egan MF et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Gauthier J et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet 130, 563–573, doi:10.1007/s00439-011-0975-z (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Genomes Project C et al. A global reference for human genetic variation. Nature 526, 68–74, doi:10.1038/nature15393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94, doi:10.1038/nbt.4042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tukiainen T et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248, doi:10.1038/nature24265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen WG et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889, doi:10.1126/science.1086446 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Gendrel AV et al. Developmental dynamics and disease potential of random monoallelic gene expression. Dev Cell 28, 366–380, doi:10.1016/j.devcel.2014.01.016 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Garrison E & Marth G Haplotype-based variant detection from short-read sequencing. arXiv preprintarXiv:1207.3907 [q-bio.GN] (2012). [Google Scholar]
- 49.Qiu X et al. Single-cell mRNA quantification and differential analysis with Census. Nat Methods 14, 309–315, doi:10.1038/nmeth.4150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeisel A et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014e1022, doi:10.1016/j.cell.2018.06.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.