

Abstract
Objective:
This study examined the association between Macrophage Migration Inhibitory Factor (MIF) promoter polymorphisms and granulomatosis with polyangiitis (GPA), and MIF’s role in a murine model of granulomatous vasculitis.
Methods:
The human study involved 1077 patients with GPA and controls. Genotyping for the MIF −794 CATT5–8 promoter microsatellite (rs5844572) was performed by capillary electrophoresis. MIF promoter CATT-length dependent expression in response to β-glucan was assessed by gene reporter assays. Granulomatous disease was induced in wild type (WT), Mif −/−(Mif-KO), and Mif lung-transgenic (Mif lung-Tg2.1) C57BL/6 mice by injection of Candida albicans β-glucan (CAG). Mice were treated with a neutralizing anti-MIF and analyzed for lethality, pulmonary granulomas and inflammatory chemokine expression.
Results:
The percentage of individuals carrying >5 CATT repeats in each MIF allele (high MIF expressers) was 60.9% in patients with GPA and 53.7% in controls (adjusted p=0.049). Human MIF gene expression increased proportionally by CATT-length in response to granulomatous stimulation. Mif lung-Tg2.1 mice exhibited more pulmonary granulomas than WT, which in turn showed more granulomas than Mif-KO. A significantly higher percentage of Mif lung-Tg2.1 mice died when injected with CAG when compared to Mif-KO or WT, and anti-MIF protected against lethal disease. MIF-dependent neutrophil/macrophage chemokines were elevated in the bronchoalveolar lavage or plasma of Mif lung-Tg2.1 mice.
Conclusion:
Patients with GPA have an increased frequency of high-expression MIF CATT alleles. Higher MIF expression increased mortality and pulmonary granulomas while anti-MIF protected mice from lethal disease. MIF blockade in high genotypic MIF expressers may offer a selective pharmacologic therapy for GPA.
Keywords: CD74, macrophage migration inhibitory factor, granulomas, granulomatosis with polyangiitis, vasculitis
INTRODUCTION
Granulomatosis with polyangiitis (GPA; Wegener’s) is an ANCA-associated vasculitis (AAV) characterized by the presence of anti-neutrophil cytoplasmic antibodies (ANCA) (1). A major pathologic feature of GPA includes the presence of granulomatous inflammation in addition to vasculitis (2). The fundamental immunopathogenesis of granulomatous inflammation in GPA remains poorly understood.
The cytokine macrophage migration inhibitory factor (MIF) plays a central regulatory role in innate immunity by inhibiting the activation-induced apoptosis of macrophages and upregulating the expression of pattern recognition receptors. The protein is produced by monocytes/macrophages and barrier epithelial cells to promote autocrine/paracrine activation and survival signals (3, 4). MIF is expressed within granulomas in hypersensitivity pneumonitis (5), sarcoidosis (6), and tuberculosis (7), and circulating MIF levels are elevated in patients with AAV and associated with disease activity (8). Both MIF and its receptor CD74 are overexpressed in renal mesangial and parietal epithelial cells in proliferative glomerulonephritis (9), and in inflammatory lung disease (10, 11); both processes are common in AAV.
Human genetic studies have identified a four-nucleotide microsatellite in the MIF promoter, −794 CATT5–8 (rs5844572) that functionally regulates MIF transcription (12) (Figure 1). Microsatellite repeat number is associated with constitutive and inducible MIF expression such that the CATT5 repeat is a low expression allele and the CATT6–8 repeats are progressively higher expression alleles (13, 14). These MIF promoter variants occur commonly in the population (minor allele frequency >5%) and higher CATT number has been linked to the susceptibility or the clinical severity of rheumatoid and juvenile idiopathic arthritis (12, 15), sarcoidosis (6), systemic sclerosis (14), and systemic lupus erythematosus (13). A single nucleotide polymorphism (SNP) in the MIF promoter region, −173 G/C SNP (rs755662) also has been shown to be associated with autoimmune diseases likely because of linkage disequilibrium with the functional high expression CATT7 (13, 14).
Figure 1. The human MIF −794 CATT5–8 promoter polymorphism.
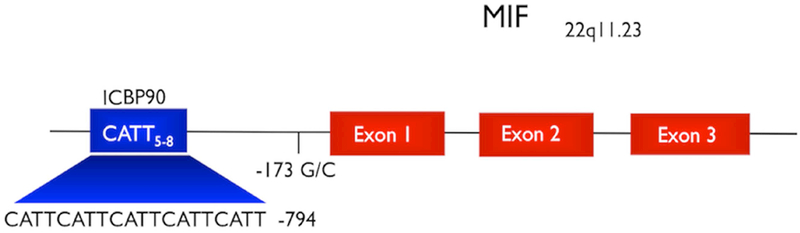
Schematic diagram of the human MIF gene with its three exons and the two studied promoter polymorphisms. The CATT5–8 promoter microsatellite is the binding site for the ICBP90 transcription factor (15).
In the present study, we sought to determine potential relationships between MIF gene polymorphisms and GPA by conducting a candidate gene study using samples from a multicenter observational study of 1077 vasculitis and control patients in the United States. We analyzed MIF genotype and MIF levels in relation to disease incidence for their correlation with two MIF promoter polymorphisms: the −794 CATT5–8 microsatellite and −173 G/C SNP. We additionally studied an experimentally-induced model of granulomatous disease using genetically engineered mice that overexpress MIF in the lung in order to mimic the elevation in MIF expression in target organs that may be evident in human high genotypic MIF expressers.
MATERIALS AND METHODS
Patients:
Our study cohort consisted of 501 patients with GPA and 576 healthy controls recruited from the Wegener’s Granulomatosis Genetic Repository (WGGER) conducted at 9 US academic centers between 2001 and 2005 Group (16). Plasma samples from patients with GPA were obtained from the Vasculitis Clinical Research Consortium (VCRC), a large, multicenter, North American observational cohort. All patients were ≥ 18 years old and fulfilled the modified American College of Rheumatology (ACR) criteria for GPA (16, 17). Ethnicity was determined by self-report and given the rarity of GPA in minority groups, the study focused on Caucasians. Disease activity was measured by the Birmingham Vasculitis Activity Score (BVAS). All relevant Institutional Review Boards approved the study.
Genotyping:
Genomic DNA was isolated from serum using the easy-DNA kit (Invitrogen, Carlsbad, CA, USA) and the two MIF promoter polymorphisms (rs5844572, rs755622) were analyzed following methodologies previously described (14). MIF −794 CATT5–8 genotyping was performed by PCR using a forward primer (5’-TGCAGGAACCAATACCCATAGG-3’) and a TET fluorescent reverse primer (TET lab5’-AATGGTAAACTCGGGGGAC-3’). The quality of the PCR product was assessed by agarose gel electrophoresis. Automated capillary electrophoresis on a DNA sequencer was performed on each sample and the CATT5–8 repeat length was identified using Genotyper 3.7 software (Applied Biosystems). MIF −173 G/C genotyping was performed using a pre-developed TaqMan assay for allelic discrimination (Applied Biosystems, Foster City, CA, USA). After real-time PCR, the genotype of each sample was automatically attributed using fluorescence detection in an ABI Prism 7900HT instrument (Applied Biosystems). Allelic discrimination was analyzed with SDS 2.1.1 software (Applied Biosystems). Although at least 8 additional polymorphisms have been identified within the human MIF locus, these additional variants (all SNPs) are rare and have a low likelihood of functionality given their location in introns or within the 3’-untranslated region (18).
Plasma MIF:
Plasma samples were obtained at the time of study entry. Circulating MIF levels were measured by enzyme-linked immunosorbant assay (ELISA) using specific antibodies (19). A native sequence and fully active recombinant human MIF was used as a standard (20). Plasma MIF levels were compared and culled from patients with GPA identified as having the high expression, −794 CATT6–8 and −173 C containing genotypes or the low expression, −794 CATT5 and −173 G/G genotypes.
Granulomatous Vasculitis Mouse Model:
A murine model of granulomatous vasculitis induced by the injection of Candida albicans β-glucan (Sigma-Aldrich, St Louis, MO, USA) into mice (C57BL/6, female, aged 8–12 weeks) was used for the experimental studies (21, 22). Discrete and multiple granulomatous lesions appear in lungs within 48 hours after the intravenous injection of β-glucan. All procedures were performed in accordance with the Institutional Animal Care and Use Committee protocols of Yale University.
We developed tissue conditional transgenic mouse lines that overexpress MIF in lung epithelium and selected a founder line that showed a two-fold increase in Mif mRNA expression (designated Mif lung-Tg2.1) (23). Briefly, a 0.4 Kb DNA fragment containing mouse Mif cDNA was inserted into an expression plasmid under the control of the pulmonary Type II epithelial-cell specific CC10 promoter and transfected into Bruce4 (C57BL/6) ES cells for the establishment of founder mice in the pure C57BL/6 background. Quantitative PCR (qPCR) confirmed a two-fold increase in Mif mRNA expression in lung and corresponding increases in MIF protein in bronchoalveolar lavage fluid (BAL) and alveolar lung epithelium (Figure 2). A Mif-KO mouse strain created previously in the pure C57/BL6 background was used for comparison studies (24).
Figure 2. MIF overexpression in Mif-lung Tg2.1 founder mice and regulation transcription by The MIF −794 CATT5–8 promoter polymorphism in response to β-glucan in a CATT-length dependent manner.
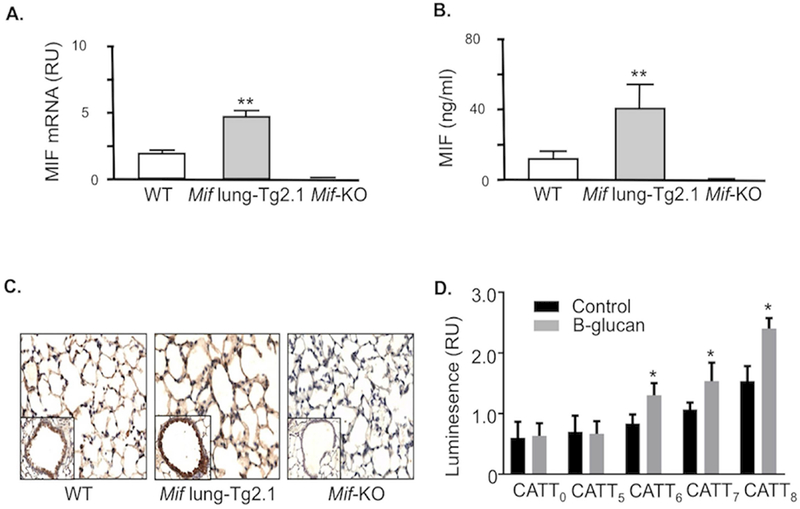
(A) Quantitative PCR analysis of Mif mRNA in lung tissue harvested from 3 representative wild type (WT), Mif lung-Tg2.1, and Mif-KO mice at 6 weeks of age. (B) ELISA analysis of MIF content in bronchoalveolar lavage fluid obtained from three mice of indicated strains. (C) Immunohistochemistry analysis of mouse lung tissue stained with anti-MIF pAb showing prominent staining in alveolar epithelium and increased MIF content in Mif lung-Tg2.1 mice. Tissue sections from a Mif-KO mouse lung was used as an antibody specificity control (200×). Insets show a representative terminal bronchus and sections are representative of three mice per genotype. **p<0.01 by Student’s t test, two tailed. (D) Human pulmonary A549 cells transfected with MIF promoter/luciferase reporter fusion plasmids bearing 0, 5, 6, 7, and 8 CATT repeats were stimulated with 100 ng/ml β-glucan for 8 hrs and luciferase expression expression measured as luminescence relative units (RU). Mean ± SD for quadruplicate determinations, replicated twice. *p<0.05 for comparisons to 5-CATT or to β-glucan stimulation versus control by Student’s T test (two-tailed).
Following the injection of β-glucan into tail veins, lungs were resected from dead or sacrificed mice (48 hours post-injection) and examined histologically for pulmonary pathology. Additional cohorts of mice were observed for mortality and sacrificed when judged to be agonal. After initial dose-ranging experiments, responses to β-glucan were assessed in a blinded fashion. The granuloma density (number of granulomas per mm2) of at least ten random lung sections was calculated and compared between experimental groups by an observer blinded to the experimental groups. For lung immunohistochemistry, formalin-fixed and paraffin-embedded sections were stained with polyclonal anti-MIF (11b, 1:1500 dilution) or non-immune serum and developed with immunoperoxidase. Bronchoalveolar lavage (BAL) was sampled (25) and plasma cytokines of (IL-1, IL-6, IL-10, IL-17, TNF, KC, MCP-1, VEGF and MIP-2) were measured using a cytokine multiplex panel (Invitrogen, Carlsbad, CA, USA) and levels expressed as mean ± SD (pg/ml). Selected mice also received treatment with a neutralizing anti-MIF mAb (clone NIH.IIID9) (26) or an anti-IgG1 isotype control (20 mg/kg, i.p.).
The ability of β-glucan to stimulate MIF expression in a −794 CATT0–8 length-dependent manner was assessed in human pulmonary epithelial A549 cells co-transfected with expression plasmids engineered to contain the −794 CATT0,5,6,7,8 MIF promoter sequences ligated to a luciferase gene together with a β-actin promoter luciferase control. These studies utilized the high efficiency Amaxa nucleofactor platform (Lonza, Ltd, Switzerland) with 1 μg of each plasmid per transfection reaction (12). The - 173 SNP status of these MIF constructs was in accordance with the most commonly occurring genotypes: e.g., 5G, 6G, 7C, and 8G (13). The transfected cells were incubated at 37°C for 24 hours followed by stimulation with 100 ng/ml of β-glucan before luminescence measurement.
Statistical analysis:
The Pearson chi-square test was used to analyze Hardy-Weinberg equilibrium. Genotypic frequencies were compared using the chi-square test and (when appropriate) Fisher’s exact test. We compared high MIF expression > −794 CATT5 containing genotypes (−794 CATT6/6, −794 CATT6/7, −794 CATT7/7) with low MIF expression −794 CATT5 containing genotypes (−794 CATT5/5, −794 CATT5/6, −794 CATT5/7) and −173 C containing genotypes (−173 C/C, −173 G/C) with −173 G/G genotypes between patients and healthy controls. The −794 CATT8 allele was found rarely (<1 %) (27) and was excluded from analysis. The difference between the 2 groups was adjusted for age and sex.
To account for population stratification and its effect on genotype differences, we used formulas established by Lee et al. to gauge the impact of population stratification bias (28). The population stratification bias can be quantified by the Confounding Rate Ratio (CRR), which equals the biased relative rate divided by the true relative rate. Lee and Wang showed that CRR is always bound above by “U” and below by “1/U”, where “U” is the highest odds ratio that can be attributed to the population stratification bias and “1/U” is the lowest odds ratio. “U” takes into account the highest and lowest reported prevalence of a disease in a population affected by stratification and the highest and lowest frequency of the genotype in question reported in the same population. If the observed odds ratio is above the highest calculated odds ratio “U” or lower than the lowest calculated odds ratio “1/U”, then these observed odds ratio cannot just be explained by population stratification.
Differences in circulating plasma MIF levels between groups were compared using the Student’s t test. Similar MIF expression categories were used: High MIF expression, > −794 CATT5 and −173 C containing genotypes versus low MIF expression, −794 CATT5 containing and −173 G/G genotypes. Genetic statistical analyses were performed using STATA version 13. All significance levels reported are 2-tailed. P values ≤ 0.05 were considered statistically significant.
Mice survival curves were compared using Log-rank (Mantel-Cox) test. Lung granuloma densities and cytokine levels were compared using t test. Statistical analyses for the animal studies were performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). P values ≤0.05 were considered statistically significant.
RESULTS
MIF gene polymorphisms.
At total of 1077 subjects were studied (501 patients and 576 controls). Table 1 describes the baseline demographics of the participants and the frequencies of the −794 CATT5–8 and −173 G/C MIF genotypes. The studied populations did not deviate from the Hardy-Weinberg equilibrium and the allelic distributions are similar to those previously reported in the general population (14, 15).
Table 1.
Baseline demographics of study participants (patients with granulomatosis with polyangiitis and healthy controls) and percentages of MIF genotypes in each group.
| Cases (n=501) | Controls (n=576) | p-value | |
|---|---|---|---|
| Female % | 52 | 40 | p<0.001 |
| Age (Mean yrs ± SD) | 52.8 ± 15.6 | 47.2 ± 15.7 | p<0.001 |
| − 173 GG (%) | 65.7 | 59.8 | |
| − 173 GC (%) | 30.8 | 36.2 | |
| − 173 CC (%) | 3.5 | 4.0 | |
| −173 C containing genotypes | 34.2 | 40.2 | 0.210* |
| −794 CATT5/5 (%) | 5.6 | 8.2 | |
| −794 CATT5/6 (%) | 26.9 | 29.0 | |
| −794 CATT5/7 (%) | 6.8 | 8.9 | |
| −794 CATT5/8 (%) | 0.4 | 0.0 | |
| −794 CATT6/6 (%) | 41.0 | 36.6 | |
| −794 CATT6/7 (%) | 17.5 | 15.7 | |
| −794 CATT7/7 (%) | 0.2 | 0.5 | |
| −794 CATT7/8 (%) | 1.6 | 1.1 | |
| > −794 CATT5 containing genotypes | 60.2 | 53.9 | 0.049* |
p adjusted for age and sex.
The percentage of patients carrying > −794 CATT5 repeats in their genotypes was 60.2% in patients with GPA and 53.9% in controls (odds ratio: 1.38 (1.08–1.76), unadjusted p=0.030 and p=0.049 adjusted for sex and age). There was no significant difference in the −173 C containing genotypes between the GPA and healthy control groups (34.2% in patients versus 40.2% in controls, unadjusted p=0.050 and p=0.210 adjusted for age and sex).
The prevalence of GPA in the United States is 26 to 90 per 100,000 (29), and the frequency of the > −794 CATT5 containing genotypes in US Caucasians is reported to be 49.7–57.1% (14, 15), which is similar to our reported frequencies. Based on the prevalence of GPA and the frequency of > −794 CATT5 containing genotypes, we calculated the highest odds ratio “U” and the lowest “1/U” that can be attributed to population stratification. The highest odds ratio was “U”=1.10 and the lowest was “1/U”=0.91. Our measured odds ratio of 1.38 is higher than the highest calculated odds ratios, indicating that the observed association between GPA and > −794 CATT5 genotype cannot be attributed to population stratification.
MIF plasma levels.
We measured MIF levels in plasma obtained at the time of study entry in a subset of patients with GPA, and in age- and sex-matched healthy controls. The mean plasma MIF level in patients was significantly higher than in disease-free controls (15.9 ± 10.4 ng/ml in patients [n=73] versus 6.7 ± 5.0 ng/ml in controls [n=45]; mean ± SD, p<0.001) (Figure 3). There were no differences in the mean MIF plasma levels between female and male patients (15.1 ± 11.1 ng/ml for females [n=45] vs. 17.2 ± 9.2 ng/ml for males [n= 28], p=0.405). Also, there was no correlation between disease activity as measured by the BVAS and plasma MIF levels.
Figure 3. MIF plasma levels in patients with granulomatosis with polyangiitis (GPA).
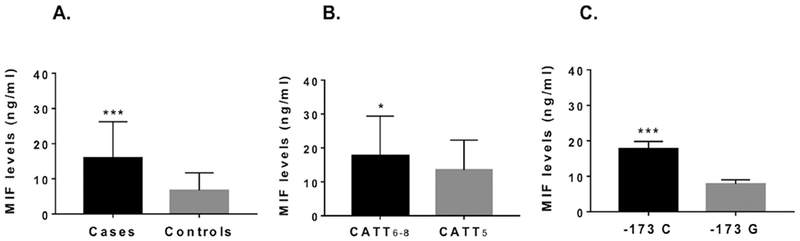
(A) Mean MIF plasma levels (ng/ml) in patients with GPA (n=73) and healthy controls (n=45). (B) Mean MIF plasma levels (ng/ml) in patients with GPA carrying high MIF expression - 794 CATT6–8 containing genotypes (n=35) and patients carrying low MIF expression −794 CATT5 containing genotypes (n=38). (C) Mean MIF plasma levels (ng/ml) in patients with GPA carrying high MIF expression −173 C containing genotypes (n=23) and low MIF expression −173 G containing genotypes (n=50).
To determine whether there was a relationship between MIF polymorphisms and MIF levels in patients with GPA, we compared plasma MIF levels in high genotypic (>−794 CATT5) and low genotypic (−794 CATT5) MIF expressers (Figure 3). The mean MIF plasma levels were higher in GPA patients with high MIF expression −794 CATT6–8 containing genotypes (17.7 ± 11.7 ng/ml, n=35) than in those with low MIF expression −794 CATT5 containing genotypes (13.5 ± 8.8 ng/ml, n=38; mean ± SD, p=0.040). A similar elevation in MIF plasma levels was observed in patients with −173 C containing genotypes (17.7 ± 2.1 ng/ml, n=23) when compared with patients with −173 G genotypes (7.8 ± 1.2 ng/ml, n=50; mean ± SD, p<0.001) (Figure 3).
Granulomatous Disease Model Studies.
To examine potential roles for MIF in GPA, we examined the impact of MIF expression in a mouse model of granulomatous vasculitis. MIF is known to be expressed constitutively in lung epithelium, and both mRNA and protein expression increase as a consequence of inflammatory stimulation (10, 30). While the precise stimuli underlying granulomatous inflammation in GPA is unknown, we confirmed the ability of β-glucan to stimulate MIF expression in a −794 CATT length-dependent manner by measuring transcriptional activation in human lung epithelial cells transfected with MIF −794 CATT0–8 promoter-luciferase reporter plasmids. As shown in Figure 2, β-glucan stimulation increased MIF expression, with −794 CATT5 showing the lowest gene transcription and −794 CATT8 showing the highest transcription.
We studied C57BL/6 mice that were wild type (WT), MIF gene deficient (Mif-KO) (24), or engineered to transgenically express a 2-fold increase in MIF mRNA in lung alveolar epithelium (Mif lung-Tg2.1) in order to mimic the > CATT5 human MIF promoter phenotype (Figure 2). Age-matched cohorts of mice were administered a single 1 mg intravenous injection of β-glucan and mortality assessed over time. As shown in Figure 4, Mif lung-Tg2.1 mice showed greater mortality than WT mice (15% vs. 75% survival, p=0.009), which in turn showed a trend toward improved survival over Mif-KO mice (75% vs. 100% survival, p=.070). The enhanced lethality attributable to Mif overexpression in the lung was abrogated by administration of a neutralizing anti-MIF monoclonal antibody (Figure 4).
Figure 4. MIF overexpression enhances β-glucan lethality and pulmonary granuloma.
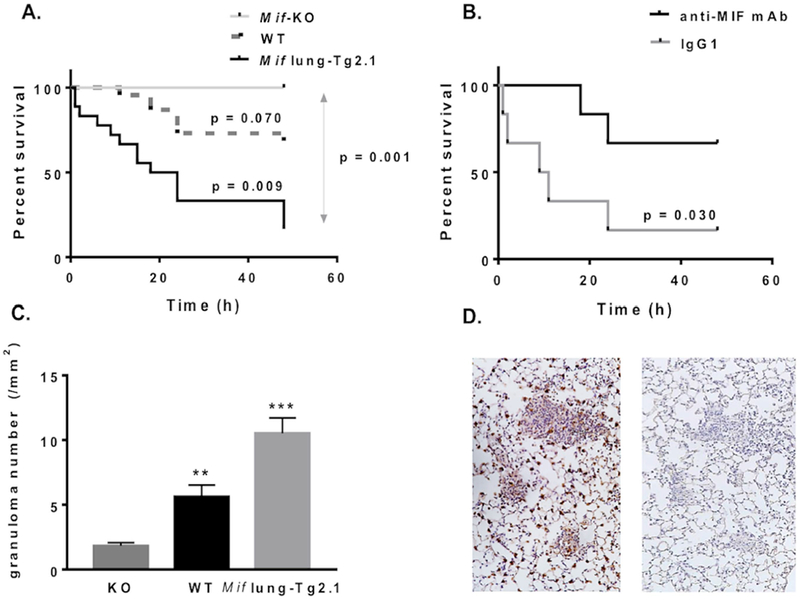
(A) Kaplan-Meyer survival analysis of Mif lung-Tg2.1 transgenic mice, wild-type (WT), and Mif-knockout (Mif-KO) mice after intravenous injection of β-glucan (1 mg per mouse, iv). (B) Survival of Mif lung-Tg2.1 transgenic mice treated with anti-MIF mAb versus an isotypic control immunoglobulin (IgG1). Mice received a single i.p. injection of antibody (20 mg/kg, ip) at time 0. Data are compiled from 4 independent experiments, with A and B each replicated. A total of 44 mice were studied. (C) Granuloma density measured by histologic enumeration of mouse lungs obtained from Mif-KO, WT and Mif lung-Tg2.1 transgenic mice. Values represent 2 high power fields per section in >10 representative lung sections obtained from 36 mice. Values represent means ± SEM. (D) Representative lung sections showing immunostaining for MIF in alveolar epithelial cells and infiltrating mononuclear cells in pulmonary granulomas in Mif lung-Tg2.1 mice. Sections were obtained 48 hrs after β-glucan injection and stained with anti-MIF (left panel) or control antibody (right panel). Magnification = 100×. P values by Student’s t test (two-tailed).
To determine the effect of MIF on pulmonary granuloma formation in this model of experimental vasculitis, lungs were resected 48 hours following the injection of β-glucan and examined histologically. The Mif lung –Tg2.1 mice, which showed the highest mortality in this model, exhibited more pulmonary granulomas than WT or Mif-KO mice (Mif lung-Tg2 = 10.5 ± 1.2 /mm2 vs, WT = 5.6 ± 0.9 /mm2 [p=0.002] vs. Mif-KO = 1.8 ± 0.2/mm2 [p<0.0001]) (Figure 4). To examine whether MIF is overexpressed in the pulmonary granulomas, lung sections from Mif lung-Tg2 mice were obtained 48 hours after β-glucan injection and stained with anti-MIF or control Ig. Representative lung sections demonstrating MIF immunostaining in alveolar epithelial cells and mononuclear cells within pulmonary granulomas (Figure 4).
Neutrophils are considered to play an important role in the pathogenesis of GPA (31). Because MIF attracts neutrophils via the non-cognate MIF receptor CXCR2 (32) and in some disease setting promotes the downstream expression of specific neutrophil chemo-attractants (10, 33) we measured plasma and bronchoalveolar (BAL) neutrophil and monocyte chemokines in Mif lung –Tg2.1 and WT mice. The neutrophil chemo-attractants KC and MIP-2 (CXCL2) were significantly elevated in the plasma and BAL of Mif lung –Tg2.1 mice as compared with WT (Figure 5). The monocyte chemokine MCP-1 (CCL2), which is MIF-dependent (26, 34), also was significantly elevated in the Mif lung –Tg2.1 mice, suggesting an additional role for MIF in recruiting the monocytes/macrophages that persist in granulomatous inflammation. An elevation in the levels of IL-6 – known to be MIF-dependent (35), also was observed in BAL samples from the Mif lung –Tg2.1 mice.
Figure 5.
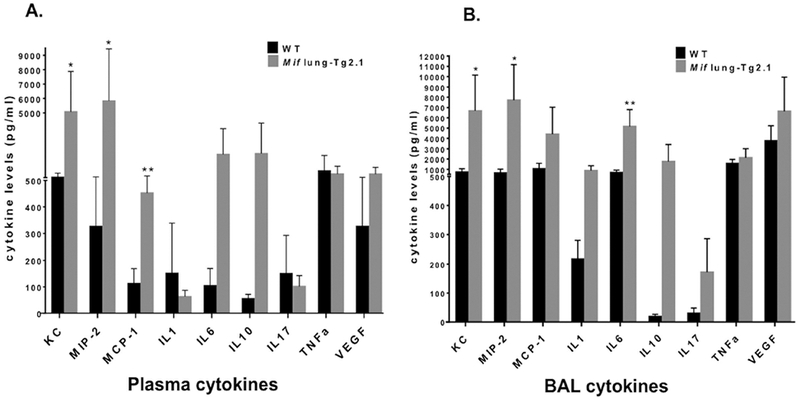
Cytokine levels (pg/ml) of KC (CXCL1), MIP-2 (CXCL2), MCP-1 (CCL2), IL-1, IL-6, IL-10, IL-17, TNF, and VEGF in wild type (WT) and Mif lung-Tg2.1 mice in plasma (A) and bronchoalveolar lavage (BAL) fluid (B). Samples were obtained 24–48 hrs after β-glucan injection. Data are mean ± SD of cytokine levels in blood and BAL samples harvested from 8 WT mice and 4 Mif lung-Tg2.1 mice. P values by Student’s t test (two-tailed).
DISCUSSION:
This study demonstrates that high MIF expression >−794 CATT5 containing genotypes are over-represented in patients with GPA as compared with healthy controls. We also show that Mif lung transgenic mice engineered to express a similar two-fold increase mRNA level as cells transfected with >−794 CATT5 human MIF promoter, have increased pulmonary granuloma formation, higher neutrophil/macrophage chemokine production, and increased mortality after β-glucan injection when compared to wild type and Mif-KO mice. Furthermore, the administration of anti-MIF monoclonal antibody to Mif lung –Tg2.1 mice improved survival. These data collectively support the role of MIF in the pathogenesis GPA.
MIF is an upstream regulatory cytokine that plays an essential role in both the expression of innate and adaptive immunity. MIF inhibits activation-induced apoptosis to sustain inflammatory signaling, prevents the egression of macrophages, and promotes the migration of neutrophils and lymphocytes into sites of inflammation (36). More recently MIF has been implicated in the formation of tissue granulomas; it is expressed in tuberculous granulomas to maintain cellular aggregation within the granuloma microenvironment (7, 25). Granuloma formation is a major component in GPA and in a relevant mouse model, Mif lung transgenic mice show higher pulmonary granuloma density than wild type or Mif-KO mice. One can hypothesize that higher MIF expression in the lungs of mice injected with β-glucan inhibits macrophage egress and promotes neutrophil and lymphocyte migration leading to higher granuloma density and higher mortality.
Neutrophils are considered a major cellular component in the pathogenesis of AAV (31). ANCA immunoglobulin G activates both neutrophils and monocytes, leading to the generation of reactive oxygen species, the degranulation of tissue-damaging proteases, and the formation of neutrophil extracellular traps (31, 37). Neutrophil and macrophage chemo-attractants such as IL-8 and MCP-1 correlate with disease activity in AAV and outcome (38). In our experimental model, Mif lung-Tg2.1 mice showed elevated production of KC and MIP-2, the murine homologues of IL-8, following β-glucan injection. In addition, the monocyte chemo-attractant MCP-1 - a key mediator for the infiltration of monocytes/macrophages, was elevated in the serum of Mif lung-Tg2.1 mice following β-glucan administration. These results suggest that MIF-dependent neutrophil and macrophage/monocytes chemokines contribute to the pathogenesis of granulomatous vasculitis in this murine model and support the potential pathogenic role of high expression MIF alleles in human AAV.
The mechanism(s) underlying the high mortality observed in the Mif lung-Tg2.1 mice after β-glucan injection remain to be elucidated but may be due to higher pulmonary granuloma burden and an elevation in the systemic inflammatory response leading to shock. β-glucan administration to mice has been reported to result in an anaphylactoid reaction (39). That lethality was abrogated by anti-MIF mAb is noteworthy and warrants further study given the ongoing clinical testing of humanized anti-MIF and anti-MIF receptor antibodies (40).
Our genetic data indicate that patients with GPA have a greater prevalence of high expression (i.e., > – 794 CATT5) MIF genotypes when compared to healthy controls. These findings mirror the associations that have been reported previously between MIF polymorphisms and disease susceptibility or clinical severity in several autoimmune diseases, including systemic lupus erythematosus (13, 41), scleroderma (14), rheumatoid arthritis (15, 42), autoimmune liver disease (43), and celiac disease (44), as well as in granulomatous conditions such as sarcoidosis (6) and tuberculosis (25, 45). Furthermore, higher MIF plasma levels were noted in patients with GPA when compared to healthy controls, a finding previously reported in ANCA-associated vasculitis and other granulomatous diseases (8, 46, 47). Recent genome-wide association studies have identified several gene polymorphisms that are associated with susceptibility to GPA. These analyses have been limited to the study of selected SNPs however, and have not examined potentially important structural variants such as promoter microsatellites (48, 49). While patients with GPA had a greater prevalence of high expression MIF genotypes when compared to healthy controls, healthy controls also had a fair percentage of high expression MIF genotypes. We suspect that other genetic predispositions contribute to higher susceptibility for GPA by interacting with the MIF gene.
This study has several strengths and weaknesses. While there is no validation cohort for this candidate gene study, the sample size is large considering the rarity of GPA, and the results confirm an a priori hypothesis derived from a functional locus defined previously in studies of autoimmunity (13, 14, 50) and of unrelated granulomatous conditions (6, 25). Importantly, this study also provides the first concordance in findings between a GPA genetic susceptibility and a relevant mouse model of vasculitis. The exact cellular composition and molecular expression profile of the pulmonary granulomas observed in the mouse model remains to be examined to better define MIF-dependent pathways of granulomatogenesis.
In conclusion, MIF appears to play an important role in the susceptibility and pathogenesis of GPA. Therapies aimed at blocking MIF-directed pathogenesis pathways may offer a promising and precision medicine approach to treating vasculitis in high genotypic MIF expressers.
Acknowledgments
Funding: This work was supported by The Vasculitis Clinical Research Consortium (VCRC) (U54 AR057319) which is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Science (NCATS). The VCRC is funded through collaboration between NCATS, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and has received funding from the National Center for Research Resources (U54 RR019497). Additional funding was provided by NIAMS R01 AR047799 and AR049610. Dr. Sreih was the recipient of a Scientist Development Award from the Research and Education Foundation of the American College of Rheumatology. Gratitude is expressed to our patients for contributing to this study.
Abbreviations
- GPA
granulomatosus with polyangiiitis
- MIF
macrophage migration inhibitory factor
- MIF
human gene for macrophage migration inhibitory factor
- Mif
mouse gene for macrophage migration inhibitory factor
- SNP
single-nucleotide polymorphism
Footnotes
COI: Dr. Bucala is listed as an inventor of Yale patents for MIF antagonists.
REFERENCES
- 1.Cartin-Ceba R, Peikert T, Specks U. Pathogenesis of ANCA-associated vasculitis. Current rheumatology reports. 2012;14(6):481–93. [DOI] [PubMed] [Google Scholar]
- 2.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis and rheumatism. 2013;65(1):1–11. [DOI] [PubMed] [Google Scholar]
- 3.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. The Journal of experimental medicine. 1994;179(6):1895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(15):7849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suga M, Yamasaki H, Nakagawa K, Kohrogi H, Ando M. Mechanisms accounting for granulomatous responses in hypersensitivity pneumonitis. Sarcoidosis, vasculitis, and diffuse lung diseases: official journal of WASOG / World Association of Sarcoidosis and Other Granulomatous Disorders. 1997;14(2):131–8. [PubMed] [Google Scholar]
- 6.Amoli MM, Donn RP, Thomson W, Hajeer AH, Garcia-Porrua C, Lueiro M, et al. Macrophage migration inhibitory factor gene polymorphism is associated with sarcoidosis in biopsy proven erythema nodosum. The Journal of rheumatology. 2002;29(8):1671–3. [PubMed] [Google Scholar]
- 7.Wang D, Zhou W, Lu S, Wang Q, Feng Y, Zhu G, et al. Increased density of macrophage migration inhibitory factor (MIF) in tuberculosis granuloma. Experimental and molecular pathology. 2012;93(2):207–12. [DOI] [PubMed] [Google Scholar]
- 8.Becker H, Maaser C, Mickholz E, Dyong A, Domschke W, Gaubitz M. Relationship between serum levels of macrophage migration inhibitory factor and the activity of antineutrophil cytoplasmic antibody-associated vasculitides. Clinical rheumatology. 2006;25(3):368–72. [DOI] [PubMed] [Google Scholar]
- 9.Djudjaj S, Lue H, Rong S, Papasotiriou M, Klinkhammer BM, Zok S, et al. Macrophage Migration Inhibitory Factor Mediates Proliferative GN via CD74. Journal of the American Society of Nephrology: JASN. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly SC, Haslett C, Reid PT, Grant IS, Wallace WA, Metz CN, et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nature medicine. 1997;3(3):320–3. [DOI] [PubMed] [Google Scholar]
- 11.Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, et al. Proinflammatory Signature of the Dysfunctional Endothelium in Pulmonary Hypertension. Role of the Macrophage Migration Inhibitory Factor/CD74 Complex. American journal of respiratory and critical care medicine. 2015;192(8):983–97. [DOI] [PubMed] [Google Scholar]
- 12.Yao J, Leng L, Sauler M, Fu W, Zheng J, Zhang Y, et al. Transcription factor ICBP90 regulates the MIF promoter and immune susceptibility locus. The Journal of clinical investigation. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sreih A, Ezzeddine R, Leng L, LaChance A, Yu G, Mizue Y, et al. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis and rheumatism. 2011;63(12):3942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu SP, Leng L, Feng Z, Liu N, Zhao H, McDonald C, et al. Macrophage migration inhibitory factor promoter polymorphisms and the clinical expression of scleroderma. Arthritis and rheumatism. 2006;54(11):3661–9. [DOI] [PubMed] [Google Scholar]
- 15.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes and immunity. 2002;3(3):170–6. [DOI] [PubMed] [Google Scholar]
- 16.Group WR. Design of the Wegener’s Granulomatosis Etanercept Trial (WGET). Controlled clinical trials. 2002;23(4):450–68. [DOI] [PubMed] [Google Scholar]
- 17.Bloch DA, Michel BA, Hunder GG, McShane DJ, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Patients and methods. Arthritis and rheumatism. 1990;33(8):1068–73. [DOI] [PubMed] [Google Scholar]
- 18.Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, et al. The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. Faseb J. 2009;23(8):2403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grigorenko EL, Han SS, Yrigollen CM, Leng L, Mizue Y, Anderson GM, et al. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics. 2008;122(2):e438–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry. 1994;33(47):14144–55. [DOI] [PubMed] [Google Scholar]
- 21.McClintock SD, Barron AG, Olle EW, Deogracias MP, Warner RL, Opp MR, et al. Role of interleukin-6 in a glucan-induced model of granulomatous vasculitis. Experimental and molecular pathology. 2007;82(2):203–9. [DOI] [PubMed] [Google Scholar]
- 22.Ohno N A murine model of vasculitis induced by fungal polysaccharide. Cardiovascular & hematological agents in medicinal chemistry. 2008;6(1):44–52. [DOI] [PubMed] [Google Scholar]
- 23.Sun H, Choo-Wing R, Sureshbabu A, Fan J, Leng L, Yu S, et al. A critical regulatory role for macrophage migration inhibitory factor in hyperoxia-induced injury in the developing murine lung. PloS one. 2013;8(4):e60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das R, Koo MS, Kim BH, Jacob ST, Subbian S, Yao J, et al. Macrophage migration inhibitory factor (MIF) is a critical mediator of the innate immune response to Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(32):E2997–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leng L, Chen L, Fan J, Greven D, Arjona A, Du X, et al. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. Journal of immunology. 2011;186(1):527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong XB, Leng L, Beitin A, Chen R, McDonald C, Hsiao B, et al. Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic acids research. 2005;33(13):e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee WC, Wang LY. Simple formulas for gauging the potential impacts of population stratification bias. American journal of epidemiology. 2008;167(1):86–9. [DOI] [PubMed] [Google Scholar]
- 29.Mahr AD, Neogi T, Merkel PA. Epidemiology of Wegener’s granulomatosis: Lessons from descriptive studies and analyses of genetic and environmental risk determinants. Clinical and experimental rheumatology. 2006;24(2 Suppl 41):S82–91. [PubMed] [Google Scholar]
- 30.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, et al. Migration inhibitory factor expression in experimentally induced endotoxemia. The American journal of pathology. 1997;150(1):235–46. [PMC free article] [PubMed] [Google Scholar]
- 31.Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nature reviews Rheumatology. 2014;10(8):463–73. [DOI] [PubMed] [Google Scholar]
- 32.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature medicine. 2007;13(5):587–96. [DOI] [PubMed] [Google Scholar]
- 33.Santos LL, Fan H, Hall P, Ngo D, Mackay CR, Fingerle-Rowson G, et al. Macrophage migration inhibitory factor regulates neutrophil chemotactic responses in inflammatory arthritis in mice. Arthritis and rheumatism. 2011;63(4):960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoi AY, Hickey MJ, Hall P, Yamana J, O’Sullivan KM, Santos LL, et al. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. Journal of immunology. 2006;177(8):5687–96. [DOI] [PubMed] [Google Scholar]
- 35.de Jong YP, Abadia-Molina AC, Satoskar AR, Clarke K, Rietdijk ST, Faubion WA, et al. Development of chronic colitis is dependent on the cytokine MIF. Nature immunology. 2001;2(11):1061–6. [DOI] [PubMed] [Google Scholar]
- 36.David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proceedings of the National Academy of Sciences of the United States of America. 1966;56(1):72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreiber A, Choi M. The role of neutrophils in causing antineutrophil cytoplasmic autoantibody-associated vasculitis. Current opinion in hematology. 2015;22(1):60–6. [DOI] [PubMed] [Google Scholar]
- 38.Ohlsson S, Bakoush O, Tencer J, Torffvit O, Segelmark M. Monocyte chemoattractant protein 1 is a prognostic marker in ANCA-associated small vessel vasculitis. Mediators of inflammation. 2009;2009:584916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinohara H, Nagi-Miura N, Ishibashi K, Adachi Y, Ishida-Okawara A, Oharaseki T, et al. Beta-mannosyl linkages negatively regulate anaphylaxis and vasculitis in mice, induced by CAWS, fungal PAMPS composed of mannoprotein-beta-glucan complex secreted by Candida albicans. Biological & pharmaceutical bulletin. 2006;29(9):1854–61. [DOI] [PubMed] [Google Scholar]
- 40.Wallace DJ, Wegener WA, Horne H, Goldenberg DM. CT-01|Phase IB study of IMMU-115 (humanised ANTI-CD74 antibody) targeting antigen presenting cells in patients with systemic lupus erythematosus (SLE). Lupus Science & Medicine. 2016;3(Suppl 1):A37–A8. [Google Scholar]
- 41.De la Cruz-Mosso U, Bucala R, Palafox-Sanchez CA, Parra-Rojas I, Padilla-Gutierrez JR, Pereira-Suarez AL, et al. Macrophage migration inhibitory factor: association of −794 CATT5–8 and −173 G>C polymorphisms with TNF-alpha in systemic lupus erythematosus. Human immunology. 2014;75(5):433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llamas-Covarrubias MA, Valle Y, Bucala R, Navarro-Hernandez RE, Palafox-Sanchez CA, Padilla-Gutierrez JR, et al. Macrophage migration inhibitory factor (MIF): genetic evidence for participation in early onset and early stage rheumatoid arthritis. Cytokine. 2013;61(3):759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, et al. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology. 2014;59(2):580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nunez C, Rueda B, Martinez A, Lopez-Nevot MA, Fernandez-Arquero M, de la Concha EG, et al. Involvement of macrophage migration inhibitory factor gene in celiac disease susceptibility. Genes and immunity. 2007;8(2):168–70. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Yuan T, Lu W, Chen M, Cheng X, Deng S. Association of tuberculosis and polymorphisms in the promoter region of macrophage migration inhibitory factor (MIF) in a Southwestern China Han population. Cytokine. 2012;60(1):64–7. [DOI] [PubMed] [Google Scholar]
- 46.Wendt M, Borjesson O, Avik A, Bratt J, Anderstam B, Qureshi AR, et al. Macrophage migration inhibitory factor (MIF) and thyroid hormone alterations in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). Molecular medicine. 2013;19:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohwatari R, Fukuda S, Iwabuchi K, Inuyama Y, Onoe K, Nishihira J. Serum level of macrophage migration inhibitory factor as a useful parameter of clinical course in patients with Wegener’s granulomatosis and relapsing polychondritis. The Annals of otology, rhinology, and laryngology. 2001;110(11):1035–40. [DOI] [PubMed] [Google Scholar]
- 48.Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA-associated vasculitis. The New England journal of medicine. 2012;367(3):214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of Functional and Expression Polymorphisms Associated With Risk for Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis. Arthritis & rheumatology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, et al. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis and rheumatism. 2005;52(10):3020–9. [DOI] [PubMed] [Google Scholar]