

Abstract
Metabolic alterations are increasingly recognized as important novel anti‐cancer targets. Among several regulators of metabolic alterations, fructose 2,6 bisphosphate (F2,6BP) is a critical glycolytic regulator. Inhibition of the active form of PFKFB3ser461 using a novel inhibitor, PFK158 resulted in reduced glucose uptake, ATP production, lactate release as well as induction of apoptosis in gynecologic cancer cells. Moreover, we found that PFK158 synergizes with carboplatin (CBPt) and paclitaxel (PTX) in the chemoresistant cell lines, C13 and HeyA8MDR but not in their chemosensitive counterparts, OV2008 and HeyA8, respectively. We determined that PFK158‐induced autophagic flux leads to lipophagy resulting in the downregulation of cPLA2, a lipid droplet (LD) associated protein. Immunofluorescence and co‐immunoprecipitation revealed colocalization of p62/SQSTM1 with cPLA2 in HeyA8MDR cells uncovering a novel pathway for the breakdown of LDs promoted by PFK158. Interestingly, treating the cells with the autophagic inhibitor bafilomycin A reversed the PFK158‐mediated synergy and lipophagy in chemoresistant cells. Finally, in a highly metastatic PTX‐resistant in vivo ovarian mouse model, a combination of PFK158 with CBPt significantly reduced tumor weight and ascites and reduced LDs in tumor tissue as seen by immunofluorescence and transmission electron microscopy compared to untreated mice. Since the majority of cancer patients will eventually recur and develop chemoresistance, our results suggest that PFK158 in combination with standard chemotherapy may have a direct clinical role in the treatment of recurrent cancer.
Keywords: ovarian and cervical cancer, Chemoresistance, PFKFB3, lipid droplet, lipophagy
Short abstract
What's new?
Ovarian and cervical cancer patients experience high rates of chemoresistance and tumor recurrence. To improve patient outcome, greater understanding of mechanisms behind these phenomena is needed. Here, activity of PFKFB3, a glycolytic regulator overexpressed in cancer, was found to be positively correlated with chemoresistance and lipid droplet (LD) biogenesis in ovarian and cervical cancer cells. PFK‐158, a PFKFB3 inhibitor, sensitized chemoresistant cells to drug‐induced cytotoxicity by simultaneously targeting both glycolytic and lipogenic pathways to inhibit tumor growth and LDs in a drug‐resistant xenograft model. The findings warrant further investigation of PFK158 as a treatment for recurrent gynecological malignancy.
Abbreviations
- AA
arachidonic acid
- ACC
acetyl co‐A carboxylase
- ACLY
ATP citrate lyase
- BafA
bafilomycin A
- CBPt
carboplatin
- CE
cholesterol esters
- CI
combination index
- DRI
dose reduction index
- F2,6BP
Fructose 2,6bis‐phosphate
- FASN
fatty acid synthase
- FTE
fallopian tube epithelial
- IFC
immunofluorescence
- IHC
immunohistochemistry
- LCFA
long chain fatty acids
- LD
lipid droplet
- OC
ovarian cancer
- PFKFB3
6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase 3
- PTX
paclitaxel
- TAG
triacylglycerol
- TG
triglycerides
Introduction
Ovarian Cancer (OC) and cervical cancer are the two most lethal gynecological cancers with an incidence rate of 22,000 and 12,990 new diagnoses, respectively, in United States.1 Although most patients initially respond to chemotherapy, 70% of the patients recur with tumors typically resistant to chemotherapy.2 Among, the various mechanisms of chemo‐resistance, increasing evidence indicate that metabolic alterations involving the glycolytic and lipid pathways play a vital role in the induction of drug resistance.3 Understanding what molecular alterations lead to increased glycolytic flux is important to successfully target this pathway toward limiting the progression of the disease and/or response to chemotherapy.
A critical factor in controlling glycolytic rate is Fructose 2,6 bis‐phosphate (F2,6BP) levels. F2,6BP is generated mainly by the action of 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase 3 enzyme (PFKFB3)4 which is overexpressed in cancer and is regulated by HIF‐1α or by phosphorylation by AMPK, MAPK and other kinases.5, 6 The roles of PFKFB3 and F2,6BP in cell cycle regulation and angiogenesis is well documented.7, 8 Importantly, upregulation of PFKFB3 activity is directly associated with enhanced glycolytic flux, while genetic or pharmacological inhibition has been shown to reduce glycolytic flux and inhibit tumor growth in vivo.9 Moreover, in case of aggressive cancers, augmented glycolysis is functionally linked to enhanced lipogenesis.10 Enhanced lipogenesis is the result of higher activity of lipogenic enzymes such as fatty acid synthase (FASN), acetyl Co‐A carboxylase (ACC) and ATP citrate lyase (ACLY). The higher amount of fatty acids generated by the action of these enzymes are then esterified and stored as triacylglycerol (TAG) and cholesterol esters (CE) in lipid droplets (LD).11Under nutrient‐deprived conditions, the free long‐chain fatty acids (LCFA) and cholesterols are enzymatically released from LDs by the sequential action of lipases such as ATGL, HSL and MAGL.12 The released FAs are oxidized to provide more energy to rebuild the cells and the excess FA is re‐esterified to be stored in LDs to prevent lipotoxicity. Thus, cancer cells utilize both lipogenic and lipolytic pathways to acquire fatty acids13 which, in turn, promote cancer cell proliferation and survival.14, 15 Interestingly, LD‐rich cancer cells also exhibit resistance to chemotherapeutic agents which suggests a crucial role of lipids in drug resistance.16, 17
In OC, very little is known about the expression, activation and role of PFKFB3. In the present study, we demonstrated a higher PFKFB3 activity in OC and cervical cancer which correlated with enhanced chemoresistance. Further, we used a potent and selective inhibitor of PFKFB3, PFK158,18 and showed that inhibition of PFKFB3 activity is associated with chemo‐sensitization to standard chemotherapy. Additionally, we report a novel crosstalk between the glycolytic and lipogenic pathways modulated by PFK158 in gynecologic cancer.
Materials and Methods
Materials
PFK158 is developed and supplied by Advanced Cancer Therapeutics (ACT). For the in vivo studies specifically formulated PFK158 was dissolved in PBS and for in vitro studies PFK158 was dissolved in DMSO. The materials, antibodies and reagents used in our study are listed in Table S1, Supporting Information.
Cell culture
The human OC cell lines HeyA8, HeyA8MDR and OVCAR5 cells were grown in DMEM medium (4.5 g/l glucose) supplemented with 10% fetal bovine serum, whereas cervical cancer cell lines OV2008 and C13 were grown in RPMI‐1640 medium (4.5 g/l glucose). OV90 cells were grown in OSE media consisting of 50:50 medium 199:105 supplemented with 15% fetal bovine serum, and sodium bi‐carbonate (2.2 g/l).
Cell viability and drug combination assay
Cells were seeded in 96‐well plates, treated with increasing concentrations of drugs for 48 h and inhibitory concentration 50% (IC50) values were determined by MTT assays as previously described.19 To determine if PFK158 acts synergistically with cisplatin and carboplatin, constant ratio synergy studies were performed using the Chou–Talalay method20 and the combination index (CI) and dose reduction index (DRI) were calculated using CalcuSyn software (Biosoft, USA).To determine the role of autophagy in PFK158‐mediated sensitization of chemoresistant cells, cells were pretreated with 50 nM Bafilomycin A (BafA) for 2 h and were further treated with increasing concentrations of Carboplatin (CBPt) plus PFK158. After 24 h, cell viability was assessed by MTT.
Apoptosis assay
Briefly, cells (1 × 106) were treated with PFK158, CBPt, Paclitaxel (PTX) alone and in combination, and Phosphatidyl‐serine externalization was analyzed by double staining the cells with FITC‐Annexin V and PI (5 μg/mL). Cells were acquired and analyzed by CellQuest Pro software (BD FACSCalibur) as previously described.21
Cell imaging using 2‐NBDG
Glucose uptake of the live cells was measured using 2‐NBDG as previously described.22 Briefly, cells were treated with 5 μMPFK158 in glucose‐free medium for 30 min along with 2‐NBDG and then examined under Zeiss‐LSM 510 fluorescence microscope. The fluorescent intensities were calculated using Image J software as previously reported.22
2‐[1‐14C]‐Deoxy‐d‐glucose uptake assay
Cells seeded overnight were treated with increasing concentration of PFK158 (0–15 μM) for 1 h, washed with PBS and placed in glucose‐free media for 30 min. 2‐[1‐14C]‐deoxy‐d‐glucose (0.05 μCi/mL; Perkin Elmer) was added and cells were further incubated for 1 h. After washing with PBS, cell lysates were prepared in 500 μL 0.5% SDS and 400 μL lysate was added to 5 mL scintillation fluid and counts were measured on a Tri‐Carb 2,910 liquid scintillation analyzer (Perkin‐Elmer). Counts were then normalized to protein concentration measured by Bradford assay and data are represented as percent control of untreated cells.
Immunoprecipitation assay
Cell lysates containing 500 μg of protein were incubated with 2 μg of anti‐cPLA2 or anti‐p62/SQSTM1 antibody at 4°C for 24 h, and then 10 μL of 50% protein A‐agarose beads were added and mixed at 4 °C for 24 h. The immunoprecipitates were collected and washed thrice with lysis buffer, subjected to electrophoresis on 4–20% SDS‐PAGE and blotted using an anti‐p62/SQSTM1 or anti‐cPLA2 or anti‐pcPLA2 antibody.
Generation of PFKFB3 downregulated stable clones
PFKFB3 downregulation was performed in HeyA8MDR, C13, OV90 and OVCAR5 cells with ShPFKFB3[Sh55:CGGGTGCATGATTGTGCTTAA (targeting 3′UTR), Sh59:CCACCAATACTACTAGAGAGA (targeting 5′ UTR)] using standard transfection protocol and reagents.23
Bodipy staining
Cells were fixed in 4% paraformaldehyde for ten minutes and stained with Bodipy as previously.24 Stained cells were then examined under Zeiss‐LSM 510 fluorescence microscope.
Immunofluorescence (IFC)
after transient transfection in HeyA8MDR cells for 48 h with Cherry‐GFP‐LC3B, cells were treated with increasing concentrations (0‐15 μM) of PFK158. Induction of autophagy by PFK158 resulted in an increase in the mCherry positive signals in the treated cells compared to untreated cells. Quantification of red fluorescent protein (RFP) was measured using Image J software.
Cells were grown on coverslips overnight, and then fixed with 100% methanol followed by blocking with 1% BSA in PBS and IFC was performed as previously described.25 Stained samples were visualized using a Zeiss‐LSM 510 fluorescence microscope.
Tumor xenograft
Female athymic nude mice (nu/nu) (5–6 weeks old) were randomized in six groups (n = 10) and HeyA8MDR cells (3 × 106) were injected intraperitoneally (i.p). Seven days after i.p., mice were treated with (i) PBS for control group, (ii) 25 mg/kg of PFK158 every 3rd day, (iii) 51 mg/kg of CBPt every 3rd day, (iv) PTX (15 mg/kg) every 5th day, (v) combination of CBPt (51 mg/kg) and PFK158 (15 mg/kg) once a week, and (vi) PTX (5 mg/kg) and PFK158 (25 mg/kg) once a week. The treatments were continued until the end of the study (28 days), however, control mice were sacrificed during week 3 as the tumor burden exceeded 10% of their body weight. Tumors and tissues were excised and preserved either in formalin or −80 °C. Mayo Clinic Animal Care and Use Committee approved all procedures. All experimental use of animals complied with the guidelines of the Animal Care and Use Committee at the Mayo Foundation, in accordance with approved protocols.
Immunohistochemistry
Immunohistochemical staining using Ki67 antibody on paraffin‐embedded sections from the treated and untreated xenografts and frozen tissue sections were stained with Bodipy to detect LDs as previously described.23
TUNEL staining
Terminal dUTP Nick End‐Labeling (TUNEL) assay on frozen tissue sections of tumors from control, PFK158, CBPt and PFK158 plus CBPt‐treated mice was performed using In Situ Apoptosis Detection Kit according to the manufacturer's instructions.
Statistical analyses
All results were expressed as mean ± standard deviation. Data were obtained from three separate experiments. All statistical analyses were performed using the GraphPad Prism software (San Diego, CA). Data were analyzed using t‐test as appropriate. A level of P less than 0.05 was considered statistically significant unless otherwise noted.
Results
Increased activity of PFKFB3 in chemoresistant cells
To determine the differential expression and activation of PFKFB3, we performed immunohistochemistry (IHC) on formalin‐fixed paraffin embedded tissues (FFPE) using both PFKFB3 and a phospho‐specific antibody against Ser461 (PFKFB3ser461). Activation of PFKFB3 by phosphorylation at Ser461 is a crucial event in regulation of glycolysis in different cancers.26 Our results demonstrated that total PFKFB3 (t‐PFKFB3) is expressed both in the normal ovaries and OC, while p‐PFKFB3ser461 showed tumor‐specific expression (Fig. 1 a and Fig. S1, Supporting Information). Consistent with this, western blot analysis showed negligible expression of p‐PFKFB3ser461 in two fallopian tube epithelial (FTE) cell lines (Fig. 1 b). Interestingly, immunoblot analysis further revealed increased p‐PFKFB3 levels in isogenic resistant C13 and HeyA8MDR cells compared to their sensitive counterparts, OV2008 and HeyA8, respectively (Fig. 1 c). Moreover, consistent with the increased p‐PFKFB3 level, we found a two‐fold increase in F2,6BP in C13 cells compared to OV2008 cells (8.87 vs 4.6 pmol/mg protein) (Fig. 1 d) and enhanced PFK activity in resistant C13 and HeyMDR cells compared to their isogenic sensitive counterparts (Fig. 1 e).
Figure 1.
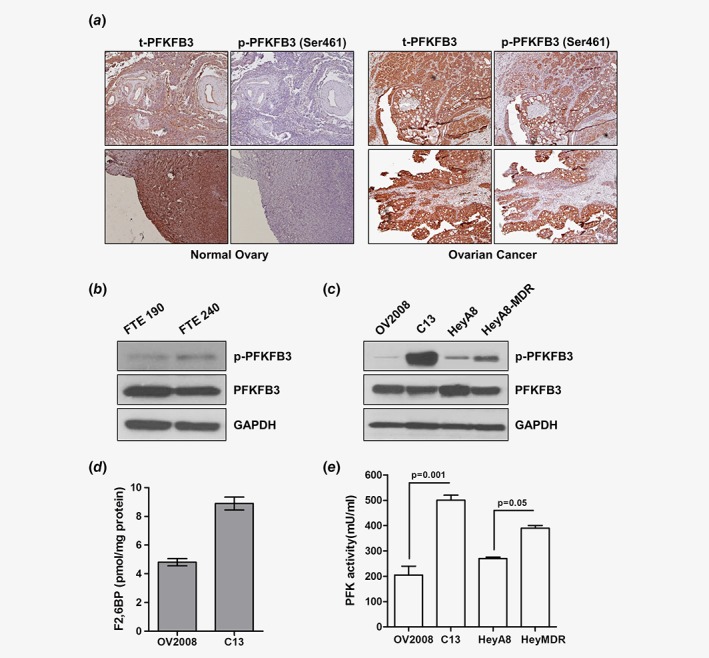
Ovarian tumors and cells express high PFKFB3 activity. (a) Representative images of immunohistochemical staining of phospho (p)‐PFKFB3 (ser461) and total (t)‐PFKFB3 performed in ovarian tumor tissue and normal ovary. p‐PFKFB3 showed tumor‐specific expression. Immunoblot analysis of p‐PFKFB3 and t‐PFKFB3 in FTE (fallopian tube epithelial) cells (b) and in OV2008, C13, HeyA8 and HeyA8MDR cells (c) showed that the resistant cell lines have higher levels of p‐PFKFB3. (d) F2,6BP assay performed in OV2008 and C13 cells as described in the methods section. (e) Total PFK activity in OV2008, C13, HeyA8 and HeyA8MDR cells were determined with PFK activity assay. All experiments were repeated at least three times. Data are shown as mean ± SD of 3 replicates per cell line. *p < 0.05; **p < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
PFKFB3 expression directly correlates with higher glycolysis in chemoresistant cells
In order to assess whether chemoresistant cells exhibit higher glycolytic rate, we determined glucose uptake in the paired isogenic sensitive and resistant cell lines. Results showed that the resistant C13 and HeyA8MDR cells had higher glucose uptake as determined using 2NBDG and 14C 2‐Deoxy‐D Glucose analog (Fig. 2 a, b and c). Along with the higher glucose uptake, chemoresistant cells exhibited enhanced ATP levels (Fig. 2 d), lactate production (Fig. 2 e) and higher lactate dehydrogenase activity (Fig. 2 f) compared to its sensitive counterparts OV2008 and HeyA8, suggesting a higher glycolytic rate in chemoresistant cells. Consistent with this observation, resistant C13 and HeyA8MDR cells exhibited a lower oxygen consumption rate (OCR) compared to their sensitive counterparts OV2008 and HeyA8 (Fig.S2A‐B, Supporting Information) suggesting compromised mitochondria function in resistant cells.
Figure 2.
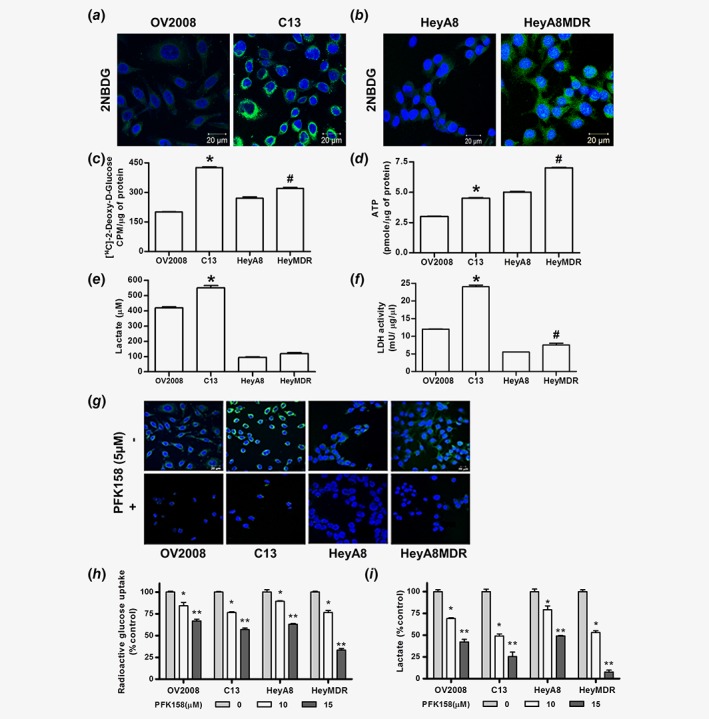
Chemoresistant cells manifest increased glucose uptake, lactate release, intracellular ATP and LDH activity which is inhibited by PFK158 treatment. Fluorescence images of glucose uptake using 2‐NBDG in OV2008 and C13 (a), HeyA8 and HeyA8MDR (b) cells. Glucose uptake using [1‐14C] 2‐deoxy‐d‐glucose (c), intracellular ATP (d), lactate release (e) and LDH activity (f) were measured in OV2008, C13, HeyA8 and HeyA8MDR cells, where *p < 0.01 and #p < 0.05. Glucose uptake using 2‐NBDG dye (g), 14C‐2‐DG glucose (h) and lactate release (i) in the presence and absence of PFK158 in OV2008, C13, HeyA8 and HeyA8MDR cells. All experiments were repeated at least three times. Data are shown as mean ± SD for 3 replicates per cell line. *p < 0.05; **p < 0.01. [Color figure can be viewed at wileyonlinelibrary.com]
To confirm whether the expression level of PFKFB3 has any impact on the higher glycolytic rate of chemoresistant cells, we further investigated glucose uptake after genetic manipulation. ShRNA‐mediated downregulation of PFKFB3 (Fig.S2C, Supporting Information) reduced glucose uptake in C13 cells (Fig.S2D, Supporting Information), while enhanced expression (Fig.S2E, Supporting Information) in OV2008 cells promoted more glucose uptake compared to vector‐transfected cells (Fig.S2F, Supporting Information), suggesting a direct correlation between PFKFB3 expression and glycolysis.
Next, to elucidate the role of the PFKFB3 inhibitor, PFK158, on glycolysis, we examined glucose uptake and lactate production after PFK158 treatment. Results showed that PFK158 treatment significantly decreased glucose uptake (Fig. 2 g and h, respectively) and lactate production (Fig. 2 i) in a dose‐dependent manner. Interestingly, the dose‐dependent reduction was more pronounced in the resistant cells compared to their sensitive counterparts. Collectively, these results suggest that 1) PFKFB3 is a crucial regulator of glycolysis, 2) chemoresistant cells have higher glycolytic rate and 3) pharmacological inhibition with PFK158 exerts stronger inhibitory effects on glycolysis in resistant cells.
PFK158 treatment sensitizes chemoresistant cells and induces cell death
To determine if PFK158 will synergize with cisplatin and/ or CBPt, constant ratio synergy studies were performed in the paired isogenic chemosensitive and chemoresistant cells by treating them with a combination of cisplatin with PFK158 as well as CBPt with PFK158 in an equipotent ratio (IC50:IC50). C13 cells are cisplatin resistant and originally derived from OV2008 cells.16 Our data indicate that PFK158 has synergistic anti‐proliferative effects in vitro when combined with cisplatin in C13 and HeyA8MDR cells compared to OV2008 and HeyA8, respectively (Fig. S3A‐C and E‐G, Supporting Information). Similar results were obtained with CBPt (Fig.S3D and H, Supporting Information, respectively). While the mechanistic basis for the differences in synergy in the sensitive and resistant cells is unknown, we propose that the differences in the glycolytic rate (Fig. 2) could be, at least in part, a contributing factor.
Next, we sought to investigate whether PFK158 in combination with CBPt or PTX could induce a synergistic effect in cell death. We thus determined the rate of apoptosis induced by PFK158 alone and in combination with CBPt and PTX in sensitive and resistant cells. Flow cytometry analysis of Annexin V/PI staining showed that the combined treatment of PFK158 and CBPt resulted in significant increase in apoptosis in C13 (45%) compared to OV2008 cells (24.6%) (Fig. 3 a and b). Similar analysis using a combination of PFK158 and PTX showed a marked increase in apoptosis in the HeyA8MDR (70%) cells compared to HeyA8 (48%), respectively (Fig. 3 c and d).
Figure 3.
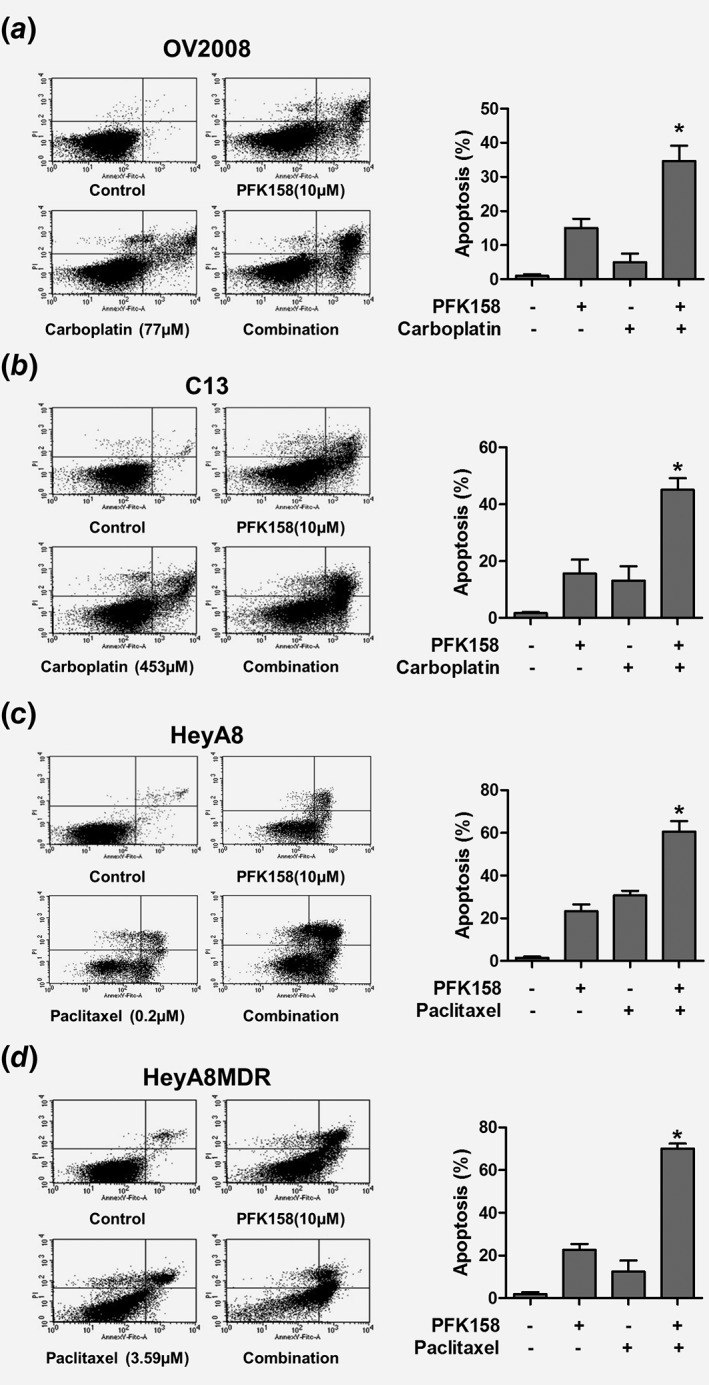
Combined treatment of PFK158 and chemotherapeutic agents enhances cell death. AnnexinV/Propidium Iodide (PI) staining of OV2008 (a), C13 (b), HeyA8 (c) and HeyA8MDR (d) cells treated with PFK158 and/or cotreated with CBPt or PTX for 24 h shows that the combination of PFK158 with chemotherapeutic agents was more effective in inducing cell death in the resistant cells. Data are representative of three independent experiments where *P < 0.001.
PFK158 modulates LD biogenesis
Since elevated glucose utilization in cancer supports lipogenesis at multiple levels13 and considering that it could be another contributing factor toward chemoresistance, we checked whether PFK158 treatment could modulate lipid pathways. Results showed that the C13 and HeyA8MDR cells had more LDs compared to the chemosensitive cells (Fig. 4 a and b) and PFK158 treatment significantly reduced the number of LDs in these cells (Fig. 4 c and d). Of interest, genetic downregulation of PFKFB3 in C13 and HeyA8MDR cells (Fig. 4 e and g) also resulted in a reduction of LDs (Fig. 4 f and h). Activated cPLA2 (p‐cPLA2ser505) is an important LD‐associated protein and crucially implicated in the biogenesis of LDs.27 To understand if cPLA2 was involved in PFKFB3‐mediated LD biogenesis, we checked its expression by immunoblot analysis. Results showed there was a dose‐dependent decrease in p‐PFKFB3ser461 and p‐cPLA2ser505 in C13 and HeyA8MDR (Fig. 4 i and j). Moreover, transient overexpression of PFKFB3 in OV2008 and HeyA8 cells resulted in increased LDs (Fig. 4 k‐n). These results indicated an intrigue involvement of PFKFB3 in LD biogenesis. To further establish this, we used a high‐grade serous cell line OV90. Results demonstrated a dose‐dependent decrease in p‐PFKFB3, p‐cPLA2 and LD levels after PFK158 treatment (Fig. S4A and B, Supporting Information). Also, genetic downregulation of PFKFB3 in OV90 cells resulted in the reduction of LDs (Fig. S4C and D, Supporting Information).
Figure 4.
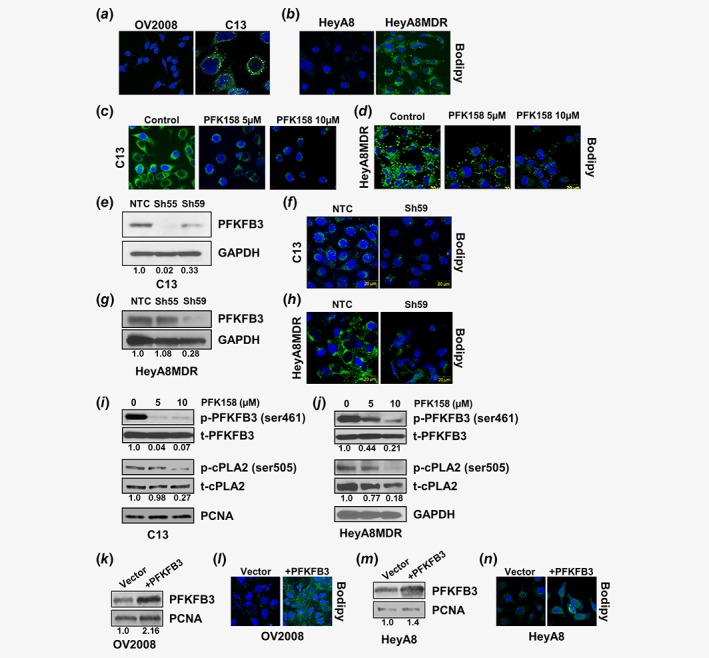
Inhibition of PFKFB3 triggers degradation of LDs. Chemosensitive (OV2008, HeyA8) and chemoresistant (C13, HeyA8MDR) cells were labeled with Bodipy 493/503 (green) and DAPI (Blue) to determine the cytoplasmic LDs (a‐b) and nuclei, respectively. Bodipy staining performed in C13 (c) and HeyA8MDR (d) cells to detect LDs in PFK158 (0‐10 μM) treated cells. Bodipy staining performed to assess LDs after transient downregulation of PFKFB3 (confirmed by western blot; e, g) in C13 (f) and HeyA8MDR (h) cells. Protein expression of p‐PFKFB3, t‐PFKFB3, p‐cPLA2, t‐cPLA2 and GAPDH are detected by western blotting in PFK158 treated (0‐10 μM) C13 (i) and HeyA8MDR (j) cells. Bodipy staining of LDs in OV2008 (l) and HeyA8 (n) cells after transient transfection with either empty vector or PFKFB3 (verified by western blot) (k, m). [Color figure can be viewed at wileyonlinelibrary.com]
PFK158 significantly increases autophagic flux to promote lipophagy
Total intracellular LDs are regulated by a lysosomal degradative pathway of autophagy known as lipophagy.28 As increased LDs were associated with chemoresistance, we evaluated whether PFKFB3 inhibition could induce lipophagy by reducing the number of LDs. Results showed a dose‐dependent downregulation of p62/SQSTM1 and upregulation of LC3BII, two markers of autophagy induction, in both C13 and HeyA8MDR cells (Fig. 5 a and b). Moreover, PFK158 treatment reduced the numbers of LDs and inhibition of autophagolysosome formation by BafA nullified the effect of PFK158 in both cell lines (Fig. 5 c and d) suggesting a selective role of lipophagy in PFK158‐mediated loss of LDs. Consistent with this observation, immunoblot analysis showed that the PFK158‐induced upregulation of LC3B‐II was significantly more increased in the presence of BafA (Fig. 5 e and f). Although the increase in LC3B‐II is indicative of autophagic flux, autophagic flux is reliably monitored with the use of fluorescent‐tagged Cherry‐GFP‐LC3B construct after autophagy induction.29 Since the pH‐sensitive GFP signal is quenched in the lysosomes, the autophagosomes light up as both red and green, while autolysosomes light up as mostly red. In HeyA8MDR, increasing concentration of PFK158 showed induction of autophagy as confirmed by an increase in the mCherry positive signals in the treated cells (Fig. 5 g, panels 2–4) compared to untreated control cells (Fig. 5 g, panel 1) which clearly indicate that activation of autophagic flux is both dose‐ (Fig. 5 h) and time‐dependent (Fig. 5 i).
Figure 5.
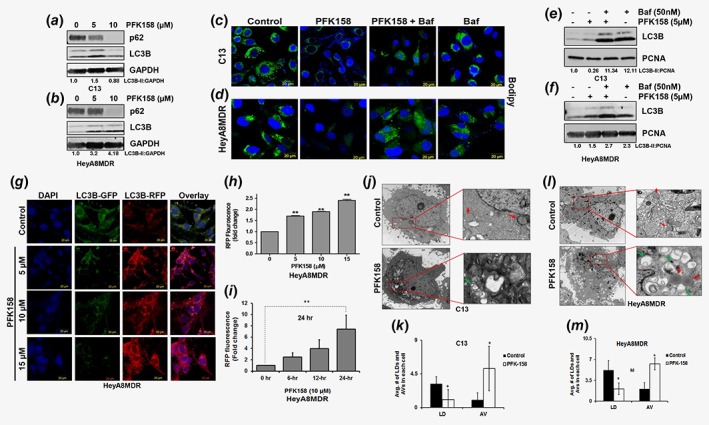
PFK158 treatment triggers autophagy and degrades LDs in an autophagy‐dependent manner. (a–b) C13 and HeyA8MDR cells were treated with PFK158 (0‐10 μM) for 24 h and cell lysates were evaluated with immunoblot analysis using anti‐p62/SQSTM1, anti‐LC3B and anti‐GAPDH antibodies. (c–d) Bodipy staining of LD in C13 and HeyA8MDR cells treated with PFK158 (5 μM) for 24 h with or without co‐treatment of bafilomycin A (50 nM) for the last 12 h. (e–f) Immunoblot analysis of LC3B in C13 and HeyA8MDR cells treated with PFK158 (5 μM) for 24 h with or without bafilomycin A (50 nm) treatment for the last 12 h. PCNA is used as loading control. (g) Autophagic flux in 24 h PFK158 (0‐15 μM) treated HeyA8MDR cells after transient expression of Cherry‐GFP‐LC3B (48 h). Confocal microscopy analysis showed changes in the autolysosome formation showing enhancement of autophagy is both drug‐ and time‐dependent (h, i). The fold changes in each concentration of PFK158 compared to control are statistically significant, where **p < 0.01. (j, l). Trans‐electron microscopic images of LD (red arrow) and autophagic vesicles (green arrow) are shown in untreated and PFK158 treated C13 and HeyA8MDR cells. Quantification of LD and autophagic vesicles counted in 25 cells are shown in (k–m) (*p = <0.05), respectively. [Color figure can be viewed at wileyonlinelibrary.com]
Additionally, we analyzed PFK158‐treated C13 and HeyA8MDR cells for autophagic vesicles formation and quantified the number of LDs by transmission electron microscopy analysis. Results showed that while PFK158 treatment significantly reduced the number of LDs in C13 and HeyA8MDR cells (red arrows) (Fig. 5 j and l, respectively), it resulted in significantly higher numbers of autophagic vesicles (AVs; green arrows) when compared to untreated cells (insets in Fig. 5j and l, lower panels). Quantification of LDs and AVs in 25 cells after PFK158 treatment (Fig. 5 k and m) further confirmed the trend and suggests that PFKFB3 inhibition induces lipophagy.
Inhibition of PFK158‐induced lipophagy reverses PFK158‐induced chemosensitivity
There are reports that showed LD‐rich cancer cells exhibit resistance to chemotherapeutic agents.30 Therefore, to understand whether induction of lipophagy by PFK158 is essential for sensitizing cancer cells to CBPt/PTX‐induced cytotoxicity, we pretreated cells with BafA for 2 h and measured the cytotoxicity. Results showed that attenuation of autophagy by BafA diminished the synergistic cytotoxic effect of PFK158 plus CBPt and that this was more pronounced in the chemoresistant C13 cells compared to the chemosensitive OV2008 cells (Fig. S5A and B, Supporting Information). These results suggest that PFK158‐induced autophagy may be one of the factors promoting synergy with standard chemotherapeutic agents in the chemoresistant cells.
To further confirm the role of PFKFB3 in chemoresistance, we transiently over expressed PFKFB3 in OV2008 and HeyA8 cells (western blots in Fig. S5, Supporting Information) and determined that the synergy we observed between PFK158 and CBPt in resistant cells was autophagy‐dependent. Our results showed pretreatment with 50 nM BafA for 2 h resulted in increased resistance to CBPt in PFKFB3 overexpressing OV2008 and HeyA8 cells compared to cells not treated with BafA (Fig. S5C and D and E and F, Supporting Information, respectively).These results clearly suggest that the PFK158‐induced autophagy sensitizes PFKFB3 overexpressing cells to drug‐induced cytotoxicity.
Our results demonstrated that PFK158 treatment downregulated p‐cPLA2 levels in C13 and HeyA8MDR cells (Fig. 4 i and j). As a determinant of cPLA2 activity after PFK158 treatment, we assessed arachidonic acid (AA) release in the presence and absence of BafA. As shown in Figure S6, Supporting Information, the reduction in AA by PFK158 treatment in both C13 and HeyA8MDR cells, was rescued by BafA treatment. These results suggest that PFK158‐mediated inhibition of cPLA2 activity and degradation of LDs is autophagy‐dependent.
Mobilization of LDs by lipophagy has been proposed as one of the mechanisms by which LDs are degraded and triglycerides are utilized. In the initial step of lipolysis, ATGL catalyzes the hydrolysis of triglycerides to release free FA in the cytoplasm.31 To understand the status of released triglycerides, we performed metabolite profiling of OVCAR5 cells with genetic KD of PFKFB3 and of PFK158‐treated parental OVCAR5 cells (Fig.S7A and B, Supporting Information). Diminution of cholesteryl ester and total triacylglycerols in both PFKFB3 KD and parental OVCAR5 cells suggests that both genetic and chemical inhibition of PFKFB3 resulted in marked reduction in neutral lipids (Fig. S7Ci‐iv, Supporting Information).
cPLA2 interacts with p62/SQSTM1 on LDs
It is reported that during lipid overload, p62 mediates LD catabolism by associating with PLIN2 (ADRP) on the LDs and aids in LD turnover.32 To investigate whether p62 is associated with LD, our IFC analysis demonstrated co‐localization of p62‐HA with LDs in OVCAR5 cells (Fig. S8‐top panel, Supporting Information). Moreover, increased autophagic flux upon EBSS treatment attenuated both LDs and p62 (Fig. S8, middle panel, Supporting Information), while treatment with BafA rescued the phenotype (Fig. S8, bottom panel, Supporting Information).
There are reports that show cPLA2 is bound to the surface of LDs.33 To confirm whether p62 indeed colocalizes with LDs and interacts with cPLA2, we performed immunoprecipitation (IP) of endogenous p62/SQSTM1 with cPLA2. Results demonstrated that p62/SQSTM1 associates with cPLA2 at the basal level (Fig. S9A, Supporting Information). This was further confirmed by reverse IP where we immune‐precipitated endogenous cPLA2 and blotted for p62 (Fig. S9A, Supporting Information). Consistent with these results, IFC analysis also revealed colocalization of p62 with cPLA2 (Fig. S9B, Supporting Information).
PFK158 alone suppresses tumor growth and ascites in vivo
The efficacy of PFK158 alone and in combination with CBPt was evaluated on primary tumor growth and metastasis in HeyA8MDR‐bearing nude mice i.p. A marked reduction of tumor growth was observed in the combination treatment (Fig. 6 a‐b). At necropsy, tumor weight (Fig. 6 c), ascites volume (Fig. 6 d), and Ki67 staining (Fig. 6 e) showed that the combination treatment was more effective in reducing cancer progression compared to all other treatment groups. No significant body weight loss was observed in the control or any of the drug‐treatment groups (data not shown). Consistent with these data, the western blot analysis revealed decreased expression of p‐PFKFB3 and increased levels of the apoptotic marker cleaved‐caspase 3 in the combination treatment group (Fig. 6 f). Representative images of Bodipy (Fig. 6 g) and TUNEL staining(Fig. 6 h) from frozen sections of xenografts from the untreated controls as well as treatment groups showed that PFK158 treatment was more effective in reducing LDs while CBPt had little effect in inhibiting LDs. Similarly, there was more cell death in the combination group as assessed by TUNEL staining. These data demonstrate that PFK158 plus CBPt treatments lead to significantly enhanced antitumor activity in a gynecologic cancer mouse model.
Figure 6.
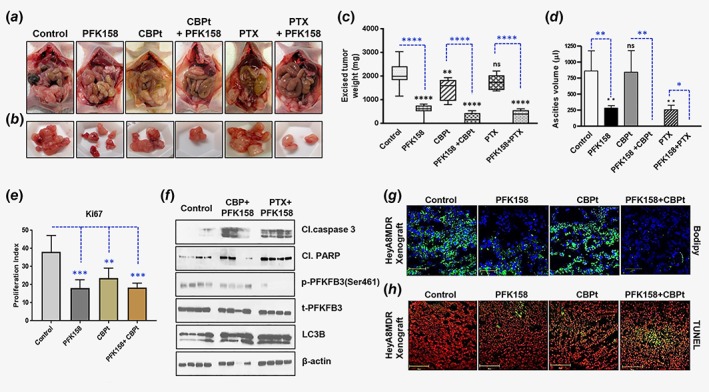
PFK158 alone and in combination with carboplatin/paclitaxel suppresses tumor growth and induces apoptosis in vivo. Randomized HeyA8MDR tumor‐bearing mice (n = 10), were treated with water, PFK158 (10 mg/kg), CBPt (51 mg/kg), PTX (15 mg/kg), a combination of PFK158 with CBPt, or combination of PFK158 with PTX for 4 weeks. After 28 days mice were euthanized. Images of tumors from representative animals from each group are shown in (a) and (b). (c) Excised tumor weight from different groups are shown in a bar diagram. Blue asterisk *, comparison between the groups; Black asterisk *, comparison with control (**p < 0.01, ****p < 0.0001 and ns = nonsignificant) (d) Ascites volume from mice from the different treatment groups and the untreated group is plotted in a bar diagram, blue, comparison between the groups; Black asterisk *, comparison with control (*p < 0.05, **p < 0.01 and ns = nonsignificant). (e) Bar graph showing the Ki67 proliferation index (**p < 0.01 and ***p < 0.001) as measured using GraphPad Prism 7. (f) Immunoblot analysis of cleaved caspase‐3, cleaved PARP, p‐PFKFB3, t‐PFKFB3 and LC3BI/II in lysates from untreated, CBPt plus PFK158, and PTX plus PFK158 treated xenografts with beta‐actin as a loading control. Representative images show Bodipy staining (g) and TUNEL staining (h) of frozen section of xenografts from different treatment groups. Green fluorescence (h) indicates TUNEL and red fluorescence indicates propidium iodide (PI) staining. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
It is increasingly recognized that, in addition to providing energy in the form of ATP, a more important role of glycolysis is to provide metabolic intermediates as building blocks for the macromolecular synthesis of carbohydrates, proteins, lipids, and nucleic acids to meet the increasing demand of cancer cells.34 In this context, altered lipid metabolism characterized by increased FA synthesis is a common feature of aggressive cancers and is functionally related to glycolysis.10 This direct coupling of increased glycolysis with excessive lipogenesis opens up a window of therapeutic opportunity to simultaneously target both pathways. A key step in controlling glycolytic rate is fructose 2, 6 bisphosphate levels which in turn is controlled by PFKFB3. While several studies have identified the role of PFKFB3 in promoting glycolysis, cell cycle progression, angiogenesis, and modulation of the Wnt signaling pathway in numerous cancers, we are the first to investigate the impact of PFKFB3 depletion in OC and cervical cancer as well as how this simultaneously affects both glycolysis and lipid biogenesis.
While previous reports suggest that total PFKFB3 has increased tumor‐specific overexpression,8, 35 our data indicate that the total PFKFB3 is expressed equally in normal fallopian tube epithelium, which, of note, is increasingly recognized as the origin of OC, normal ovarian surface epithelial cells as well as ovarian tumors. However, the activated form p‐PFKFB3ser461 shows tumor‐specific expression. Interestingly, the highly chemoresistant cell lines C13 and HeyA8MDR showed higher p‐PFKFB3 compared to their isogenic sensitive cells OV2008 and HeyA8. Inhibiting PFKFB3 activity resulted in decreased glucose uptake, lactate production and ATP levels in both OC and cervical cancer cells. One of the unique aspects of our study is that we showed that PFK158 treatment synergized with cisplatin, carboplatin and paclitaxel more in the chemoresistant cells compared to their isogenic chemosensitive counterparts to promote apoptosis. This synergy could, at least partly, be due to the increased glycolytic rate (consistent with increased p‐PFKFB3 levels) seen in the chemoresistant cells. While we were trying to identify additional factors that may account for this synergy in the resistant cells, we uncovered that resistant cells had a higher level of LDs and that PFK158 treatment reduced the number of LDs as well as the level of cPLA2, one of the LD‐associated proteins.
Mechanistically, PFK158‐induced LD inhibition involved increased autophagic flux promoted by downregulation of p62/SQSTM1 as well as increased levels of the lipidated form of LC3BII also the levels of p‐cPLA2 was attenuated. Inhibition of autophagy with BafA rescued cPLA2, LC3BII and LD levels implicating lipophagy as one of the mechanisms for the mobilization of LDs by PFK158. The reciprocal relationship between LDs and autophagy was first described by Singh et al. where they showed that the presence of exogenous lipids leads to inhibition of autophagy and accumulation of triglycerides promoting lipotoxic‐mediated cell death.32, 36 This is in contrast to what we observed in our study. Metabolite profiling of OVCAR5 cells treated with PFK158 and PFKFB3 knockdown cells resulted in decrease in the total triacylglycerols and cholesteryl esters, suggesting that genetic or chemical inhibition of PFKFB3 induced marked reduction in neutral lipids. These findings are consistent with other studies indicating that PFKFB3 knockdown in adipose tissue leads to less glucose incorporation into lipids.37
Reduction in the AA release is consistent with the downregulation of p‐cPLA2 by PFK158 treatment resulting in the inhibition of LDs in C13 and HeyA8MDR cells. In contrast to other studies involving lipophagy, inhibition of autophagy with BafA rescued cPLA2 and LDs and prevented PARP cleavage. These results add to the growing list of evidence that supports the idea that autophagy is both cell‐ and context‐dependent and thus can be either a survival mechanism or a cell death‐inducing phenomenon.38
Since the initial finding that autophagy regulates lipophagy, additional studies have highlighted the role of chaperone‐mediated autophagy to clear lipids. In one of the seminal studies by Kaushik et al., the authors showed that the ATGL‐catalyzed lipolysis in the liver involved the degradation of the LD proteins perilipin 2 and 3 (PLIN2 and 3) through CMA.39 Another report highlighted the role of ATGL‐mediated LD catabolism in the liver that involved SIRT1 as the primary mechanism for lipophagy and fatty acid oxidation.40 Pertinent to our observation, a recent study highlighted the role of p62/SQSTM1 in promoting lipophagy by binding to ADRP on the LD.32 In our study, we showed that p62/SQSTM1 interacts with cPLA2 and LC3B on LDs. Although the presence of p62/SQSTM1, LC3B and cPLA2 on LDs has been well documented, we showed here a previously unobserved p62–LD interaction with cPLA2 on the LDs. We hypothesize the after scenario: under autophagy‐inducing conditions, such as glucose starvation as mimicked by PFK158 treatment, p62/LC3‐bound autophagosomes directly interact with LD‐associated cPLA2 to promote lipophagy. This is consistent with the current concept that LD removal by autophagy involves LD‐associated proteins serving as recognition sites for promoting lipophagy.31 While the functional consequences of lipophagy are context‐ and cell‐type dependent, we surmise that lipophagy induced by this glycolytic inhibitor in OC and cervical cancer cells leads to apoptosis and inhibition of tumorigenesis.
Several studies support the idea that de novo lipogenesis in cancer cells is associated with chemoresistance at multiple levels.41 Other investigators have shown that high levels of stearoyl‐CoA desaturase expression, which is commonly encountered in several types of tumors, catalyzes the Δ9 desaturation of 12–19 carbon‐saturated FA to mono‐unsaturated FA resulting in altered membrane fluidity and resistance to chemotherapy.42 Several reports lend evidence to the concept that LD‐mediated drug resistance is multifactorial. It has also been reported that, in colon cancer, the expression of cancer stem marker of CD133/Wnt/β‐catenin signaling pathway correlates with LDs.17 In fact, increase in LDs, commonly seen in breast, prostate and OC cells, has been associated with resistance to paclitaxel43 and platinum agents.16, 30 We recently reported that loss of HSulf‐1 expression leads to a higher number of LDs and thus a lipogenic phenotype22 and is associated with worse overall survival.44 All these data suggest that targeting LDs alone or in combination with standard chemotherapy may be a novel way to treat cancer patients.41 Here, we clearly show that PFK158 treatment induces lipophagy and also sensitizes chemoresistant cells to chemotherapy‐induced cytotoxicity both in vitro and in vivo. Importantly, we also showed that inhibiting autophagy with BafA reverses the PFK158‐induced chemosensitivity to carboplatin more in the resistant than the sensitive cells. While the mechanism underlying the LD‐mediated resistance to chemotherapy is not well defined and could be multifactorial, our data implicate the higher number of LDs observed in the chemoresistant cells as a disease marker and an important anticancer target.
In conclusion, this is one of the first studies to show that PFK158, a specific inhibitor of PFKFB3, simultaneously targets both the glycolytic and lipogenic pathways, two pathways that are very active in cancer, and promotes lipophagy to inhibit tumor growth. Since resistance to chemotherapy is a major impediment in prolonging survival cancer patients, it is imperative to identify therapeutic strategies to overcome chemoresistance. Toward this aim, the synergy that we observed between PFK158 and carboplatin in chemoresistant cells has tremendous clinical relevance as it could represent a novel therapeutic option for patients with recurrent chemoresistant disease.
Supporting information
Figure S1: Higher p‐PFKFB3 in OV tumors
Representative images of immunohistochemical staining of phospho (p)‐PFKFB3 (ser461) and total (t)‐PFKFB3 performed in three ovarian tumor samples.
Figure S2A‐C. Mitochondrial respiration is compromised in resistant cells.
A‐B. Oxygen consumption rate (OCR) was measured with an XF24 extracellular flux analyzer (Seahorse Bioscience) after protocols recommended by the manufacturer. Resistant cells (C13 and HeyMDR) showed lower OCR compared to OV2008 and HeyA8. Data represent mean ± SD, two‐tailed unpaired t‐test, **p ≤ 0.01.
Figure S2C‐F. PFKFB3 regulates glucose uptake.
Glucose uptake was measured using 2‐NBDG after PFKFB3 knockdown (D) in C13 cells and PFKFB3 overexpression in OV2008 cells (F) after confirming PFKFB3 expression by western blot in these cells (C and E, respectively).
{kind=link}
Figure S3. PFKFB3 synergizes with chemoresistant ovarian cancer cells.
Combination of PFK158 with cisplatin and with carboplatin in equipotent combinations (IC50 over IC50 ratio) was assessed for synergy using the Chou‐Talalay methodology. The cells were exposed to each drug alone and in combination per protocol for 48 h. The combination indices (CI), fraction affected (Fa) in OV2008 and C13 (A and B), in Hey A8 and HeyA8MDR (E and F) were generated by the Calcusyn software and plotted with the use of GraphPad. CI values at 25, 50, 75 and 90% FA are presented in the tables below the graphs (C, D, G, H) with CI values at 75% FA highlighted in blue and red. CI values between 0.3–0.7 indicate strong synergism, 0.7–0.85 moderate synergism, 0.85–0.9 slight synergism, 0.9–1.10 nearly additive effect, and greater than 1.10 antagonism.
Figure S4. PFK158 treatment inhibits LD biogenesis.
A. OV90 cells were treated with PFK158 (0‐10 μM) followed by Bodipy staining to detect LDs. B. Immunoblot analysis shows the protein expression of p‐PFKFB3, t‐PFKFB3, p‐cPLA2 and t‐cPLA2 after PFK158 (0‐10 μM) treatment in OV90 cells. (C‐D) Transient downregulation of PFKFB3 in OV90 cells shows a reduced number of LDs.
Figure S5. Autophagy inhibition confers resistance to PFK158 plus carboplatin‐mediated synergy.
Cell viability assays were performed with a combination of increasing concentrations of carboplatin with 1x IC50 of PFK158 with and without bafilomycin A (BafA) pretreatment in replicates of 4. Cells were pretreated with 50 nM BafA for 2 h followed by drug treatment. Cell viability was assessed by MTT assays 48 h later. Pretreatment with BafA inhibited the combined PFK158 plus carboplatin‐induced cytotoxicity more effectively in C13 cells (B) compared to OV2008 (A), and in PFKFB3 overexpressed OV2008 and HeyA8 cells (D and F) compared to empty vector‐transfected (EV) OV2008 and HeyA8 cells (C and E), respectively. *p < 0.05; ***p = 0.01.
Figure S6. PFK158 mediated inhibition of cPLA2 activity and degradation of LDs is autophagy‐dependent.
The arachidonic acid release was evaluated in C13 and HeyA8MDR cells in the presence of 5 μM PFK158 along with 50 nM Bafilomycin with untreated cells as controls. Cells were incubated with 3H‐AA under serum‐starved condition for 24 h. Fresh medium was added to the cells after washing and aliquots of growth medium were measured for radioactivity shown as counts per minute (CPM)/ml after 24 h.
Figure S7. Reduction in neutral lipids in PFK158 treated and PFKFB3 knockdown OVCAR5 cells.
OVCAR5 cells stably downregulated with shRNA‐PFKFB3 (A) or treated with 5 μM of PFK158 for 12 h and 24 h (B) were subjected to analysis for neutral lipids including cholesteryl ester and triacylglycerols The samples were extracted using Metabolon's standard solvent extraction method from cells with five biological replicates for each sample and distributed into equal parts for analysis on the GC/MS and LC/MS/MS platforms. Cholesteryl ester and TAG levels in non‐treated controls (NTC), Sh55, Sh59 and PFK158 treated are demonstrated in bar diagram (C).
Figure S8. p62 co‐localizes with LDs in ovarian cancer cells.
Co‐localization of p62 and LD were evaluated by immuno‐fluorescence analysis in OVCAR5 cells. Co‐localization of p62‐HA with Bodipy (Fig.S8, second row) is attenuated with EBSS treatment (Fig.S8, third row), while treatment with bafilomycin rescued the phenotype (Fig.S8, last row).
Figure S9. p62 associates with cPLA2 in ovarian cancer cells.
A. HeyA8MDR cell lysates were co‐immunoprecipitated with either p62/SQSTM1 or t‐cPLA2 antibody and subsequently immunoblotted to determine the protein expression of t‐cPLA2, and p62/SQSTM1. B. Immunofluorescence analysis shows co‐localization of t‐cPLA2 with p62/SQSTM1.
Table S1: Antibodies and Reagents
Acknowledgments
We would like to acknowledge Dr. Daniel Billadeau, Mayo Clinic, Rochester, MN for providing the RFP‐GFP‐LC3B plasmid. The FTE cells were obtained from Dr. Ronny Drapkin at Dana‐Farber Cancer Institute, Inc. on an MTA. We would like to acknowledge the contributions of the Pathology Research Core laboratory, the Microscopy Core laboratory, and the Flow Cytometry Facility, Mayo Clinic Rochester.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 2003;3:502–516. [DOI] [PubMed] [Google Scholar]
- 3. Madhok BM, Yeluri S, Perry SL, et al. Targeting glucose metabolism: an emerging concept for anticancer therapy. Am J Clin Oncol 2011;34:628–635. [DOI] [PubMed] [Google Scholar]
- 4. Okar DA, Manzano A, Navarro‐Sabate A, et al. PFK‐2/FBPase‐2: maker and breaker of the essential biofactor fructose‐2,6‐bisphosphate. Trends Biochem Sci 2001;26:30–35. [DOI] [PubMed] [Google Scholar]
- 5. Marsin AS, Bouzin C, Bertrand L, et al. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP‐activated protein kinase and inducible 6‐phosphofructo‐2‐kinase. J Biol Chem 2002;277:30778–30783. [DOI] [PubMed] [Google Scholar]
- 6. Novellasdemunt L, Bultot L, Manzano A, et al. PFKFB3 activation in cancer cells by the p38/MK2 pathway in response to stress stimuli. Biochem J 2013;452:531–543. [DOI] [PubMed] [Google Scholar]
- 7. De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell 2013;154:651–663. [DOI] [PubMed] [Google Scholar]
- 8. Yalcin A, Clem BF, Imbert‐Fernandez Y, et al. 6‐Phosphofructo‐2‐kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1‐mediated phosphorylation of p27. Cell Death Dis 2014;5:e1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clem B, Telang S, Clem A, et al. Small‐molecule inhibition of 6‐phosphofructo‐2‐kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 2008;7:110–120. [DOI] [PubMed] [Google Scholar]
- 10. Costello LC, Franklin RB. 'Why do tumour cells glycolyse?': from glycolysis through citrate to lipogenesis. Mol Cell Biochem 2005;280:1–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koizume S, Miyagi Y. Lipid droplets: a key cellular organelle associated with cancer cell survival under Normoxia and hypoxia. Int J Mol Sci 2016;17:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science (New York NY) 2004;306:1383–1386. [DOI] [PubMed] [Google Scholar]
- 13. Zaidi N, Lupien L, Kuemmerle NB, et al. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 2013;52:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuemmerle NB, Rysman E, Lombardo PS, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther 2011;10:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nomura DKLJ, Niessen S, Hoover HS, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell Cycle 2010;140:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montopoli M, Bellanda M, Lonardoni F, et al. "Metabolic reprogramming" in ovarian cancer cells resistant to cisplatin. Curr Cancer Drug Targets 2011;11:226–235. [DOI] [PubMed] [Google Scholar]
- 17. Rappa G, Le TT, Corbeil D, et al. An intriguing relationship between lipid droplets, cholesterol‐binding protein CD133 and Wnt/β‐catenin signaling pathway in carcinogenesis. Stem Cells 2015;33:1366–1370. [DOI] [PubMed] [Google Scholar]
- 18. Clem BF, O'Neal J, Tapolsky G, et al. Targeting 6‐Phosphofructo‐2‐kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 2013;12:1461–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mondal S, Mandal C, Sangwan R, et al. Withanolide D induces apoptosis in leukemia by targeting the activation of neutral sphingomyelinase‐ceramide cascade mediated by synergistic activation of c‐Jun N‐terminal kinase and p38 mitogen‐activated protein kinase. Mol Cancer 2010;9:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chou TC. Drug combination studies and their synergy quantification using the Chou‐Talalay method. Cancer Res 2010;70:440–446. [DOI] [PubMed] [Google Scholar]
- 21. Mondal S, Bhattacharya K, Mallick A, et al. Bak compensated for Bax in p53‐null cells to release cytochrome c for the initiation of mitochondrial signaling during Withanolide D‐induced apoptosis. PloS one 2012;7:e34277. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Mondal S, Roy D, Camacho‐Pereira J, et al. HSulf‐1 deficiency dictates a metabolic reprograming of glycolysis and TCA cycle in ovarian cancer. Oncotarget 2015;6:33705–33719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khurana A, Roy D, Kalogera E, et al. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget 2015;6:36354–36369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy D, Mondal S, Wang C, et al. Loss of HSulf‐1 promotes altered lipid metabolism in ovarian cancer. Cancer Metab 2014;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roy D, Mondal S, Khurana A, et al. Loss of HSulf‐1: the missing link between autophagy and lipid droplets in ovarian cancer. Sci Rep 2017;7:41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bando H, Atsumi T, Nishio T, et al. Phosphorylation of the 6‐phosphofructo‐2‐kinase/fructose 2,6‐bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res 2005;11:5784–5792. [DOI] [PubMed] [Google Scholar]
- 27. Guijas C, Rodriguez JP, Rubio JM, et al. Phospholipase A2 regulation of lipid droplet formation. Biochim Biophys Acta 2014;1841:1661–1671. [DOI] [PubMed] [Google Scholar]
- 28. Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013;20:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farkas T, Hoyer‐Hansen M, Jaattela M. Identification of novel autophagy regulators by a luciferase‐based assay for the kinetics of autophagic flux. Autophagy 2009;5:1018–1025. [DOI] [PubMed] [Google Scholar]
- 30. Pan X, Wilson M, McConville C, et al. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metabolomics 2013;9:722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dong H, Czaja MJ. Regulation of lipid droplets by autophagy. Trends in endocrinology and metabolism: TEM 2011;22:234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lam T, Harmancey R, Vasquez H, et al. Reversal of intramyocellular lipid accumulation by lipophagy and a p62‐mediated pathway. Cell Death Discov 2016;2:16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wooten RE, Willingham MC, Daniel LW, et al. Novel translocation responses of cytosolic phospholipase A2alpha fluorescent proteins. Biochim Biophys Acta 2008;1783:1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York NY) 2009;324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hue L, Rousseau GG. Fructose 2,6‐bisphosphate and the control of glycolysis by growth factors, tumor promoters and oncogenes. Adv Enzyme Regul 1993;33:97–110. [DOI] [PubMed] [Google Scholar]
- 36. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huo Y, Guo X, Li H, et al. Disruption of inducible 6‐phosphofructo‐2‐kinase ameliorates diet‐induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J Biol Chem 2010;285:3713–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klarer AC, O'Neal J, Imbert‐Fernandez Y, et al. Inhibition of 6‐phosphofructo‐2‐kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab 2014;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaushik S, Cuervo AM. Degradation of lipid droplet‐associated proteins by chaperone‐mediated autophagy facilitates lipolysis. Nat Cell Biol 2015;17:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/Lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep 2017;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beloribi‐Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogene 2016;5:e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peck B, SA . Lipid desaturation—the next step in targeting lipogenesis in cancer? FEBSJ 2016;283:2767–2778. [DOI] [PubMed] [Google Scholar]
- 43. Zietkowski D, Nagy E, Mobberley MA, et al. Comparison of NMR lipid profiles in mitotic arrest and apoptosis as indicators of paclitaxel resistance in cervical cell lines. Magn Reson Med 2012;68:369–377. [DOI] [PubMed] [Google Scholar]
- 44. Liu P, Khurana A, Rattan R, et al. Regulation of HSulf‐1 expression by variant hepatic nuclear factor 1 in ovarian cancer. Cancer Res 2009;69:4843–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Higher p‐PFKFB3 in OV tumors
Representative images of immunohistochemical staining of phospho (p)‐PFKFB3 (ser461) and total (t)‐PFKFB3 performed in three ovarian tumor samples.
Figure S2A‐C. Mitochondrial respiration is compromised in resistant cells.
A‐B. Oxygen consumption rate (OCR) was measured with an XF24 extracellular flux analyzer (Seahorse Bioscience) after protocols recommended by the manufacturer. Resistant cells (C13 and HeyMDR) showed lower OCR compared to OV2008 and HeyA8. Data represent mean ± SD, two‐tailed unpaired t‐test, **p ≤ 0.01.
Figure S2C‐F. PFKFB3 regulates glucose uptake.
Glucose uptake was measured using 2‐NBDG after PFKFB3 knockdown (D) in C13 cells and PFKFB3 overexpression in OV2008 cells (F) after confirming PFKFB3 expression by western blot in these cells (C and E, respectively).
Figure S3. PFKFB3 synergizes with chemoresistant ovarian cancer cells.
Combination of PFK158 with cisplatin and with carboplatin in equipotent combinations (IC50 over IC50 ratio) was assessed for synergy using the Chou‐Talalay methodology. The cells were exposed to each drug alone and in combination per protocol for 48 h. The combination indices (CI), fraction affected (Fa) in OV2008 and C13 (A and B), in Hey A8 and HeyA8MDR (E and F) were generated by the Calcusyn software and plotted with the use of GraphPad. CI values at 25, 50, 75 and 90% FA are presented in the tables below the graphs (C, D, G, H) with CI values at 75% FA highlighted in blue and red. CI values between 0.3–0.7 indicate strong synergism, 0.7–0.85 moderate synergism, 0.85–0.9 slight synergism, 0.9–1.10 nearly additive effect, and greater than 1.10 antagonism.
Figure S4. PFK158 treatment inhibits LD biogenesis.
A. OV90 cells were treated with PFK158 (0‐10 μM) followed by Bodipy staining to detect LDs. B. Immunoblot analysis shows the protein expression of p‐PFKFB3, t‐PFKFB3, p‐cPLA2 and t‐cPLA2 after PFK158 (0‐10 μM) treatment in OV90 cells. (C‐D) Transient downregulation of PFKFB3 in OV90 cells shows a reduced number of LDs.
Figure S5. Autophagy inhibition confers resistance to PFK158 plus carboplatin‐mediated synergy.
Cell viability assays were performed with a combination of increasing concentrations of carboplatin with 1x IC50 of PFK158 with and without bafilomycin A (BafA) pretreatment in replicates of 4. Cells were pretreated with 50 nM BafA for 2 h followed by drug treatment. Cell viability was assessed by MTT assays 48 h later. Pretreatment with BafA inhibited the combined PFK158 plus carboplatin‐induced cytotoxicity more effectively in C13 cells (B) compared to OV2008 (A), and in PFKFB3 overexpressed OV2008 and HeyA8 cells (D and F) compared to empty vector‐transfected (EV) OV2008 and HeyA8 cells (C and E), respectively. *p < 0.05; ***p = 0.01.
Figure S6. PFK158 mediated inhibition of cPLA2 activity and degradation of LDs is autophagy‐dependent.
The arachidonic acid release was evaluated in C13 and HeyA8MDR cells in the presence of 5 μM PFK158 along with 50 nM Bafilomycin with untreated cells as controls. Cells were incubated with 3H‐AA under serum‐starved condition for 24 h. Fresh medium was added to the cells after washing and aliquots of growth medium were measured for radioactivity shown as counts per minute (CPM)/ml after 24 h.
Figure S7. Reduction in neutral lipids in PFK158 treated and PFKFB3 knockdown OVCAR5 cells.
OVCAR5 cells stably downregulated with shRNA‐PFKFB3 (A) or treated with 5 μM of PFK158 for 12 h and 24 h (B) were subjected to analysis for neutral lipids including cholesteryl ester and triacylglycerols The samples were extracted using Metabolon's standard solvent extraction method from cells with five biological replicates for each sample and distributed into equal parts for analysis on the GC/MS and LC/MS/MS platforms. Cholesteryl ester and TAG levels in non‐treated controls (NTC), Sh55, Sh59 and PFK158 treated are demonstrated in bar diagram (C).
Figure S8. p62 co‐localizes with LDs in ovarian cancer cells.
Co‐localization of p62 and LD were evaluated by immuno‐fluorescence analysis in OVCAR5 cells. Co‐localization of p62‐HA with Bodipy (Fig.S8, second row) is attenuated with EBSS treatment (Fig.S8, third row), while treatment with bafilomycin rescued the phenotype (Fig.S8, last row).
Figure S9. p62 associates with cPLA2 in ovarian cancer cells.
A. HeyA8MDR cell lysates were co‐immunoprecipitated with either p62/SQSTM1 or t‐cPLA2 antibody and subsequently immunoblotted to determine the protein expression of t‐cPLA2, and p62/SQSTM1. B. Immunofluorescence analysis shows co‐localization of t‐cPLA2 with p62/SQSTM1.
Table S1: Antibodies and Reagents