

Abstract
Hereditary mixed polyposis syndrome (HMPS) is a hereditary syndrome that is characterized by multiple colon polyps of mixed pathologic subtypes and an increased risk for colorectal cancer. A 40kb duplication in the 5′ regulatory region of the GREM1 gene was recently found to be the causal mutation in a subset of Ashkenazi Jewish families with HMPS. Given this discovery, the GREM1 5′ regulatory region is now analyzed on many different multi-gene cancer panels, however the data on duplications distinct from the 40kb duplication remains minimal. Herein we report a novel 24kb tandem duplication of the 5′ regulatory region of GREM1 in a patient without Ashkenazi Jewish heritage, who had a family history that was concerning for Lynch syndrome and satisfied Amsterdam II criteria. This is only the third reported GREM1 duplication separate from the 40kb Ashkenazi Jewish duplication, and is the only reported duplication to selectively involve exon 1 of GREM1. This finding supports comprehensive testing of the GREM1 regulatory region in families of all ethnicities with multiple colon polyps or colon cancer and when Lynch syndrome is suspected.
Keywords: hereditary mixed polyposis syndrome, GREM1, colon polyps
Introduction
Hereditary mixed polyposis syndrome (HMPS) was described originally as an autosomal dominantly inherited syndrome associated with colonic polyposis consisting of multiple different polyp types including tubular/villous/tubulovillous adenomas, serrated adenomas, hyperplastic polyps, and hamartomatous polyps [1, 2]. Robust clinical diagnostic criteria for HMPS have yet to be firmly established, although HMPS is associated with increased colorectal cancer risk [2]. However, at this time it remains uncertain whether there are any extra-colonic cancer risks associated with this syndrome.
A genetic etiology for HMPS was first described in 2012, when a 40kb duplication in the 5′ regulatory region of the GREM1 gene was identified as the causal mutation in Ashkenazi Jewish families with shared haplotypes, suggesting a potential founder effect [3]. Subsequently, two additional GREM1 duplications have been reported: a 16kb duplication in the regulatory region of GREM1 was identified in a non-Ashkenazi Jewish family with attenuated polyposis [4] and a 57kb duplication of the entire GREM1 gene was identified in an individual with early onset colon cancer [5]. Given the association of GREM1 with HMPS, analysis of the GREM1 5′ regulatory region is now available and included in many hereditary cancer gene panels offered by commercial genetic testing laboratories.
In HMPS, duplication of the predicted transcriptional enhancer of GREM1 has been associated with greatly increased, allele-specific GREM1 expression, which is in contrast to other polyposis conditions such as familial adenomatous polyposis (FAP) and MUTYH-associated polyposis (MAP) where causative mutations lead to dysfunctional protein products [6]. Increased GREM1 expression leads to disruption of the bone morphogenetic protein (BMP) pathway, which can both promote colonic polyp development and increase risk for neoplastic transformation of colon polyps that is outside of the stem cell niche [6]. Dysregulation of the BMP pathway is also known to be an important mechanism for polyp development in juvenile polyposis [7].
Given limited data in the field, medical management of patients with HMPS remains a topic that continues to evolve. Recently, guidelines from the National Comprehensive Cancer Network (NCCN) have started to include recommendations for individuals with a GREM1 duplication: initiate colonoscopy screening at age 25-30, repeating every 2-3 years if no polyps are found, and repeating every 1-2 years if polyps are detected [8]. However, these guidelines are based largely upon data from the 40kb duplication reported in Ashkenazi Jewish families, and it remains unclear whether individuals with other duplications should be managed in a similar manner.
Methods
Clinical Presentation
A 63-year-old female, with no Ashkenazi Jewish ancestry and no personal history of cancer, was referred for a genetics evaluation given concern for Lynch syndrome. The patient had a strong maternal family history of cancer including colon cancer diagnosed in her mother (age 56), two maternal aunts (at unknown ages) and maternal grandfather (mid 50′s), as well as a maternal aunt who was diagnosed with endometrial cancer in her early 40′s (Figure 1). Although none of the affected relatives had genetic testing performed, prior to her arrival at our program the patient was being managed as if she had Lynch syndrome based upon her family fulfilling Amsterdam II criteria. The patient had her first colonoscopy at the age of 46, with a total of 8 colonoscopies performed over the next 17 years, during which time she had a total of 20 colon polyps that were removed. Pathology reports were available from 16 of her polyps, which showed 11 tubular adenomas and 5 hyperplastic polyps. Given the high clinical suspicion of Lynch syndrome, this patient had also undergone several upper endoscopies, which were normal, and had a risk reducing total hysterectomy and bilateral salpingo-oophorectomy at age 62.
Figure 1.
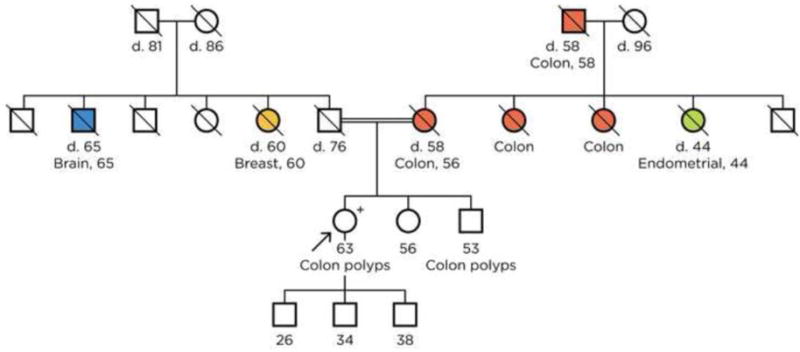
Proband’s family pedigree. The proband’s paternal great grandfather and maternal great grandfather are brothers.
Genetic Evaluation
After presentation to our program, the patient’s personal and family history was elicited by a genetic counselor and genetic testing was recommended. She was consented and provided a sample for multi-gene panel testing, which was performed by Color Genomics using a next-generation sequencing based 30-gene panel for hereditary cancer risk that included the following genes: APC, ATM, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, GREM1, MITF, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, POLE, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53. Copy number changes were assessed based on read depth analysis. A dedicated split read detection algorithm was used to identify structural variants. Subsequent Sanger sequencing was used to confirm the presence of detected variants.
Genetic Test Results
This multi-gene panel identified a novel 24kb tandem duplication of the 5′ regulatory region of GREM1 spanning Chr15:g.32987207-g.33011059 (23,853bp) and a pathogenic BRCA1 mutation (E1373X). For secondary confirmation of the duplication, an amplicon was designed that is specific to this in-tandem duplication using NCBI Primer-BLAST (forward primer -TGAGATGGAGACTGGCAAGG aligning at Chr15:g.33010905-33010924; reverse primer - AGTGGCACCTACCAACTACC aligning at Chr15:g.32988251-32988270), which rendered the appropriate 1219bp PCR product supporting the presence of the in-tandem duplication. In addition, the Sanger sequence based on the forward primer confirmed the breakpoints as derived from NGS. The identified 24kb in-tandem duplication (chr15:g.32987207_33011059dupinsTG) partially overlaps with the previously published 16kb, 40kb and 57kb duplications in the upstream regulatory region of GREM1, and includes the enhancer element thought to be responsible for overexpression of GREM1 (Figure 2). Based on the identification of this GREM1 duplication, the patient will continue to undergo colonoscopic screening but will no longer receive other Lynch syndrome surveillance, such as upper endoscopy. At this time, no other living members of the proband’s family have pursued genetic testing, all of the proband’s older relatives have already passed away without having prior genetic testing or blood banking, and it is not feasible to obtain tumor tissue from any of the proband’s relatives affected with cancer. Therefore, the lineage of these two mutations has not been established.
Figure 2.
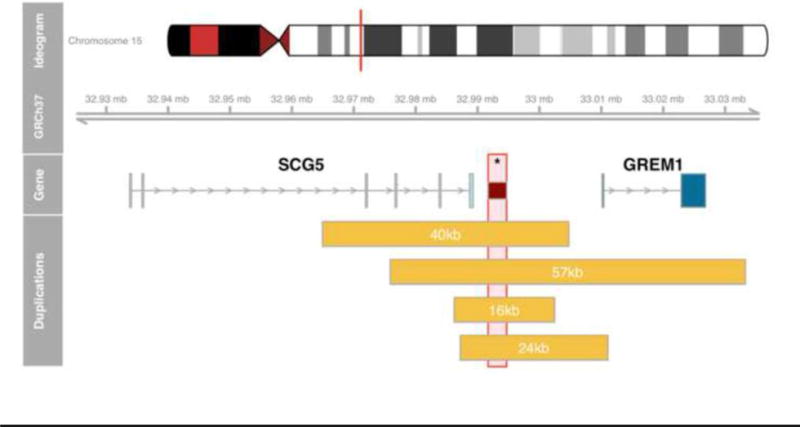
Schematic overview of published duplications overlapping the GREM1 putative enhancer region as defined in [1] (dark red box, marked with asterisk). The 16kb and 40kb duplications do not include any GREM1 coding sequence; the novel 24kb variation duplicates GREM1 exon 1; and, the 57kb variation constitutes a whole gene duplication.
Discussion
To our knowledge, this is only the third report of a copy number variation in the regulatory region of GREM1 distinct from the 40kb Ashkenazi Jewish founder mutation, and is the first report of this specific 24kb duplication in the GREM1 regulatory region. Both the 40kb [3] and 16kb duplications [4] have been shown to significantly increase GREM1 expression. While the 57kb [5] and this novel 24kb duplication completely overlap the GREM1 enhancer region and support the clinical phenotype, the impact upon expression level resulting from these duplications has not been assessed explicitly. Interestingly, the proband also has a germline BRCA1 mutation. Although some data has pointed toward a possible increased CRC risk in young BRCA1 carriers [9], current guidelines do not support increased CRC screening in BRCA1 carriers [10], and to our knowledge there is no known association between pathogenic BRCA1 mutations and colonic polyposis. Additionally, we are not aware of concurrent GREM1 (chromosome 15q13) and BRCA1 (chromosome 17q21) mutations, nor is there, to our knowledge, evidence that these mutations are influencing each other at the germline level or having epistatic interactions. Additional research is needed to determine if coexisting mutations in BRCA1 and GREM1 could potentially have additive effects on cancer risk.
The paucity of knowledge about the phenotype of HMPS, especially outside of the Ashkenazi Jewish population, makes clinical screening recommendations challenging. Although current NCCN guidelines recommend screening colonoscopy beginning as early as age 25, there are reports of earlier colonic findings in GREM1 mutation carriers, including a tubulovillous adenoma in a 13-year-old [11], and metastatic colon cancer in a 21-year-old [12]. It is difficult to determine if the aforementioned families represent rare outliers or if polyp formation in adolescents should be included in the clinical spectrum of HMPS. Nonetheless, several groups have advocated for colonoscopy surveillance starting at age 18 or earlier for those with a GREM1 mutation [11, 12]. What also remains unclear is whether all GREM1 duplications have the same colon cancer risk, and ultimately additional research on the phenotypic spectrum of GREM1 associated HMPS is necessary to make appropriate medical management recommendations.
With the recent identification of GREM1 duplications and their association with colonic polyp development and increased colon cancer risk, it is conceivable that these copy number variations (CNVs) in the regulatory region of GREM1 are present in families with colonic polyposis without a known genetic cause or in kindred with familial colorectal cancer type X [13] who may have had genetic testing prior to recognition of the significance of GREM1. Additionally, CNVs of regulatory regions are not typically analyzed during whole exome sequencing (WES), and therefore, changes in the regulatory region of GREM1 may not be detected in polyposis families who have previously undergone WES. Furthermore, as illustrated by this case, GREM1 testing should be pursued in families where there is concern for Lynch syndrome due to potential phenotypic overlap.
The two main limitations of this report include lack of co-segregation data, which would be helpful in understanding the associated phenotype of this duplication, as well as tumor testing demonstrating increased protein expression resulting from this duplication. Nonetheless, given the paucity of data in the field, this report contributes an important addition to GREM1 literature, and provides support for full sequencing of the GREM1 promoter region when indicated for cancer risk assessment.
Conclusion
In summary, we present a non-Ashkenazi Jewish female with a personal history of multiple colon polyps and a strong family history of colon cancer, who was found to have a novel 24kb duplication in the regulatory region of GREM1. The identification of this novel duplication associated with colon polyps provides further evidence to support testing for GREM1 mutations in individuals with polyposis or a clinical suspicion of Lynch syndrome, independent of their ethnicity.
Acknowledgments
Funding:
The Lustgarten Family Colon Cancer Research Fund (AR) and NIH/NIDDK grant 1K08DK106489 (BK).
References
- 1.Jaeger EE, Woodford-Richens KL, Lockett M, et al. An ancestral Ashkenazi haplotype at the HMPS/CRAC1 locus on 15q13-q14 is associated with hereditary mixed polyposis syndrome. Am J Hum Genet. 2003;72(5):1261–7. doi: 10.1086/375144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitelaw SC, Murday VA, Tomlinson IP, et al. Clinical and molecular features of the hereditary mixed polyposis syndrome. Gastroenterology. 1997;112(2):327–34. doi: 10.1053/gast.1997.v112.pm9024286. [DOI] [PubMed] [Google Scholar]
- 3.Jaeger E, Leedham S, Lewis A, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. 2012;44(6):699–703. doi: 10.1038/ng.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohlin A, Eiengard F, Lundstam U, et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes, chromosomes & cancer. 2016;55(1):95–106. doi: 10.1002/gcc.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkatachalam R, Verwiel ET, Kamping EJ, et al. Identification of candidate predisposing copy number variants in familial and early-onset colorectal cancer patients. International journal of cancer Journal international du cancer. 2011;129(7):1635–42. doi: 10.1002/ijc.25821. [DOI] [PubMed] [Google Scholar]
- 6.Davis H, Irshad S, Bansal M, et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nature medicine. 2015;21(1):62–70. doi: 10.1038/nm.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batts LE, Polk DB, Dubois RN, Kulessa H. Bmp signaling is required for intestinal growth and morphogenesis. Dev Dyn. 2006;235(6):1563–70. doi: 10.1002/dvdy.20741. [DOI] [PubMed] [Google Scholar]
- 8.National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Colorectal (Version 2.2017) 2017 Sep 1; [Google Scholar]
- 9.Sopik V, Phelan C, Cybulski C, Narod SA. BRCA1 and BRCA2 mutations and the risk for colorectal cancer. Clin Genet. 2015;87(5):411–8. doi: 10.1111/cge.12497. [DOI] [PubMed] [Google Scholar]
- 10.National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Breast and Ovarian (Version 2.2017) 2017 Novemver; [Google Scholar]
- 11.Lieberman S, Walsh T, Schechter M, et al. Features of Patients With Hereditary Mixed Polyposis Syndrome Caused by Duplication of GREM1 and Implications for Screening and Surveillance. Gastroenterology. 2017;152(8):1876–80 e1. doi: 10.1053/j.gastro.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Ziai J, Matloff E, Choi J, Kombo N, Materin M, Bale AE. Defining the polyposis/colorectal cancer phenotype associated with the Ashkenazi GREM1 duplication: counselling and management recommendations. Genet Res (Camb) 2016;98:e5. doi: 10.1017/S0016672316000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293(16):1979–85. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]