

Abstract
Background
Cold-inducible RNA-binding protein (CIRP) is a novel damage-associated molecular pattern (DAMP) that causes inflammation. C23, a short peptide derived from CIRP, has demonstrated efficacy in blocking CIRP’s activity. We hypothesize that C23 reduces inflammation and tissue injury induced by intestinal ischemia-reperfusion (I/R).
Methods
Male C57BL/6 mice were subjected to 60-min of intestinal ischemia by clamping the superior mesenteric artery. Immediately after reperfusion, either normal saline (vehicle) or C23 peptide (8 mg/kg BW) was injected intraperitoneally. Four hours after reperfusion, blood, intestinal and lung tissues were collected for the analysis of inflammatory and tissue injury parameters.
Results
CIRP levels in intestinal were significantly increased following intestinal I/R. Histological examination of the intestine revealed a significant reduction in injury score in the C23 group by 48% as compared to the vehicles after intestinal I/R. The serum levels of LDH and AST were increased in the vehicle-treated intestinal I/R animals, while C23 treated animals exhibited a significant reduction by 48% and 53%, respectively. The serum and intestinal tissue levels of TNF-α were elevated in the vehicle-treated intestinal I/R mice, but decreased by 72% and 69%, respectively, in C23-treated mice. IL-6 mRNA levels in the lungs were reduced by 86% in the C23-treated group in comparison to the vehicle-treated group after intestinal I/R. The expression of MIP-2 and the level of MPO activity in the lungs were dramatically increased after intestinal I/R and significantly reduced by 91% and 25%, respectively, in the C23-treated group.
Conclusion
C23 has potential to be developed into a possible therapy for reperfusion injury after mesenteric ischemia and reperfusion.
Keywords: CIRP, C23, Intestinal I/R, Inflammation, Tissue injury
Graphical Abstract
C23 can function as an inhibitor of the damage associated molecular pattern CIRP and mitigate inflammatory damage after intestinal reperfusion injury. The importance of this report is providing evidence of C23's therapeutic potential in humans.
Introduction
Intestinal ischemia-reperfusion injury (I/R) is an inflammatory condition caused by the restoration of blood flow to previously ischemic bowel. This condition is seen in a myriad of clinical scenarios including acute thrombo-embolic mesenteric ischemia, low-flow shock states, volvulus, trauma, intestinal transplantation, abdominal aortic surgery, cardiopulmonary bypass, and strangulated hernias.1, 2 With prompt diagnosis and intervention, bowel can be salvaged with correction of occlusive disease or volume status. However, despite advances in the treatment of this disease, the mortality rate for intestinal ischemia remains between 60% and 80% and the incidence is on the rise.3, 4
After intervention for intestinal ischemia, the restoration of perfusion triggers multiple complex acute inflammatory cascades that parallel the host response to microorganisms. Resident cells within the intestinal tissue that are metabolically stressed during ischemia release damage-associated molecular patterns (DAMPs), which are molecules that are normally located intracellularly but when released from the cell serve as an alarm to the body’s immune system that injury has occurred.5, 6 DAMPs bind to Toll-like receptors (TLRs), which in turn activate the transcription factor nuclear factor-κB (NF-kB) that potentiates the production of cytokines and chemokines to further activate and recruit innate immune cells, such as macrophages and neutrophils.7, 8 These innate immune cells infiltrate the reperfused tissue and produce further inflammation, ultimately inflicting more cellular injury than the original ischemic insult primarily by disrupting local microcirculation.9
The inflammatory response during the reperfusion phase is not only damaging to the original ischemic tissue, but can also potentiate activation of inappropriate inflammation in remote organs, particularly in the lungs.10 Multiple organ failure, particularly acute lung injury (ALI), is a common complication of intestinal I/R that contributes to the high mortality rate in intestinal I/R.11, 12 ALI is caused by a systemic inflammatory response due to the release of proinflammatory cytokines and bacterial endotoxins that are released from reperfused ischemic intestine.13 Therefore, the development of therapies targeting the host response to DAMPs during intestinal I/R has clinical value in the treatment or prevention of both local and remote organ injury.
We have identified a new DAMP named cold-inducible ribonucleic acid (RNA) binding protein (CIRP).14 It is expressed constitutively at low levels in various tissues and normally functions as an RNA chaperone protein during translation.15 During the hypoxic conditions of shock states, macrophages release CIRP, which induces the expression of pro-inflammatory cytokines, such as TNF-α and interleukin IL-6, proteins that significantly contribute to an inappropriate and damaging immune response.14 In both animal and human models, CIRP has been shown to play a deleterious role in various critical care scenarios, such as hemorrhagic shock, sepsis, and kidney ischemia reperfusion.16–19
CIRP acts as a DAMP through its binding to TLR4 and myeloid differentiation factor 2 (MD2) complex, which in turn potentiates the expression of pro-inflammatory cytokines.(14) We previously showed that CIRP knock-out (CIRP−/−) mice undergoing intestinal I/R exhibited reduced local, systemic, and remote organ inflammation and injuries when compared to their wild-type (WT) counterparts.20 With this confirmation of CIRP’s damaging role in intestinal I/R, we aimed to block CIRP’s DAMP effects in a way that could be translated into a human therapy. Many short sequences within the 172 amino-acid CIRP protein were screened for their binding affinity to TLR4-MD2 complex using surface plasmon resonance. A 15-mer peptide with the highest binding affinity for TLR4-MD2 complex was identified and named C23.14
In previous work, our lab has demonstrated the efficacy of the CIRP-derived peptide, C23, in downregulating recombinant murine CIRP-induced TNF-α production by human macrophages in vitro.21 In this study, we demonstrated C23’s ability to mitigate inflammation and injury both locally and remotely in the lungs in mice undergoing intestinal I/R.
Materials and Methods
Animal model of intestinal I/R
Male, C57BL/6 WT mice (7–9 weeks old, 20–25 g BW) purchased from Charles River (Kingston, NY) were housed in temperature-controlled environments and were fed a standard laboratory mouse diet. The animal model for intestinal I/R began with anesthetic induction with 2.5% inhalational isoflurane, followed by preparation of their abdomen with 10% povidoneiodine wash. Mice anesthesia was maintained with 2% isoflurane inhalation. An upper midline laparotomy was performed to expose the abdomen and the superior mesenteric artery (SMA) was isolated. The SMA was then occluded with a vascular clip. On completion of 60 min of ischemia, the vascular clip was removed to allow reperfusion. After closure, all mice were resuscitated with 1 mL of normal saline administered subcutaneously. At the time of reperfusion, mice designated randomly to the treatment group received 8 mg of C23 per kg of body weight of the mouse through intraperitoneal injection (i.p.) in 200 μl of total volume adjusted with normal saline. We injected mice with normal saline i.p. at 200 μl of total volume to serve as vehicle control group of mice. We euthanized the mice 4 h after the i.p. injection of C23 for the treatment group and normal saline for the vehicle group. Sham mice were not operated on and underwent organ and blood harvest while under anesthesia. The sample size for each experimental group was 5 mice. All experiments were performed in accordance with the guidelines for the use of experimental animals by the National Institutes of Health (Bethesda, MD) and were approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
Histologic analysis
Samples of the small intestine non-necrotic areas of the distal ileum selected based on the color of the small intestine segment were fixed in 10% formalin and embedded in paraffin. The terminal ileum was chosen to ensure uniformity in segment selection among animals and prevent an observer bias. In our previous studies on intestinal I/R injury model in mice we studied terminal illium to demonstrate inflammation and injury. Tissue blocks were sectioned at a thickness of 5 mm, transferred to glass slides, and stained with hematoxylin/eosin. Morphologic examinations were performed in a blinded fashion using light microscopy, and intestinal I/R injury severity was scored using an established injury scoring system developed by Stallion et al.22 The injury severity scale ranged from 0–4. In the scoring method 0 represents normal villus to crypt ratio, minimal number of lymphocytes and presence of tall columnar surface epithelial cells; 1 represents epithelial cell degenerative changes (cuboidal, vacuolated), and mild increase of lymphocytes in lamina propria; 2 represents decreased villus height, epithelial cell necrosis, erosions, infiltration of neutrophils and glandular dilatation; 3 represents affected villi (flat surface), epithelial cell necrosis, erosions, pseudomembrane on surface, glandular destruction, and inflammation extending deep to muscle layer, and 4 represents transmural changes together with all of the above parameters and change in muscle layer.22
ELISA
Intestinal tissue was homogenized in a lysis buffer (10 mM Tris-HCl, pH 7.5, 120 mM NaCl, 1% sodium deoxycholate, and 0.1 % sodium dodecyl sulfate) containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein concentration was determined by the BioRad protein assay reagent (Hercules, CA). TNF-α levels in the serum and intestinal tissue were determined with a mouse enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences, San Diego, CA). CIRP level in the serum were determined using ELISA kit provided by LifeSpan Biosciences, Inc (Seattle, WA).
Ly6G/C staining of intestinal tissue
Intestinal tissue samples were de-waxed, incubated with proteinase K, stained using a green fluorescent-tagged Ly6G/C Ab, counterstained with propidium iodide, and examined under a fluorescence microscope. Ly6G/C cells appeared green fluorescent on a red background staining and were blindly counted per visual field at a 200x magnification.
Analysis of serum injury markers
Blood samples were drawn prior to euthanasia from the inferior vena cava and centrifuged at 8,000 × g for 10 min. The supernatant containing the serum was collected and then analyzed immediately for lactate dehydrogenase (LDH) and aspartate aminotransferase (AST) to serve as tissue injury markers. LDH and AST levels were measured in serum using assay kits from Pointe Scientific (Canton, MI) by following the protocol provided in the kits’ literature and analyzed the kinetic values using a spectrophotometer (Synergy H1 Hybrid Reader, BioTek, South San Francisco, CA) at 340 nm wavelength.
Quantitative real-time polymerase chain reaction (PCR) for determining the expression of IL-6 and MIP-2 mRNAs in lung
Total RNA was extracted from lung tissue using a Trizol reagent (Invitrogen, Carlsbad, CA) and was reverse-transcribed into cDNA using reverse transcriptase enzyme (Applied Biosystems, Foster City, CA). A quantitative real-time PCR assay was carried-out in 20 μl of a final volume containing 0.08 μmol of each forward and reverse primer, cDNA (2 μl of 10× diluted cDNA), and 10 μl SYBR Green PCR master mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems Step One Plus real-time PCR machine. The level of mouse β-actin mRNA was used for normalization. Relative expression of mRNA was expressed as the fold change in comparison with the sham tissues. The primers used for quantitative real-time PCR are: IL-6: (NM_000600.3257) ‘5-CAAATTCGGTACATCCTC-3’, ‘5-CTGGCTTGTTCCTCACTA-3’; MIP-2: (NM_009140) 5’-CCCTGGTTCAGAAAATCATCCA-3’, ‘5-GCTCCTCCTTTCCAGGTCAGT-3’; β-actin: (NM_031144) ‘5-GTGAAAAGATGACCCAGATCA-3’, ‘5-TGGTACGACCAGAGGCATACAG-3’.
Measurement of myeloperoxidase (MPO) activity
Lung tissue samples were homogenized in potassium phosphate (KPO4) buffer containing 0.5% hexa-decyl-trimethyl-ammonium bromide. After centrifugation, the supernatant was diluted in a reaction solution, and the rate of change in optimal density for 30 seconds was measured at 460 mm to calculate the MPO activity.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM) and compared by one-way analysis of variance (ANOVA) and Student-Newman-Keuls (SNK) test for multiple group comparisons. T-test was used for comparison of CIRP serum levels between sham and vehicle mice. Significance was considered if P < 0.05 between the experimental groups. Our group sizes (n = 5 per group) were comparable to our previous work with intestinal ischemia reperfusion and ultimately were justified by statistical significance.
Results
CIRP is increased in serum after intestinal I/R
We first demonstrated that the levels of CIRP in serum were significantly elevated in mice 4 h after undergoing intestinal I/R in the vehicle mice (266 ± 101.4 pg/mL) as compared to sham mice, which were not detectable (p < 0.05) (Fig. 1). This result ensures that our intestinal I/R model created enough damage to produce extracellular CIRP and thus an appropriate model for testing whether C23 could antagonize CIRP. We did not include CIRP levels in C23-treated mice as C23 is protective against inflammation and would interrupt the positive feedback loop that increases CIRP level. Thus, the C23-treated mice may have a diminished CIRP level that is not indicative of severity of intestinal I/R in those mice.
Fig. 1. Serum level of CIRP in vehicle and sham mice.

Serum was collected at 4 h after reperfusion following intestinal IR and used to measure level of CIRP in the sham and intestinal IR mice (n=5 mice/group). Data are expressed as mean ± SEM and compared with ANOVA. *, p < 0.05 vs. sham. IR, ischemia-reperfusion; SEM, standard error of the mean.
Levels of serum and intestinal TNF-α attenuated in C23-treated mice
We next measured the level of the pro-inflammatory cytokine TNF-α in both the serum and intestinal tissue lysate for our experimental groups. The level of TNF-α in the serum was significantly elevated by 25-fold in the vehicle group as compared to the sham group (12.4 ± 5.0 vs 316 ± 76.9 pg/mL; p<0.05). The C23 group experienced a 71% decrease in serum TNF-α as compared to the vehicle mice (316 ± 76.9 vs 89.3 ± 10.8 pg/mL; p<0.05) (Fig. 2A). Likewise, in the intestine the level of TNF-α in the vehicle group was elevated 3-fold compared to the sham group (75.3 ± 9.4 vs 220 ± 29.0 pg/mg; p<0.05). We observed a significant 69% decrease in the TNF-α level in the intestine from the C23 treatment group as compared to the vehicle mice (220 ± 29.0 vs 68.3 ± 11.8 pg/mg; p<0.05) (Fig. 2B).
Fig. 2. Assessment of serum and intestinal TNF-α.

SSerum (2A) and intestine (2B) were harvested 4 h after reperfusion and used to measure levels of TNF-α using ELISA. (n=5 mice/group) Data are expressed as mean ± SEM and compared with one way ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. IR, ischemia-reperfusion; SEM, standard error of the mean; TNF-α, tumor necrosis factor-α; ELISA, enzyme-linked immunosorbent assay.
C23 treatment mitigates organ injury after intestinal I/R
We next sought to detect differences in organ injury between our experimental groups using the surrogates AST and LDH in serum. The levels of AST (Fig. 3A) and LDH (Fig. 3B) in the serum of vehicle mice (312 ± 85.1 IU/L and 939 ± 175.0 IU/L, respectively) were elevated 5-fold and 76-fold, respectively, when compared to the sham group (54 ± 3.6 IU/L and 12 ± 1.5 IU/L, respectively). In contrast, the C23 treatment group observed a 53% reduction in AST (147 ± 10.7 IU/L) and a 48% reduction in LDH (488 ± 102.0 IU/L) relative to the vehicle group (p < 0.05).
Fig. 3. Assessment of serum AST and LDH.

Serum was collected 4 h after reperfusion for measurement of (A) AST and (B) LDH (n=5 mice/group). Data are expressed as mean ± SEM and compared with one way ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. I–R, ischemia-reperfusion; SEM, standard error of the mean; AST, aspartate aminotransferase; LDH, lactate dehydrogenase.
C23 reduces intestinal tissue damage and myeloid/granulocyte infiltration in the intestine from intestinal I/R
In order to ascertain the effect of C23 on intestinal tissue after I/R, we examined and scored randomized hematoxylin and eosine sections of intestine from our experimental groups. In Fig 4A, the intestinal tissue sections of the vehicle group showed severe villus blunting and transmural myonecrosis as compared to the C23-treated intestinal tissue sections, which reveal less villus blunting and no transmural necrosis. The vehicle group had an average intestinal injury score of 2.5 ± 0.09 out of a maximum score of 4, a 10-fold increase in damage as compared to sham mice (0.26 ± 0.05) (p<0.05). The intestinal sections from the C23-treated mice, with an average score of 1.3 ± 0.08, exhibited a 48% reduction in histology injury score as compared to the vehicle-treated mice (p< 0.05) (Fig. 4B).
Fig. 4. Intestinal histology injury score and myeoloid-derived cell (Ly6C/G) infiltration into intestinal tissue.

Intestinal tissue from sham, vehicle, and C23-treated mice were harvested 4 h after reperfusion and subjected to histologic analysis with hematoxylin-eosin staining. (A) Representative images were chosen from each group. Magnification 200 ×. (B) Histologic injury score measuring severity on scale of 0–4, as described in Materials and Methods (n =5 mice/group). Data are expressed as mean ± SEM and compared with ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. IR, ischemia-reperfusion; SEM, standard error of the mean. Intestinal tissue from sham, vehicle, and C23-treated mice were harvested 4 h after reperfusion and subjected to immunohistochemistry with anti-Ly6G/C Ab staining (Green) and propidium iodide counterstain of cell nuclei (Red). (C) Representative images were chosen from each group. Magnification 400 ×. (D) Ly6G/C positive cells per hpf infiltrating cells were counted blindly and averaged. (n=5mice/group) Data are expressed as mean ± SEM and compared with ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. IR, ischemia-reperfusion; hpf, high power field; SEM, standard error of the mean.
The innate immune cells that are most often responsible for the inflammatory damage after ischemia reperfusion are neutrophils and macrophages. In order to demonstrate the infiltration of these cells in our intestinal I/R model, we stained the intestinal sections using anti-Ly6G/C Abs for each of our experimental groups (Fig. 4C). We observed a significant increase in the numbers of Ly6G/C positive cells in the vehicle sections (68 ± 6.8 cells/hpf) as compared to sham (10 ± 0.9 cells/hpf; Fig. 4D). Furthermore, we observed a significant decrease in the number of Ly6G/C stained cells in the C23 treated mice intestinal sections (28 ± 2.8 cells/hpf) as compared to the vehicle group (p<0.05).
Lung IL-6 gene expression is attenuated in C23-treated mice in intestinal I/R
In order to determine the remote effects of CIRP-induced inflammation in mice undergoing intestinal I/R, we measured the gene expression of IL-6 in the lung tissue lysate from our three experimental groups. The expression of IL-6 at mRNA level was increased by 36-fold in the vehicle group as compared to sham mice (Fig. 5). The IL-6 expression in the C23-treated animals was diminished significantly by 85% in comparison to the vehicle group (p < 0.05) (Fig. 5).
Fig. 5. Assessment of lung IL-6 Gene expression.
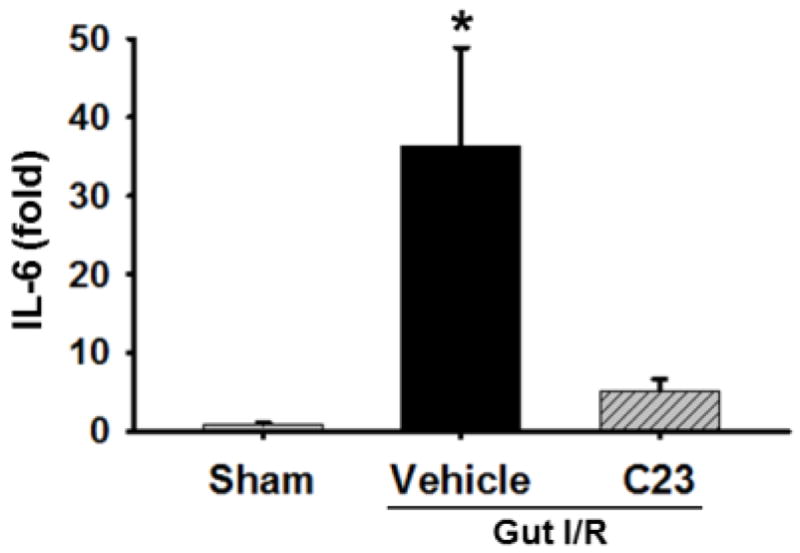
Lung tissue was collected 4 h after reperfusion and used for measurement of IL-6 gene expression using real-time PCR. (n=5 mice/group). Data are expressed as mean ± SEM and compared with one way ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. IR, ischemia-reperfusion; SEM, standard error of the mean; PCR, polymerase chain reaction; IL-6, interleukin 6.
Decreased levels of MIP-2 and MPO in lungs of C23-treated mice following intestinal I/R
We finally examined the remote lung tissue for signs of activity of a key immune cell involved in I/R injury, the neutrophil. We used gene expression of the neutrophil chemokine MIP-2 and the level of MPO activity as surrogates for the presence of neutrophils. MIP-2 gene expression was elevated 163-fold in the vehicle compared to the sham (p< 0.05; Fig. 6A). The MIP-2 expression in C23 treated mice (15-fold above the sham) exhibited a 91% decrease compared to the vehicle-treated mice (p<0.05). Similarly, MPO activity was increased 13-fold in the vehicle group as compared to the sham mice (Fig. 6B). MPO activity in the C23-treated mice (10-fold above sham) was reduced significantly by 25% as compared to the vehicle mice (p<0.05; Fig. 6B).
Fig. 6. Lung MIP-2 and MPO expression assessment.

Lung tissue was collected 4 h after reperfusion and used for measurement of (A) MIP-2 gene expression via real-time PCR and (B) MPO activity. (n=5 mice/group) Data are expressed as mean ± SEM and compared with one way ANOVA. *, p < 0.05 vs. sham; #, p < 0.05 vs. vehicle. IR, ischemia-reperfusion; SEM, standard error of the mean; MIP-2, macrophage inflammatory protein-2; MPO, myeloperoxidase.
Discussion
In the present study, we found that following intestinal I/R serum levels of CIRP were significantly induced in mice. Our previous study showed that the deficiency of CIRP in CIRP−/− mice protected animals from intestinal I/R injuries.20 Here, we implemented a reliable, safe, and clinically relevant approach to antagonize CIRP by C23 and evaluated the beneficial outcomes in intestinal I/R injuries. Our data clearly showed the inhibition of serum levels of pro-inflammatory cytokine TNF-α and inflammation and injury parameters in local (intestine) and remote organ (lung) after C23 treatment in intestinal I/R injury model in mice.
In the present intestinal I/R injury model, we did not notice significant increase in the TNF-α levels in lung tissues at 4 h after intestinal I/R injury. As such, the treatment of C23 also did not show noticeable inhibition in the TNF-α levels in lung following intestinal I/R injuries. The severity of intestinal I/R injury model and the timing of sample collection after intestinal I/R in mice might play an important role in optimal TNF-α production in lung. In line with this finding, Soares et al. reported that TNF-α is not associated with intestinal I/R-induced lung inflammation.23 They have shown that after intestinal I/R injury in mice, several cytokines were upregulated in lung, in particular IL-6, but not TNF-α. Furthermore, neutralization of TNF-α by antibodies did not affect neutrophil recruitment to the lung, the cytokine response, and pulmonary injury. The inflammatory response in the lung in the absence of TNF-α or of TNF receptors was not significantly different from WT controls upon intestinal I/R injury.23 That study did however demonstrate that TNF-α is elevated in intestinal tissue after intestinal I/R and appears to be responsible in part for intestinal inflammation. The importance of IL-6 in lung inflammation seen after intestinal I/R has been further demonstrated by Yuan et al.24 They revealed that IL-6 knockdown ameliorates ALI induced by intestinal I/R in rats demonstrating that IL-6 is a significant driver of lung inflammation in intestinal I/R.24 It is for these reasons that we focused on the measurement and difference of IL-6 in the lung tissue as a marker for inflammatory activation in the lung tissue instead of TNF-α, which was used as a marker for inflammation in the intestinal tissue.
After the initial activation of the innate immune response, adhesion of innate immune cells to endothelial cells within the intestine and the subsequent infiltration of these leukocytes into intestinal tissue represents the next step towards local damage during the maladaptive response of reperfusion. Leukocyte-vessel wall adhesion and plugging of microcirculation with leukocytes is an established mechanism of intestinal I/R damage.10, 25 Riaz et al. revealed that by blocking neutrophil adhesion with anti-P-selectin antibody, neutrophil rolling and adhesion were reduced, which lead to significantly less intestinal injury following intestinal I/R. Once innate cells enter intestinal parenchyma, they cause further damage through release of cytotoxic molecules, such as elastase and reactive oxygen species.26 Macrophages have been previously shown to express TNF-α when exposed in vitro to CIRP.14 Thus, once these cells enter previously ischemic intestinal tissue, we hypothesize that macrophages produce further inflammation in part through CIRP stimulation. Ly6G and Ly6C was applied to our intestinal sections to stain neutrophils and macrophages, respectively, in order to measure adhesion and infiltration of these cells into the intestinal parenchyma. C23 significantly reduced the presence of these damaging innate immune cells. We applied an established intestinal injury scoring system to our H&E intestinal sections to demonstrate the therapeutic effect of C23 treatment on ameliorating intestinal tissue injury.22 Ultimately, by reducing the activation, adhesion, and infiltration of innate immune cells, C23 treatment significantly reduced the histologically evident damage as compared to the vehicle mice.
Intestinal I/R not only causes local tissue inflammation but a robust systemic inflammatory response.27 In our study we used serum TNF-α levels to reveal a significant elevation in systemic inflammation compared to sham mice. This elevation was accompanied by an elevation in AST and LDH, circulating markers of general cellular injury present in higher quantity in the vehicle mice relative to sham. C23 treatment significantly reduced the levels of TNF-α as well as AST and LDH levels relative to vehicle, demonstrating C23’s ability to reduce systemic inflammation and injury in intestinal I/R.
Following intestinal I/R we noticed significant decrease of the tissue level of MPO activity and MIP2 gene expression in C23-treated mice. While no significant difference in histologically evident damage was seen after 4 h of reperfusion between vehicle- and C23-treated mice (data not shown). The discrepancy can be explained by the timing of the lung collection. We believe that we would likely see a difference at a later collection point (20 h). Cen et al, showed significant difference in the injury score between WT and CIRP−/− mice after 20 h of intestinal I/R injuries, while they confessed that at 4 h, the lung injury score between CIRP−/− mice and WT mice undergoing intestinal I/R injury were not significantly different. Based on our previous experiences we believe the differences in lung damage requires more than 4 h to be evident histologically.
C23 is a 15-aa peptide derived from human CIRP protein.14 It has strong binding affinity to TLR4/MD2 complex, the receptor for CIRP as defined by surface plasmon resonance.14 While we have not directly demonstrated that C23 functions by binding to and antagonizing MD2-TLR4 under in vivo condition, our study demonstrated C23’s efficacy to inhibit CIRP-induced pro-inflammatory responses in intestinal I/R injuries possibly by interfering the binding of CIRP with its receptor. Since C23 has been shown to neutralize only CIRP’s binding to TLR4 while allowing other TLR4 ligands to bind, this specificity may be more advantageous than general TLR4 inhibition as TLR4 is heavily relied upon for recognition and neutralization of bacterial pathogens. Furthermore, DAMP inhibition using a peptide confers a shorter drug half-life, less immunogenicity, and less expense than inhibition with an antibody. Finally, CIRP inhibition or genetic knockout in mice has not yielded obvious side effects or increased mortality. In our previous study, we showed that the treatment of cells with C23 alone did not show any toxicity, inflammatory or immunogenic activities, indicating C23 treatment to have no apparent side effects or toxicity when delivered in vivo.21
We have previously demonstrated extracellular CIRP’s function as a DAMP in propagating inflammation and tissue damage in intestinal I/R through the use of CIRP knockout mice, which, while scientifically enlightening, is not translatable into a human treatment. This manuscript describes the study of the novel peptide C23 and the potential to be translated into a therapy for human patients experiencing intestinal I/R injury. The clinical relevance of our intestinal I/R model is most directly applicable to a surgical scenario in which the patient has return of perfusion with concomitant C23 treatment 60 min after the ischemic insult, such as a patient with vaso-occlusisve disease that is recognized and treated early or a patient undergoing an elective procedure where there is a risk of intestinal I/R, such as intestinal transplantation and use of cardiopulmonary bypass during cardiac surgery or abdominal aortic clamping during vascular surgeries. Our model does not adequately represent the situation in which most vaso-occlusive patients find themselves; most vaso-occlusive patients have delayed presentation and treatment beyond 1 hour leading to a prolonged ischemic time. Future study of C23’s benefit will include intestinal I/R models with a delay before C23 treatment is administered. Still, we have shown that the C23 peptide lowers intestinal inflammation and damage as well as systemic and remote organ inflammation after 60 minutes of ischemia. We thus recommend further study with this novel therapy.
Acknowledgments
This study was supported by NIH grants R01 HL076179 and R35 GM118337 (P.W.).
Footnotes
Author Contributions
P.W. conceived idea; JTM, FZ performed the experiments; JTM, MA, WLY analyzed the data; JTM, JTM wrote the manuscript, MA revised the manuscript; JMN, GFC, PW helped in manuscript preparation and experimental design; PW supervised the whole project. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grace PA. Ischaemia-reperfusion injury. Br J Surg. 1994;81(5):637–47. doi: 10.1002/bjs.1800810504. [DOI] [PubMed] [Google Scholar]
- 2.Clair DG, Beach JM. Mesenteric Ischemia. New England Journal of Medicine. 2016;374(10):959–68. doi: 10.1056/NEJMra1503884. [DOI] [PubMed] [Google Scholar]
- 3.Kassahun WT, Schulz T, Richter O, Hauss J. Unchanged high mortality rates from acute occlusive intestinal ischemia: six year review. Langenbeck’s Archives of Surgery. 2008;393(2):163–71. doi: 10.1007/s00423-007-0263-5. [DOI] [PubMed] [Google Scholar]
- 4.Park WM, Gloviczki P, Cherry KJ, Hallett JW, Bower TC, Panneton JM, et al. Contemporary management of acute mesenteric ischemia: factors associated with survival. Journal of vascular surgery. 2002;35(3):445–52. doi: 10.1067/mva.2002.120373. [DOI] [PubMed] [Google Scholar]
- 5.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(48):20388–93. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 7.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32(1):4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 9.Eltzschig HK, Eckle T. Ischemia and reperfusion [mdash] from mechanism to translation. Nature medicine. 2011;17(11):1391–401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190(3):255–66. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Schoots IG, Koffeman GI, Legemate DA, Levi M, van Gulik TM. Systematic review of survival after acute mesenteric ischaemia according to disease aetiology. British Journal of Surgery. 2004;91(1):17–27. doi: 10.1002/bjs.4459. [DOI] [PubMed] [Google Scholar]
- 12.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 13.Bellingan GJ. The pulmonary physician in critical care * 6: The pathogenesis of ALI/ARDS. Thorax. 2002;57(6):540–6. doi: 10.1136/thorax.57.6.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiang X, Yang W-L, Wu R, Zhou M, Jacob A, Dong W, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nature medicine. 2013;19(11):1489–95. doi: 10.1038/nm.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishiyama H, Higashitsuji H, Yokoi H, Itoh K, Danno S, Matsuda T, et al. Cloning and characterization of human CIRP (cold-inducible RNA-binding protein) cDNA and chromosomal assignment of the gene. Gene. 1997;204(1):115–20. doi: 10.1016/s0378-1119(97)00530-1. [DOI] [PubMed] [Google Scholar]
- 16.Cen C, Yang W-L, Yen H-T, Nicastro JM, Coppa GF, Wang P. Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery. 2016;160(2):473–83. doi: 10.1016/j.surg.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godwin A, Yang WL, Sharma A, Khader A, Wang Z, Zhang F, et al. Blocking Cold-Inducible RNA-Binding Protein (CIRP) Protects Liver from Ischemia/Reperfusion Injury. Shock. 2015;43(1):24–30. doi: 10.1097/SHK.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan MM, Yang WL, Brenner M, Bolognese AC, Wang P. Cold-inducible RNA-binding protein (CIRP) causes sepsis-associated acute lung injury via induction of endoplasmic reticulum stress. Sci Rep. :72017. doi: 10.1038/srep41363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang W-L, Sharma A, Wang Z, Li Z, Fan J, Wang P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Scientific reports. 2016:6. doi: 10.1038/srep26571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cen C, McGinn J, Aziz M, Yang WL, Cagliani J, Nicastro JM, et al. Deficiency in cold-inducible RNA-binding protein attenuates acute respiratory distress syndrome induced by intestinal ischemia-reperfusion. Surgery. 2017;162(4):917–27. doi: 10.1016/j.surg.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 21.McGinn J, Zhang F, Aziz M, Yang WL, Nicastro J, Coppa GF, et al. The Protective Effect of a Short Peptide Derived from Cold-Inducible Rna-Binding Protein in Renal Ischemia-Reperfusion Injury. Shock. 2017 doi: 10.1097/SHK.0000000000000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stallion A, Kou TD, Latifi SQ, Miller KA, Dahms BB, Dudgeon DL, et al. Ischemia/reperfusion: a clinically relevant model of intestinal injury yielding systemic inflammation. J Pediatr Surg. 2005;40(3):470–7. doi: 10.1016/j.jpedsurg.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 23.Soares AL, Coelho FR, Guabiraba R, Kamal M, Vargaftig BB, Li L, et al. Tumor necrosis factor is not associated with intestinal ischemia/reperfusion-induced lung inflammation. Shock. 2010;34(3):306–13. doi: 10.1097/SHK.0b013e3181cdc585. [DOI] [PubMed] [Google Scholar]
- 24.Yuan B, Xiong LL, Wen MD, Zhang P, Ma HY, Wang TH, et al. Interleukin-6 RNA knockdown ameliorates acute lung injury induced by intestinal ischemia reperfusion in rats by upregulating interleukin-10 expression. Mol Med Rep. 2017;16:2529–37. doi: 10.3892/mmr.2017.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beuk RJ, Tangelder GJ, Maassen RL, Quaedackers JS, Heineman E, Oude Egbrink MG. Leucocyte and platelet adhesion in different layers of the small bowel during experimental total warm ischaemia and reperfusion. Br J Surg. 2008;95(10):1294–304. doi: 10.1002/bjs.6320. [DOI] [PubMed] [Google Scholar]
- 26.Riaz AA, Wan MX, Schaefer T, Schramm R, Ekberg H, Menger MD, et al. Fundamental and Distinct Roles of P-Selectin and LFA-1 in Ischemia/Reperfusion-Induced Leukocyte-Endothelium Interactions in the Mouse Colon. Ann Surg. (2362002) :777–84. doi: 10.1097/00000658-200212000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu H, Deng YY, Liu L, Tan QH, Wang CH, Guo MM, et al. Intestinal ischemia-reperfusion of macaques triggers a strong innate immune response. World J Gastroenterol. 2014;20(41):15327–34. doi: 10.3748/wjg.v20.i41.15327. [DOI] [PMC free article] [PubMed] [Google Scholar]