

Abstract
Inflammation contributes to neonatal brain injury. Pro-inflammatory cytokines represent key inflammatory meditators in neonatal hypoxic-ischemic (HI) brain injury. The high mobility group box-1 (HMGB1) protein is a nuclear protein with pro-inflammatory cytokine properties when it is translocated from the nucleus and released extracellularly after stroke in adult rodents. We have previously shown that HMGB1 is translocated from the nucleus to cytosolic compartment after ischemic brain injury in fetal sheep. In the current study, we utilized the Rice-Vannucci model to investigate the time course of HMGB1 translocation and release after HI injury in neonatal rats. HMGB1 was located in cellular nuclei of brains from sham control rats. Nuclear to cytoplasmic translocation of HMGB1 was detected in the ipsilateral-HI hemisphere as early as zero h after HI, and released extracellularly as early as 6 h after HI. Immunohistochemical double staining detected HMGB1 translocation mainly in neurons along with release from apoptotic cells after HI. Serum HMGB1 increased at 3 h and decreased by 24 h after HI. In addition, rat brains exposed to hypoxic injury alone also exhibited time dependent HMGB1 translocation at 3, 12 and 48 h after hypoxia. Consequently, HMGB1 responds similarly after HI injury in the brains of neonatal and adult subjects. We conclude that HMGB1 is sensitive early indicator of neonatal HI and hypoxic brain injury.
Keywords: brain, hypoxia, HMGB1, ischemia, neonate
Introduction
Hypoxia-ischemia (HI) and hypoxic-ischemic encephalopathy (HIE) represent common causes of neurological injury in preterm and full term infants with birth related complications (Fatemi, et al., 2009, Scafidi, et al., 2009). Neonates exposed to HI injury can have poor neurological and behavioral outcomes including increased risk and incidences of learning deficits (Stephens, et al., 2010). However, the underlying mechanisms by which HI and HIE result in brain injury have not been completely elucidated. Although recent studies have revealed numerous mechanisms predisposing to HI injury, these mechanisms are complex and new molecules are continually being identified. Hypothermia is the only approved intervention for HIE, which unfortunately is only partially protective (Perrone, et al., 2012, Shankaran, et al., 2010, Shankaran, et al., 2012). Consequently, there is a critical need to identify additional mechanism(s) underlying HI in order to develop novel neuroprotective strategies.
HI is characterized by brain injury caused by hypoxia and/or reduced cerebral blood flow to the brain resulting in neuronal damage from intracellular accumulation of sodium, water, and calcium, inter-synaptic glutamate accumulation, activation of nitric oxide, and synthesis of free radicals (Arundine and Tymianski, 2003, Back, et al., 2007, Lipton, et al., 1993). These cytotoxic processes result in various forms of cell death, such as cellular necrosis, apoptosis and autophagy (Balduini, et al., 2009, Balduini, et al., 2012, Pulera, et al., 1998). These events can induce a robust inflammatory reaction by activation of endogenous microglia (Weinstein, et al., 2010). Inflammation has also been implicated in neuronal and white matter injury after neonatal HI (McAdams and Juul, 2012). Pro-inflammatory cytokines have been described as key contributors to inflammation after HI brain injury (Liu, et al., 1994, McAdams and Juul, 2012, Saito, et al., 1996). Findings in preterm and full term infants also suggest that elevations in pro-inflammatory cytokines are important in the pathogenesis of HI brain injury (McAdams and Juul, 2012, Silverstein, et al., 1997).
The high mobility group box-1 (HMGB1) protein is a ubiquitously abundant nuclear protein that functions as a structural co-factor for proper transcriptional regulation in the nucleus. HMGB1 binds to the minor groove with moderate affinity and folds DNA into a spiral (Ulloa and Messmer, 2006). In the developing brain under normal conditions, HMGB1 can also function as a neurite growth factor (Merenmies, et al., 1991, Zhang, et al., 2011). However, during injury, activated immune and damaged cells release HMGB1 into the extracellular space, where HMGB1 functions as a pro-inflammatory mediator and contributes to the pathogenesis of inflammation related brain injury (Wang, et al., 1999). Numerous studies have demonstrated that ischemic brain injury results in HMGB1 translocation from the neuronal nucleus into the brain parenchyma in adult subjects (Kim, et al., 2006, Muhammad, et al., 2008, Zhang, et al., 2011). The release of HMGB1 can occur as early as 2 h after ischemic reperfusion related brain injury in adult rats (Zhang, et al., 2011). Extracellular HMGB1 stimulates glutamate release, inflammatory responses and blood-brain barrier injury in adult subjects (Zhang, et al., 2011). Most importantly, several in vivo studies have shown that anti-HMGB1 treatment of ischemic injury attenuates brain inflammation and suppresses cell death in the early phase of ischemic injury (Liu, et al., 2007, Muhammad, et al., 2008, Yang, et al., 2010, Zhang, et al., 2011). However, there is very limited information regarding its characteristics after HI or hypoxia in the immature brain.
We have previously reported that HMGB1 is translocated from the nucleus to the cytosolic compartment in the cerebral cortex after ischemic brain injury in the fetal brain (Zhang, et al., 2016). In our previous work, HMGB1 was localized primarily to the cellular nuclei and partially to the cytoplasmic compartment in the cerebral cortex of sham operated control fetal sheep. Ischemia increased the amount of neuronal cells that demonstrated cytoplasmic HMGB1 staining (Zhang, et al., 2016). In addition, Western immunoblots revealed that nuclear HMGB1 expression decreased and cytosolic HMGB1 increased in the brains of the fetal sheep exposed to ischemia compared with the sham control treatment (Zhang, et al., 2016). Our findings in the fetal sheep brain are consistent with previous findings in adult rodents after ischemic injury, and suggest that alterations in HMGB1 expression and localization after ischemia in fetal brain could contribute to ischemia-related inflammation. Other studies are also consistent with our findings suggesting cytosolic localization of HMGB1 in neurons and astrocytes in the normal fetal sheep brain (Frasch and Nygard, 2017). However, systemic acidosis induced by multiple umbilical cord occlusions caused subtle and brain region-specific reverse shifts in neuronal HMGB1 patterns in cortical gray matter (Frasch and Nygard, 2017, Frasch, et al., 2016) Differences among the studies summarized above are most likely a result of differences in the animal models and methodology (Frasch and Nygard, 2017, Frasch, et al., 2016). Nonetheless, these studies have not examined potential sequential changes in HMGB1 in the immature brain after hypoxic ischemic injury.
In the current study, we utilized the well characterized Rice-Vannucci model of neonatal HI (Rice, et al., 1981) to determine the time course of the HMGB1 translocation and release in neonatal rat brain. In addition, we identified cell specific translocation and release of HMGB1 and the relationship between HMGB1 release and cell death, along with the time course of alterations in HMGB1 in serum and brain by Western immunoblot and ELISA after neonatal HI brain injury. Additionally, we examined the effects of hypoxia without ischemia on HMGB1 expression in neonatal rat brain.
Materials and Methods
This study was conducted after approval by the Institutional Animal Care and Use Committees of the Alpert Medical School of Brown University and Women & Infants Hospital of Rhode Island and in accordance with the National Institutes of Health Guidelines for the use of experimental animals.
Animal preparation, study groups, and experimental study design
The subjects were neonatal rats born to time-mated dams (Charles River Laboratories; Wilmington, MA) in the Animal Care Facility at Brown University. Pregnant Wistar rats on embryonic day 15 (E15) or E16 were shipped and then housed in a 12-hour light/dark cycled facility with ad libitum access to food and water in the Animal Care Facility at Brown University. After the delivery date was confirmed for each pregnant dam, the date upon which the rat pups were born was designated as postnatal day 0 (P0). Pups from different litters born on the same day were then culled and balanced with regard to sex differences so that each dam had no more than 10 pups. On P7, the pups were randomly assigned to sham operated control, sham operated hypoxia alone, and HI exposed groups. HI was induced in the pups by carotid artery ligation on the right side along with exposure to 8% oxygen for 2 hours using previously described methods. (Rice, et al., 1981)
Anesthesia was induced in the pups with 3-4% isoflurane and maintained with 1-2% isoflurane during the procedure. Body temperature was maintained at 36°C during surgery with an isothermal heating pad. A skin incision was made at the midline of the neck overlying the trachea using sterile surgical scissors. The right common carotid artery (RCCA) was separated from the trachea and the surrounding nerves and double ligated using 5-0 silk sutures. The incision was closed and sterilized with betadine and alcohol. Sham treated subjects were exposed to the same procedure except the RCCA was not ligated. The pups were sutured and labeled with neonatal tattooing system (Neo-9, Animal Identification & Marking Systems, Inc., Hornell, NY, USA). The pups were returned to their dams for 1.5–3 h for feeding and recovery from surgery. They were then placed in a hypoxia chamber with 8% humidified oxygen and balanced nitrogen for 2 hours with a constant temperature of 36°C. Sham control subjects were exposed to room air for 2 hours. The sham treated subjects that were exposed to 8% oxygen for 2 hours are hereafter designated as the hypoxia alone group. The rats in each study group were spread among different litters to account for the potential inter-litter variability. (Rice, et al., 1981) Animals in the HI or hypoxia alone groups were sacrificed at various time points after exposure to HI or hypoxia.
Brain collection and immunohistochemical staining
Brains were obtained from the neonatal rat pups that were exposed to carotid artery ligation and hypoxia (HI), or sham treatment with and without hypoxia. Brains were collected, paraffin-embedded, and sectioned for immunostaining from the sham operated control (n=25) and from neonatal rats at zero (n=8), 3 (n=8), 6 (n=7), 12 (n=14), 24 (n=15), and 48 h (n=14) after exposure to HI. Brains were also collected and similarly treated for immunostaining from the sham operated control group (n=9) and from neonatal rats at zero (n=9), 3 (n=9), 12 (n=8), 48 h (n=9) after exposure to hypoxia alone. For the purpose of this report, zero, 3, and 6 h were considered as the early phases of recovery from HI or hypoxia alone, and 24 and 48 h were considered as the later phases of recovery from HI or hypoxia alone.
The pups were first sedated with an intraperitoneal injection of a mixture of ketamine (74 mg/kg) and xylazine (4 mg/kg). The injection was followed by a quick pinch on the hind leg of the animal to ensure adequate sedation. Blood samples were collected from the left ventricle, and brains were perfused with cold saline and 4% paraformaldehyde (PFA) at a flow rate of 3 ml/min. Brains were removed and post fixed in PFA for 24 h. The paraffin-embedded brain tissues were sectioned in coronal planes at 6 microns thicknesses. One coronal section containing the dorsal hippocampus per brain (bregma −3.12 ± 0.6mm) was utilized to standardize the immunohistochemical analyses (Paxinos and Watson, 2017) using previously reported methodologies (Zhang, et al., 2016, Zhang, et al., 2011). Briefly, the sections were processed in an antigen retrieval buffer of 0.1 M sodium citrate and heated in an autoclave at 120°C for 10 minutes, and blocked with 1% bovine serum albumin (BSA, Sigma, St. Louis, MO, USA) and 5% normal goat serum (Vector Laboratories, Burlingame, CA, USA) in tris-buffered saline (TBS). The sections were then incubated overnight with a primary antibody solution with mouse anti-HMGB1 monoclonal antibody (R&D Systems Inc., Minneapolis, MN, USA) at a dilution of 1:500 in combination with either a rabbit anti-NeuN polyclonal antibody (Abcam, Cambridge, MA, USA) at a dilution of 1:1000, or rabbit anti-Iba-1 monoclonal antibody (Dako, Carpinteria, CA, USA) at a dilution of 1:1000, or a rabbit anti-GFAP polyclonal antibody (Dako, Carpinteria, CA, USA) at a dilution of 1:1000. Rabbit anti-LC3II polyclonal antibody (Abcam, Cambridge, MA, USA) was used at a dilution of 1:1000 to detect autophagy. The secondary antibodies were Alexa-555 labeled anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) and Alexa-488 labeled anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA) both at a dilution of 1:1000. Coverslips were applied to the slides with anti-fading mounting reagent and DAPI (Vector Laboratories, Burlingame, CA, USA). Stained slides were visualized under a Zeiss Axio Imager M2 imaging system microscope (Carl Zeiss, Inc., Jena, Germany) connected with computer software Stereo Investigator 10.0 (MicroBrightField, Inc., Williston, VT, USA) for image analysis.
HMGB1 translocation and release
The slides from animals studied were randomly chosen from each study group (n=4 for each group except n=3~4 for the 48-h group) to visualize or quantify HMGB1 translocation and release. The slides were coded and labeled by an independent third party to ensure that the slides were evaluated without knowledge of the treatment protocol or study group. Image acquisition and quantification were carried out according to previous publications with modifications as follows (Gross, et al., 2016, Malaeb, et al., 2009, Ortiz-Lopez, et al., 2017). Briefly, separate images with a 400 x magnification of HMGB1 staining (cy3), NeuN, GFAP, or Iba-1 staining (FITC), and DAPI filters were obtained from five random fields per slide from the cerebral cortex of the neonatal rats exposed to HI or hypoxia alone. The number of immunoreactive cells was manually quantified with the count tool in Adobe Photoshop CC (Adobe, San Jose, CA, USA). Double-fluorescent labeling for HMBG1 and NeuN (neuronal marker), GFAP (astrocytic marker), and Iba-1 (microglial marker) were obtained to quantify the number of positive stained cells. HMGB1 translocation from the nuclear compartment to the cytosol and release from the cell were defined as follows. The cells with HMGB1-positive staining within only nuclear compartment were considered HMGB1 positive in nucleus (nuclear HMGB1-positive staining). Reductions or absence of HMGB1 staining within the nuclear compartment along with positive HMGB1 staining within the cytosolic compartment was considered HMGB1 translocation from the nucleus to the cytosolic compartment (cytoplasmic HMGB1-positive staining), whereas, the absence of nuclear HMGB1 staining and presence of HMGB1 outside of the cells in the extracellular matrix or extracellular space was considered release from the cell (negative HMGB1 staining). The HMGB1 positive cells in nuclear and cytosol compartments and HMGB1 negative cells were calculated as a percentage to the total number of cells, which was determined by counting the DAPI positive nuclei using Image J software (NIH, Bethesda, MD, USA). The minimum size of particles was set to 4 μm2 to avoid false positive staining of cell fragments (Threlkeld, et al., 2014).
Double-fluorescent labeling for in situ DNA fragmentation for apoptosis and HMGB1
Brains for double-fluorescent labeling for in situ DNA fragmentation for apoptosis and HMGB1 staining were obtained from the sham operated control (n=6) and from neonatal rats at 48 h (n=6) after exposure to HI. The TdT-mediated dUTP-biotin nick end labeling (TUNEL) assay was used to detect of the apoptosis 48 h after exposure to HI. A subset of paraffin-embedded brain sections were used for the TUNEL assay with the ApopTag® fluorescein detection method using an in situ apoptosis kit according to the protocols described by the manufacturer (Millipore, Billerica, MA, USA). The coronal brain sections of the cerebral cortex were outlined as previously described (Zhang, et al., 2016). The number of apoptotic cells in cerebral cortex was quantified without knowledge of the treatment group using the Fractionator probe of Stereo Investigator 10.0 software (MBF Bioscience). Sections of the cerebral cortex were traced under 100 x magnification. An automated stage paired with the Stereo Investigator was used to step systematically through the counting frames (Fractionator probe) (Threlkeld, et al., 2014). Cells that were ApopTag® positive and did not exhibit HMGB1 staining were considered to be cells undergoing apoptosis that had been depleted of HMGB1 or were HMGB1 negative (ApopTag+/HMGB1−). Cells that were ApopTag® negative and HMGB1 negative were considered to be cells that were not undergoing apoptosis but were depleted of HMGB1 (ApopTag−/HMGB1−). The data were expressed as a percentage of counted cells (ApopTag+, ApopTag+/HMGB1−, ApopTag−/HMGB1−) to the total number of cells counter-stained with DAPI.
Enzyme-linked immunosorbent assay (ELISA)
Serum for HMGB1 analysis by ELISA was obtained from the sham control rats and from neonatal rats at 0, 3, 6, 12, 24, and 48 h after exposure to HI. Serum levels of HMGB1 were determined on whole blood samples that were collected from the left ventricle before the transcardial perfusion without using an anticoagulant reagent. Blood samples were centrifuged immediately at 2,300 g at room temperature. Clots were removed and the supernatant collected in a separate tube and placed on ice for 2 h. Any additional clots that formed were also removed and the supernatant was saved at −80°C freezer until analysis. Before analysis, the samples were centrifuged again and the supernatant was used for the ELISA assay. HMGB1 concentration was determined using an HMGB1 ELISA kit (Shino-Test, Tokyo, Japan) following the manufacture’s protocol. (Liu, et al., 2007, Zhang, et al., 2011)
Western immunoblot for analysis of HMGB1
The ipsilateral-HI cerebral cortical hemispheres for Western immunoblot analysis of HMGB1 were obtained from the sham operated control and from neonatal rats at 3, 12 and 48 h after exposure to HI. Rat brains for protein analyses were perfused with 0.9% NaCl without PFA fixative. Brain homogenates were prepared in T-PER (Thermo Fisher, Waltham, MA, USA) buffer extraction reagent with a cock-tail proteinase inhibitor (Roche, Basel, Switzerland). Equal amounts of 10 μg of protein per well were fractionated for the Western immunoblots using the ready-to-use 4-12% BIS TRIS SDS-polyacrylamide gel (Invitrogen) electrophoresis. After electrophoresis, proteins on the gel were transferred onto polyvinylidene diflouride (PVDF) membrane (PVDF, 0.2 micron, Bio-Rad Laboratories, Hercules, CA) using a semi-dry transfer technique. Membranes were blocked with 10% skim milk (Bio-Rad) prepared in TBS with 0.1% Tween-20 solution (TBST), and washed three times in TBST. The membrane was probed overnight with peroxidase conjugated rat anti-HMGB1 monoclonal antibody (mAb) at 4°C (Zhang, et al., 2011). This antibody detects a band of HMGB1 approximately 28 kDa (predicted molecular weight: 25 kDa) in adult rat brain tissue. Membranes were washed on the following day four times with TBST and incubated with enhanced chemiluminescence solution (Amersham Pharmacia Biotech, Piscataway, NJ, USA) before exposure to autoradiography film (Daigger, Vernon Hills, IL, USA). β-Actin was probed with a mouse β-Actin antibody (1:10000 dilution, Santa Cruz Biotechnology, Inc, Dallas, TX, USA) overnight at 4°C to serve as the loading control, and a HRP-conjugated goat anti-mouse IgG (ThermoFisher Scientific, Carlsbad, CA, USA) was used to visualize the immunoblots.
All experimental samples were normalized to a protein extract obtained from a homogenate pool from the cerebral cortex of a single adult rat brain. As we have previously described, these protein samples served as internal control samples (Sadowska, et al., 2010). The use of the internal control standard allows accurate quantification among different groups and Western immunoblots. The optical densities of total HMGB1 were expressed as a ratio to the internal control values thus facilitating normalized comparisons among different experimental groups and immunoblots.
Statistical analysis
Statistical significance was evaluated using a one-way analysis of variance (ANOVA) followed by post hoc comparisons using the Fisher least significance difference (LSD) test. Two-way ANOVA was also used to compare differences in HMGB1 cytoplasmic translocation in cerebral cortical cells for the different time periods between HI- and hypoxia alone-treated groups. Values are expressed as mean ± standard deviation (SD). Differences with a probability value of <0.05 were considered to be significant.
Results
Time-dependent HMGB1 translocation and release
Immunofluorescent staining of HMGB1 revealed that HMGB1 was expressed ubiquitously in the cells of the brains in the sham control neonatal rats (Fig. 1A, Sham, white arrows). HMGB1 expression was localized mainly in the nuclear compartment in the sham control neonatal rats. Immediately after exposure to HI at the zero h time point, the cerebral cortices exhibited clear HMGB1 translocation in the ipsilateral-HI hemisphere showing HMGB1 cytosolic staining (Fig. 1A, 0 h, yellow arrows). In addition, similar findings were detected at three hours after HI (Fig. 1A, 3 h, yellow arrows). Six hours after HI, the HMGB1 stain exhibited punctate structures along the cellular somata on the HI side of the brain (Fig. 1A, 6 h, thick white arrow). At 12 h after HI, the HMGB1 stain was detected both in the cytosol (Fig. 1A, 12 h, yellow arrow) and in the extracellular space (Fig. 1A, 12 h, thick white arrows). At 24 h after HI, the positive HMGB1 staining exhibited speckled appearing structures with strong signals in the extracellular matrix and along the neuronal somata and axons (24 h thick white arrows) suggesting the extracellular release of HMGB1. A similar phenomenon was also observed at 48 h after HI (Fig. 1, 48 h). Fig. 1 (right corner) shows a schematic representation of the brain regions that exhibited HMGB1 translocation and release. A major portion of the ipsilateral-HI hemisphere exhibited translocation and/or release within 6 h after exposure to HI.
Fig. 1. Immunohistochemical staining and quantification of HMGB1 in neonatal rat brain with and without HI.

(A) Representative images of neonatal rat brains obtained from sham operated and zero, 3 6, 12, 24 and 48 h after 2 h of hypoxia and carotid artery ligation. A sham operated neonatal rat (P7) shows nuclear HMGB1 staining in most cells (white arrows) of the cerebral cortical region. Immediately at 0 h after HI, HMGB1 exhibited translocation from the cellular nuclei into the cytosol (0, 3, and 12 h, yellow arrows) in the cerebral cortex of the ipsilateral HI brain hemisphere. At 6 24, and 48 h after HI, HMGB1 exhibited punctate structures with strong signals in extracellular matrix and along neuronal cell somata and axons (6, 12, 24, and 48 h, thick white arrows). A schematic illustration showing the brain region (highlighted in gray) that exhibited HMGB1 translocation and release (right corner). All images are from the HI region of the cerebral cortex. Scale bar = 50 μm. (B) Quantification of total HMGB1 cytoplasmic translocation in the cerebral cortical cells of sham-operated, zero, 3, 12, and 48h after HI in the neonatal rats. HMGB1 cytoplasmic translocation was increased at zero, 3, and 12 h after exposure to HI compared to the sham group. Mean ± SD, n= 4 for each group. *P<0.05 versus Sham, ANOVA, Fisher LSD.
Cells with HMGB1 cytoplasmic translocation as a percent of the total number of cells counted in the cerebral cortex are plotted for the sham control group and the groups immediately (0 h) and 3, 12 and 48 h after exposure to HI (Fig. 1B). The ipsilateral HI-injured cerebral cortices exhibited a higher percent of cells with HMGB1 cytoplasmic translocation at zero, 3 and 12 h after exposure to HI compared with those in the sham-operated neonatal rat brains (Fig. 1B, ANOVA, P < 0.001). However, cells with cytosolic staining of HMGB1 were markedly reduced 48 h after HI (Fig. 1B, ANOVA, P = 0.88) most likely because HMGB1 had been released into extracellular space as a result of damage to the brain cells at this late stage of injury.
Cell specific translocation and release of HMGB1
Double immunohistochemical staining of HMGB1 with different cellular markers was performed in the brain including Fox3/neuronal nuclei (NeuN) for neurons, fibrillary acidic protein (GFAP) for astrocytes, and the ionized calcium binding adaptor molecule 1 (Iba-1) for microglia to identify the specific cell types.
Fig. 2 contains representative images of HMGB1 translocation and release in the NeuN-positive cells for the sham control group and the groups immediately (0 h) and 3, 6, 12, 24, and 48 h after exposure to HI. The sham control neonatal rats show HMGB1 localized in the nucleus of the NeuN positive neurons (Merged, white arrows). Immediately at 0 h and 3 h after HI, cytosolic HMGB1 translocation was identified in the NeuN positive neurons (Fig. 2, 0 and 3 h, yellow arrows). At 6 h after HI, HMGB1 translocation and HMGB1 cellular release along with extracellular staining was detected in the NeuN positive neurons (Fig. 2, 6 h, yellow arrows show translocation; thick white arrows show release and extracellular staining). Similar findings were observed in the NeuN positive neurons at 12 and 24 h after HI (Fig. 2, 12 and 24 h, yellow arrows show translocation; thick white arrows indicate release and extracellular staining). At 48 h after HI, most of the NeuN positive neurons exhibited HMGB1 cellular depletion along with extracellular staining (Fig. 2, 48 h, yellow arrows show translocation; thick white arrows indicate release and extracellular staining).
Fig. 2. Immunohistochemical double staining of HMGB1 with a neuronal marker NeuN in the neonatal rat cerebral cortex at zero, 3, 6, 12, 24, and 48 h after exposure to HI injury.

HMGB1 translocation was observed as early as zero and 3 h after HI injury (zero and 3 h, yellow arrows). HMGB1 release was detected at 6 h after HI injury (6 h, thick white arrows). Major release of HMGB1 into extracellular compartment was observed at 48 h after HI injury (48 h, thick white arrows). Scale bar = 20 μm.
Fig. 3 contains quantification for nuclear staining of HMGB1 (HMGB1+/NeuN+, A), cytoplasmic translocation of HMGB1 (HMGB1+/NeuN+, B) and the negative staining of HMGB1 (HMGB1−NeuN+, C) as a percent of the DAPI+ cells for the NeuN-positive neurons in the sham control neonatal rats and the groups 3, 12 and 48 h after exposure to HI. The percent of cells with HMGB1+/NeuN+ staining in the nuclei decreased at 3, 12, and 48 h after HI compared with the sham control group (Fig. 3A, ANOVA, P < 0.01). The percent of cells with cytoplasmic HMGB1+/NeuN+ increased at 3 and 12 h after exposure to HI compared with the sham control neonatal rats (Fig. 3B, ANOVA, P < 0.001). The percent of cells that were negative for HMGB1 (HMGB1−-NeuN+) increased in the NeuN-positive neurons at 12 and 48 h after HI compared with the sham control group (Fig. 3C, ANOVA, P < 0.05) suggesting extracellular release of HMGB1 at these time points after HI.
Fig. 3. Quantification of HMGB1 translocation and release in cerebral cortical neurons after exposure to HI injury.

(A) The percent of nuclear staining of HMGB1 and NeuN positive cells to total DAPI positive cells decreased at 3, 12, and 48 h after HI injury. (B) The percent of cytoplasmic double staining of HMGB1 and NeuN positive cells to total DAPI positive cells increased at 3 and 12 h, but decreased at 48 h after HI injury. (C) The percent of HMGB1 negative and NeuN positive cells to total DAPI stained cells increased at 12 and 48 h after HI injury. Mean ± SD, n = 4 for each group except n = 3 for the 12 h group. * P < 0.05 versus Sham, ANOVA, Fisher LSD.
Fig. 4 contains representative images of HMGB1 in the GFAP-positive astrocytes for the sham control and the groups immediately (0 h) and 3, 6, 12, 24, and 48 h after exposure to HI. The sham control neonatal rats show HMGB1 localized in the nucleus of the GFAP-positive astrocytes (Merged, white arrow). At 0, 3, 6, 12, 24, and 48 h after HI, nuclear HMGB1 staining was detected in the GFAP-positive astrocytes (Fig. 4, 0, 3, 6, 12, 24 and 48 h, white arrows). Cytosolic HMGB1 translocation was not identified in the GFAP stained astrocytes. Three hours after exposure to HI, DNA damage was observed in some astrocytes on the border of the corpus callosum and the cerebral cortex with ring-like structures that co-localized with HMGB1 (Fig. 4, 3 h, white arrowheads). Similar staining was observed during the late phase of recovery after HI at 24 and 48 h. In addition, some GFAP-positive cells showed negative staining of HMGB1 at 48 h after HI injury (Fig. 4, 48 h, white arrowheads) suggesting that HMGB1 could have been translocated and released from astrocytes in the late phase after HI injury.
Fig. 4. Immunohistochemical double staining of HMGB1 with astrocyte marker (GFAP) in the neonatal rat brain cerebral cortex at zero, 3, 6, 12, 24, and 48 h after exposure to HI injury.

The HMGB1 expression was mainly in the nucleus of astrocytes in the sham-operated, and at zero, 3, 6, 12, and 24 h after HI. HMGB1 translocation and release was not observed in the astrocytes. DNA damage with ring-like structures co-localized with HMGB1 was observed in some astrocytes (3 h, white arrowhead). Furthermore, some astrocytes showed negative nuclear staining of HMGB1 at 48 h after HI injury (48 h, white arrowhead). White arrows show the representative nuclear localization of HMGB1 in GFAP positive cells. Scale bar = 20 μm.
Table 1 contains the values for GFAP positive cells with nuclear HMGB1-positive, cytoplasmic HMGB1-positive, and HMGB1-negative cells as percent of the DAPI-positive cells in the cerebral cortex of sham control group, and 3, 12, and 48 h after exposure to HI. The total GFAP-positive cells as a percent of the DAPI-positive cells was increased 12 and 48 h after HI compared with the sham control group (ANOVA, P < 0.01) suggesting potential astrogliosis due to tissue injury. The nuclear staining of HMGB1+/GFAP+ astrocytes as a percent of the total DAPI-positive cells was increased (ANOVA, P < 0.05) at 12 and 48 h after HI compared with the sham control group. Differences were not observed in the cytoplasmic staining of HMGB1 in the GFAP positive cells among the study groups (ANOVA, P = 0.34). The number of GFAP positive cells that did not stain for HMGB1 was increased 48 h after HI (ANOVA, P < 0.01). Therefore, HMGB1 translocation may not be a major pathological hallmark of astrocytes in the neonates with HI injury except in the late phase after HI (Fig. 4 and Table 1).
Table 1.
HMGB1 translocation and release in GFAP positive cells after HI injury in neonatal rats.
| Percent GFAP+ ratio to total DAPI+ cells |
Sham | 3 h | 12 h | 48 h |
|---|---|---|---|---|
| Total GFAP+ cells | 5.53 ± 1.67 | 11.04 ± 3.84 | 14.29 ± 3.41* | 26.57 ± 5.63*+‡ |
| Nuclear staining of HMGB1+- GFAP+ cells |
5.08 ± 1.52 | 10.76 ±3.81 | 13.72 ± 2.82* | 24.80 ±6.38*+‡ |
| Cytoplasmic staining of HMGB1+- GFAP+ cells |
0.14 ± 0.17 | 0.04 ± 0.08 | 0.35 ± 0.35 | 0.23 ± 0.27 |
| Negative staining of HMGB1−- GFAP+ cells |
0.31 ± 0.30 | 0.24 ± 0.31 | 0.22 ± 0.26 | 1.53 ± 0.86*+‡ |
Values are expressed as mean ± SD,
P < 0.05 vs. Sham,
P < 0.01 vs. 3 h,
P < 0.01 vs. 12 h
Fig. 5 contains representative images of HMGB1 in the Iba-1-positive cells for the sham control and the groups immediately (0 h) and 3, 6, 12, 24, and 48 h after exposure to HI. The sham control neonatal rats show HMGB1 localized in the nucleus of the Iba-1-positive microglial cells (Merged, white arrows). At zero, 3, 6, 12, 24, and 48 h after HI, HMGB1 localization was mainly identified in the nucleus of Iba-1-positive microglial cells (Fig. 5, 0, 3, 6, 12, 24 and 48 h, white arrows). Cytosolic HMGB1 translocation was not observed in microglial cells at any time point examined after HI (data not shown).
Fig. 5. Immunohistochemical double staining of HMGB1 with microglia marker (Iba-1) in the neonatal rat brain cerebral cortex at zero, 3, 6, 12, 24, and 48 h after exposure to HI.

The HMGB1 expression was detected in the nucleus of the microglial cells in the sham-operated, and in the zero, 3, 6, 12, and 24 h after HI. HMGB1 translocation and release were not observed in microglia. White arrows show the representative nuclear localization of HMGB1 in the Iba-1 positive cells. Scale bar = 20 μm.
HMGB1 translocation and release and cell death
HMGB1 has previously been shown to be released during apoptosis into apoptotic cell-derived membranous vesicles (Schiller, et al., 2013). However, translocation and release of HMGB1 in cells undergoing apoptosis after HI injury has not been previously reported in the brain. In this study, we examined HMGB1 cellular translocation and release in brain cells undergoing apoptosis using double staining of HMGB1 along with a late phase apoptotic cell marker, ApopTag® (TUNEL assay). Fig. 6A shows representative images of the cerebral cortex on the ipsilateral HI-hemisphere of a neonatal rat brain 48 h after HI. Translocation and release of HMGB1 was examined in conjunction with apoptosis 48 h after HI because the density of apoptotic cells in the cerebral cortex has been shown to be elevated between 12 h to 7 days after HI (Nakajima, et al., 2000). Abundant TUNEL positive cells were detected suggesting that apoptosis represented the majority of cellular death 48 h after HI injury (Fig. 6A ApopTag) but not at the earlier time points (data not shown). A substantial number of cells undergoing apoptosis exhibited HMGB1 depletion suggesting that HMGB1 was completely released from the apoptotic cells (Fig. 6A, Merge, white arrows).
Fig. 6. Immunohistochemical staining of HMGB1 and different cell death markers in the neonatal rat brain after exposure to HI brain injury.

(A) Double immunohistochemical staining of HMGB1 and ApopTag 48 h after exposure to HI: ApopTag+/HMGB1− cells shown white arrows; ApopTag− /HMGB1− cells shown by thick white arrows. Scale bar = 50 μm. (B) Quantification of apoptotic cells (ApopTag+ in Sham and 48 h after HI, blue bar), apoptotic positive cells with depleted HMGB1 (ApopTag+/HMGB1−, red bar), and non-apoptotic cells with depleted HMGB1 (ApopTag−/HMGB1−, green bar) after HI injury. Bar graphs represent the percent of apoptotic cells, which have released HMGB1 48 h after HI injury. Mean ± SD, n = 6, three cerebral cortical regions counted for each rat brain. *P<0.001 by ANOVA, Fisher LSD. (C) (a) 2,3,5-triphenyltetrazolium chloride (TTC) staining of brain section. Dashed line shows HI region examined. (b) Double immunohistochemical staining for HMGB1 and LC3-II as an autophagic cellular marker. HMGB1 translocation is co-localized with the autophagic cells in a circumscribed region of the cerebral cortex; Scale bar =10 μm.
Fig. 6B contains quantification of the immunohistochemical analysis as the percent of cells from the total number of cells counted on the Y-axis plotted for the Sham control group showing the ApopTag positive (+) cells, and the HI exposed group 48 h after HI as ApopTag positive (+) cells (blue bar), ApopTag positive (+)/HMGB1 negative (−) cells (red bar) and Apop Tag negative (−)/HMGB1 negative (−) cells (green bar). Quantification showed that 48 h after HI approximately 68% of the total number of cells in the ipsilateral-HI hemisphere were undergoing apoptosis (Fig. 6A, ApopTag, and 6B, blue bar). More than 90% of the cells undergoing apoptosis exhibited HMGB1 depletion suggesting that HMGB1 was released mostly from the apoptotic cells (Fig. 6A Merge, white arrows, and 6B, red bar). In contrast, only two percent of cells that did not undergo apoptosis exhibited release of HMGB1 suggesting that most of the cells undergoing apoptosis in the later phase after HI injury had released HMGB1 into the extracellular space (Fig. 6A Merge, thick white arrows, and 6B, green bar).
Autophagy is another form of programmed cell death that occurs in the brain exposed to HI injury (Balduini, et al., 2009, Balduini, et al., 2012). Although it has been shown that tumor cells dying via autophagy selectively release HMGB1 (Thorburn, et al., 2009) this phenomenon has not been examined after HI related brain injury. Autophagic pathways have been shown to be present after HI brain injury (Carloni, et al., 2008). Consequently, we performed double immunohistochemical staining for HMGB1 and cellular autophagy using the microtubule-associated protein 1 light chain (LC3-II), an autophagic marker to determine if cells undergoing autophagy release HMGB1 24 h after HI brain injury. LC3-II positive autophagic cells were detected 24 h after HI in a circumscribed area of the cerebral cortex confined to cortical layers 2 to 3 (Fig. 6C-a, area shown by the dashed line). HMGB1 translocation was clearly detected in all cells undergoing autophagy and co-localized with the LC3-II stain (Fig. 6C-b), which indicates that HI induced autophagic cells also release HMGB1.
Serum concentrations and brain expression of HMGB1
HMGB1 concentrations in serum samples collected from sham operated and HI exposed neonatal rats at zero, 3, 6, 12, 24, and 48 h after HI were determined by ELISA. Serum concentrations of HMGB1 were significantly elevated at 3 h and remained elevated up to 12 h after exposure to HI. A rapid decrease in serum HMGB1 was detected between 12 and 24 h after HI and returned to levels similar to those in the sham control group at 24 and 48 h after HI (Fig. 7A).
Fig. 7. Serum concentrations and brain expression of HMGB1 after HI brain injury in neonatal rats.
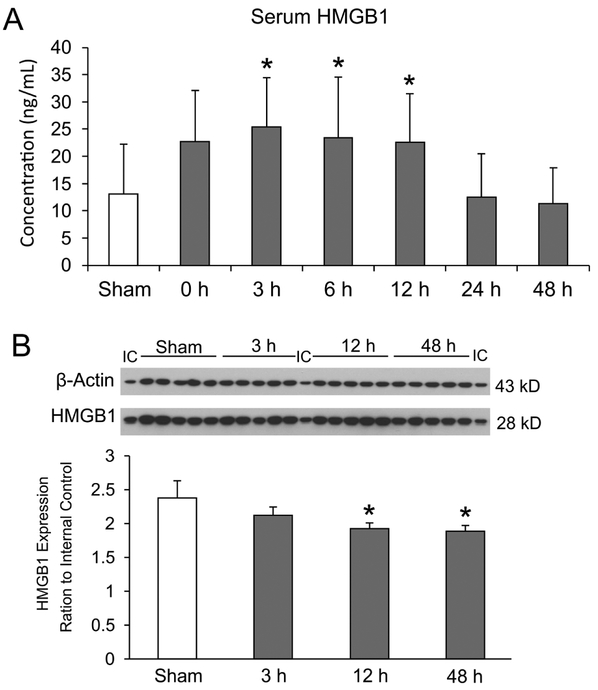
(A) Serum collected from sham operated (n=17) and HI rats immediately 0 h (n=5), 3 h (n=12), 6 h (n=14), 12 h (n=12), 24 h (n=11), and 48 h (n=15) h after exposure to HI. Mean ± SD, *P<0.05, ANOVA, Fisher LSD. (B) Representative Western immunoblots for β-Actin (43KD) and HMGB1 (28 kD) shown for the internal control protein (IC), in Sham control, and 3, 12 and 48 h after HI. Total protein extracted from ipsilateral-HI cerebral cortical hemisphere from rat brain of sham treated, 3, 12, and 48 h after exposure to HI (each group: n=5). Total HMGB1 expression in rat brain, plotted as ratio to internal control (IC). Mean ± SD, * P<0.05, ANOVA, Fisher LSD.
Expression of HMGB1 in sham control and 3, 12, and 48 h after HI in neonatal rat brain was examined by Western immunoblot analysis using the total protein extraction from the brains of each group (Fig. 7B). Fig. 7B (upper panel) shows representative immunoblots of β-Actin as a loading control, and HMGB1 from the internal control sample (IC) and the brains of sham control and of neonatal rats 3, 12 and 48 h after exposure to HI. HMGB1 protein expression in the neonatal rat brain was detected as a single band at approximately 28 kDa, which is consistent with a previous study in adult rats (Zhang, et al., 2011). In the lower panel, HMGB1 expression is plotted as a ratio to the internal control values on the Y-axis for the sham control group and the groups 3, 12 and 48 h after exposure to HI on the x-axis. Quantitative analysis of the densities of the HMGB1 bands demonstrated a time-dependent decrease in the amount of HMGB1 in the neonatal rat brain with significant reductions appearing 12 and 48 h after HI (Fig. 7B). The reduction in HMGB1 expression within the HI-cerebral cortical tissue supports the contention that HMGB1 was translocated from the cellular to the extracellular compartment at 12 and 48 h after exposure to HI.
Hypoxia alone induced HMGB1 translocation
Neonatal rats exposed to hypoxia alone were exposed to 8% oxygen for 2 h after sham surgery and the brains collected zero, 3, 12 and 48 h after hypoxia. Fig. 8 shows that HMGB1 was mostly located in the nucleus of the sham treated control animals (8A, white arrows). In contrast, significant numbers of cells demonstrated HMGB1 translocation from the nucleus to the cytoplasmic compartment 12 h after exposure hypoxia alone (Fig. 8A, yellow arrows). Similarly, 48 h after exposure to hypoxia alone NeuN labelled neuronal nuclei exhibited translocation of HMGB1, whereas release of HMGB1 from the cellular somata was not apparent (Fig. 8A, yellow arrows).
Fig. 8. HMGB1 immunohistochemical staining after exposure to hypoxia alone.

(A) Representative images of HMGB1 double immunochistochemical staining in sham treated and 12 and 48 h after hypoxia alone. Double immunohistochemical staining of HMGB1 and the neuronal marker (NeuN) from rat cerebral cortex 48 h after hypoxia. (B) Quantification of the percent of cells exhibiting HMBG1 translocation from the nucleus to the cytosolic compartment in after 2 h of hypoxia alone. Mean ± SD, n=4 for each group except n=3 for the 48 h group. *P<0.05 versus Sham, ANOVA, Fisher LSD.
Fig. 8B contains quantification for cells exhibiting HMGB1 cytoplasmic translocation as a percent of the total number of cells counted in the cerebral cortex of the rat brain. HMGB1 translocation from the nuclear to the cytoplasmic compartment was significantly greater in both of the cerebral hemispheres at 3, 12 and 48 h hypoxia compared with the sham control treated animals. Therefore, based upon these findings, HMGB1 is a very sensitive marker for hypoxic brain injury in neonatal subjects.
Inspection of Figs. 1B and 8B suggested the potential that HI was associated with a larger percent of cells exhibiting cytoplasmic HMGB1 translocation than with hypoxia alone. Figure 9 contains the number of cells exhibiting HMGB1 cytoplasmic translocation as a percent of the total number of DAPI+ cells for the sham control groups and the groups immediately (0 h) and 3, 12 and 48 h after exposure to HI (Fig. 9, closed bars) and hypoxia alone (Fig. 9, gray bars). There were no differences in the number of cells exhibiting HMGB1 cytoplasmic translocation as a percent of the total number of cells counted between the two different sham control groups. The numbers of cells exhibiting HMGB1 cytoplasmic translocation was significantly greater at zero, 3, and 12 h after HI than for the groups exposed to hypoxia alone (Fig. 9, ANOVA, P < 0.001). In contrast, HMGB1 cytoplasmic translocation in cerebral cortices was lower at 48 h after HI than after hypoxia alone (Fig. 9, ANOVA, P < 0.001). HI-induced brain cell damage most likely resulted in greater HMGB1 release into extracellular space than hypoxia alone in the neonatal rats.
Fig. 9. Comparison of HMGB1 cytoplasmic translocation in cerebral cortical cells during different time periods after exposure to HI and hypoxia alone.
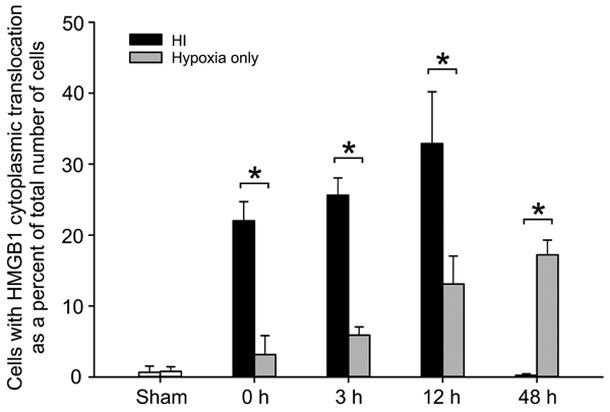
HMGB1 cytoplasmic translocation was significantly higher at zero, 3, and 12 h in Hi-treated group (closed bar) than in hypoxia alone group (gray bar). Due to HMGB1 release, HMGB1 cytoplasmic translocation was lower at 48 h in HI treated group than in hypoxia alone group. Mean ± SD, n=4 for each group except n=3 for the 48 h hypoxia group. *P<0.05, two-way ANOVA, Fisher LSD.
Discussion
HMGB1 translocation and release have been reported in numerous studies in adult rodents with ischemic brain injury (Kim, et al., 2006, Qiu, et al., 2008, Zhang, et al., 2011). However, there is limited information regarding the localization and expression of HMGB1 after brain injury in the perinatal period. Our recent study demonstrated that HMGB1 also exhibited translocation from the nucleus to the cytosolic compartment after ischemic brain injury in fetal sheep (Zhang, et al., 2016). Similarly, there is very little information regarding the effects of HI on HMGB1 expression in the neonatal brain. Furthermore, the time course of changes in HMGB1 localization and expression after neonatal HI brain injury remains to be determined. The present series of experiments are the first to examine time dependent expression of HMGB1 after neonatal HI injury with regard to its cellular localization, cell type specific expression, and expression as a function of specific types of cellular death, and protein expression by Western Immunoblot. In addition, the time course of HMGB1 translocation was also examined after exposure to a hypoxic stress without ischemia.
HMGB1 has been shown to be a very sensitive and early marker of brain injury in the adult (Kim, et al., 2006, Qiu, et al., 2008, Zhang, et al., 2011). It is located in the nuclear compartment in the normal adult rat brain. (Zhang, et al., 2011) In our former study, we detected HMGB1 mainly in cellular nuclei along with a small quantity of cells that exhibited cytosolic HMGB1 staining in the cerebral cortical cells of the non-ischemic fetal sheep brain (Zhang, et al., 2016). In the current study, although HMGB1 was localized primarily to the nucleus in the normal neonatal rat brain (Fig. 1), approximately one percent of the cerebral cortical cells exhibited a cytosolic distribution of HMGB1 (Figs. 1B, 8B, 9, Sham, open bars). Previous work has similarly shown that the normal developing mouse brain on embryonic day 16 exhibited one percent of cells with cytosolic HMGB1 localization (Guazzi, et al., 2003). These findings are consistent with our previous work in fetal sheep, in which we found that five percent of cerebral cortical cells in normal fetal brain sheep exhibited cytoplasmic HMGB1 (Zhang, et al., 2016), suggesting there could be small differences in HMGB1 localization between the normal developing and the adult brain.
In previous studies of stroke in adult rats, typical translocation of HMGB1 into the cytosolic compartment was evident in the ischemic core at two hours after ischemia followed by release and depletion of HMGB1 from the cell at 24 and 48 hours after brain injury. The findings in the current study are consistent with results in adult rodents (Qiu, et al., 2008, Zhang, et al., 2011) revealing similar phenomena detected immediately after HI injury (Fig. 1B) with release of HMGB1 from the cell beginning at 6 h and lasting up to 48 h after HI (Fig. 1, 6, 24, and 48 h) (Kim, et al., 2006, Muhammad, et al., 2008, Zhang, et al., 2011). Therefore, HMGB1 is also a sensitive marker of HI brain injury in neonates.
The findings in the neonatal rats exposed to the hypoxic stress indicates that hypoxia alone results in sufficient injury to the brain to result in HMGB1 translocation and release in neurons of neonatal rats (Fig. 8). Moreover, significant increases in translocation of HMGB1 were observed at 3, 12 and 48 h after the end of hypoxia (Fig. 8B). In contrast, HMGB1 translocation was observed immediately at zero h after exposure to HI (Fig. 1A, 1B). These findings suggest that the HMGB1 translocation potentially could be delayed after the exposure to hypoxia alone compared with the translocation immediately after HI. Although it has been suggested that hypoxia as such does not result in major brain injury in the Rice-Vannucci model (Rice, et al., 1981, Vannucci, et al., 1998), a recent publication suggested that neonatal rats exposed to 8% oxygen for 90 minutes without ischemia showed delayed abnormalities in neural function and histopathological injury suggesting that oxygen deprivation alone is capable of injuring the neonatal brain 24 h after an hypoxic insult (Zhang, et al., 2013). Consistent with these findings, we found that the hypoxic stress alone resulted in increased numbers of cells with HMGB1 translocation as early as 3 h after the termination of hypoxia (Fig. 8) suggesting HMGB1 translocation represents a relatively early event that potentially begins before histopathological abnormalities can be detected. However, comparison of HMGB1 nuclear to cytoplasmic translocation after HI and hypoxia alone (Fig. 9) suggests that the quantity of translocation was greater after HI than hypoxia at zero, 3, and 12 h after the insults suggesting that HI represents the greater stimulus for HMGB1 translocation than hypoxia. In contrast, HMGB1 translocation was lower at 48 h after HI than hypoxia alone suggesting that after HI HMGB1 release from the cells was potentially greater.
Release of HMGB1 was visualized as punctate structures located in the extracellular space as early as 6 h after HI that continued increasing during the later phase at 24 to 48 h after HI injury (Fig. 1A). Similar punctate collections of extracellular HMGB1 have also been reported in our previous study of adult rats with ischemic brain injury (Zhang, et al., 2011). Although the mechanism and precise composition of these punctate collections has not yet been defined, Bonaldi et.al reported similar structures that accumulated in the cytosol and were released into the extracellular space in association with a lysosomal type of HMGB1 release in an in vitro study (Bonaldi, et al., 2003). Additional work is required to determine whether lysosomal release of HMGB1 occurs by a comparable process in vivo in the developing brain after HI injury. Nonetheless, our findings suggest that the responses and biologic functions of HMGB1 during ischemic brain injury are similar in the brain of neonatal and adult subjects.
Previous studies in the adult show that neuronal cells represent the major cell type exhibiting HMGB1 translocation and release after ischemic injury in the brain (Kim, et al., 2008, Kim, et al., 2006, Zhang, et al., 2011). Our recent study in fetal sheep demonstrated similar findings showing that the translocation of HMGB1 was mainly located in neurons. Similar to previous findings, neurons were the main cell type to exhibit this phenomenon especially at the early stages of the HI injury (Figs. 2-5). Several studies have demonstrated the release of HMGB1 from cultured astrocytes as a result of exposure to various chemical stimuli by the detection of HMGB1 release into the culture media. However, translocation of HMGB1 from the nuclear to cytosolic compartment was not observed consistent with our findings (Faraco, et al., 2007, Hayakawa, et al., 2012, Li, et al., 2014, Passalacqua, et al., 1998). In addition, HMGB1 has been detected as late as two days after reperfusion in astrocytes (Kim, et al., 2008). However, we were not able to detect a clear change in HMGB1 localization in astrocytes in neonatal rats after HI. This may be a result of limitations in the use of paraffin-embedded brain sections to detect the release of proteins from the cells in the brain. Compared with neurons, the astrocytes and microglia have relatively smaller cell bodies with smaller cytosolic compartments, and therefore, the smaller cytosol could potentially render it more challenging to detect a clear shift of HMGB1 from the nucleus to the cytosol. Hence, it would be important to utilize an in vitro cell culture system in future studies to determine cell-specific translocation and release of HMGB1 in response to HI injury.
HI can result in cell death by a variety of mechanisms including necrosis, apoptosis and autophagy (Balduini, et al., 2009, Balduini, et al., 2012, Pulera, et al., 1998). Previous work has shown that the maximum number of TUNEL-positive cells were present 24 to 72 h after HI in the ipsilateral HI-hemisphere in neonatal rodents (de Torres, et al., 1997, Pulera, et al., 1998). Consistent with these findings, we detected abundant ApopTag positive cells 48 h after HI. In addition, the ApopTag positive cells also exhibited HMGB1 release 48 h after HI in the neonatal rat brain (Fig. 6A and 6B). Therefore, HMGB1 release is associated with apoptosis after HI in the neonatal rodent brain.
Autophagy has a protective role in neonatal HI by bulk degradation of cellular constituents (Carloni, et al., 2008). Autophagosome activity was detected by LC3-II staining in a circumscribed area of the cerebral cortex along with HMGB1 co-localization and release 24 h after HI (Fig. 6Ca and Cb). These findings may suggest that autophagy is associated with the translocation of HMGB1 without resulting in cell membrane lysis or necessarily cellular death (Thorburn, et al., 2009). The differential HMGB1 release during cell death by a variety of processes may reflect different chromatin structural mechanisms resulting in HMGB1 release (Bell, et al., 2006).
Serum HMGB1 concentrations were increased at 3, 6 and 12 h after HI brain injury but returned to levels that were similar to the sham control subjects by 24 and 48 h after HI (Fig. 7A), even though cerebral cortical cellular HMGB1 release was most prominent in the later phases after HI. The differences in the patterns of HMGB1 response between serum concentration changes and cerebral cortical cellular HMGB1 release suggest that the source of the serum increases may not be mainly from brain. Rather, the origin of the serum HMGB1 increases could result from peripheral responses to HI brain injury, and/or the effects of systemic hypoxia on other organs. Nevertheless, elevated plasma levels of HMGB1 have been shown to correlate with the severity of stroke in adult patients (Huang, et al., 2013). Umbilical artery levels of HMGB1 were elevated in infants with HIE before exposure to therapeutic hypothermia and the levels decreased after institution of therapeutic hypothermia (Nakamura, et al., 2013). Our findings along with these studies support the contention that serum levels of HMGB1 could be an important biomarker of brain injury and of other adverse perinatal events.
The changes detected by immunohistochemistry (Figs. 1-5) were consistent with the findings by Western immunoblot analysis (Fig. 7B). HMGB1 expression was detected as a solitary band at 28 kD in the neonatal rat brain with and without exposure to HI injury similar to previous findings in the adult rat (Zhang, et al., 2011). However, the current findings in neonatal rats after exposure to HI differ somewhat from our earlier findings in the fetal sheep after exposure to brain ischemia (Zhang, et al., 2016). In the previous study, we examined the HMGB1 expression separately in the cytosolic and nuclear fractions using the same specific rat anti-bovine HMGB1 monoclonal antibody and found that HMGB1 was detected as two bands in the cytosolic and one band in the nuclear fraction both in fetal sheep exposed to the sham treatment and brain ischemia with reperfusion for 48 and 72 h (Zhang, et al., 2016). In contrast, HMGB1 expression was examined only on total protein in the brains of the neonatal rats after exposure to HI. Nonetheless, reductions in the expression of HMGB1 measured on the total protein in the neonatal rats were consistent with the reductions that we observed in the nuclear fraction of fetal sheep 48 h after brain ischemia (Zhang, et al., 2016) and our previous work in the adult rat brain after experimental stroke (Zhang, et al., 2011). In addition, reductions in the expression of HMGB1 measured in total protein from the neonatal rat brain was consistent with the immunohistochemical findings showing that HMGB1 was released from the neonatal rat brain after exposure to HI.
HMGB1 is found diffusely in extracellular space after the initial disruption of necrotic cells in areas of the brain that are severely damaged (Scaffidi, et al., 2002). Extracellular HMGB1 then binds to RAGE receptors resulting in further release of inflammatory mediators that can predispose to inflammation in the brain (Liu, et al., 2007, Qiu, et al., 2008). In addition, anti-HMGB1 monoclonal antibodies have been shown to be effective in the treatment of ischemic brain injury, traumatic brain injury, and seizures in adult rodent models most likely by neutralizing extracellular HMGB1 (Fu, et al., 2017, Okuma, et al., 2012, Zhang, et al., 2011). Therefore, based upon our findings in neonatal rats after HI, targeting HMGB1 along with its signaling molecules involved in HMGB1 translocation from cytosol to extracellular space, cell specific targeting of damage-vulnerable neurons and inhibition of downstream cytokine pathways of HMGB1 could represent potential strategies in the treatment of neonatal HI.
HI brain injury is the one of leading causes of neurodevelopmental morbidities in preterm and full term infants (Fatemi, et al., 2009, Scafidi, et al., 2009). Although many factors could contribute to its pathogenesis, perinatal hypoxia contributes to abnormal brain development (Gonzalez-Rodriguez, et al., 2014, Li, et al., 2012, Novak, et al., 2018, Schmid, et al., 2015, Tomalski and Johnson, 2010). Recent studies have suggested that hypoxia increases the vulnerability of the neonatal brain to HI injury and could predispose to a variety of neurological deficits (Gonzalez-Rodriguez, et al., 2014, Li, et al., 2012). HMGB1 serum levels have been demonstrated to be increased in neonates with asphyxia-related brain damage within 30 min after birth, suggesting that HMGB1 is useful for the early biomarker of brain injury caused by perinatal asphyxia (Zhang, et al., 2017). Similarly, in the present study, we detected HMGB1 translocation from the neuronal nuclear to the cytoplasmic compartment at as early as 3 h after a hypoxic insult (Fig. 8). Based upon our findings, HMGB1 could be a sensitive biomarker of hypoxic brain injury in neonates.
There are several limitations to the present study because HMGB1 translocation and release was primarily evaluated using immunohistochemical methodology. In future studies, translocation and release of HMGB1 could also be determined by separating the nuclear and cytosolic fractions extracted from brain tissue and then examining the expression of HMGB1 by Western immunoblot as we have previously reported in the sheep brain (Zhang, et al., 2016). However, although this method would give us more quantitative information regarding HMGB1 translocation in the total brain tissue, such quantification could not separate the different cell types. Another consideration could be to examine cytoplasmic and nuclear fractions of proteins extracted from isolated cell cultures to further examine cell-specific HMGB1 translocation and release after exposure to HI brain injury in vitro. Nonetheless, although there are limitations to the use of paraffin-embedded brain sections for the detection of proteins released from cells in the brain, we have identified the time course of nuclear to cytosolic translocation and release of HMGB1 from zero to 48 h after HI and hypoxic insults to the neonatal brain.
In summary, immunohistochemical double staining identified HMGB1 translocation primarily in neurons along with release from apoptotic cells 48 h after HI. Serum HMGB1 increased 3 h after HI and decreased by 24 h after HI. In addition, rat brains exposed to hypoxic injury also exhibited time dependent early HMGB1 translocation. Consequently, HMGB1 responds similarly after HI injury in the brains of neonatal and adult subjects. We conclude that HMGB1 is sensitive early indicator of neonatal HI and hypoxic brain injury and potentially could represent a therapeutic target. In addition, it also could represent a sensitive biomarker in the treatment of and diagnosis of neonatal HI and HIE.
Highlights.
HMGB1 translocation and release occur as early as zero and 6 h after HI injury.
HMGB1 translocation occurs mainly in neurons.
Apoptosis was associated with HMGB1 release from cells.
Hypoxia alone results in time-dependent HMGB1 translocation.
HMGB1 is a sensitive early indicator of neonatal HI and hypoxic brain injury.
Acknowledgements
Research reported in this publication was supported by the American Heart Association Postdoctoral Fellowship Award: 13POST16860015, National Institute of General Medical Sciences of the National Institutes of Health under award numbers 1R01-HD-057100, R21-NS096525, an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 RR018728 and P20GM103537. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosure/conflict of interests
The authors have no duality or conflicts of interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arundine M, and Tymianski M, 2003. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34, 325–337. [DOI] [PubMed] [Google Scholar]
- Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK, Allcock N, and Fern R, 2007. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab 27, 334–347. [DOI] [PubMed] [Google Scholar]
- Balduini W, Carloni S, and Buonocore G, 2009. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy 5, 221–223. [DOI] [PubMed] [Google Scholar]
- Balduini W, Carloni S, and Buonocore G, 2012. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med 25 Suppl 1, 30–34. [DOI] [PubMed] [Google Scholar]
- Bell CW, Jiang W, Reich CF 3rd, and Pisetsky DS, 2006. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291, C1318–1325. [DOI] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, and Bianchi ME, 2003. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22, 5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, and Balduini W, 2008. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 32, 329–339. [DOI] [PubMed] [Google Scholar]
- de Torres C, Munell F, Ferrer I, Reventos J, and Macaya A, 1997. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett 230, 1–4. [DOI] [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, and Chiarugi A, 2007. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem 103, 590–603. [DOI] [PubMed] [Google Scholar]
- Fatemi A, Wilson MA, and Johnston MV, 2009. Hypoxic-ischemic encephalopathy in the term infant. Clin Perinatol 36, 835–858, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch MG, and Nygard KL, 2017. Location, Location, Location: Appraising the Pleiotropic Function of HMGB1 in Fetal Brain. J Neuropathol Exp Neurol 76, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch MG, Szynkaruk M, Prout AP, Nygard K, Cao M, Veldhuizen R, Hammond R, and Richardson BS, 2016. Decreased neuroinflammation correlates to higher vagus nerve activity fluctuations in near-term ovine fetuses: a case for the afferent cholinergic anti-inflammatory pathway? J Neuroinflammation 13, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Liu K, Wake H, Teshigawara K, Yoshino T, Takahashi H, Mori S, and Nishibori M, 2017. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci Rep 7, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez PJ, Xiong F, Li Y, Zhou J, and Zhang L, 2014. Fetal hypoxia increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: role of glucocorticoid receptors. Neurobiol Dis 65, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P, Honnorat N, Varol E, Wallner M, Trappanese DM, Sharp TE, Starosta T, Duran JM, Koller S, Davatzikos C, and Houser SR, 2016. Nuquantus: Machine learning software for the characterization and quantification of cell nuclei in complex immunofluorescent tissue images. Sci Rep 6, 23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzi S, Strangio A, Franzi AT, and Bianchi ME, 2003. HMGB1, an architectural chromatin protein and extracellular signalling factor, has a spatially and temporally restricted expression pattern in mouse brain. Gene Expr Patterns 3, 29–33. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Pham LD, Katusic ZS, Arai K, and Lo EH, 2012. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A 109, 7505–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JM, Hu J, Chen N, and Hu ML, 2013. Relationship between plasma high-mobility group box-1 levels and clinical outcomes of ischemic stroke. J Crit Care 28, 792–797. [DOI] [PubMed] [Google Scholar]
- Kim JB, Lim CM, Yu YM, and Lee JK, 2008. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res 86, 1125–1131. [DOI] [PubMed] [Google Scholar]
- Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, and Lee JK, 2006. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26, 6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Sun L, Luo Y, Xie C, Pang Y, and Li Y, 2014. High-mobility group box 1 released from astrocytes promotes the proliferation of cultured neural stem/progenitor cells. Int J Mol Med 34, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gonzalez P, and Zhang L, 2012. Fetal stress and programming of hypoxic/ischemic-sensitive phenotype in the neonatal brain: mechanisms and possible interventions. Prog Neurobiol 98, 145–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, and Stamler JS, 1993. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626–632. [DOI] [PubMed] [Google Scholar]
- Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N, Adachi N, Yoshino T, and Nishibori M, 2007. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21, 3904–3916. [DOI] [PubMed] [Google Scholar]
- Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, and Feuerstein GZ, 1994. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 25, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Malaeb SN, Hovanesian V, Sarasin MD, Hartmann SM, Sadowska GB, and Stonestreet BS, 2009. Effects of maternal antenatal glucocorticoid treatment on apoptosis in the ovine fetal cerebral cortex. J Neurosci Res 87, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams RM, and Juul SE, 2012. The role of cytokines and inflammatory cells in perinatal brain injury. Neurol Res Int 2012, 561494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, and Rauvala H, 1991. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J Biol Chem 266, 16722–16729. [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, and Schwaninger M, 2008. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28, 12023–12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, and Johnston MV, 2000. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20, 7994–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yamada S, and Yoshioka T, 2013. Brain hypothermic therapy dramatically decreases elevated blood concentrations of high mobility group box 1 in neonates with hypoxic-ischemic encephalopathy. Dis Markers 35, 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak CM, Ozen M, and Burd I, 2018. Perinatal Brain Injury: Mechanisms, Prevention, and Outcomes. Clin Perinatol 45, 357–375. [DOI] [PubMed] [Google Scholar]
- Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, Yoshino T, Ohtsuka A, Otani N, Tomura S, Shima K, Yamamoto Y, Yamamoto H, Takahashi HK, Mori S, and Nishibori M, 2012. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol 72, 373–384. [DOI] [PubMed] [Google Scholar]
- Ortiz-Lopez L, Gonzalez-Olvera JJ, Vega-Rivera NM, Garcia-Anaya M, Carapia-Hernandez AK, Velazquez-Escobar JC, and Ramirez-Rodriguez GB, 2017. Human neural stem/progenitor cells derived from the olfactory epithelium express the TrkB receptor and migrate in response to BDNF. Neuroscience 355, 84–100. [DOI] [PubMed] [Google Scholar]
- Passalacqua M, Patrone M, Picotti GB, Del Rio M, Sparatore B, Melloni E, and Pontremoli S, 1998. Stimulated astrocytes release high-mobility group 1 protein, an inducer of LAN-5 neuroblastoma cell differentiation. Neuroscience 82, 1021–1028. [DOI] [PubMed] [Google Scholar]
- Paxinos G, and Watson C, 2017. The Rat Brain in Stereotaxic Coordinates: Compact. Elsevier Science. [Google Scholar]
- Perrone S, Stazzoni G, Tataranno ML, and Buonocore G, 2012. New pharmacologic and therapeutic approaches for hypoxic-ischemic encephalopathy in the newborn. J Matern Fetal Neonatal Med 25 Suppl 1, 83–88. [DOI] [PubMed] [Google Scholar]
- Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, and Wasterlain CG, 1998. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke 29, 2622–2630. [DOI] [PubMed] [Google Scholar]
- Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, and Moskowitz MA, 2008. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab 28, 927–938. [DOI] [PubMed] [Google Scholar]
- Rice JE 3rd, Vannucci RC, and Brierley JB, 1981. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9, 131–141. [DOI] [PubMed] [Google Scholar]
- Sadowska GB, Malaeb SN, and Stonestreet BS, 2010. Maternal glucocorticoid exposure alters tight junction protein expression in the brain of fetal sheep. Am J Physiol Heart Circ Physiol 298, H179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Suyama K, Nishida K, Sei Y, and Basile AS, 1996. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett 206, 149–152. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, and Bianchi ME, 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. [DOI] [PubMed] [Google Scholar]
- Scafidi J, Fagel DM, Ment LR, and Vaccarino FM, 2009. Modeling premature brain injury and recovery. Int J Dev Neurosci 27, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller M, Heyder P, Ziegler S, Niessen A, Classen L, Lauffer A, and Lorenz HM, 2013. During apoptosis HMGB1 is translocated into apoptotic cell-derived membranous vesicles. Autoimmunity 46, 342–346. [DOI] [PubMed] [Google Scholar]
- Schmid MB, Hopfner RJ, Lenhof S, Hummler HD, and Fuchs H, 2015. Cerebral oxygenation during intermittent hypoxemia and bradycardia in preterm infants. Neonatology 107, 137–146. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Laptook AR, Poole WK, and Eunice Kennedy Shriver NNRN, 2010. Hypothermia for perinatal asphyxial encephalopathy. N Engl J Med 362, 1051–1052; author reply 1052. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, Gustafson KE, Leach TM, Green C, Bara R, Petrie Huitema CM, Ehrenkranz RA, Tyson JE, Das A, Hammond J, Peralta-Carcelen M, Evans PW, Heyne RJ, Wilson-Costello DE, Vaucher YE, Bauer CR, Dusick AM, Adams-Chapman I, Goldstein RF, Guillet R, Papile LA, Higgins RD, and Eunice Kennedy Shriver NNRN, 2012. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 366, 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein FS, Barks JD, Hagan P, Liu XH, Ivacko J, and Szaflarski J, 1997. Cytokines and perinatal brain injury. Neurochem Int 30, 375–383. [DOI] [PubMed] [Google Scholar]
- Stephens BE, Liu J, Lester B, Lagasse L, Shankaran S, Bada H, Bauer C, Das A, and Higgins R, 2010. Neurobehavioral assessment predicts motor outcome in preterm infants. J Pediatr 156, 366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, and Thorburn A, 2009. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ 16, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlkeld SW, Gaudet CM, La Rue ME, Dugas E, Hill CA, Lim YP, and Stonestreet BS, 2014. Effects of inter-alpha inhibitor proteins on neonatal brain injury: Age, task and treatment dependent neurobehavioral outcomes. Exp Neurol 261, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomalski P, and Johnson MH, 2010. The effects of early adversity on the adult and developing brain. Curr Opin Psychiatry 23, 233–238. [DOI] [PubMed] [Google Scholar]
- Ulloa L, and Messmer D, 2006. High-mobility group box 1(HMGB1) protein: friend and foe. Cytokine Growth Factor Rev 17, 189–201. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Towfighi J, and Vannucci SJ, 1998. Hypoxic preconditioning and hypoxic-ischemic brain damage in the immature rat: pathologic and metabolic correlates. J Neurochem 71, 1215–1220. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, and Tracey KJ, 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP, and Moller T, 2010. Microglia in ischemic brain injury. Future Neurol 5, 227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QW, Wang JZ, Li JC, Zhou Y, Zhong Q, Lu FL, and Xiang J, 2010. High-mobility group protein box-1 and its relevance to cerebral ischemia. J Cereb Blood Flow Metab 30, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Klufas D, Manalo K, Adjepong K, Davidson JO, Wassink G, Bennet L, Gunn AJ, Stopa EG, Liu K, Nishibori M, and Stonestreet BS, 2016. HMGB1 Translocation After Ischemia in the Ovine Fetal Brain. J Neuropathol Exp Neurol 75, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, and Nishibori M, 2011. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ding Y, Yao Y, Yu Y, Yang L, and Cui H, 2013. Creating rat model for hypoxic brain damage in neonates by oxygen deprivation. PLoS One 8, e83589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Zhang BL, Guo SM, Wang P, and Yang JW, 2017. Clinical significance of dynamic measurements of seric TNF-alpha, HMGBl, and NSE levels and aEEG monitoring in neonatal asphyxia. Eur Rev Med Pharmacol Sci 21, 4333–4339. [PubMed] [Google Scholar]