

Abstract
Genetic imprinting is the process of epigenetic labelling or silencing of particular genes, based on the maternal or paternal origin of the gene, in a heritable pattern. The incidence of imprinting disorders has become a growing concern due to the potential association between these congenital syndromes and assisted reproductive technologies (ARTs). This review presents a general summary of the imprinting process as well as the current knowledge surrounding the genetic and epigenetic underpinnings of the most prevalent imprinting disorders: Beckwith-Wiedemann syndrome (BWS), Silver-Russell syndrome (SRS), Prader-Willi syndrome (PWS), and Angelman syndrome (AS). As research continues to elucidate the molecular pathways that characterize genetic imprinting, efforts have been made to establish guidelines that incorporate phenotypic manifestations as well as genetic testing to ensure safe and effective management of symptoms. While these efforts are likely to benefit future clinical management, their efficacy cannot yet be generalized to all patients diagnosed with these syndromes, as many of the genetic abnormalities and the associated phenotypic manifestations have yet to be characterized. Furthermore, future advances in the knowledge of epigenetic processes and genetic loci involved in the development of these syndromes may allow for the development of curative therapies.
Keywords: Epigenetics, imprinting, beckwith-wiedemann syndrome, silver-russell syndrome, prader-willi syndrome, angelman syndrome
Introduction
Epigenetic reprogramming refers to processes that allow cell differentiation via activation or inactivation of genes within different cell populations [1]. One type of reprogramming, genetic imprinting, is the epigenetic labelling or silencing of particular genes depending on the maternal or paternal origin of that gene in a tissue-specific manner [2]. The presumption that parental alleles are expressed equally by both parental chromosomes in accordance with Mendelian genetics was first challenged in the mid-1980s with nuclear transplantation experiments [1]. While eggs receiving a male pronucleus developed to term, diploid studies evaluating same sex pronuclei failed to complete normal embryogenesis, introducing the concept that factors outside of a complete set of genes were necessary for fetal development [1]. These studies ultimately suggested that specific genetic loci contributed to development in a manner that was dependent upon parental chromosomal origin in a process later termed genetic imprinting [3].
Individuals inherit two alleles of each gene, one copy from the maternal chromosome, and the other allele from the paternal chromosome [1]. For some genes, however, only one of these alleles is expressed. The allele that is not expressed is silenced via epigenetic regulation. Epigenetic processes are heritable changes in gene expression that alter the expression of a DNA segment or segments without changing the sequence. DNA methylation is one such process that refers to the reversible addition of a methyl group (-CH3) to cytosine bases located at 5’-cytosine-guanine-3’ regions in DNA sequences. These CpG regions are referred to as CpG islands. In genetic imprinting, DNA methylation at certain CpG islands silences either the maternal or paternal allele for a particular gene. Once either the maternal or paternal allele for the gene is silenced, the gene is said to be imprinted. As the embryo continues to grow and expand through sequential cellular divisions, the epigenetic imprinting is maintained through the cell line. If the ability of the cell to imprint is compromised or the non-silenced allele is deleted, numerous syndromes can arise depending on which DNA sequence or gene harbors the error [1].
Based on reported incidence rates, congenital syndromes associated with imprinting disorders are relatively uncommon [4]. The increasing use of assisted reproductive technologies (ARTs) as a potential solution to infertility, however, has increased the incidence of imprinting disorders and is a growing concern [5]. Since 1978 more than 5 million births have been the product of ARTs worldwide. Furthermore, the number of infants conceived in this manner has since grown substantially [5]. While the association between ARTs and imprinting abnormalities has been highly contested in the scientific community over the last decade, a meta-analysis released within the last 5 years indicates increasing incidence of imprinting disorders in children conceived through ARTs [6]. However, contradictory evidence exists [1], and the factors underlying these potential associations is an area of ongoing research [6].
Here, we present a general summary of the imprinting process as well as the current knowledge surrounding the genetic and epigenetic underpinnings of the most prevalent imprinting disorders: Beckwith-Wiedemann syndrome, Silver-Russell syndrome (alternatively called Russell-Silver syndrome), Prader-Willi syndrome, and Angelman syndrome. The increased use of ARTs and their association with the increasing prevalence of imprinting disorders present an upcoming challenge for scientists and clinicians, as research continues to investigate potential risk factors for the development of these syndromes. Given the heterogeneous nature of both the molecular abnormalities involved as well as the clinical presentations of these syndromes, it has proven difficult to fully elucidate how the implicated genetic pathways lead to the variety of phenotypic manifestations of imprinting syndromes, as well as to uncover the full spectrum of genetic abnormalities that may lead to imprinting syndrome development [4,7,8]. While recent efforts have been made to optimize clinical management based on genetic and epigenetic subtyping [9,10], further research is required to advance current knowledge of associated genetic loci and epigenetic regulation mechanisms in order to improve current symptom management as well as move toward potential curative therapies.
Genetic imprinting: origins, mechanisms, and pathways toward disease
Genetic imprinting is likely to have arisen during the divergence of placental mammals as a competition between the evolutionary interests of the maternal and paternal genes in a developing fetus [11]. The father, equally capable of impregnating the female as other male contenders, wants a large baby capable of carrying on his genes. In theory, a larger offspring equates to a better chance of survival and a further transfer of the father’s genes by his progeny. The mother, on the other hand, must be capable of bearing multiple offspring in order to propagate her genes in subsequent generations. Thus, it would be advantageous for her to have smaller offspring who demand fewer maternal resources and therefore would grant the mother additional opportunities to bear subsequent offspring. This parental conflict hypothesis is consistent with the trend that maternally imprinted genes, which are transcriptionally silent on maternal chromosome but expressed on the paternal chromosome, enhance growth, while paternally imprinted genes repress growth [11].
The process of genetic imprinting occurs during maternal and paternal gametogenesis [12,13]. When Primordial Germ Cells (PGCs) migrate into the genital ridge (the future testis or ovary) the entire genome is demethylated in a process called erasure; more specifically, imprinting and non-imprinting methylation patterns are removed at this stage [13]. Following this event, de novo methylation occurs in a sex-specific fashion in a process known as establishment. In other words, although the maternal and paternal imprinted loci help develop the fetus through their maintenance and expression in somatic cells, the gametic imprinting pattern that the fetus will pass on to its offspring is established in that fetus in utero in males and after sexual maturation in females. The PGCs of a female offspring will undergo erasure and establishment such that mature gametes will bear only the maternal imprinting patterns. Similarly, the mature gametes of a male fetus will only express paternal imprinting patterns. Following fertilization of an oocyte with sperm, the resulting zygote will have an imprinting profile that contains both maternally and paternally imprinted genes [13]. As the zygote begins replication and cleavage, the entire genome except those imprinted gene clusters undergoes demethylation, resulting in totipotency. Near the period of implantation, genome-wide de novo methylation occurs; an event that marks the differentiation of cells in embryological development. From this point on, the developing embryo maintains imprinted gene regions in the transcriptionally active somatic cells. The entire process repeats when the PGCs in the fetus migrate to the gonadal ridge and establish a biparental imprinting pattern [12].
Abnormalities in imprinted genes result in imprinting disorders [1]. Certain imprinting disorders are associated with abnormalities on specific chromosomal loci. Chromosomes 11 and 15 are contain loci commonly responsible for Beckwith-Wiedemann syndrome (BWS) and Silver-Russell syndrome (SRS), as well as Prader-Willi syndrome (PWS) and Angelman syndrome (AS) respectively. Furthermore, the same imprinting disorder can have multiple genetic and epigenetic etiologies, each with varying prevalence. These etiologies include epigenetic mechanisms such as hypomethylation or hypermethylation at specific loci, or genetic mechanisms including deletions or loss-of-function mutations of the normally-expressed parental allele of an imprinted gene [1]. For example, one genetic etiology associated with AS is a mutation in the maternal UBE3A gene, which leads to a nonfunctional protein product [14]. Similarly, a more significant loss of genetic information can occur with a failure in chromosomal separation. This process, called uniparental disomy (UPD), yields two chromosomes from a single parent and is one of the major etiologies implicated in PWS [15]. As for aberrant alterations in the methylation process, maternal hypermethylation and paternal hypomethylation of an imprinting region at the same genetic locus can lead to BWS and SRS respectively [7,8]. A table was created to compare and contrast these syndromes on a genetic basis, based on the references provided (Table 1). These four major imprinting disorders, including their range of genetic and epigenetic etiologies, phenotypic manifestations, and current efforts to improve their diagnostic and clinical management, are discussed below.
Table 1.
Chromosome regions and genetic mechanisms implicated in BWS, SRS, PWS, and AS
| Syndrome | Chromosome Region Affected | Gene(s) Affected | Examples of Epigenetic and Genetic Mechanisms That Can Lead to Syndrome | Reference |
|---|---|---|---|---|
| Beckwith-Wiedemann Syndrome (BWS) | 11p15 | ICR1 domain (H19, IGF2) ICR2 domain (KCNQ1, CDKN1C) | Imprinting Defects (ie hypermethylation ICR1; hypomethylation ICR2), Paternal UPD, Heterozygous Deletion | [4,7,8,18-23] |
| Silver Russel Syndrome (SRS) | 11p15 | ICR1 domain (H19, IGF2) | Imprinting Defects (ie hypomethylation ICR1), Maternal UPD, Clinical Heterogeneity | [7,8,24] |
| Prader-Willi Syndrome (PWS) | 15q11-q13 | SNRPN, NDN | Imprinting Defects, Maternal UPD, Paternal deletion of chromosome region | [15,29,30] |
| Angelman Syndrome (AS) | 15q11-q13 | UBE5A | Imprinting Defects, Paternal UPD, Maternal Deletion, Mutations in the UBE5A gene | [31,32] |
Beckwith-wiedemann syndrome
Beckwith-Wiedemann syndrome (BWS) is the most common overgrowth imprinting disorder, with recent reports indicating the incidence to be 1:10,340 live births [16]. BWS is characterized by macrosomia and cancer predisposition as well as its variable association with anatomical anomalies including macroglossia, abnormal facies, abdominal wall defects, renal anomalies, organomegaly, and islet cell hyperplasia and hypoglycemia [17]. Given its diverse constellation of presentations, diagnosis is dependent on three or more major findings, or two major findings and at least one minor finding. Once a clinical diagnosis has been made, molecular testing for genetic and epigenetic alterations on chromosome 11p15 may be used to confirm the diagnosis [18].
While BWS has been linked to small clusters of growth regulatory DNA on chromosome 11p15 [18,19], a variety of associated genetic loci and epigenetic mechanisms have been identified [7,8]. Chromosome 11p15 contains two gene imprinting domains each regulated by discrete imprinting control regions (ICR1 and ICR2). ICR1 (alternatively called DMR1) regulates the H19/IGF2 genes within Domain 1 and is paternally methylated, while ICR2 (alternatively called DMR2 or KvDMR) regulates the KCNQ1/CDKN1C genes within Domain 2 and is maternally methylated [7]. Methylation of ICR1 blocks the promoter region of H19 and allows the transcription of IGF2, a fetal growth factor. Normally, H19 is maternally expressed and IGF2 is paternally expressed [20]. It is estimated that greater than 10% of patients with BWS demonstrate isolated hypermethylation of the maternal H19 allele [4]. As H19 is normally expressed on the maternal chromosome, this indicates BWS is associated with hypermethylation of maternal ICR1 [7]. Methylation of ICR2 results in maternal expression of KCNQ1, which mediates cell cycle arrest [21] and CDKN1C, a tumor suppressor [18]. Hypomethylation of the maternal allele results in the loss of KCNQ1, as well as biallelic expression of KCNQ1OT1, which is maternally methylated and paternally expressed in undiseased states [22]. It is estimated that greater than 40% of patients with BWS demonstrate isolated hypomethylation of the maternal ICR2 allele, and up to 60% demonstrate maternal ICR2 hypomethylation in conjunction with additional epigenetic and genetic abnormalities [4]. Genetic analysis has shown that 25% of BWS patients with hypomethylated maternal ICR2 alleles also demonstrate hypomethylation of imprinted gene on additional chromosomes, including ZAC1 on chromosome 6q24 and PEG1/MEST on chromosome 7q23, although the patients do not show symptoms of other clinical disorders [7].
In addition to epigenetic abnormalities, BWS has been associated with certain genetic alterations including paternal uniparental disomy (UPD), which refers to the inheritance of two paternal copies of a chromosomal region, and CDKN1C mutations [18]. Furthermore, a recent case study found an association between a heterozygous deletion of the KCNQ1 gene and BWS [23]. A 24-year-old woman pregnant with twins presented with a history of three prior unsuccessful pregnancies complicated by fetal development of omphaloceles. Intrauterine death of one twin occurred at 14 weeks gestation, and termination of the other occurred at 22 weeks due to poor postnatal prognosis, with a tentative diagnosis of BWS based upon the presence of an omphalocele and placental mesenchymal dysplasia. Molecular genetic analysis of the twins revealed normal methylation at ICR1, but a total loss of methylation at ICR2 and a deletion at the 5’ region of the KCNQ1 gene as well as another deletion telomeric to the gene. This same genetic pattern was found in the mother, although her epigenetic methylation patterns were normal, and both the father’s and maternal grandparents’ methylation patterns and gene dosages were normal. The mother was determined to be a healthy carrier of a genetic rearrangement, which was located on her paternal chromosome, and she had passed this on to her twins. Given the deletion of KCNQ1 and the complete loss of methylation on the maternal allele, it was concluded that the mechanism was likely a failure in the initial imprint establishment in the maternal germline, rather than a defect in the postzygotic maintenance of the methylation pattern. Additionally, the preservation of ICR2 may suggest that transcription of KCNQ1 is required for the initial imprint establishment in the maternal gamete [23].
Management of BWS includes surgical repair of anatomical anomalies as well as screening for hypoglycemia and tumor development [18]. Genetic testing, including karyotype and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA), can be implemented to provide genetic counseling to affected individuals, particularly in regard to recurrence rates and potential familial screening. The risk of recurrence varies by the molecular etiology, with UPD having a low level of recurrence due to the postzygotic nature of somatic recombination. However, genetic abnormalities carry a higher risk, and genetic screening of family members is advised. Prenatal genetic testing is also possible via chorionic villus sampling or amniocentesis [18].
As genetic analysis continues to advance, studies have targeted genetic and epigenetic-based management procedures. A recent study evaluating a large cohort of 318 BWS patients were categorized based on the four major etiological subclasses: hypomethylation of ICR2 (IC2-LoM), hypermethylation of ICR1 (IC1-GoM), chromosome 11p15 paternal UPD, and CDKN1C mutations. Each cohort was associated with a different symptomatology [9]. The IC2-LoM group was associated with postnatal overgrowth, and both the IC2-LoM and CDKN1C group were associated with omphalocele, ear anomalies and nevus flammeus, preterm birth, as well as the lowest overall risk of cancer. The IC1-GoM group was associated with neonatal macrosomia, renal defects and urethral malformations, and Wilms’ tumor, and carried the highest overall risk of cancer. The UPD group was associated with hemihyperplasia, renal defects, Wilms tumor, a high risk of hepatoblastoma, and an overall intermediate cancer risk. These results indicate that BWS can be thought of as four separate conditions with unique molecular etiologies and different patterns of phenotypic abnormalities. The varying patterns of disease presentations may warrant revisions in the guidelines for clinical care of BWS patients, particularly for cancer screening schedules [9].
Silver-russell syndrome
The most prevalent dysregulated genes implicated in Silver-Russell syndrome (SRS) are harbored within the same genetic loci as BWS [24]. However, despite typically involving the same gene clusters, SRS is considered a mirror opposite of BWS in that it primarily leads to diminished intrauterine and postnatal growth. An illustration comparing the phenotypes of BWS and SRS was created based on clinical presentations described in Eggermann et al., 2010 [26] and Weksberg et al., 2010 [18] (Figure 1). Aside from the characteristic fetal growth restriction observed in SRS, the condition is considered heterogeneous and spectral with regards to the disorder severity, the imprinting abnormality locale, and the clinical presentation [24]. The wide variety of clinical presentations in the initial reports of the disease include dysmorphia or atypia in nearly every organ system [25]. Given this variety, the simplest and most widely accepted method for diagnosis of SRS is Netchine-Harbison clinical scoring system, in which the presence of at least four out of six total diagnostic criteria indicates patients are likely to have the condition, although recent recommendations are that prominent forehead and relative macrocephaly should be present if a diagnosis is to be made in the absence of a positive molecular genetic test [10]. As the clinical diagnosis of SRS has proven difficult, worldwide estimates of the incidence of SRS range from 1:30,000 to 1:100,000 births; a number that is likely underestimated due to the array of presentations and the disorder’s spectrum of severity [10]. Following a suspicion of SRS using the aforementioned clinical scoring system, robust methylation studies can be ordered to detect the known genetic pathways associated with SRS. A diagnostic algorithm for the genetic testing of clinically suspicious individuals has been proposed within this decade [26].
Figure 1.
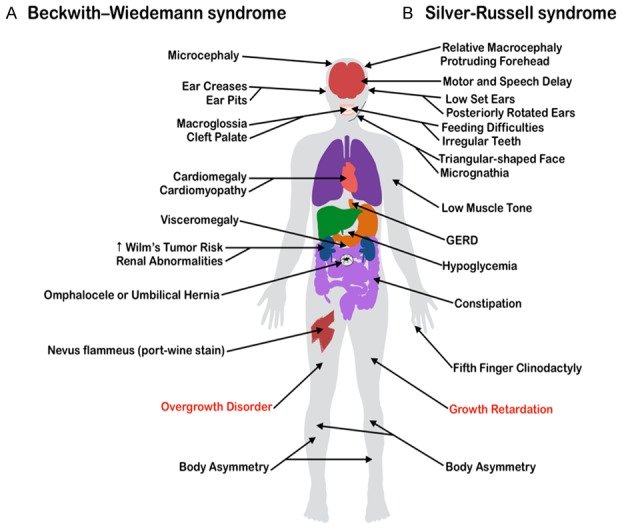
The clinical manifestations of two imprinting disorders often associated with a genetic or epigenetic [etiology] in a common imprinted gene [cluster] on the short arm of chromosome 11. The disease characteristics displayed are displayed are not exhausting or unique to every diagnosed case of each [syndrome]. Common clinical manifestations of (A) Beckwith-Wiedemann Syndrome (BWS), displayed on the left side of the body, are placed in contrast with those of (B) Silver-Russell Syndrome (SRS), shown on the right.
Approximately 40% of SRS cases are due to a hypomethylation of ICR1 (DMR1) at 11p15; an epimutation that nearly mirrors the hypermethylation of this region observed in BWS [8]. Around 10% of SRS cases are a result of maternal uniparental disomy of chromosome 7 and around 1-2% are segmental duplication of the mothers 11p15 chromosomal region. The remaining cases are sparse and can be attributed to the genetic and clinical heterogeneity of the disease [8]. Currently, treatment of SRS almost exclusively involves the management of the patient symptoms [10]. Hypoglycemia is managed through strict monitoring of urine ketone levels and substituting high molecular weight glucose to infants during night time feeds in order to prevent nocturnal hypoglycemia. The growth restriction observed in SRS is treated through growth hormone administration, which has proven effective in increasing the adult height of children diagnosed with SRS. The benefits of limb lengthening surgery has been questioned given the delayed healing response observed in many children diagnosed with SRS [10].
Similar to BWS, a link between the genetics of SRS and its phenotypic profile has been documented, with abnormal facies and muscular hypotonia particularly prevalent in SRS due to maternal UPD of chromosome 7 [26]. A recently published consensus statement from a collaboration of experts regarding the clinical implications of such epigenetic-phenotypic relationships has might serve as a framework for establishing future diagnostic guidelines for similar imprinting abnormalities [10]. The recommendations put forth in the consensus statement are aimed toward enabling clinicians to provide optimal care based on the genetic and epigenetic etiology of SRS in the future. Careful sequential genetic testing must be completed to properly manage symptoms, as inaccurate or incomplete diagnoses can have drastically adverse effects. For example, while Growth Hormone (GH) treatment is standard for SRS patients, administration of GH to patients with SHORT syndrome, a similar congenital anomaly associated with short stature, has been linked to the development of insulin resistance [10]. Although the authors of the consensus statement have proposed an SRS subtype-based approach to clinical management, noting certain patterns such as the earlier onset of puberty and epiphyseal plate fusion in SRS patients with 11p15 LoM, only 60% of patients presenting with the clinical symptoms of SRS have a diagnosis confirmed with the current molecular testing available [10], indicating that many of the genetic pathways have yet to be discovered. The heterogeneity of SRS has made molecular diagnosis as well as prenatal diagnosis exceptionally challenging [27]. Future research and molecular testing of SRS patients with presently-unconfirmed genetic abnormalities may enable further subtype stratification of the syndrome, and perhaps advance current efforts in association with prenatal testing [27].
Prader-willi syndrome
Chromosome 15 also harbors genetic domains implicated in the development of imprinting disorders. Prader-Willi syndrome (PWS) is characterized by cognitive disabilities, psychiatric complications, weak muscle tone, oxytocin deficits, and insatiable appetite [15,28,29]. With an estimated prevalence ranging anywhere from 1:15,000 to 1:30,000, PWS can be the result of various pathophysiologies that result in the suppression of the 15q11-q13 region on the paternal chromosome [15,29]. Specifically, PWS has been associated with the lack of expression of the SNRPN and NDN genes from the 15q11-q13 region, which are normally methylated and suppressed on the maternal chromosomal copy and expressed from the paternal copy [15,30]. Mechanisms of suppression include imprinting defects in the paternal copy of 15q11-q13 in around 5% of cases, maternal UPD in just under 30% of cases, or, as in 70% of cases, the deletion of the paternal 15q11-q13 region [15,29].
Treatment of PWS relies on symptomatic management, including growth hormone and oxytocin replacement therapy [15,29]. Current research is being done to investigate how to maximize the efficacy of these treatments in determining optimal onset for treatment and duration of treatment course [29]. One method aims to optimize oxytocin treatment, as due to the characteristic lack of oxytocin-producing neurons in the hypothalamus, PWS often manifests in infancy with an inability to suckle and breastfeed. A recent Phase 2 clinical trial investigated the effects of intranasal administration of oxytocin in 18 PWS patients less than six months of age. The results of the study were promising, as infants who received this intervention had improved breastfeeding outcomes and engaged in more meaningful maternal-infant interactions throughout the three weeks following the last intranasal oxytocin administration [29]. Future studies with larger sample sizes should bring more clarity to the long-term impact of this intervention, as well as to the timing, duration, and dose concentrations that yield the greatest clinical benefit. A second study investigated the effects of growth hormone administration on the cognitive development of PWS patients [28]. The results demonstrated that patients who began growth hormone treatment before one year of age reached higher IQ levels than children who began treatment later. Nevertheless, growth hormone treatment increased IQ levels across all age groups compared to the control group [28].
While current research has led to potential techniques for symptom management of PWS, an area of research that remains lacking is therapies that target the chromosomal abnormalities associated with the development of PWS. A permanent treatment option would offer clear benefits as compared to symptomatic treatment of PWS, which requires chronic administration of the intervention [28,29].
To investigate the growing concern of ART and its potential association with PWS, a study compared two groups of PWS patients who were either conceived naturally or via ART [15]. No significant association was found between the PWS and ART as compared to the general population. However, a sub-analysis comparing PWS within the ART population based on the genetic etiology of their disease indicated that imprinting defects and maternal UPD were more frequent etiologies in the PWS-ART group than the naturally-conceived group. This study suggests a potential association of imprinting abnormalities and ART, although it should be noted that the authors themselves indicate their study power and small population warrants future follow-up studies [15]. As more is learned regarding epigenetic and genetic patterns of dysregulation, the goal should remain to develop treatments that targets the aberrant genetic pathways to avoid the syndrome altogether.
Angelman syndrome
The diagnosis of Angelman Syndrome (AS), a developmental disorder affecting the nervous system, is dependent on the presence of four major symptoms and at least three minor symptoms [31,32]. The major symptoms are developmental disabilities, speech impairment, movement or balance disorders, and certain behavioral characteristics, and minor symptoms include postnatal deceleration of head growth, seizures, abnormal EEG, sleep disturbances, an abnormal fascination with reflective objects, and drooling [32]. An illustration comparing the phenotypes of PWS and AS was created based on clinical presentations described in Gold et al., 2014 [15] and Tan et al., 2011 [32] (Figure 2).
Figure 2.
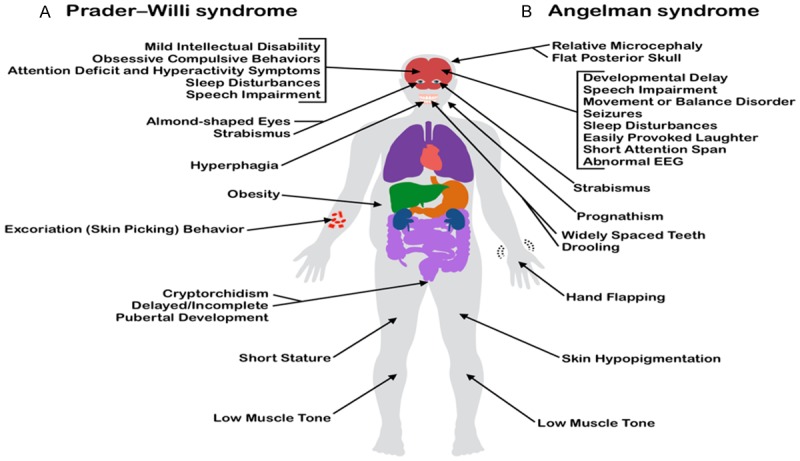
The clinical manifestations of two imprinting disorders often associated with a [genetic] or epigenetic etiology in a common imprinting gene [cluster] [on] the long arm of chromosome 15. The disease characteristics displayed are not exhausting or unique to every diagnosed case of each syndrome. Common clinical manifestations of (A) Prader-Willi Syndrome (PWS), displayed on the left side of the body, are placed in contrast to [with] those of (B) Angelmman syndrome (AS) shown on the right.
AS and PWS have similar genetic etiologies, with patients sharing identical deletions on chromosome 15q11-13 [15,32]. Whereas paternal expression of 15q11-13 is abnormally absent in PWS, maternal expression of this same locus is abnormally absent in AS [32]. Imprinting defects in the maternal copy of chromosome 15q account for 3% of AS cases [31]. It is estimated that 70% of AS cases are caused by maternal deletions on Chromosome 15q11.2-q13, while 2% are caused by paternal UPD of the same chromosome. Mutations in the UBE3A gene itself account for a portion of the remaining percentage of cases [31]. Similar to the other congenital syndromes discussed in this review, ARTs have been linked to the development of epigenetic abnormalities implicated in AS. Specifically, analysis of imprinting defects in children with diagnosed AS conceived through intracytoplasmic sperm injection (ICSI) has indicated that ICSI may alter maternal imprinting in the early stages of fertilization [33].
Without a cure [34], management of AS would currently be limited to symptomatic treatment, including behavioral therapy, communication therapy, physical therapy, and anti-seizure medications, based on the clinical manifestations reported [32]. Similar to PWS, it will be essential to create treatments that correct the imprinting defect, rather than tone down the symptoms that arise from this disease. While only a minority of cases of AS arise from imprinting defects, as is the case with PWS, ART nevertheless represents a target for reducing the incidence of these disorders. Finding solutions to potential ART-related imprinting errors should be a priority as research continues to characterize the biochemical mechanisms through which ART may cause these errors.
Conclusion
As research continues to elucidate the genetic and epigenetic pathways that underpin imprinting disorders, strides have been made toward characterizing the phenotypic expression of aberrant alterations at specific genetic loci. Additionally, methods of molecular genetic testing continue to advance, which has allowed for familial screening and accurately assessment of recurrence risk based on the presently-known genetic etiology of these syndromes [10,18]. These advances have culminated in recent efforts to stratify these heterogeneous syndromes based on symptom presentation, as well as establish systematic diagnosis guidelines that incorporate phenotypic manifestations as well as genetic testing to ensure safe and effective management of symptoms [16,18,29]. As additional aberrant genetic loci are found in association with imprinting disorders, the screening and clinical management of patients will continue to improve.
Despite the improvement in the diagnosis and clinical management of imprinting disorders, the discrepancy surrounding potential risk factors, namely ARTs, remains at the forefront. The timing of procedural manipulation of oocytes and sperm associated with ARTs overlaps with the timing of natural epigenetic reprogramming and imprinting, making the association between imprinting disorders and ART an area of intense investigation [1]. While there is strong evidence supporting the association between imprinting disorders and ARTs, potential causative agents have yet to be characterized [6], and contradictory evidence exists [1]. Some research has described an association specifically between epigenetic etiologies of imprinting disorders and ART, with no difference observed in the overall incidence of imprinting disorders in ART-conceived population compared to the general population [15]. ICSI has been specifically linked to the alteration of maternal imprinting in the oocyte [33]. However, the effects of ART on DNA methylation patterns independent of infertility have not been demonstrated outside of animal studies [1]. The uncertainty surrounding the impact of ARTs necessitates continued research and greater characterization of how these methods might affect the imprinting process. As epigenetic modifications are reversible to an extent [1], it is worth exploring the implications of ART on genetic imprinting in order to create safe options for reproductive technology.
Furthermore, certain well-characterized epigenetic abnormalities associated with imprinting disorders have been investigated as targets for therapy, with preliminary results demonstrating success in reversing the methylation of silenced genes and improving symptoms [14]. An approach to therapy that targets imprinting loci has the potential to shift the paradigm from symptomatic management of syndromes like BWS, SRS, PWS, and AS to reversal and prevention of symptom manifestation. Ultimately, as understanding of the epigenetic regulation of genes advances and additional genetic loci implicated in the development of imprinting disorders are characterized, the potential exists for curative interventions for imprinting disorders.
Disclosure of conflict of interest
None.
References
- 1.Uyar A, Seli E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr Opin Obstet Gynecol. 2014;26:210–221. doi: 10.1097/GCO.0000000000000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sapienza C, Peterson AC, Rossant J, Balling R. Degree of methylation of transgenes is dependent on gamete of origin. Nature. 1987;328:251–254. doi: 10.1038/328251a0. [DOI] [PubMed] [Google Scholar]
- 3.Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature. 1985;315:496–498. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- 4.Gaston V, Le Bouc Y, Soupre V, Burglen L, Donadieu J, Oro H, Audry G, Vazquez M, Gicquel C. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2001;9:409–418. doi: 10.1038/sj.ejhg.5200649. [DOI] [PubMed] [Google Scholar]
- 5.Whitelaw N, Bhattacharya S, Hoad G, Horgan GW, Hamilton M, Haggarty P. Epigenetic status in the offspring of spontaneous and assisted conception. Hum Reprod. 2014;29:1452–1458. doi: 10.1093/humrep/deu094. [DOI] [PubMed] [Google Scholar]
- 6.Lazaraviciute G, Kauser M, Bhattacharya S, Haggarty P, Bhattacharya S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum Reprod Update. 2014;20:840–852. doi: 10.1093/humupd/dmu033. [DOI] [PubMed] [Google Scholar]
- 7.Azzi S, Rossignol S, Steunou V, Sas T, Thibaud N, Danton F, Le Jule M, Heinrichs C, Cabrol S, Gicquel C, Le Bouc Y, Netchine I. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (russell silver and beckwith wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18:4724–4733. doi: 10.1093/hmg/ddp435. [DOI] [PubMed] [Google Scholar]
- 8.Netchine I, Rossignol S, Dufourg M, Azzi S, Rousseau A, Perin L, Houang M, Steunou V, Esteva B, Thibaud N, Raux Demay M, Danton F, Petriczko E, Bertrand A, Heinrichs C, Carel J, Loeuille G, Pinto G, Jacquemont M, Gicquel C, Cabrol S, Le Bouc Y. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical russell-silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab. 2007;92:3148–3154. doi: 10.1210/jc.2007-0354. [DOI] [PubMed] [Google Scholar]
- 9.Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet. 2015;89:403–415. doi: 10.1111/cge.12635. [DOI] [PubMed] [Google Scholar]
- 10.Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, Canton AP, Chrzanowska KH, Davies JH, Dias RP, Dubern B, Elbracht M, Giabicani E, Grimberg A, Grønskov K, Hokken-Koelega AC, Jorge AA, Kagami M, Linglart A, Maghnie M, Mohnike K, Monk D, Moore GE, Murray PG, Ogata T, Petit IO, Russo S, Said E, Toumba M, Tümer Z, Binder G, Eggermann T, Harbison MD, Temple IK, Mackay DJ, Netchine I. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2016;13:105–124. doi: 10.1038/nrendo.2016.138. [DOI] [PubMed] [Google Scholar]
- 11.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 12.Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–550. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- 13.Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med. 2013;34:826–840. doi: 10.1016/j.mam.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. doi: 10.1038/nature13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gold J, Ruth C, Osann K, Flodman P, McManus B, Lee H, Donkervoort S, Khare M, Roof E, Dykens E, Miller JL, Driscoll DJ, Butler MG, Heinemann J, Cassidy S, Kimonis VE. Frequency of Prader-Willi syndrome in births conceived via assisted reproductive technology. Genet Med. 2014;16:164–169. doi: 10.1038/gim.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, Richiardi L, Larizza L, Cirillo Silengo M, Riccio A, Ferrero G. Prevalence of Beckwith-Wiedemann syndrome in north west of italy. Am J Med Genet A. 2013;161A:2481–6. doi: 10.1002/ajmg.a.36080. [DOI] [PubMed] [Google Scholar]
- 17.Pettenati MJ, Haines JL, Higgins RR, Wappner RS, Palmer CG, Weaver DD. Wiedemann-Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet. 1986;74:143–154. doi: 10.1007/BF00282078. [DOI] [PubMed] [Google Scholar]
- 18.Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14. doi: 10.1038/ejhg.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ping AJ, Reeve AE, Law DJ, Young MR, Boehnke M, Feinberg AP. Genetic linkage of Beckwith-Wiedemann syndrome to 11p15. Am J Hum Genet. 1989;44:720–723. [PMC free article] [PubMed] [Google Scholar]
- 20.Felsenfeld G, Bell AC. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 21.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 22.Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, Feinberg AP. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KvLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci U S A. 1999;96:5203–5208. doi: 10.1073/pnas.96.9.5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beygo J, Joksic I, Strom TM, Lüdecke H, Kolarova J, Siebert R, Mikovic Z, Horsthemke B, Buiting K. A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016;24:1280–1286. doi: 10.1038/ejhg.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartholdi D, Krajewska-Walasek M, Ounap K, Gaspar H, Chrzanowska KH, Ilyana H, Kayserili H, Lurie IW, Schinzel A, Baumer A. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet. 2009;46:192–197. doi: 10.1136/jmg.2008.061820. [DOI] [PubMed] [Google Scholar]
- 25.Bliek J, Terhal P, van den Bogaard MJ, Maas S, Hamel B, Salieb-Beugelaar G, Simon M, Letteboer T, vad der Smagt J, Kroes H, Mannens M. Hypomethylation of the H19gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet. 2006;78:604–614. doi: 10.1086/502981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eggermann T, Begemann M, Binder G, Spengler S. Silver-russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010;5:19. doi: 10.1186/1750-1172-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, Mackay DJ, Grønskov K, Riccio A, Linglart A, Netchine I. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. 2015;7:123. doi: 10.1186/s13148-015-0143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dykens EM, Roof E, Hunt-Hawkins H. Cognitive and adaptive advantages of growth hormone treatment in children with prader-willi syndrome. J Child Psychol Psychiatry. 2017;58:64–74. doi: 10.1111/jcpp.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tauber M, Boulanouar K, Diene G, Çabal-Berthoumieu S, Ehlinger V, Fichaux-Bourin P, Molinas C, Faye S, Valette M, Pourrinet J, Cessans C, Viaux-Sauvelon S, Bascoul C, Guedeney A, Delhanty P, Geenen V, Martens H, Muscatelli F, Cohen D, Consoli A, Payoux P, Arnaud C, Salles JP. The use of oxytocin to improve feeding and social skills in infants with prader-willi syndrome. Pediatrics. 2017;139 doi: 10.1542/peds.2016-2976. [DOI] [PubMed] [Google Scholar]
- 30.Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, Jenkins NA, Copeland NG, Brannan CI. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat Genet. 1998;19:25–31. doi: 10.1038/ng0598-25. [DOI] [PubMed] [Google Scholar]
- 31.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 32.Tan W, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Gentile JK, Glaze DG, Horowitz LT, Kothare SV, Lee H, Nespeca MP, Peters SU, Sahoo T, Sarco D, Waisbren SE, Bird LM. Angelman syndrome: mutations influence features in early childhood. Am J Med Genet A. 2011;155A:81–90. doi: 10.1002/ajmg.a.33775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox GF, Bürger J, Lip V, Mau UA, Sperling K, Wu B, Horsthemke B. Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am J Hum Gen. 2002;71:162–164. doi: 10.1086/341096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dagli A, Buiting K, Williams CA. Molecular and clinical aspects of angelman syndrome. Mol Syndromol. 2012;2:100–112. doi: 10.1159/000328837. [DOI] [PMC free article] [PubMed] [Google Scholar]