

Abstract
The alternative splicing plays an important role to generate protein diversity. Recent studies have shown alterations in alternative splicing, resulting in loss, gain or changes of functions in the resulting protein. Specific products of alternative splicing are known to contribute in cancer-related mechanisms, such as angiogenesis, migration, adhesion and cell proliferation, among others. We using high-density microarrays reported a CENP-E as a one of significant transcript expressed and potentially is alternatively spliced in cancer. We focus in validate alternative splicing of CENP-E transcript using RT-PCR and sequencing in different cancer cell lines. We performed RT-PCR using specific primers designed to delimit the non-reported alternative splicing in CENP-E transcript. Our results showed the co-expression of the variant one and two of CENP-E in all cell lines evaluated. We detected more expression of variant one than two. Moreover, we identify an alternative 5’splice site of CENP-E in the exon 38 and was observed in RoVa cell line. Additionally, we characterized alternative skipping from exon 20 (NAT-CENP-E), these alternative splicing was observed in all cell lines evaluated except RoVa. Finally, we corroborate alternative mRNA splicing in leukemia patients using quantitative RT-PCR, in 71.8% of the patients NAT-CENP-E is downregulated and 28.2% is overexpressed.
Keywords: Alternative splicing, CENP-E, cancer cell lines, gene expression, luekemia
Introduction
The alternative splicing (AS) is an important mechanism to generate large number of mRNA and protein diversity. Approximately 75% of human genes are subject to alternative splicing [1-4], however, little is known about the diversity of mRNA isoforms expressed in health and human diseases. The AS mechanisms are not completely clear, however, a large number of molecules including: DNA, RNA and proteins, interacting and promote AS. The spliceosome recognize specific sequences that delimit constitutive transcript [5,6]. Additionally, SR, hnRNP proteins and specific RNA sequences (ESS, ESE, ISS, ISE) promote the AS [6,7]. In cancer, several factors are involved in aberrant AS such as: mutation in ESS, ESE, ISS, ISE; changes in gene expression and proteins diversity that plays some roles in AS regulation. The consequence mRNA could be alternatively processed such as: alternative exon skipping, intronic retention, premature stop, alternative capping and polyadenylation [8]. The aberrant mRNA splice is associated to development and cancer progression [9,10], treatment resistance, among others [11-13].
Recent evidence has shown that AS of mRNA is modulated according tissue type [14], suggesting that specific mRNA isoforms are expressed tissue-type [15]. The alternative mRNA splicing in cancer can confer cellular advantages such as: proliferation, migration, angiogenesis, among others [16]. The cancer represents the most complex human disease and involves several cellular characteristics including: evading growth suppressor, sustaining proliferative signals, angiogenesis, enabling replicative immortality, migration, metastasis, and chromosomal instability, among others [17-19]. However, little is known about the diversity of mechanisms of alternative mRNA splicing in cancer disease. Nowadays, several studies in cancer has showed diversity transcripts that are alternatively spliced and its contribution with evading growth suppressors (TP53, AXA7), sustaining proliferative signalling (KRAS, RAC1), avoiding immune destruction (IL7, HLA-G), enabling replicative immortality (TERT), Tumor-promoting inflammation (RAC1, CD44), activating invasion and metastasis (CD44, FGFR2), angiogenesis (VEGFA), genome instability and mutation (RAC1), resisting cell death (TP53, BCL2L1) and deregulation cellular energetics (PKM, GLS) [19]. This events of aberrant splicing in different cancer have an important impact as potential biomarkers of diagnosis, prognosis or therapeutic targets.
The acute lymphoblastic leukemia (ALL) is the most common childhood malignance in the world wide [20] several congenital disorders are associated to susceptibility [21], which could contribute with changes in gene expression as well as alternative splicing, Sun L, et al. identify aberrant expression of Ikaros coding sequence between 6 and 7 yielded the wild type sequence, that contribute to leukemia [22]. However, little is known about the diversity of alternative mRNA splicing in leukemia.
In a previous study, we performed analysis of transcriptome using Affymetrix GeneChip 1.0. The analysis included healthy and ovarian cancer and ovarian [1], cervical, breast, head and neck and leukemia cell lines (data no was shown). Our results revealed that 207 transcripts expressed alternative mRNA isoforms, including: Centromere-associated protein E (CENP-E), p-value = 1.87E-03, AS p-value = 5.00E-05, fold change = 4.3574 [1].
CENP-E is expressed before mitosis and is accumulated in G2 phase, its role is implicated in promote chromosome movement and spindle elongation [23], consequently, correct chromosomal segregation [24]. CENP-E has been associate with aneuploidies, chromosome instability [25,26] and cancer develop [27]. The mechanism is not clear, however chromosome instability could promoter loss of tumour suppressor genes and cellular transformation. In addition, the aneuploidy is a very common characteristic in several types of cancer including: breast [26], colorectal [25,28] and represent risk for specific cancer, including leukemia [29]. Moreover, CENP-E expression is consider as tumour suppressor and oncogene [30].
In order to identify AS in CENP-E transcript, in this study we designed specific primers to delimit the potential alternative splicing, CENP-E expression was evaluated in 22 cancer cell lines using RT-PCR and sequencing. Additionally, we quantify the expression of new alternative transcript of CENP-E using Quantitative RT-PCR in 72 leukemia patients (LP). Our results demonstrate a new alternative mRNA splice of CENP-E was expressed in cancer cell lines and leukemia patients.
Materials and methods
Microarray data mining
We used CELL files of microarray that previously reported. The analysis was performed using Partek Genomics Suite v6.6 according to a previous study [1], the probe exon ID were identified and mapping using NetAffy http://www.affymetrix.com/estore/ and UCSC Genome Browser http://genome.ucsc.edu/ and Integrated Genome Browser v8.2.3.
Cell lines
We used 22 cancer cell lines, that include: ovarian (NIH-OVECAR-3, SK-OV-3, TOV-21G, TOV-112D) provide by PhD. Laura Diaz; cervical (HeLa, SiHa, CaLo, RoVa, ViBo, C-33A, ViPa, INBL, Caski, MS-751, HaCaT) provided by PhD. Alberto Monroy; breast and leukemia (MCF-7, MDA-MB231, Jurkat) provided by provide by PhD. Alejandro Zentella; Leukemia (REH, K-562) provided by PhD. Patricia Pérez Vera and PhD. Fabian Arechavaleta provided Hep-G2, Hek-293T. The adherent cell lines were cultured at 37°C, 5% of CO2, Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Life Technologies CA. USA), supplemented with 10% of Fetal bovine serum (Gibco, Life Technologies CA. USA) and 1% Penicillin-Streptomycin. Leukemia cell lines (REH, K-562) were cultured al 37°C, 5% of CO2, 10% Fetal bovine serum (Gibco, Life Technologies CA. USA), 1% Penicilin-Streptomycin, 1% L-Glutamine and 1% non-essential amino acids.
Leukemia bone marrow
All investigations were performed in accordance with the Declaration of Helsinki and approval by research and ethics committee of Instituto Nacional de Pediatria, SSA number CONBIOETICA09CEI02420130507. After informer consent was obtain. We obtain ~1 ml of bone marrow of the LP, the sample was collected in EDTA tube, after that was added Lymphoprep (Stem cell technology) and were obtain the mononuclear cell. The total RNA was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to manufacturer’s instructions. Besides, white blood cells of healthy donator were obtained.
cDNA synthesis and RT-PCR amplification
The cDNA was synthesized; in brief 5 ug of total RNA was digested using 1 u DNase, 1x DNase buffer (Thermo Scientific). The master mix was incubated for 30 min at 37°C. After that, was added 1 ul EDTA 5 mM and was incubated at 65°C for 10 min. Subsequently, the samples were place in master mix containing: 0.2 μg of random hexamer primer, 40 u M-MLV, 10 mM DNTP’s mix, 40 U RNase inhibitor, 0.2 M DTT, 1x M-MLV buffer. The mix was incubated at 25°C 10 min, 37°C 60 min and 70°C 10 min (Invitrogen).
We evaluated housekeeping RPL4 expression as reference control; the cDNA was dilute 1:10. The PCR amplification was performed using 2 ul of dilute cDNA, Dream Taq Green DNA Polymerase 0.125 U, Buffer Dream Taq 1x, dNTPs Mix 0.4 mM (Thermo Scientific, USA), primer 5’ CGAATGAGAGCTGGCAAAGGCAAA 3’ Fw and 5’ ACGCCAAGTGCCGTACAATTCATC 3’ Rev 0.2 μM final concentration. The mix reactions were initially incubated at 95°C 2 min, and then ware run 35 cycles 95°C 15 s, 60°C 15 s and 72°C 15 s, finally 72°C 5 min.
CENP-E amplification was performed using three sets of primers; we used the same conditions of mix reaction Set1 5’ CCAACTCAAGGAAAGCCTGCAAGA 3’ Fw, 5’ TTCCATGGAGCATCTCTGGTTTGC 3’ Rev (NM_001286734.1; 490 pb, NM_001813.2; 778 pb); Set2 5’ AGCTGCTTAGAGAAAAGGAAGACC 3’ Fw, 5’ GCAAAATGACTTCTTCCCGCA 3’ Rev (NM_001286734.1 and NM_001813.2; 492 pb); Set3 5’ GAAGGAGAAAATGATTTGCTCTG 3’ Fw, 5’ GCAAAATGACTTCTTCCCGCA 3’ Rev (118 pb). The mix reactions were initially incubated at 95°C for 2 min, then ware run for 35 cycles at 95°C for 15 s, 59.5°C for 15 s and 72°C for 15 s, finally 72°C for 5 min, the Tm of Set2 was 56.5 and Set3 53.5°C, all amplifications were performed using 2720 Thermal Cycler (Applied Biosystems).
DNA purification and sequencing
PCR products were separated by electrophoresis gel using 2.5% agarose, the DNA were purified using QIAquick Gel Extraction (Qiagen, Valencia, CA, USA) according to the standard protocol. After, amplicons were subjected to Sanger sequencing in ABI Prism 3130 Genetic Analyzer (Applied Biosystems) using the BigDye Terminator 3.1 cycle sequencing Kit (Applied Biosystems). Subsequently, the sequences were assembled using annotated sequences in National Center for Biotechnology Information (NCBI).
Quantitative RT-PCR
The cDNA of LP was performed as follows: one ug of total RNA was digested using 1 u DNase, 1x DNase buffer (Thermo Scientific). The master mix was incubated for 30 min at 37°C. After that, was added 1 ul EDTA 5 mM and was incubated at 65°C for 10 min. Subsequently, the samples were place in master mix containing: 0.2 μg of random hexamer primer, 40 u M-MLV, 10 mM DNTP’s mix, 40 U RNase inhibitor, 0.2 M DTT, 1x M-MLV buffer. The mix was incubated at 25°C for 10 min, 37°C for 60 min and 70°C for 10 min (Invitrogen). For quantitative PCR we using KAPA SYBR Fast Universal qPCR Kit according to the manufacture’s recommendations. The cDNA was diluted 1:10 to perform real time RT-PCR, the master mix contain 5 ul of Kapa Syber, 2 ul of cDNA diluted, 0.2 of ROX dye, 1.8 ul of H2O and 0.5 ul primers 10 uM (set3 5’ GAAGGAGAAAATGATTTGCTCTG 3’ Fw, 5’ GCAAAATGACTTCTTCCCGCA 3’ Rev) or housekeeping RPS18 5’ CAGCCAGGTCCTAGCCAATG 3’ Fw, 5’ CCATCTATGGGCCCGAATCT 3’ Rev. The amplification of RPS18 and CENP-E was performed as follows: 95°C for 20 seconds, for 30 cycles at 95°C for 3 seconds, 57°C seconds. The melting curve was performed at 95°C for 15 seconds and the ramp was 55°C to 95°C, the temperature was increased +0.3°C. The amplification was carried out in a StepOne AB (Applied Biosystems). CENP-E expression was determined by relative quantification, which was calculated using 2 e (-ΔΔCt) methods, where ΔCt = Ct NAT-CENP-E - Ct RPS18 and ΔΔCt = ΔCt leukemia - ΔCt heatly [31].
Statistical analysis
We performed a non-parametric Kruskal-Wallis test, the significant was consider with P < 0.05 and was carried using Prism 6.
Results
Analysis of exon expression in CENP-E transcript
Previous results in ovarian cancer showed significant and differential expression in CENP-E transcript [1]. We performed analysed using high-density microarrays Affymetrix GeneChip 1.0 the bioinformatics analysis was performed using Partek Genomics suite v6.6. 50 probes set that measure expression in 49 exons of CENP-E Figure 1. The plot showed over-expressed in Tumors (T) and Cell Lines (CL) vs Healthy Tissue (HT) samples. The heat map of CENP-E expression showed in almost six targets low expression, suggesting AS. However, we focused in the evaluation of two regions in CENP-E transcript, indicating by blue arrow, those targets showed the most consistent suppression in all samples analysed Figure 1.
Figure 1.

Exons expressed in CENP-E transcript. The plot showed individual expression of all exons that constitute centromere-associated protein E transcript. A. CENP-E is transcribed in antisense chain, two alternative mRNA splice are retrieved from UCSC Genome Browser. B. Plot depicting expression of probe set in healthy tissue (HT, blue), Tumors (T, green) and Ovarian Cell Lines (CL, red). C. Heat map contain individual expression from each sample and exon.
PCR amplification
The cDNA synthesis was evaluated using a housekeeping gene RPL4 according a previous report [1]. The RPL4 expression in cancer cell lines is depicted in the Figure 2A. After that, we evaluated expression of CENP-E using consensus primers that amplified variant one and two (Set1). Set1 primers, amplify the exon 36-40; the PCR products were predicted using primer blast https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome (778 pb and 490 pb products for CENP-E variant one and two, respectively). CENP-E was evaluated in cancer cell lines and the PCR showed the co-expression of both variants, as expected the size correspond at transcript variant one and two. However, the expression was inconspicuous in the variant one in contrast to variant two Figure 2B. In some cell lines the variant one was not observed in: C33, HaCaT, REH and Jurkat cell lines. Unexpectedly, the ViBo cells showed an amplicon in ~600 pb and in order to reveal the identity of the PCR product, the DNA was purified and sequencing Figure 2C. After that, the sequence resulting was aligned to reference mRNA (NM_001813.2 and NM_001286734.1) the alignment showed a loss of 159 nucleotides in the end 5’ of exon 38 Figure 2D.
Figure 2.
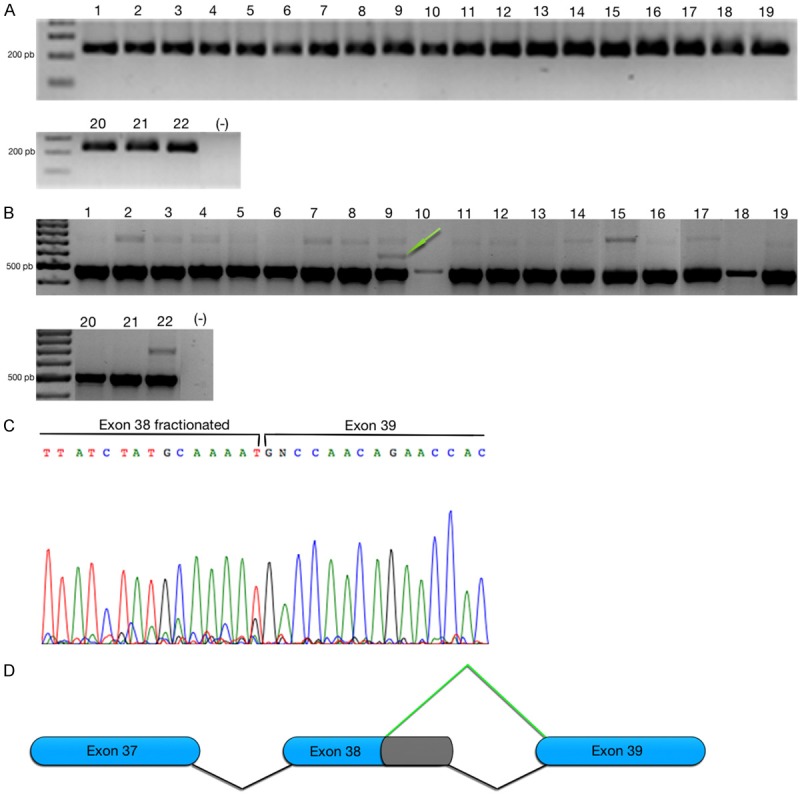
CENP-E transcript variant one and two are expressed in cancer cell lines. A. Agarose gel showed expression in housekeeping gene RPL4 in 22 cancer cell lines that include: 1 (NIH-OVCAR-3), 2 (SK-OV-3), 3 (TOV-21G), 4 (TOV-112D), 5 (HeLa), 6 (SiHa), 7 (CaLo), 8 (RoVa), 9 (ViBo), 10 (C-33A), 11 (Vipa), 12 (INBL), 13 (Caski), 14 (MS-751), 15 (MDA-MB-231), 16 (MCF-7), 17 (Hep-G2), 18 (HaCat), 19 (Hek-293T), 20 (REH), 21 (Jurkat), 22 (K-562). B. The agarose gel is showing the co-expression of two CENP-E transcripts using consensus primers in 22 cancer cell lines. C. Electropherogram shows the sequence of alternative splice, the top lines indicate the boundaries of partial alternative skipping of exon 38. D. Model of alternative 3’ splice site form exon 38 in CENP-E.
On the other hand, we design specific primers to amplify exon 18-21 (Set2), the design exclusively amplify a consensus sequence of the transcript one/two (492 pb). We observed two additional amplicons in ~600 and ~400 pb, however, the sequences were not corresponded to CENP-E transcript. We observed strong expression in the constitutive mRNA variant one/two of CENP-E (PCR fragment of ~492 pb) Figure 3A, and the sequence showed the expression for CENP-E transcript one/two, as expected Figure 3B. Additionally, we could observe in eight cell lines (OVECAR-3, SK-OV-3, TOV-112D, C33-A, Hep-G2, Jurkat, K-562, MS751) inconspicuously amplicon in ~300 pb indicated in Figure 3A, the amplicon was purified and sequence. The sequence revealed a novel mRNA transcript that is expressed in cancer cell lines; the sequence and alignment showed skipping from exon 20 Figure 3C, 3E.
Figure 3.
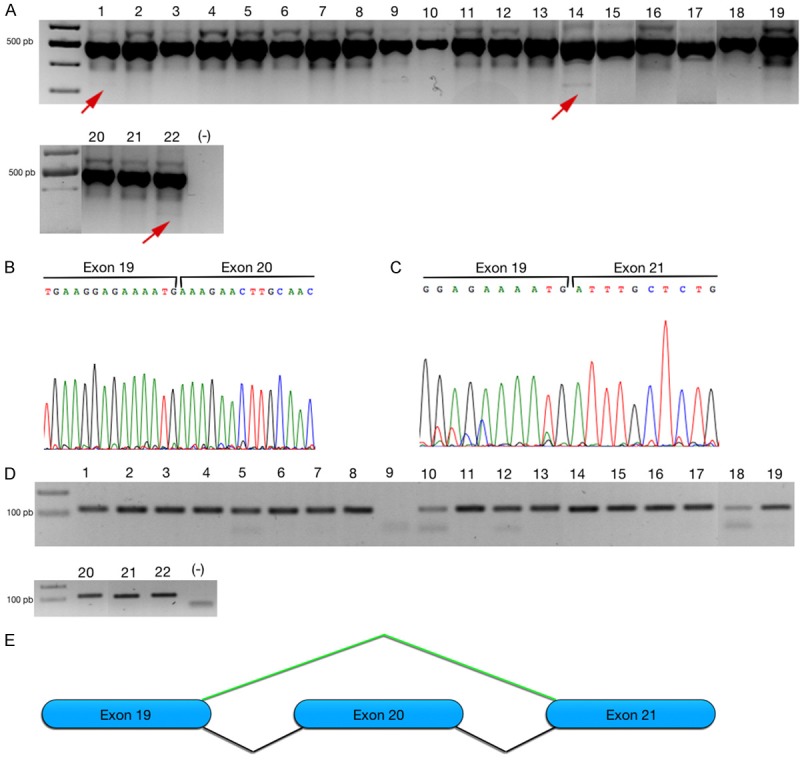
Alternative mRNA splice of CENP-E reveal exon skipping form exon 20. Expression of CENP-E in 22 cell lines included 1 (NIH-OVCAR-3), 2 (SK-OV-3), 3 (TVO-21G), 4 (TVO-112D), 5 (HeLa), 6 (SiHa), 7 (CaLo), 8 (RoVa), 9 (ViBo), 10 (C-33A), 11 (Vippa), 12 (INBL), 13 (Caski), 14 (MS-751), 15 (MDA-MB231), 16 (MCF-7), 17 (Hep-G2), 18 (HaCat), 19 (Hek-293T), 20 (REH), 21 (Jurkat), 22 (K-562). A. Agarose gel showed expression of consensus primers to amplified transcript variant one/two of CENP-E. In some cell lines was observed inconspicuous expression on ~300 pb showed with red arrow. B. Electropherogram showed the expression of constitutive CENP-E transcript, the top line indicated the boundary sites of exon 19 and 20. C. Electropherogram showed the boundaries sites of exon 19 and 21, indicating the alternative exon skipping of exon 20. D. In agarose gel showed specific amplification of NAT-CENP-E. E. Model of alternative exon skipping of NAT-CENP-E.
Our results indicated the alternative exon skipping in eight cell lines evaluated. In order to evaluate the new alternative splicing in CENP-E, we designed specific primer in forward that include boundary exons 19 and 21, the reverse primer used was the same that the previous analysis (Set3). The PCR showed only one amplicon of ~100 pb, name NAT-CENP-E Figure 3D. The expression observed is less than constitutive transcripts (one or two). Our results showed the skip of exon 20 in almost all cell lines evaluated. Interestingly, the ViBo cell line did not express this transcript.
Alternative mRNA splice of CENP-E is expressed in acute lymphoblastic luekemia
The amplification of the new alternative transcript of CENP-E (NAT-CENP-E) showed an expression in different types of cell lines, include leukemia. These results suggest that NAT-CENP-E could be expressed in many types of cancer. In addition, our results showed the specific expression of NAT-CENP-E. In order to identify the expression of NAT-CENP-E, we evaluated in healthy controls (HC) and LP using RT-PCR end point Figure 4. We evaluated NAT-CENP-E expression in 10 HC and 10 LP, interestingly we observed inconstant expression in HC Figure 4A as well as LP Figure 4B.
Figure 4.
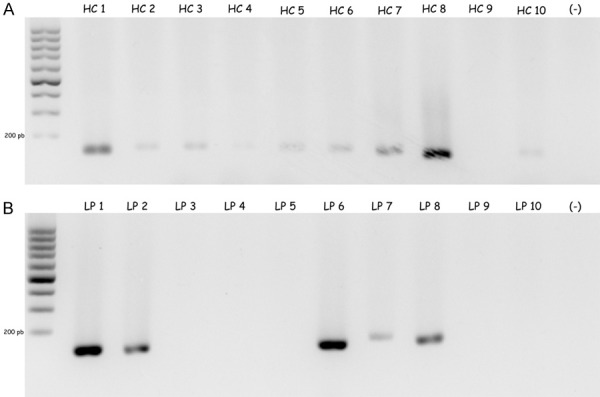
Acute Lymphoblastic Leukemia expressed NAT-CENP-E. Agarose gel at 1.5% showed the expression NAT-CENP-E. A. 10 healthy controls are shown, the expression were heterogenous in all samples, HC1 to HC10. B. 10 Acute Lymphoblastic Leukemia patients were evaluated, LP1 to LP10. We observer some patients overexpressed NAT-CENP-E and some patients no expressed NAT-CENP-E.
In order to quantify level expression, we performed quantitative RT-PCR using housekeeping gene RPS18, we performed standardization of our primer design that include (standard and melting curves for both of them transcripts and their validation Supplementary Figure 1 according to Livak K et. al., 2001 [31]. Then was evaluated NAT-CENP-E in 10 HC and 71 LP, our results showed expression in HC as well as patients. Then, we grouped the patients as low expression (lower expression that the normalized control group, n = 51) and high expression (more expression that the normalized control group, n = 20). Finally, we compared HC expression vs patients with low expression and HC vs high expression. In both comparison, we observed significant differences P = 0.0001 and P = 0.0016, respectively Figure 5.
Figure 5.
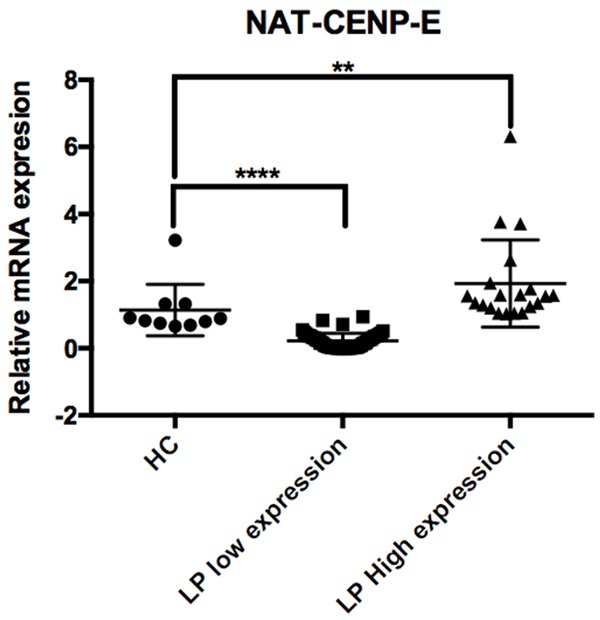
Relative quantification of NAT-CENP-E in Acute Lymphoblastic Leukemia. Evaluation of NAT-CENP-E using Real Time RT-PCR. The plot showed the relative expression of 10 healthy donator and 71 LP. The LP were fractionated in low (n = 51, less to one-fold in gene expression P = 0.0001) and high expression (n = 20, more of one-fold in gene expression P = 0.0016) and were compared against to reference control.
Discussion
The cancer is a complex disease with several cellular processes involved in malignance development. The great challenge in cancer is the identification of specific molecular markers for diagnosis, prognosis and therapeutic targets. However, several tumour type shows heterogeneity molecular profile such as: medulloblastoma [32,33], melanoma [34], leukemia [35] among other, and several subtypes of cancer showed different prognosis. Approximately, the cancer is characterized by marks including: sustaining proliferation, angiogenesis, resisting cell death, invasion and metastasis, among others [17]. Sustaining proliferation signalling is consequence of alteration in control of cell cycle, by means of over expression of oncoproteins such as: RAS, MYC, RAF [36]. On the other hand, down regulation of tumour suppressor including: TP53, RB; this proteins plays a central role in cell cycle control [18]. Recent studies have shown complex gene expression profile as well as alternative mRNA splice is cancerassociated.
The AS constitutes an important key of control in gene expression. It allows generate RNA diversity, consequently large number of protein expressed, this enable high capacity of a single gene increase transcriptional potential [37]. In fact, the proteins result of AS allowed changes in protein topology, location, function. Different reports has shown the expression of mRNA splice cancer-associated, such as: GLS MAX (deregulating cellular energetics) [38,39]; VEGFA (inducing angiogenesis) [40,41]; BCL2L1, CASP2, CASP8, MDM2, TP53 (resisting cell death) [42-45]; BRAF, EGFR, FGFR2 (sustaining growth suppressors) [39,46,47]; CD44, FGFR2, RAC1 (activating invasion and metastasis) [47-49]. On the other hands, the proteins product of alternative splicing could be therapeutic target [19]. However, little it is known about of diversity of mRNA splice in human diseases. Although, the bioinformatics analysis has shown that up to 70% of human genes could generate at least two transcripts from a single gene [50] there are little mRNA transcript variants identify and characterized.
The acute lymphoblastic leukemia is the most common childhood malignance in the world wide and the second leading cause died in Mexico [51]. Approximately 20% of patients relapse with poor prognostic, probably by treatment resistance [35]. Several aberrant alternative splicing has been involved in different cancer type such as: GLS, LDHC, AIMP2, CEACAM1, TERT, CD44, RAC1, FGFR2 [19], however, is little know of the diversity of alternative mRNA splicing in leukemia.
Centromere-associated protein E is very important for transition from metaphase to anaphase [52] contribute to alignment [53] and accurate chromosome segregation mediated to mitotic spindle. The crucial role of CENP-E is mediated the motor domain, which is essential to moves toward microtubules, in addition other kinesin-related motor proteins are required to drive correct function [54]. Our findings have showed the significant over expression of CENP-E ovarian cancer-associated [1] and cancer cell lines, including leukemia (data no shown). CENP-E was one of most significant alternatively splice transcript [1]. Our experimental findings showed expression of CENP-E in all cancer cell lines evaluated. Moreover, all cell lines expressed the new alternative mRNA spliced of CENP-E, except RoVa cell line. Additionally, we quantify the expression in leukemia patients, these results showed that NAT-CENP-E is over expressed in 20 patients.
CENP-E is a protein essential for maintenance of chromosomal stability by means of stabilization of microtubule capture at kinetochores. CENP-E has been reported as suppressor gene. However, in several types of cancer CENP-E has shown to be up-regulated, including in pituitary tumours [55], breast [56,57], ovarian [58], and lung cancers [59], and gliomas [60], among others. Our results showed two groups of patients, one that NAT-CENP-E is suppressed that correspond ~80%, and the remaining patients over expressed NAT-CENP-E. Interestingly, the group that over expressed NAT-CENP-E is the 20%. Approximately, the same percentage of patients in México showed relapse with poor prognosis. Different reports have shown contradictory results on CENP-E, suggesting two roles in cancer as tumour suppressor and oncogene. Probably the capacity as a suppressor and oncogene is the result of alternative mRNA splice of CENP-E transcript. The classic example of antagonic function of proteins consequence of alternative splicing is Bcl-xL with pro-survival and Bcl-xS pro-apoptotic functions [42,61]. TNR6 is subject to alternative splicing [62] producing a short protein, the consequence is loss the transmembrane domain and the protein resulting is soluble and inhibit FAS-mediated cell death [63].
On the other hand, the cancer cells exhibit genetic alterations that include deletions, amplifications, polyploidy, aneuploid, among others. Probably the over expression of CENP-E and alternative mRNA transcripts of this gene plays a role in genetic alterations in some types of cancer, including leukemia. In childhood leukaemia some patients showed high percent of aneuploid and the better prognosis. Probably the expression of CENP-E and their transcripts contribute with the course of the disease.
In this study, we identify new alternative mRNA splice of CENP-E transcript in several cancer cell lines and demonstrated the expression of new alternative mRNA transcript of the CENP-E gene in Childhood Acute Lymphoblastic Leukemia. Although our bioinformatics and experimental analysis in cancer cell lines a leukemia patients have shown the expression of NAT-CENP-E, future experiments are required to demonstrate the function and impact of transcriptional variants of CENP-E in cancer.
Conclusion
In summary, our study we demonstrated by RT-PCR and sequencing the expression of new alternative mRNA splice of CENP-E transcript, resulting from exon 20 is skip. Only one cell line expressed alternative 5’ splice site in exon 38. We consider that over expression of NAT-CENP-E in patients could play an important role in several types of cancer.
Acknowledgements
This work was supported by a Basic Science grant from SEP-CONACyT México 243233, FOSISS grant 272633 and Federal Funds INP 2017.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Juarez-Mendez S, Zentella-Dehesa A, Villegas-Ruiz V, Perez-Gonzalez OA, Salcedo M, Lopez-Romero R, Roman-Basaure E, Lazos-Ochoa M, Montes de Oca-Fuentes VE, Vazquez-Ortiz G, Moreno J. Splice variants of zinc finger protein 695 mRNA associated to ovarian cancer. J Ovarian Res. 2013;6:61. doi: 10.1186/1757-2215-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan T, Benovoy D, Dias C, Gurd S, Serre D, Zuzan H, Clark TA, Schweitzer A, Staples MK, Wang H, Blume JE, Hudson TJ, Sladek R, Majewski J. Heritability of alternative splicing in the human genome. Genome Res. 2007;17:1210–1218. doi: 10.1101/gr.6281007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29:2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–185. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu M, Conzen SD, Cole CN, Arrick BA. Characterization of functional messenger RNA splice variants of BRCA1 expressed in nonmalignant and tumor-derived breast cells. Cancer Res. 1996;56:4578–4581. [PubMed] [Google Scholar]
- 9.Philips AV, Cooper TA. RNA processing and human disease. Cell Mol Life Sci. 2000;57:235–249. doi: 10.1007/PL00000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nissim-Rafinia M, Kerem B. Splicing regulation as a potential genetic modifier. Trends Genet. 2002;18:123–127. doi: 10.1016/s0168-9525(01)02619-1. [DOI] [PubMed] [Google Scholar]
- 11.Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, Lucier JF, Thibault P, Rancourt C, Tremblay K, Prinos P, Chabot B, Elela SA. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16:670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 12.Moran-Jones K, Grindlay J, Jones M, Smith R, Norman JC. hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Res. 2009;69:9219–9227. doi: 10.1158/0008-5472.CAN-09-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orban TI, Olah E. Emerging roles of BRCA1 alternative splicing. Mol Pathol. 2003;56:191–197. doi: 10.1136/mp.56.4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007;8:349–357. doi: 10.1016/S1470-2045(07)70104-3. [DOI] [PubMed] [Google Scholar]
- 15.Bonomi S, Gallo S, Catillo M, Pignataro D, Biamonti G, Ghigna C. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol. 2013;2013:962038. doi: 10.1155/2013/962038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52:720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35:2413–2427. doi: 10.1038/onc.2015.318. [DOI] [PubMed] [Google Scholar]
- 20.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, Pei D, Scheet P, Burchard EG, Eng C, Huntsman S, Torgerson DG, Dean M, Winick NJ, Martin PL, Camitta BM, Bowman WP, Willman CL, Carroll WL, Mullighan CG, Bhojwani D, Hunger SP, Pui CH, Evans WE, Relling MV, Loh ML, Yang JJ. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105:733–742. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L, Goodman PA, Wood CM, Crotty ML, Sensel M, Sather H, Navara C, Nachman J, Steinherz PG, Gaynon PS, Seibel N, Vassilev A, Juran BD, Reaman GH, Uckun FM. Expression of aberrantly spliced oncogenic ikaros isoforms in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 1999;17:3753–3766. doi: 10.1200/JCO.1999.17.12.3753. [DOI] [PubMed] [Google Scholar]
- 23.Yen TJ, Li G, Schaar BT, Szilak I, Cleveland DW. CENP-E is a putative kinetochore motor that accumulates just before mitosis. Nature. 1992;359:536–539. doi: 10.1038/359536a0. [DOI] [PubMed] [Google Scholar]
- 24.Ding X, Yan F, Yao P, Yang Z, Wan W, Wang X, Liu J, Gao X, Abrieu A, Zhu T, Zhang J, Dou Z, Yao X. Probing CENP-E function in chromosome dynamics using small molecule inhibitor syntelin. Cell Res. 2010;20:1386–1389. doi: 10.1038/cr.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 26.Vaclavicek A, Bermejo JL, Wappenschmidt B, Meindl A, Sutter C, Schmutzler RK, Kiechle M, Bugert P, Burwinkel B, Bartram CR, Hemminki K, Forsti A. Genetic variation in the major mitotic checkpoint genes does not affect familial breast cancer risk. Breast Cancer Res Treat. 2007;106:205–213. doi: 10.1007/s10549-007-9496-9. [DOI] [PubMed] [Google Scholar]
- 27.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Rao CV, Yamada HY, Yao Y, Dai W. Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis. 2009;30:1469–1474. doi: 10.1093/carcin/bgp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganmore I, Smooha G, Izraeli S. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet. 2009;18:R84–93. doi: 10.1093/hmg/ddp084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527–539. doi: 10.1038/nrc3310. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Castillo-Rodriguez RA, Davila-Borja VM, Juarez-Mendez S. Data mining of pediatric medulloblastoma microarray expression reveals a novel potential subdivision of the Group 4 molecular subgroup. Oncol Lett. 2018;15:6241–6250. doi: 10.3892/ol.2018.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, Agnihotri S, Thompson YY, Kuzan-Fischer CM, Farooq H, Isaev K, Daniels C, Cho BK, Kim SK, Wang KC, Lee JY, Grajkowska WA, Perek-Polnik M, Vasiljevic A, Faure-Conter C, Jouvet A, Giannini C, Nageswara Rao AA, Li KKW, Ng HK, Eberhart CG, Pollack IF, Hamilton RL, Gillespie GY, Olson JM, Leary S, Weiss WA, Lach B, Chambless LB, Thompson RC, Cooper MK, Vibhakar R, Hauser P, van Veelen MC, Kros JM, French PJ, Ra YS, Kumabe T, Lopez-Aguilar E, Zitterbart K, Sterba J, Finocchiaro G, Massimino M, Van Meir EG, Osuka S, Shofuda T, Klekner A, Zollo M, Leonard JR, Rubin JB, Jabado N, Albrecht S, Mora J, Van Meter TE, Jung S, Moore AS, Hallahan AR, Chan JA, Tirapelli DPC, Carlotti CG, Fouladi M, Pimentel J, Faria CC, Saad AG, Massimi L, Liau LM, Wheeler H, Nakamura H, Elbabaa SK, Perezpena-Diazconti M, Chico Ponce de Leon F, Robinson S, Zapotocky M, Lassaletta A, Huang A, Hawkins CE, Tabori U, Bouffet E, Bartels U, Dirks PB, Rutka JT, Bader GD, Reimand J, Goldenberg A, Ramaswamy V, Taylor MD. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31:737–754. e736. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 35.Tang X, Guo X. [Research progress in Phlike childhood acute lymphoblastic leukemia] . Zhongguo Dang Dai Er Ke Za Zhi. 2017;19:1213–1218. doi: 10.7499/j.issn.1008-8830.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 38.van den Heuvel AP, Jing J, Wooster RF, Bachman KE. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther. 2012;13:1185–1194. doi: 10.4161/cbt.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, Zhu S, Gu Y, Villa GR, Akhavan D, Nathanson D, Gini B, Mareninov S, Li R, Camacho CE, Kurdistani SK, Eskin A, Nelson SF, Yong WH, Cavenee WK, Cloughesy TF, Christofk HR, Black DL, Mischel PS. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013;17:1000–1008. doi: 10.1016/j.cmet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ladomery MR, Harper SJ, Bates DO. Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 2007;249:133–142. doi: 10.1016/j.canlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 41.Shehadeh LA, Yu K, Wang L, Guevara A, Singer C, Vance J, Papapetropoulos S. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PLoS One. 2010;5:e9104. doi: 10.1371/journal.pone.0009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 43.Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57–68. doi: 10.2147/OTT.S53876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci U S A. 2008;105:15708–15713. doi: 10.1073/pnas.0805569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himeji D, Horiuchi T, Tsukamoto H, Hayashi K, Watanabe T, Harada M. Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood. 2002;99:4070–4078. doi: 10.1182/blood.v99.11.4070. [DOI] [PubMed] [Google Scholar]
- 46.Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, Hatanpaa KJ, Mickey B, Madden C, Maher E, Boothman DA, Furnari F, Cavenee WK, Bachoo RM, Burma S. EGFRvIII and DNA doublestrand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69:4252–4259. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai A, Meetze K, Vo NY, Kollipara S, Mazsa EK, Winston WM, Weiler S, Poling LL, Chen T, Ismail NS, Jiang J, Lerner L, Gyuris J, Weng Z. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010;70:7630–7639. doi: 10.1158/0008-5472.CAN-10-1489. [DOI] [PubMed] [Google Scholar]
- 48.Nagano O, Okazaki S, Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene. 2013;32:5191–5198. doi: 10.1038/onc.2012.638. [DOI] [PubMed] [Google Scholar]
- 49.Zhou C, Licciulli S, Avila JL, Cho M, Troutman S, Jiang P, Kossenkov AV, Showe LC, Liu Q, Vachani A, Albelda SM, Kissil JL. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene. 2013;32:903–909. doi: 10.1038/onc.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genomewide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 51.Rivera-Luna R, Correa-Gonzalez C, Altamirano-Alvarez E, Sanchez-Zubieta F, Cardenas-Cardos R, Escamilla-Asian G, Olaya-Vargas A, Bautista-Marquez A, Aguilar-Romo M. Incidence of childhood cancer among Mexican children registered under a public medical insurance program. Int J Cancer. 2013;132:1646–1650. doi: 10.1002/ijc.27771. [DOI] [PubMed] [Google Scholar]
- 52.Yen TJ, Compton DA, Wise D, Zinkowski RP, Brinkley BR, Earnshaw WC, Cleveland DW. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 1991;10:1245–1254. doi: 10.1002/j.1460-2075.1991.tb08066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mao Y, Desai A, Cleveland DW. Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J Cell Biol. 2005;170:873–880. doi: 10.1083/jcb.200505040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood KW, Sakowicz R, Goldstein LS, Cleveland DW. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell. 1997;91:357–366. doi: 10.1016/s0092-8674(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 55.Wierinckx A, Auger C, Devauchelle P, Reynaud A, Chevallier P, Jan M, Perrin G, Fevre-Montange M, Rey C, Figarella-Branger D, Raverot G, Belin MF, Lachuer J, Trouillas J. A diagnostic marker set for invasion, proliferation, and aggressiveness of prolactin pituitary tumors. Endocr Relat Cancer. 2007;14:887–900. doi: 10.1677/ERC-07-0062. [DOI] [PubMed] [Google Scholar]
- 56.Agarwal R, Gonzalez-Angulo AM, Myhre S, Carey M, Lee JS, Overgaard J, Alsner J, Stemke-Hale K, Lluch A, Neve RM, Kuo WL, Sorlie T, Sahin A, Valero V, Keyomarsi K, Gray JW, Borresen-Dale AL, Mills GB, Hennessy BT. Integrative analysis of cyclin protein levels identifies cyclin b1 as a classifier and predictor of outcomes in breast cancer. Clin Cancer Res. 2009;15:3654–3662. doi: 10.1158/1078-0432.CCR-08-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corson TW, Gallie BL. KIF14 mRNA expression is a predictor of grade and outcome in breast cancer. Int J Cancer. 2006;119:1088–1094. doi: 10.1002/ijc.21954. [DOI] [PubMed] [Google Scholar]
- 58.Theriault BL, Pajovic S, Bernardini MQ, Shaw PA, Gallie BL. Kinesin family member 14: an independent prognostic marker and potential therapeutic target for ovarian cancer. Int J Cancer. 2012;130:1844–1854. doi: 10.1002/ijc.26189. [DOI] [PubMed] [Google Scholar]
- 59.Corson TW, Zhu CQ, Lau SK, Shepherd FA, Tsao MS, Gallie BL. KIF14 messenger RNA expression is independently prognostic for outcome in lung cancer. Clin Cancer Res. 2007;13:3229–3234. doi: 10.1158/1078-0432.CCR-07-0393. [DOI] [PubMed] [Google Scholar]
- 60.Wang Q, Wang L, Li D, Deng J, Zhao Z, He S, Zhang Y, Tu Y. Kinesin family member 14 is a candidate prognostic marker for outcome of glioma patients. Cancer Epidemiol. 2013;37:79–84. doi: 10.1016/j.canep.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Willimott S, Merriam T, Wagner SD. Apoptosis induces Bcl-XS and cleaved Bcl-XL in chronic lymphocytic leukaemia. Biochem Biophys Res Commun. 2011;405:480–485. doi: 10.1016/j.bbrc.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 62.Cascino I, Fiucci G, Papoff G, Ruberti G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol. 1995;154:2706–2713. [PubMed] [Google Scholar]
- 63.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.