

Abstract
CDK4/6 inhibitors represent a new treatment standard for hormone receptor-positive (HR+), HER2-negative advanced breast cancer (BC) patients. Although efficacious, resistance to these agents is universal. Here, we profiled a large panel of HR+ BC cell lines with conditioned resistance to the CDK4/6 inhibitor palbociclib, and analyzed cell cycle-related markers by gene expression profiles (GEP) and western blot (WB). GEP showed high molecular heterogeneity among the models, with E2F targets being significantly enriched both during treatment and at the time of resistance. By both WB and GEP, a common molecular feature at the time of palbociclib resistance was the concomitant overexpression of cyclin E1 and down-regulation of Rb. CCNE1 was the only significantly up-regulated gene among E2F targets at resistance with CCNE1 genomic amplification being observed in two resistant models. Rb was downregulated in all resistant models; a reduction of RB1 copy number was observed in three resistant cell lines. In silico analyses showed that CCNE1/RB1 ratio correlated with palbociclib IC50 in different datasets of both breast and non-breast cancer cell lines, performing better than CCNE1 or RB1 taken separately. Finally, the CCNE1/RB1 ratio was shown to be an adverse prognostic factor in patients with ER+ BC and to be able to discriminate palbociclib-sensitive versus resistant among patients enrolled in the NeoPalAna trial, a neoadjuvant trial testing palbociclib, performing better than CCNE1 or RB1 alone. Our data suggest that the CCNE1/RB1 ratio may be a viable biomarker of palbociclib resistance, warranting further clinical validation.
Biomarkers: Prognostic indicator for resistance to cell cycle–targeted drug
The expression levels of two genes involved in controlling the cell cycle offers a promising predictive indicator of whether women with hormone receptor–positive breast cancer are likely to develop resistance to the drug palbociclib. A team led by Luca Malorni and Ilenia Migliaccio from the Hospital of Prato, Italy, analyzed gene and protein profiles in a panel of breast cancer cell lines with differential sensitivity to palbociclib. The researchers found that, compared to palbociclib-sensitive cell lines, those that were drug-resistant tended to have elevated expression of one cell-cycle–related gene, called CCNE1, and reduced expression of another, called Rb. The ratio of CCNE1:Rb expression proved to be an accurate prognostic factor among patients enrolled in a clinical trial of palbociclib, performing better than either CCNE1 or Rb levels on their own.
Introduction
Palbociclib is a highly specific, orally active CDK4/6 inhibitor currently approved for the treatment of hormone receptor-positive, HER2-negative (HR+/HER2neg) advanced breast cancer (BC) in combination with the endocrine agents letrozole or fulvestrant.1–3 Given the efficacy and the tolerability shown by CDK4/6 inhibitors, utilization of these drugs in clinical practice is common in patients with HR+/HER2neg advanced BC. However, acquired resistance to these agents is universal, and results from clinical trials indicate that approximately 10 to 15% of patients may exhibit de novo resistance.1–4
CDK4 and CDK6 are kinases activated by binding to D-type cyclins, bearing a crucial role in cell proliferation through the regulation of cell cycle entry.5 The primary target of CDK4/6 action is the retinoblastoma susceptibility gene product (Rb) and other Rb family members (such as p107 and p130).6 Phosphorylation of Rb (pRb) by active cyclin-CDK complexes leads to release of transcription factors of the E2F family, and transcription of genes required for S-phase entry.6 Constraint upon CDK activity and G1 progression is provided by the universal CDK inhibitors of the Cip-Kip family, including p21 and p27, and the specific CDK4/6 inhibitors of the Ink family typified by p16.7
Preclinical data have already shown that deregulation of cell cycle genes and proteins is associated with resistance to palbociclib, including overexpression of cyclin E1 and loss of Rb.8–10 However, these events have been observed only in a limited number of in vitro and in vivo models, and these data have not been fully validated in the clinical setting. Therefore, the aim of our study is to further investigate if common cell cycle alterations, especially in the Rb/E2F pathway, could be observed in palbociclib-resistant models using a large panel of HR+ BC cell lines.
Results
Functional characterization of palbociclib-resistant (PDR) and palbociclib-sensitive (PDS) BC cell lines
We challenged eight BC cell lines with palbociclib including T47D, ZR75-1, MCF7, MCF7 Estrogen Deprivation Resistant (MCF7 EDR), MCF7 Tamoxifen Resistant (MCF7 TamR), CAMA1, MDA MB 361, and BT474. We developed PDR derivatives for all BC cell lines with the exception of CAMA1, which failed to restore growth with 350 nM palbociclib over two separate attempts. The time to develop resistance for all other models ranged from 10 to 27 weeks.
Proliferation of all PDS cells was significantly inhibited by palbociclib (P-value < 0.05), while PDR derivatives did not show significant differences in cell growth when treated with palbociclib 1 µM compared to vehicle-treated cells (Fig. 1a,b). Additionally, PDR cells showed IC50 values 6–30 times higher than their PDS counterparts (Supplementary Table s1). CDK4/6 inhibitors are cytostatic drugs, which induce a senescence-like phenotype, typically characterized by high β-galactosidase expression and a flattened cellular shape.11 With the exception of the MDA MB 361 cell line, our PDR models showed significantly (P-value < 0.05) lower proportions of β-galactosidase positive cells during palbociclib treatment compared to their PDS counterparts, demonstrating the ability to revert the palbociclib-induced senescence-like phenotype (Fig. 1c and Supplementary Fig. s1).
Fig. 1.

Functional characterization of PDR and PDS BC cell lines. a Proliferation of PDS models treated with palbociclib 1 µM, palbociclib STC or 0.01% DMS0 (vehicle). b Proliferation of PDR models treated with palbociclib 1 µM or 0.01% DMS0 (vehicle). Growth rate was assessed at day 6 by methylene blue assay. Growth fold changes were computed as ratio, dividing growth rate of treated cells by their controls treated with drug vehicle. Mean value of growth fold change of three independent experiments ±standard error of the mean (SEM) was plotted (*P-values < 0.05, two–way ANOVA with Sidak’s multiple comparisons test). c β-galactosidase activity of PDS and PDR models treated with palbociclib 1 µM or 0.01% DMS0 (vehicle). Positive and negative cells were counted in six randomly selected 20X fields and the percentage of positive cells was calculated. Mean percentage value of β-galactosidase positive cells of three independent experiments ±SEM was plotted (*P-values < 0.05, by Student’s t test)
Gene expression (GE) analysis of PDS and PDR cells reveals that modulation of E2F signaling occurs during palbociclib treatment and at the time of acquired resistance in HR+ BC cell lines
We analyzed GE data derived from PDS cells treated with drug vehicle (control, PDS) or with palbociclib starting treatment concentration (STC) for 3 days (PDS treated) and from PDR cells continuously receiving palbociclib 1 µM.
Hierarchical clustering analysis of GE profiles did not reveal common GE programs associated with palbociclib resistance; conversely, cells tended to segregate based exclusively on their cell type (Supplementary Fig s2), indicating that common characteristics at the time of resistance were not able to overcome cell-type specific features. To reduce cell-type specific signals, we performed an intra-cell type normalization of GE and calculated differential expression between PDR and PDS treated cells. The list of the top 100 differentially expressed genes is provided in Supplementary Table s2. None of the differentially expressed genes were significantly over or underexpressed when adjusted for multiple testing (Supplementary Table s2), probably due to the high heterogeneity observed among different cell lines. However, GE analysis of the cell cycle pathway revealed that CCNE1 and RB1 were consistently deregulated in all PDR models versus PDS treated cell lines (Fig. 2a). This prompted further analyses of the Rb/E2F pathway during palbociclib treatment and at the time of resistance in our models.
Fig. 2.

GE analysis of PDS and PDR cells. a Heatmap showing the top 15 most variable cell cycle pathway genes (rows) between PDR and PDS treated cells (columns). Values are the log2 of Fold Change (FC) of normalized expression in each cell lines with respect to the corresponding PDS. The different cell models are reported in the annotation track (PDS treated, PDR early, PDR late). b Box plot showing the distribution of RBsig score across the different cell models. c Combined Gene Set Enrichment Analysis (GSEA) plot of PDS treated (gray lines) and PDR (black lines) using averaged FC as ranking parameters and E2F targets as gene set. For both PDS treated and PDR, FDR ~ 0. d Averaged FC in PDR (y-axis) versus averaged FC in PDS treated for E2F target genes. The top five genes having the greatest absolute difference in FC between PDR and PDS treated models are shown. e Box plot showing the distribution of FC of CCNE1 expression in PDS treated and PDR models. **P-value < 0.01, ***P-value < 0.001; by Wilcoxon test
We first analyzed a previously developed GE signature of Retinoblastoma loss-of-function, the RBsig,12 in PDS, PDS treated and PDR models. Overall, RBsig levels were significantly reduced by palbociclib treatment in PDS treated cells (Fig. 2b). Such modulation differed depending on the cell type, with the MDA MB 361 and BT474 HER2+ cell lines showing the highest levels of downregulation (Supplementary Fig. s3). In PDR models, RBsig levels tended to return to basal levels, but remained significantly inhibited compared to PDS untreated cells (Fig. 2b).
To further investigate the role of Rb/E2F pathway during palbociclib treatment and at the time of resistance, we performed GSEA on the normalized GE levels using the “HALLMARK_E2F_TARGETS” gene set, comprising 200 genes, of which 186 were present in our expression data. Consistently with RBsig analysis, genes down-regulated during treatment with palbociclib were significantly enriched for E2F targets; (FDR~0, Enrichment Score, ES = −0.88); similarly, genes down-regulated at the time of resistance were also significantly enriched in E2F targets indicating that the pathway remains inhibited by palbociclib at the time of resistance (FDR~0, ES = −0.71) (Fig. 2c). Interestingly, only CCNE1 showed a significant increase of expression at the time of resistance (P-value < 0.0005) (Fig. 2d,e), while the majority of the E2F target genes tended to be down-regulated both during treatment and at the time of resistance (Fig. 2d).
Overall these data suggest that palbociclib effectively inhibits cell growth of HR+ BC cells through inhibition of the Rb/E2F pathway. However, at the time of resistance, signaling is only partially restored, with increased CCNE1 expression being a common event.
Analysis of cell cycle-related proteins in PDS and PDR cells confirms that alterations of cyclin E1 and Rb expression are common events at the time of palbociclib acquired resistance
To validate the role of cell cycle-related proteins during palbociclib treatment and at the time of resistance, we performed western blot (WB) analysis for selected markers in PDS cell lines treated with palbociclib for 3 and 6 days at the STCs, and in PDR models continuously receiving palbociclib 1 μM.
Overall, different cell cycle-related proteins showed a similar modulation in all models during palbociclib treatment, although extent of modulations differed among cell lines. As expected, we observed a consistent reduction of Rb and its phosphorylated Ser 807/811 form after 3–6 days of treatment (Fig. 3). Similarly, E2F1 was generally down-regulated during palbociclib treatment together with the Rb related protein p107, cyclin A1 and cyclin E2 (Fig. 3). Additionally, levels of the transcription factor FOXM1, a potential target of CDK4/6 signaling,13,14 were consistently downregulated by palbociclib. Conversely, cyclin D1 and cyclin E1 levels tended to increase during treatment. All other markers were modulated heterogeneously, or were not modulated by the treatment (Fig. 3).
Fig. 3.

Analysis of cell cycle-related proteins in PDS and PDR cells. Western blot analysis of cell lysates from PDS models treated with vehicle (CTR) or palbociclib at the STCs for 3 and 6 days (D3, D6) and from their PDR counterparts receiving palbociclib 1 μM. Samples were blotted with the indicated antibodies. GAPDH bands are representative of one blot for each cell line. Blots from a given cell line (PDS and PDR) derive from the same experiment and were processed in parallel. Uncropped images of the cyclin E1 and Rb blots are supplied as Supplementary Figure s7
When PDR cells were compared to their PDS counterparts, in accordance with the GE data, we found significant heterogeneity in the regulation of the majority of cell cycle-related proteins among models. Very few modulations were consistent in all cell lines. Compared to untreated PDS cells, Rb was downregulated in all PDR models, with T47D showing virtually complete loss of expression (Fig. 3). Similarly, phosphorylation of Rb was reduced in PDR compared to untreated PDS cells (Fig. 3). Additionally, the cell cycle inhibitor p27 showed reduced expression in PDR compared to PDS cells lines (Fig. 3), whereas cyclin E1 was consistently upregulated in all models (Fig. 3), confirming the GE observation. All other cell cycle-related proteins were differently regulated in each PDR model or not modulated (Fig. 3). Quantification of the bands for Rb and cyclin E1 is provided as supplementary table s3.
Genomic analysis of CCNE1 and RB1
A potential mechanism for cyclin E1 and Rb deregulation could be represented by genomic alterations encompassing the corresponding genes. We therefore analyzed the copy number status of CCNE1 and RB1, normalized to the reference gene EIF2C1, in all PDR and PDS models. BT474 and MCF7 TamR exhibited an increased copy number status of CCNE1, while T47D, MDA MB 361 and MCF7 TamR showed reduced copy number of RB1 at the time of resistance (Fig. 4). Indeed, the log2 of the ratios of CCNE1/EIF2C1 between the BT474 and TamR PDR cells and PDS counterparts were 4.4 and 3.08, respectively. Interestingly, these data suggest that >30 and >10 CCNE1 DNA copies are acquired at resistance in BT474 and TamR PDR cells. Conversely, the log2 of the ratios of RB1/ EIF2C1 in T47D, MDA MB 361, and MCF7 TamR PDR cells were −1.24, −0.40, and −0.36, respectively, indicating a genomic loss of RB1 in these cell lines.
Fig. 4.
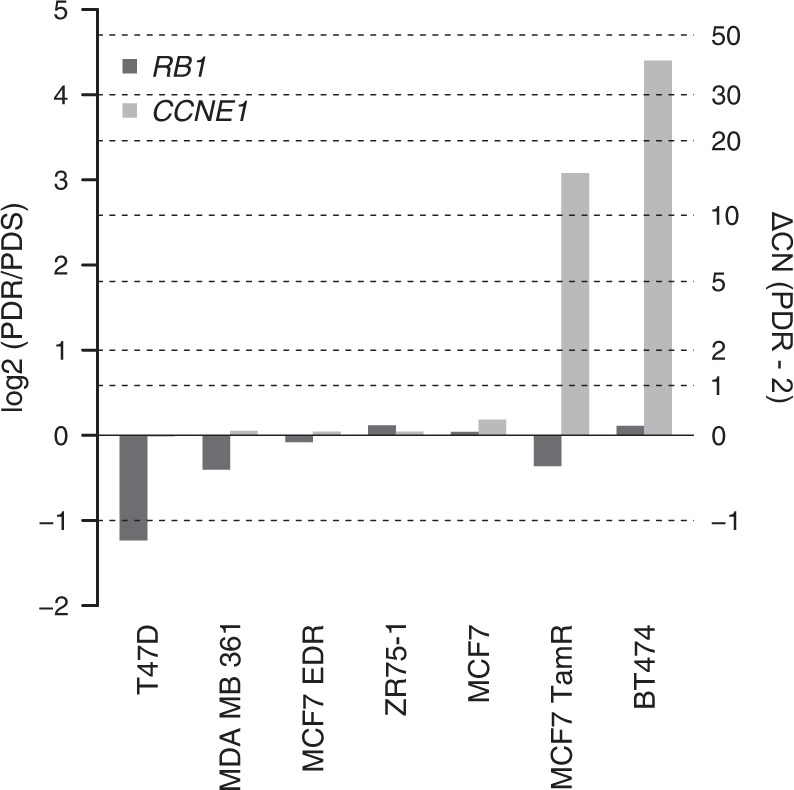
Genomic analysis of CCNE1 and RB1. Bar plot showing the log2 of the ratio of copy number estimates for RB1 (dark gray) and CCNE1 (gray) in PDR to untreated PDS models. Right vertical axis reports the estimated copy number difference between PDR and untreated PDS models, assuming 2 copies for PDS cells
CCNE1 and CCNE1/RB1 ratio as markers of de novo resistance to palbociclib
Our results demonstrated that cyclin E1 overexpression and Rb downregulation are common findings at the time of acquired resistance to palbociclib. The role of Rb in determining de novo resistance to CDK4/6 inhibitors has already been implicated in BC.15–19 Here we aimed to investigate if CCNE1 and the combined analysis of CCNE1 and RB1 as CCNE1/RB1 ratio could also represent a marker of de novo resistance to palbociclib. We therefore correlated CCNE1 and RB1 expression levels and CCNE1/RB1 ratio with palbociclib IC50 in a large dataset of cancer cell lines.20 In cell lines derived from different cancer types (pan-cancer dataset), these markers significantly correlated with higher IC50 values for palbociclib (Fig. 5a light gray bars, minimum |R| >0.27, maximum FDR = 5*10−14, correlation test) and among all drugs tested in the dataset20 palbociclib showed the strongest association with CCNE1/RB1 ratio (Fig. 5b, R = 0.41, FDR <10−30, correlation test). In addition, analyzing GE data from BC cell lines of the Iorio et al. dataset,20 but taking into account the IC50 values for palbociclib obtained from a previous BC-focused study,19 we were able to demonstrate that CCNE1, RB1, and CCNE1 /RB1 ratio correlated with higher IC50 also in BC cell lines (Fig. 5a dark gray bars, minimum |R| >0.45, maximum FDR = 0.01, correlation test). Of note, both in the pan-cancer and in the BC datasets, CCNE1/RB1 performed better than CCNE1 or RB1 alone in predicting palbociclib de novo resistance (Fig. 5a).
Fig. 5.

CCNE1 and CCNE1/RB1 ratio as markers of de novo resistance to palbociclib. a Bar plot of the absolute value of Pearson’s correlation coefficient of IC50 values versus expression of RB1, CCNE1, and CCNE1/RB1 by considering both IC50 values and expression data for 827 cell lines of different tumors from Iorio et al., 201620 (light gray); IC50 values and expression data for 31 BC cell lines from Finn et al., 200919 and Iorio et al., 201620, respectively (dark gray). b Histogram of the absolute value of Pearson’s correlation coefficient of CCNE1/RB1 versus IC50 values for 265 compounds tested in the cancer cell line dataset of Iorio et al.20 Palbociclib is the compound showing the most significant association with CCNE1/RB1. ***P < 0.001; by correlation test. c Bar plot of the absolute value of Pearson’s correlation coefficient of IC50 values versus expression of Rb, Cyclin E1, and Cyclin E1/Rb by considering IC50 values and protein expression data for 237 cell lines of different tumors from Iorio et al.,20 and MCLP21, respectively (light gray); IC50 values and expression data for 31 BC cell lines from Finn et al.19, and MCLP21, respectively (dark gray)
The correlations between protein levels of Rb1, cyclin E1, cyclin E1/Rb ratio, and palbociclib IC50 were also tested in a large dataset of cancer cell lines analyzed by reverse-phase protein arrays.21 Rb, cyclin E1, and the cyclin E1/Rb ratio correlated with palbociclib IC50, with the cyclin E1/Rb ratio performing better than cyclin E1 and Rb alone in predicting palbociclib resistance, both in the pan-cancer and the BC datasets (Fig. 5c).
CCNE1/RB1 ratio is a poor prognostic factor and a marker of palbociclib resistance in estrogen receptor-positive (ER+) BC
The prognostic role of the CCNE1/RB1 ratio in ER+ BC has previously never been investigated. As such, we analyzed survival of patients included in the METABRIC dataset22 according to the CCNE1/RB1 ratio. In the overall population, a significantly poorer overall survival (OS) was found in patients with a higher CCNE1/RB1 ratio (P-value < 0.0001, Fig. 6a). In order to exclude the potential confounder that the observed poor survival could be related to the association of the CCNE1/RB1 ratio with molecular subtypes (Supplementary Fig. s4), survival of the ER+ and ER negative (ERneg) subpopulations was analyzed separately. Interestingly, in the ER+ population the prognostic role of the CCNE1/RB1 ratio was maintained, with patients with high ratio (≥1) showing a poorer survival compared to those with a ratio <1 (P-value <0.0001, HR = 0.63, Fig. 6b). Conversely, in the ERneg population we observed no effect of the CCNE1/RB1 ratio on survival (P-value = 0.25, HR = 0.82, Fig. 6c). Results were similar when the median value of CCNE1/RB1 ratio in the ER+ population was used as cut-off (Supplementary Fig. s5a, b).
Fig. 6.

CCNE1/RB1 ratio as a poor prognostic factor and a marker of palbociclib resistance. a Kaplan–Meyer plot of METABRIC dataset22 (n = 1904). b Kaplan–Meyer plot of METABRIC ER+ samples (n = 1445). c Kaplan–Meyer plot of METABRIC ERneg samples (n = 429). Samples are divided into two groups according to the ratio of CCNE1 to RB1 expression. Cut-off is set to 1. P-values are calculated with log-rank test. d Box plot of the distributions of the ratio between gene expression level of CCNE1 to RB1 expression (CCNE1/RB1) in the NeoPalAna dataset23 for Palbociclib-sensitive (white) and resistant (gray) patients at the different time points. P-values are calculated by Wilcoxon–Mann–Whitney (WMW) test. e Bar plot showing the CCNE1/RB1 (dark gray), CCNE1 (gray), and RB1 (light gray) AUC values for discriminating between palbociclib-sensitive and resistant patients at the different time points
Further validation was sought through analysis of the publicly available NeoPalAna dataset. In the NeoPalAna trial, patients with stage II/III ER+/HER2neg primary BC received neoadjuvant palbociclib and anastrozole.23 Serial biopsies performed at baseline (C0D1), before starting palbociclib (C1D1) and after 2 weeks of palbociclib (C1D15), and surgical samples were analyzed for Ki67, GE, and mutation profiles.23 We found that CCNE1/RB1 was significantly higher in patients resistant to palbociclib at C0D1, C1D1, C1D15 (p(WMW) = 0.0048; p = 0.013; p = 0.032, respectively, Fig. 6d). Also, CCNE1/RB1 performed better than CCNE1 and RB1 alone in discriminating palbociclib-resistant versus sensitive patients at C0D1 (AUC(CCNE1/RB1) = 0.92; AUC(CCNE1) = 0.83; AUC(RB1) = 0.69), and at C1D1 (AUC(CCNE1/RB1) = 0.92; AUC(CCNE1) = 0.75; AUC(RB1) = 0.83), while showing equivalent performance compared to CCNE1 at C1D15 (AUC(CCNE1/RB1) = 0.9; AUC(CCNE1) = 0.9; AUC(RB1) = 0.55) (Fig. 6e).
Discussion
In this study, we developed a panel of HR+ BC cell lines with acquired resistance to palbociclib by chronically exposing cells to increasing doses of the drug. To account for the heterogeneity of luminal BC, we selected eight BC cell lines with different initial degrees of sensitivity to palbociclib. We deliberately challenged cell lines to palbociclib alone, rather than combined long-term estrogen deprivation (LTED) and palbociclib in order to identify markers specifically related to palbociclib resistance, thus avoiding the confounding effect of endocrine treatment. We acknowledge that the resultant models might not fully recapitulate the clinical scenario wherein CDK4/6 inhibitor resistance occurs in the setting of dual hormonal therapy and palbociclib. However, we did include two models previously resistant to hormonal therapy in order to account for the effect of combined palbociclib and endocrine resistance. Two HR+/HER2+ cell lines were also included to search for strong common mechanisms of resistance that might occur in HR+ BC despite HER2 status, given that palbociclib has also shown efficacy in HER2+ BC models.19,24–26 We obtained reliable models of acquired resistance to palbociclib, as revealed by functional analyses investigating proliferation, IC50 and β-galactosidase expression, for all cell lines except CAMA1. Yang et al. succeeded in the development of models of CAMA1 resistant to abemaciclib,10 probably due to the different approach used10 and the different inhibitory profile of abemaciclib.27
As a first step, we explored the effects of palbociclib on the Rb/E2F pathway, which is the target of CDK4/6 inhibitors.28,29 As expected, and in line with previous reports,9,30,31 we observed an inhibition of this pathway in response to palbociclib. We observed a marked reduction of the RBsig, a gene-signature of loss of RB1 function12 and found an enrichment in E2F targets within genes downregulated by palbociclib, consistent with previous observations.30 The activity of palbociclib on the Rb/E2F pathway was confirmed by WB, as revealed by decreased levels of total Rb coupled with decreased phosphorylation of Rb on S807/811 after 3 and 6 days of treatment. Together with Rb, we also observed a general decrease in p107 levels in all cell lines. As already reported by previous studies with cell lines sensitive to CDK4/6 inhibitors,9,31 our PDS models displayed cyclin D1 accumulation, reduction of E2F-regulated proteins (such as cyclins A1 and E2 and CDK2), and p130 stabilization when receiving palbociclib.
It has been demonstrated that BC cells show adaptation following CDK4/6 inhibition as early as 72 h. This has been shown to be mediated by non-canonical activation of cyclin D1/CDK2 complexes, which in turn induce a recovery of Rb phosphorylation and S-phase entry despite CDK4/6 inhibition.9 Our PDS models showed a persistent inhibition of the Rb/E2F pathway after 3 and 6 days of treatment. However, we did not analyze earlier time points, and therefore cannot exclude a partial recovery of the pathway at 72 h as suggested by Herrera-Abreu et al.9
It has already been shown that alterations in cell cycle genes and proteins are associated with acquired resistance to palbociclib.8–10,32,33 In particular, cyclin E overexpression and Rb loss have been previously indicated as possible mechanisms of acquired resistance to CDK4/6 inhibitors in some BC cell lines.8–10 Our GE and WB data showed that both cyclin E1 overexpression and Rb underexpression occur across different and heterogeneous BC cellular models, including HER2+ cells, which has not been shown previously. We also demonstrated that CCNE1 amplification and RB1 deletion might explain only partially these findings, suggesting that other mechanisms could be implicated. Intriguingly, these genomic alterations occurred in both HER2+ and HER2neg cell lines, again supporting the hypothesis that strong mechanisms of acquired resistance to palbociclib might occur in HR+ BC independent of HER2 status. This is of potential clinical relevance as CDK4/6 inhibitors are currently under investigation for patients with HR+ /HER2+ BC.34
At the time of resistance, we observed a slight reactivation of the Rb/E2F pathway as demonstrated by the increased RBsig expression in PDR cells, and by the modulation of some of the E2F targets analyzed. However, the pathway remained significantly inhibited compared to the PDS untreated cells, indicating that palbociclib treatment may still be inhibiting the pathway. This is consistent with previously published data in ovarian cancer, wherein cells resistant to palbociclib did not fully restore E2F target gene expression.32
Another common marker that we found at the time of acquired resistance is the reduced expression of p27. In a previous study, Dean et al. similarly suggested that loss of CDK inhibitors p21 and p27 may foster the ability of cells to escape from palbociclib-induced cell cycle arrest.31 Intriguingly, phosphorylation status of p27 has also been suggested as a mechanism of resistance to CDK4/6 inhibitors.35
Clinical trials with palbociclib clearly show that some patients do not benefit from CDK4/6 inhibitors.2–4,36 Loss of RB1 has been already suggested as a mechanism of de novo resistance to these drugs.15–19 Here, we identified CCNE1 as an additional potential marker of de novo resistance in a large panel of pan-cancer and BC cell lines. To date, the role of cyclin E1 as a marker of de novo resistance has been demonstrated in other tumor types such as ovarian and gastric cancer37,38 but only suggested in BC.33,39 Our data suggest that both CCNE1 and the CCNE1/RB1 ratio, which evaluates both CCNE1 and RB1, might serve as “pan-cancer” markers of de novo resistance to palbociclib and that the CCNE1/RB1 ratio might serve as better marker compared to the single biomarkers CCNE1 and RB1 used separately, both in BC and in other cancer types. In BC, this is evident for palbociclib IC50 values from Finn et al,19 but not when values from Iorio et al.20 were considered (Fig. 5a, c and Supplementary Fig. s6a, b). This discrepancy may be attributed to the different methodologies and drug concentrations used in the two studies,19,20 and might impact to a lesser extent when a larger number of heterogeneous pan-cancer cell lines are used, explaining the ability to reveal the correlations between CCNE1, CCNE1/RB1 ratio, and palbociclib IC50 when analyzing the pan-cancer dataset.
One limitation of our study is the lack of clinical validation of our findings on the role of CCNE1 and RB1 as markers of acquired resistance to palbociclib. Ideally, this should be evaluated prospectively in tissue biopsies obtained at the time of disease progression on palbociclib treatment, which was not performed as a part of this study. In lieu, we sought validation of our preclinical findings regarding CCNE1/RB1 ratio as a marker of de novo resistance to palbociclib in the NeoPalAna trial.23 Results from the NeoPalAna trial already showed that CCNE1 expression was significantly elevated in tumors resistant to the combination of palbociclib and anastozole at C1D15.23 We demonstrated that the CCNE1/RB1 ratio was significantly higher in palbociclib-resistant tumors at all time points analyzed. Of note, CCNE1/RB1 performed better than CCNE1 and RB1 alone in predicting de novo resistance to palbociclib at C0D1 and C1D1. These findings hold additional interest in light of the recently published TREnd trial,40 a phase 2 clinical study which assessed the activity of palbociclib as monotherapy, as well as in combination with the endocrine therapy (aromatase inhibitor or fulvestrant) upon which the patient had experienced previous progression as a prior line of therapy for ER+/HER2neg metastatic BC. This might be a potential additional setting to validate the role of the CCNE1/RB1 as a marker of de novo resistance to palbociclib.
Recent data have shown that expression of the cytoplasmic low-molecular-weight isoforms (LMW) of cyclin E is associated with resistance to aromatase inhibitors41 and to palbociclib in vitro.42 Similarly, overexpression of LMW cyclin E detected by immunohistochemistry has been associated with a lesser benefit from treatment with palbociclib and endocrine therapy in patients with metastatic BC.42 In our WB studies of the PDR models, LMW isoforms of cyclin E1 were not detected, suggesting that the full length is predominantly overexpressed in our models of acquired resistance. However, our in silico analyses could not exclude the possibility that LMW cyclin E may contribute to the effect on de novo resistance we described for cyclin E1, as both RNA and RPPA data cannot distinguish between the full length and the LMW isoforms.
We demonstrated that patients with early ER+ BC with a high CCNE1/RB1 ratio have poorer OS compared to patients with a ratio <1, suggesting that this may represent a population of patients with an adverse outcome, which might mandate tailored therapeutic strategies. However, more detailed studies are necessary to dissect the prognostic versus predictive value of this biomarker in early BC, which would be of particular interest since CDK4/6 inhibitors are currently being tested in the adjuvant setting.43
The strengths of our study include the size of the employed cell lines panel: to the best of our knowledge, this is the largest panel of HR+ BC cell lines ever developed to investigate acquired resistance to palbociclib. In turn, we have expanded previous knowledge on acquired resistance to palbociclib by confirming CCNE1 overexpression/amplification and reduced expression/loss of RB1 as common events that occur at the time of acquired resistance in luminal BC cell lines, independent of HER2 status. Furthermore, we have identified the CCNE1/RB1 ratio as a potential marker of de novo resistance to palbociclib both in vitro and in clinical samples.
Results of our study, if further confirmed and tested in clinical trials, might lead to the identification of new biomarkers of de novo and acquired resistance to palbociclib, which would assist to better personalize treatment of patients with HR+ BC.
Methods
Cell lines, cell culture, and reagents
T47D and ZR75-1 cell lines were obtained from Dr Livia Malorni, CNR Avellino, Italy; CAMA1 were obtained by American Type Culture Collection; MCF7 parental cells, MCF7 EDR and MCF7 TamR were previously described.44 MDA MB 361 were purchased from Sigma-Aldrich and BT474 from Interlab Cell Line Collection, Genova, Italy.
Cells were cultured at 37 °C and 5% CO2. T47D, ZR75-1, MCF7, CAMA1, MDA MB 361, and BT474 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/glucose and L-glutamine (Lonza) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone) and, 10,000 U penicillin and 10 mg steptomycin/mL solution (P/S) (Sigma-Aldrich). MCF7 EDR and MCF7 TamR were grown in DMEM with 4,5 g/glucose and without L-glutamine and phenol red (Lonza) supplemented with 10% charcoal-stripped FBS (CS-FBS) (GIBCO), and P/S. MCF7 TamR cell medium was supplemented with 100 nM final concentration of (Z)−4-Hydroxytamoxifen (Sigma-Aldrich) dissolved in 100% ethanol.
PDR were generated by long term culturing of cells with increasing concentrations of palbociclib from the STC up to 1 μM. STC was preliminary assessed on each model and defined as the concentration that, after 9 days of treatment, inhibited cells growth rate between 40 and 60%. Cells were considered resistant to palbociclib when they restored the ability to regularly grow in the presence of palbociclib 1 μM.
Palbociclib STC was 50 nM for T47D, ZR75-1, MCF7 EDR, and MCF7 TamR cell lines, 100 nM for CAMA1 and 350 nM for MCF7, MDA MB 361 and BT474 cell lines. PDR cells were then continuously cultured in their original media with the addition of palbociclib 1 μM. Palbociclib was provided by Pfizer and dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich).
In January 2016, all cell lines and their PDR derivatives have been authenticated by short tandem repeat DNA profiling analysis. This analysis was performed in service by BMR Genomics, Padova, Italy.
Proliferation and IC50 assays
For proliferation and IC50 assays a total of 3000 cells/well of PDS or PDR cell lines, cultured in their individual media, were plated in 96-well plates in triplicate, 24 h before beginning treatments. These consisted of palbociclib 1 µM, palbociclib STC or 0.01% DMS0 (vehicle) for 6 days for proliferation assays or doubling dilutions of palbociclib, from 64 µM to 0.97 nM for 9 days for the IC50 assays. Media were replaced every 72 h. Cells were fixed with 4% glutaraldehyde grade II (Sigma-Aldrich) and stained with 0.05% methylene blue (Sigma-Aldrich). The dye was subsequently extracted with 3% HCl (Carlo Erba) and absorbance measured at 655 nm. Three biological replicates for each PDS or PDR cell line were performed.
β-galactosidase assay
PDS and PDR cell lines were plated in 12-well plates in duplicate, in a range of 10,000–20,000 cells/well. Cells were plated in their individual medium which was replaced after 24 h with medium containing palbociclib 1 µM or 0.01% DMSO as a control. Beta-galactosidase activity was assessed at day 6 by Senescence detection kit (BioVision) according to the manufacturer’s instructions. Positive and negative cells were counted in six randomly selected 20X fields and the percentage of positive cells was calculated. Three biological replicates for each PDS or PDR line were performed.
Nucleic acids isolation and gene expression profiles
DNA was isolated from PDS and PDR cells grown in their individual media supplemented with 0.01% DMSO or palbociclib 1 µM, respectively. DNA extraction was performed with the Quick-gDNATM MiniPrep kit (Zymo Research), according to the manufacturer’s instructions. DNA concentrations and quality were checked by spectrophotometer.
RNA was isolated from PDS cells treated with 0.01% DMSO, as a control, or with palbociclib at the STC for 3 days, and from PDR cells at early (before or at passage 15) or late (after passage 15) stage of resistance, grown in their individual media supplemented with palbociclib 1 µM. The number of passages of PDR cells was calculated starting from the moment they restored the ability to regularly proliferate in the presence of palbociclib 1 µM. RNA was extracted from cells with the Direct-zolTM RNA MiniPrep kit (Zymo Research) according to the manufacturer’s instructions. RNA quantification and quality analysis were performed on a Bioanalyzer 2100 System (Agilent), using the RNA 6000 nano Kit (Agilent). cDNA synthesis and biotin-labeled cRNA generation were performed using the Illumina TotalPrep RNA Amplification Kit (Ambion), according to the manufacturer’s protocol using 500 ng of total RNA. Quality assessment and quantification of cRNAs were performed with Agilent RNA kits on the Bioanalyzer 2100 System. Hybridization of cRNAs (750 ng) was carried out using Illumina Human 48k gene chips (Human HT-12 v4 BeadChip). Array washing was performed using Illumina High Temp Wash Buffer for 10′ at 55 °C, followed by staining using streptavidin-Cy3 dyes (Amersham Biosciences). Probe intensity data were obtained using the Illumina GenomeStudio software.
GE data analysis
Normalization of probe intensity data (normalized expression) was performed by cubic spline algorithm implemented in the Illumina GenomeStudio software version 1.9.0. Only probes showing a detection P-value < 0.05 in at least one sample were considered for downstream analysis (n = 31016). In case of multiple probes matching to a single gene, the probe with the highest signal-to-noise ratio was taken into consideration. To mitigate the effect of cell line specific expression profiles, the log2-ratio values of normalized expression of treated PDS and PDR to the corresponding untreated PDS GE profiles were computed. (Fold Change, FC). The list KEGG_CELL_CYCLE (http://software.broadinstitute.org/gsea/msigdb/cards/KEGG_CELL_CYCLE) was used to identify cell cycle pathway genes. Top variable cell cycle genes (Fig. 2a) were defined as 25% most variable cell cycle genes across treated PDS and PDR (i.e., by standard deviation of FC) having an absolute value of average FC in PDR greater than 0.2, resulting in 15/127 genes.
E2F gene set enrichment was computed with Gene Set Enrichment Analysis (GSEA) version 2-2.2.3 by using log2-ratio values of averaged (mean) FC across PDS treated and PDR cell line models as ranking parameters and HALLMARK_E2F_TARGETS (http://software.broadinstitute.org/gsea/msigdb/cards/HALLMARK_E2F_TARGETS) as gene set. Differential expression between log2-ratio expression values was tested by Wilcoxon–Mann–Whitney (WMW) test.
Evaluation of RBsig score
For each RBsig12 gene i = 1,.,N, the median and median absolute deviation of normalized expression in untreated PDS cell lines were estimated. RBsig score of each cell line j was then calculated by the following formula:
where gij is the normalized expression of RBsig gene i in the j-th cell line.
Western blot
PDS cells treated with 0.01% DMSO, as a control, or with the STC of palbociclib for 3 or 6 days and PDR cells grown in their individual media supplemented with palbociclib 1 µM were lysed with RIPA buffer (Cell Signaling Technology), supplemented with 1% Protease Inhibitor Cocktail (Sigma-Aldrich) and 1% HaltTM Phosphatase Inhibitor Cocktail (Thermo Scientific). A volume of 15 μg of proteins were separated under denaturing conditions by electrophoresis on 10% or 7.5% polyacrylamide gels (TGX Stain-FreeTM Gels, Bio-Rad) and transferred to nitrocellulose membranes (Trans-Blot Turbo® Transfer PackTM, 0,2 µm nitrocellulose, Bio-Rad) by electroblotting (Trans-Blot® TurboTM Transfer System, Bio-Rad). The blots were checked using the ChemiDoc MP Imaging System and the Image Lab software (Bio-Rad) to confirm uniform transfer of all samples. Blots were then blocked with Tris-buffered saline (Sigma-Aldrich) containing 0.1% Tween 20 solution (Sigma-Aldrich) and 5% non-fat dry milk (Cell Signaling Technology) or 5% BSA (Santa Cruz Biotechnology) for 1 h at room temperature (RT) and then incubated with primary and secondary antibodies.
Analysis of WB bands and normalization to total proteins were performed using the Image LabTM software v 5.2.1 (Bio-rad). WB were repeated in two biological replicates. A list of primary and secondary antibodies with their working conditions is provided in Supplementary Table s4.
RB1 and CCNE1 CNV analysis by ddPCR
Copy number alterations of RB1 and CCNE1 between PDR and PDS cells were assessed with RB1, CCNE1 and the reference gene EIF2C1 CNV Assays on a QX200TM droplet digital PCR (ddPCR) system (Bio-Rad). PCR reactions were prepared with approximately 10 ng of DNA and ddPCR supermix for probes (Bio-Rad). RB1 and CCNE1 probes (Bio-Rad) were stained in FAM while EIF2C1 probe (Bio-Rad) was stained in HEX. The ratios CCNE1/EIF2C1and RB1/EIF2C1 were calculated using Quantasoft software (Bio-Rad). At least one negative control with no DNA was included in each run. Experiments were performed three times in duplicate.
Statistical analysis
For proliferation assays, growth fold changes were computed as ratio, dividing growth rate of treated cells by their controls treated with DMSO. Mean value of growth fold change of all experiments ±standard error of the mean (SEM) was plotted. Two–way ANOVA with Sidak’s multiple comparisons test was performed with GraphPad Prism version 7.03 and P-values < 0.05 were considered significant.
For IC50, the signals were converted to proportions adjusting on time zero (T0) and on untreated DMEM control values and fitted using the 4-parameter logistic regression (R package nplr version 0.1–4). The proportion values, yp, were computed as: yp = (y − T0)/(DMSO − T0), where y are the observed values. The model performance was estimated by a weighted standard error, and a weighted goodness-of-fit. Once the model was fitted, the drug 50% inhibitory concentration (IC50) could be estimated from inhibition rates by inverting the function (given a y value = 0.5 survival rate). Mean value of IC50 of all experiments ±standard error (SE) was calculated. Student’s t tests were performed and P-values < 0.05 were considered significant. Mean value of senescence percentages of all experiments ±SE was plotted. Student’s t tests were performed and P-values < 0.05 were considered significant. Calculations and plots for IC50 and senescence were performed by R (version 3.3.1).
For GE data, the two-sided Wilcoxon–Whitney test was used to check for significant differences between two distributions. The distributions of PFS and OS were estimated using the Kaplan–Meier method and compared with the log-rank test.
Code availability statement
All codes used to analyze the data and generate the figures are available upon request.
Electronic supplementary material
Acknowledgements
This research was mainly funded by Associazione Italiana per la Ricerca sul Cancro (MFAG n. 14371 to I.M. and 18880 to L.M.) and Fondazione Sandro Pitigliani per la lotta contro i tumori ONLUS, and in part funded by the Breast Cancer Research Foundation BCRF 17–143 (to R.S and C.K.O.) and BCRF 17–037 (to A.D.L.), Stand Up to Cancer Translational Grant SU2C-AACR-DT0409 (to R.S. and C.K.O.), NIH Breast Cancer Specialized Programs of Research Excellence Grants P50CA058183 and P50CA186784 (to C.K.O. and R.S.), NIH Cancer Center Grant P30CA125123 (to C.K.O.), Susan G. Komen for the Cure Foundation Promise Grants PG12221410 (to C.K.O. and R.S.) and the Cancer Prevention & Research Institute of Texas CPRIT RP 140102 - Baylor College of Medicine Comprehensive Cancer Training Program (to C.D.A.). The authors would like to thank Pfizer for providing palbociclib; Dr Gloria Capaccioli for her technical support with cell cultures; Dr Livia Malorni from Institute of Food Science (ISA), National Council of Research (CNR), Avellino, Italy, for providing cell lines; Dr. Amelia McCartney for thoughtfully revising the manuscript.
Author contributions
L.M., I.M. and A.D.L. participated to the conception and design of the study. C.G., M.Bonechi., G.B., M.Benelli., C.B., D.R. and R.V. were involved in data acquisition. C.G., M.Bonechi., G.B., M.Benelli, C.B., D.R., I.M., L.M. and A.D.L. conducted data analysis and interpretation. R.S., C.K.O. and C.D.A. contributed to data interpretation and critical revision of the manuscript. C.B., MBenelli, D.R., G.B., M.Bonechi. and C.G. performed statistical and bioinformatical analysis. I.M., L.M., C.G. and M.Bonechi. contributed to draft the paper. I.M. and L.M. edited the manuscript and served as guarantors. C.G. and M.Bonechi. contributed equally as first author; I.M. and L.M. contributed equally as last author. All authors read and approved the final version of the manuscript.
Data availability
Cell line annotations, IC50 values and pre-processed expression data from Iorio et al.20 cancer cell line dataset were downloaded from http://www.cancerrxgene.org/download. Reverse Phase Protein Array (RPPA) data generated within the MD Anderson Cell Line Project (MCLP)21 were downloaded from http://tcpaportal.org/mclp/#/download (version 1.1). IC50 values of palbociclib in breast cancer cell lines from the study of Finn et al.19 were also considered. Expression and clinical information from METABRIC study22 were downloaded from http://www.cbioportal.org.45,46 NeoPalAna GE data23 were downloaded from Gene Expression Omnibus (GSE93204) using GEOquery R Package.47GE data of our PDS and PDR models are available from the authors upon request.
Competing interests
L.M. received research support from Pfizer and is a consultant/advisory board member for Pfizer, AstraZeneca, Novartis. A.D.L. is a consultant/advisory board member for Novartis, Pfizer, AstraZeneca and Lilly. R.S. received research funding from AstraZeneca, Gilead Sciences; is a consulting/advisory committee member for Macrogenics, and Eli Lilly. C.K.O. is a consultant/advisory board member for Genentech, AstraZeneca, Tolmar Pharma; received grants from Roche, Ventana, Puma; DMC Lilly; Stock: Genentech Non-financial competing interests: R.S.: Mechanisms of action of selective estrogen receptor modulators and down-regulators; Lectured at 2018 Int. Congress on the Future of Breast Cancer, Physicians’ Educational Resource, LLC; Member: AACR, ASCO, Endocrine Society; C.K.O.: Expert Witness for O’Melvery Meyers (patient case). All other authors declare no competing interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cristina Guarducci, Martina Bonechi, Luca Malorni and Ilenia Migliaccio.
These authors jointly supervised: Luca Malorni and Ilenia Migliaccio.
Contributor Information
Luca Malorni, Phone: +39 0574802520, Email: luca.malorni@uslcentro.toscana.it.
Ilenia Migliaccio, Phone: +39 0574801110, Email: ilenia.migliaccio@uslcentro.toscana.it.
Electronic supplementary material
Supplementary information accompanies the paper on the npj Breast Cancer website (10.1038/s41523-018-0092-4).
References
- 1.Finn RS, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 2.Finn RS, et al. Palbociclib and letrozole in advanced breast cancer. New Engl. J. Med. 2016;375:1925–1936. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 3.Turner NC, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. New Engl. J. Med. 2015 doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 4.Hortobagyi GN, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. New Engl. J. Med. 2016;375:1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 5.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 6.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 7.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 8.Jansen VM, et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017 doi: 10.1158/0008-5472.can-16-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrera-Abreu MT, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–2313. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang C, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36:2255–2264. doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rader J, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013;19:6173–6182. doi: 10.1158/1078-0432.CCR-13-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malorni L, et al. A gene expression signature of Retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget. 2016;7:68012–68022. doi: 10.18632/oncotarget.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders L, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–634. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wierstra I, Alves J. Transcription factor FOXM1c is repressed by RB and activated by cyclin D1/Cdk4. Biol. Chem. 2006;387:949–962. doi: 10.1515/BC.2006.119. [DOI] [PubMed] [Google Scholar]
- 15.Fry DW, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 16.Toogood PL. Cyclin-dependent kinase inhibitors for treating cancer. Med. Res. Rev. 2001;21:487–498. doi: 10.1002/med.1021. [DOI] [PubMed] [Google Scholar]
- 17.Dean JL, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012;11:2756–2761. doi: 10.4161/cc.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012;287:29075–29087. doi: 10.1074/jbc.M112.365494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finn RS, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iorio F, et al. A landscape of pharmacogenomic interactions. Cancer Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, et al. Characterization of human cancer cell lines by reverse-phase protein arrays. Cancer Cell. 2017;31:225–239. doi: 10.1016/j.ccell.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma CX, et al. NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor positive breast cancer. Clin. Cancer Res. 2017 doi: 10.1158/1078-0432.ccr-16-3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witkiewicz AK, Cox D, Knudsen ES. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes & Cancer. 2014;5:261–272. doi: 10.18632/genesandcancer.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolai BC, et al. HER2 signaling drives DNA anabolism and proliferation through SRC-3 phosphorylation and E2F1-regulated genes. Cancer Res. 2016;76:1463–1475. doi: 10.1158/0008-5472.CAN-15-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goel S, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255–269. doi: 10.1016/j.ccell.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gelbert LM, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs. 2014;32:825–837. doi: 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Migliaccio I, Di Leo A, Malorni L. Cyclin-dependent kinase 4/6 inhibitors in breast cancer therapy. Curr. Opin. Oncol. 2014;26:568–575. doi: 10.1097/CCO.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen ES, Witkiewicz AK. Defining the transcriptional and biological response to CDK4/6 inhibition in relation to ER+ /HER2- breast cancer. Oncotarget. 2016;7:69111–69123. doi: 10.18632/oncotarget.11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 32.Taylor-Harding B, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget. 2015;6:696–714. doi: 10.18632/oncotarget.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guarducci C, et al. Mechanisms of resistance to CDK4/6 inhibitors in breast cancer and potential biomarkers of response. Breast care (Basel, Switz.) 2017;12:304–308. doi: 10.1159/000484167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corona SP, et al. CDK4/6 inhibitors in HER2-positive breast cancer. Crit. Rev. Oncol./Hematol. 2017;112:208–214. doi: 10.1016/j.critrevonc.2017.02.022. [DOI] [PubMed] [Google Scholar]
- 35.Patel P, et al. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol. Cell. Biol. 2015;35:1506–1522. doi: 10.1128/MCB.01206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finn RS, et al. Efficacy and safety of palbociclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res. 2016;18:67. doi: 10.1186/s13058-016-0721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min A, et al. Cyclin E overexpression confers resistance to the CDK4/6 specific inhibitor palbociclib in gastric cancer cells. Cancer Lett. 2018;430:123–132. doi: 10.1016/j.canlet.2018.04.037. [DOI] [PubMed] [Google Scholar]
- 38.Konecny GE, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin. Cancer Res. 2011;17:1591–1602. doi: 10.1158/1078-0432.CCR-10-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caldon CE, et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol. Cancer Ther. 2012;11:1488–1499. doi: 10.1158/1535-7163.MCT-11-0963. [DOI] [PubMed] [Google Scholar]
- 40.Malorni L, et al. Palbociclib as single agent or in combination with the endocrine therapy received before disease progression for estrogen receptor-positive, HER2-negative metastatic breast cancer: TREnd trial. Ann. Oncol. 2018;29:1748–1754. doi: 10.1093/annonc/mdy214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doostan I, et al. Cytoplasmic cyclin E mediates resistance to aromatase inhibitors in breast cancer. Clin. Cancer Res. 2017;23:7288–7300. doi: 10.1158/1078-0432.CCR-17-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vijayaraghavan S, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017;8:15916. doi: 10.1038/ncomms15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayer E, DeMichele A, Gnant M, Barry W, Pfeiler G, Metzger O, Burstein H, Miller K, Rastogi P, Loibl S, Goulioti T, Zardavas D, Fesl C, Koehler M, Huang-Bartlett C, Huang X, Piccart M, Winer E, Wolff A. Abstract OT3-05-08: PALLAS: PALbociclib CoLlaborative Adjuvant Study: A randomized phase 3 trial of palbociclib with standard adjuvant endocrine therapy versus standard adjuvant endocrine therapy alone for HR+/HER2- early breast cancer. Cancer Research. 2018;78(4 Supplement):OT3-05-08-OT3-05-08. [Google Scholar]
- 44.Fu X, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad. Sci. USA. 2016;113:E6600. doi: 10.1073/pnas.1612835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinforma. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cell line annotations, IC50 values and pre-processed expression data from Iorio et al.20 cancer cell line dataset were downloaded from http://www.cancerrxgene.org/download. Reverse Phase Protein Array (RPPA) data generated within the MD Anderson Cell Line Project (MCLP)21 were downloaded from http://tcpaportal.org/mclp/#/download (version 1.1). IC50 values of palbociclib in breast cancer cell lines from the study of Finn et al.19 were also considered. Expression and clinical information from METABRIC study22 were downloaded from http://www.cbioportal.org.45,46 NeoPalAna GE data23 were downloaded from Gene Expression Omnibus (GSE93204) using GEOquery R Package.47GE data of our PDS and PDR models are available from the authors upon request.