

Abstract
Cleft palate is a common birth defect that frequently occurs in human congenital malformations caused by mutations in components of the Sonic Hedgehog (SHH) signaling cascade. Shh is expressed in dynamic, spatiotemporal domains within epithelial rugae and plays a key role in driving epithelial-mesenchymal interactions that are central to development of the secondary palate. However, the gene regulatory networks downstream of Hedgehog (Hh) signaling are incompletely characterized. Here, we show that ectopic Hh signaling in the palatal mesenchyme disrupts oral-nasal patterning of the neural crest cell–derived ectomesenchyme of the palatal shelves, leading to defective palatine bone formation and fully penetrant cleft palate. We show that a series of Fox transcription factors, including the novel direct target Foxl1, function downstream of Hh signaling in the secondary palate. Furthermore, we demonstrate that Wnt/bone morphogenetic protein (BMP) antagonists, in particular Sostdc1, are positively regulated by Hh signaling, concomitant with downregulation of key regulators of osteogenesis and BMP signaling effectors. Our data demonstrate that ectopic Hh-Smo signaling downregulates Wnt/BMP pathways, at least in part by upregulating Sostdc1, resulting in cleft palate and defective osteogenesis.
Keywords: craniofacial abnormalities, sonic hedgehog, neural crest, bone morphogenetic proteins, micrognathism, wnt signaling pathway
Introduction
Specification, growth, elevation, adherence, and fusion of the palatal shelves are essential mechanisms involved in secondary palate formation (Mossey et al. 2009; Dixon et al. 2011). Disruption of these processes leads to cleft palate, a common congenital disorder that affects ~1:2,500 live births (Mossey et al. 2009). Cleft palate causes major morbidity through problems with feeding, speech, hearing, and social adjustment. Affected children require multidisciplinary care into adulthood at considerable cost to health care systems worldwide. The frequent occurrence and major burden imposed by cleft palate highlight the need to dissect the mechanisms underlying palatal development. Although substantial progress has been made identifying the mutations underlying syndromic forms of cleft palate, the developmental role of many of the mutated genes is unknown (Dixon et al. 2011).
Development of the mouse secondary palate mirrors that of humans; as a result, the mouse is the major model organism for analyzing palatogenesis (Bush and Jiang 2012). In mice, palatal shelves initiating from the maxillary processes on embryonic day (E) 11 grow lateral to the tongue during E12/E13 before reorientating above the tongue during E14. Subsequently, the medial edge epithelia of apposed shelves adhere to form a midline epithelial seam, which degenerates to allow mesenchymal continuity across the palate by E15. In parallel, the oral and nasal palatal epithelia differentiate into stratified, squamous, keratinizing, and pseudostratified, ciliated epithelia, respectively. Similarly, the palatal mesenchyme differentiates into bony and muscular elements forming the hard and soft palate, respectively. Reflecting these different developmental fates, gene expression studies have revealed molecular heterogeneity along both the oral-nasal and anterior-posterior axes of the palatal shelves (Hilliard et al. 2005).
Mutations in components of the Hedgehog (Hh) signaling pathway underlie several human congenital malformations that are associated with cleft palate (Mansilla et al. 2006; Cohen 2010). Shh is expressed in epithelial rugae on the oral aspect of the palate, which initially define the anterior-posterior boundary of the palatal shelves and act as signaling centers that drive epithelial-mesenchymal interactions (Rice et al. 2004; Rice et al. 2006; Pantalacci et al. 2008; Lan and Jiang 2009). Thus, Shh signaling is linked spatiotemporally to both oral-nasal and anterior-posterior patterning of the secondary palate. Recent transgenic approaches to modulate Shh signaling within cranial neural crest cells (CNCCs) (Jeong et al. 2004), facial epithelia (Rice et al. 2004; Cobourne et al. 2009; Lan and Jiang 2009; Kurosaka et al. 2014), and secondary palate mesenchyme (Lan and Jiang 2009) have demonstrated the critical importance of this pathway to normal secondary palate development. Shh signaling regulates expression of the transcription factors Foxf1, Foxf2, and Osr2 and the growth factors BMP2, BMP4, and Fgf10 in palatal mesenchyme (Lan and Jiang 2009), but the Shh-induced pathways controlling epithelial-mesenchymal crosstalk remain incompletely characterized.
In this study, we investigated how ectopic Hh signaling affects normal secondary palate development. Using a gain-of-function mouse model to activate Smoothened (Smo) signaling in the palatal mesenchyme (Osr2-IresCre;Smo+/M2), we demonstrate that ectopic Hh-Smo signaling results in fully penetrant cleft palate and defective palatine bone formation. Using transcriptional profiling and expression analyses, we demonstrate Hh-Smo signaling is expanded and disrupts oral-nasal patterning by driving the expression of several transcriptional repressors and antagonists of Hh, Wnt, and bone morphogenetic protein (BMP) signaling pathways. We show Hh-Smo signaling upregulates several Fox transcription factors, including the direct transcriptional target Foxl1. Furthermore, we reveal the dual Wnt/BMP antagonist, Sostdc1, is expressed ectopically in the nasal mesenchyme, coincident with downregulation of nasally expressed master regulators of osteogenesis (Sox9, Runx2), bone-related extracellular matrix proteoglycans (Dcn, Lum), and BMP signaling effectors (pSmad 1/5/9). Our data suggest Hh-Smo signaling negatively regulates Wnt/BMP pathways by upregulating antagonists, resulting in cleft palate and defective osteogenesis.
Materials and Methods
Detailed methods are in the Appendix. Microarray data have been deposited in Array Express with the accession E-MTAB-5518.
Results
Osr2-IresCre;Smo+/M2 Mice Have a Complete Cleft of the Secondary Palate
To investigate the gene regulatory networks downstream of the Hh-Smo signaling cascade, we used Cre/loxP to constitutively activate Smo (SmoM2) (Xie et al. 1998) in the palatal mesenchyme in vivo (Osr2-IresCre) (Appendix Fig. 1). Osr2-IresCre;Smo+/M2 embryos, hereafter referred to as mutants, displayed a wide cleft of the secondary palate. Histological analysis of E13.5 mutant mice revealed smaller, abnormally shaped palatal shelves compared with their wild-type littermates, which was most pronounced in the anterior and mid-regions, indicating reduced palatal outgrowth (Fig. 1A, B). By E14.5, wild-type palatal shelves had reorientated above the tongue while those of mutant littermates had failed to elevate and were rounded in appearance, and tooth germ development was arrested at the bud stage (Fig. 1C, D; arrowheads). These anomalies were more pronounced by E15.5, when mutant embryos displayed a fully penetrant complete cleft of the secondary palate (n = 15) compared to the fused palate of wild-type littermates (Fig. 1E, F). To investigate the cause of smaller palatal shelves, we performed cell proliferation analysis using bromodeoxyuridine (BrdU) incorporation at E13.5 and revealed a significant proliferation defect in anterior and mid-palatal regions while the posterior palate was unaffected (Appendix Fig. 2).
Figure 1.
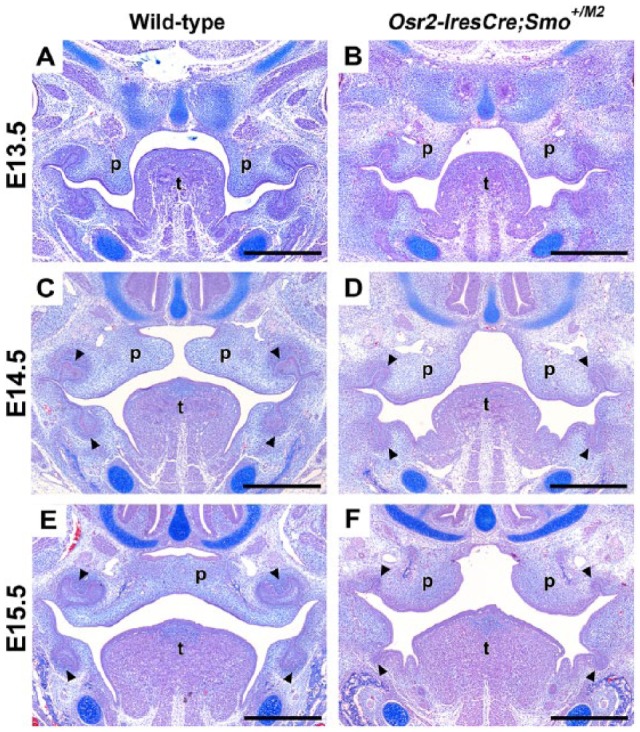
Osr2-IresCre;Smo+/M2 mice have fully penetrant cleft palate. (A–F) Histological analyses indicate mutant embryos have severe defects in secondary palate development. Mutant palatal shelves are smaller in size from E13.5, with marked reduction in vertical growth of the palatal shelves (A, B). Although mutant palatal shelves appear to reorientate by E14.5, they are abnormally shaped and far apart (C, D), resulting in a wide cleft of the secondary palate by E15.5 (E, F). Tooth germ development is also arrested at the bud stage in mutant embryos (C–F). ps, palatal shelf; t, tongue. Scale bars: A–H = 300 µm.
Osr2-IresCre;Smo+/M2 Mice Have Multiple Skeletal Defects
We analyzed alcian blue/alizarin red–stained skeletal preparations, which revealed defects in the viscerocranium of mutant embryos (Fig. 2A; E17, n = 3). The anterior midline structures of the premaxilla and posterior regions of the maxilla were absent in mutant embryos, along with the associated palatine processes, revealing the presphenoid, which is normally obscured (Fig. 2B). Mutant mandibles were shorter and showed anterior ossification defects with rudimentary condyloid processes and no defined coronoid processes (Fig. 2C).
Figure 2.

Osr2-IresCre;Smo+/M2 mice have skeletal abnormalities and lack the palatine bones. (A–C) Whole-mount skeletal preparations stained with alcian blue and alizarin red reveal multiple defects in the mutant skeleton at E17.5. (A) Skeletal abnormalities include truncated fore- and hind-limbs with cartilage and ossification defects. (B) Detailed analysis of the craniofacial skeleton reveals multiple abnormalities of the viscerocranium. Mutant embryos demonstrate reduced ossification of the maxilla (mx) and premaxilla along with absence of the palatine and palatine processes of both the premaxilla and maxilla. (C) Mutant embryos also have a shorter mandible (green arrow, wild-type; red arrow, mutant) with rudimentary angular and condyloid processes and no defined coronoid process. Mutant mandibles also show reduced ossification anteriorly. agp, angular process; als, alisphenoid; bo, basioccipital; bs, basosphenoid; cdp, condyloid process; crp, coronoid processes; eo, exoccipital; fr, frontal bone; lo, lamina obturans; md, mandible; mx, maxilla; p, palatine bone; pmx, premaxilla; ppmx, palatine process of the maxilla; ppp, palatine process of the palatine; pppmx, palatine process of the premaxilla; ps, presphenoid; ptg, pterygoid; sq, squamosal; sy, symphysis.
Transcriptional Profiling Reveals Negative Regulators of Hh, Wnt, and BMP Pathways are Upregulated in Response to Persistent Hh Signaling
To gain insight into the gene regulatory networks affected by increased Hh-Smo signaling, we compared the transcriptomes of palatal shelves dissected from E13.5 wild-type and mutant embryos. Microarray analysis identified 580 differentially expressed genes (P < 0.05) (E-MTAB-5518; Appendix Table 1; Appendix Fig. 3A), of which 327 genes were upregulated in response to increased Hh-Smo signaling. These included known direct targets (Gli1, Ptch1, Ptch2, Hhip) and several members of the Fox family of transcription factors (Foxd1, Foxd2, Foxf1, Foxf2, and Foxl1). Overrepresentation enrichment analysis indicated significant enrichment of gene ontology terms, including mesenchyme development, receptor serine/threonine kinase signaling, and cell fate commitment (Appendix Fig. 3B; Appendix Table 4). Further annotation of these gene groups revealed upregulation of several transcriptional repressors and antagonists of Hh (e.g., Ptch1, Hhip, Cdon), Wnt, and BMP (e.g., Sostdc1, Twsg1) signaling pathways.
Hh-Smo Direct and Downstream Targets Are Upregulated throughout the Palatal Mesenchyme
Subsequently, we investigated the expression of known Hh direct targets (Gli1, Ptch1) and candidate targets from the microarray analysis using a combination of whole-mount and section in situ hybridization. Gli1 and Ptch1 are normally expressed in rugae epithelium and the underlying mesenchyme on the oral side of the palate. However, their expression was expanded into the tooth germ and nasal mesenchyme of mutant embryos (Fig. 3A–H) while reduced expression was noted in epithelial rugae (Fig. 3C, D, G, H). Ectopic expression of Gli1 and Ptch1 was also observed in the mandibular mesenchyme (Fig. 3D, H; arrows), correlating with Osr2-IresCre expression (Appendix Fig. 1C).
Figure 3.

Hedgehog direct and downstream targets are upregulated in Osr2-IresCre;Smo+/M2 embryos. Whole-mount (A, B, E, F, I, J, M, N, Q, R) and in situ (C, D, G, H, K, L, O, P, S, T) hybridization sections for direct targets Gli1 (A–D) and Ptch1 (E–H) demonstrate they are associated with rugae on the oral side of E13.5 wild-type palates (A, C, E, G). Gli1 and Ptch1 are upregulated and expanded from the oral to nasal mesenchyme of mutant embryos (B, D, F, H). Foxf2 is expressed in the oral mesenchyme along the anterior-posterior length of the palate while Foxl1 is strongly expressed in the mesenchyme underlying rugae (I, K, M, O). Upregulation and expansion of both Fox factors are seen in mutant palates (J, N, L, P). Expression of Sostdc1 is observed in interrugae domains of the epithelium and in the posterior palate. In contrast, Sostdc1 is ectopically expressed in the nasal mesenchyme and upregulated in the posterior palate of mutants (Q–T). Ectopic expression of all targets is seen in the mandibular mesenchyme of mutant embryos (D, H, L, P, T; arrows). (M) Real-time quantitative polymerase chain reaction analysis of E13.5 palatal shelves confirms upregulation of all targets (*P < 0.05, **P < 0.01; Mann-Whitney U test, n = 5). md, mandible; ps, palatal shelf; t, tongue. Scale bars: C, D, G, H, K, L, O, P, S, T = 100 µm.
Members of the Fox transcription factor family, including Foxf1 and Foxf2, have been implicated downstream of Hh signaling during facial and secondary palate development (Lan and Jiang 2009; Nik et al. 2016; Xu et al. 2016). Foxf2 was expressed throughout the anterior-posterior length of the oral palatal mesenchyme in wild-type embryos with increased expression underlying rugae (Fig. 3I, K). In contrast, Foxf2 was markedly upregulated in the oral and nasal mesenchyme of mutant embryos (Fig. 3J, L). Transcriptional profiling identified Foxl1 as the highest upregulated Fox factor (Appendix Table 1; Appendix Table 2) and has not been implicated in palate development. Foxl1 was expressed in the oral palatal mesenchyme of wild-type embryos associated with rugae. However, in mutant embryos, Foxl1 was markedly upregulated and expanded from the oral to nasal palatal mesenchyme in anterior and posterior regions of the palate (Fig. 3M–P). Similar to known Hh direct targets, ectopic expression of both Fox factors was observed in the lingual aspect of the mandibular mesenchyme (Fig. 3P; arrow).
Elevated Hh-Smo signaling also upregulated several Wnt/BMP antagonists. We investigated the expression of Sostdc1, a dual Wnt/BMP secreted antagonist with reported roles in the spatial patterning of teeth and rugae (Ahn et al. 2010; Cho et al. 2011; Lee et al. 2011). Sostdc1 was expressed in interrugae domains in the anterior palatal epithelium and the posterior palate (Fig. 3Q, S) (Welsh and O’Brien 2009; Lee et al. 2011). In mutant embryos, Sostdc1 was expressed ectopically in the lingual and nasal mesenchyme of the palate while expression in the posterior palate and mandible was also upregulated (Fig. 3R). Real-time quantitative polymerase chain reaction (qPCR) confirmed significant upregulation of all these genes in the palatal shelves of mutant embryos (Fig. 3U).
Sequential Rugae Interposition Is Blocked and Shh Is Downregulated in Osr2-IresCre;Smo+/M2 Embryos
Whole-mount analysis of Shh targets indicated reduced numbers of rugae in mutant embryos at E13.5 (Fig. 3; arrowheads). Subsequently, Shh expression from E13.5 to E15.5 showed the sequential addition of up to 8 rugae in wild-type embryos (Appendix Fig. 4A–C), while mutants developed only 3 rugae (Appendix Fig. 4D–F). Furthermore, at E15.5 Shh expression was secondarily downregulated in mutant rugae, while expression persisted in wild-type embryos (Appendix Fig. 4D, H).
Extracellular Matrix Proteoglycans in the Nasal Palatal Mesenchyme Are Downregulated
Transcriptional profiling indicated that the extracellular matrix proteins decorin (Dcn), lumican (Lum), and keratocan (Kera) were amongst the most significantly downregulated genes (Appendix Table 3; Appendix Fig. 2A). These matricellular proteins are members of the small leucine-rich proteoglycan (SLRP) family with multiple roles in osteogenesis (Raouf et al. 2002; Waddington et al. 2003). Expression analyses in wild-type embryos showed Dcn and Lum were restricted to the nasal mesenchyme of the palate. In agreement with the microarray analysis, expression of both genes was downregulated in mutant palatal mesenchyme while expression elsewhere was unaffected (Fig. 4A–H). Similarly, Dlx5, a factor crucial for osteoblast differentiation (Acampora et al. 1999), is expressed in the anterior nasal mesenchyme of wild-type palatal shelves (Fig. 4I, K) but was downregulated in the palatal mesenchyme of mutant embryos (Fig. 4J, L). Real-time qPCR confirmed the reduction of Dcn and Lum, but Dlx5 was not significant (Fig. 4M).
Figure 4.

Extracellular matrix proteoglycans are downregulated in Osr2-IresCre;Smo+/M2 embryos. Whole-mount (A, B, E, F, I, J) and in situ (C, D, G, H, K, L) hybridization sections for the extracellular matrix genes Dcn (A–D), Lum (E–H), and the homeobox transcription factor Dlx5 (I–L) demonstrate Dcn and Lum are expressed in the nasal mesenchyme throughout the anterior-posterior length of the palatal shelves in E13.5 wild-type embryos, while Dlx5 is also expressed in the nasal mesenchyme in the anterior region of the palatal shelves (A, C, E, G, I, K). Mutant embryos show loss of expression of these genes in the palatal shelves (B, D, F, H, J, L). (M) Real-time quantitative polymerase chain reaction data confirm significantly reduced levels of messenger RNA for Dcn and Lum (*P < 0.05, Mann-Whitney U test, n = 4). md, mandible; ps, palatal shelf; t, tongue. Scale bars: C, D, G, H, K, L = 100 µm.
Master Regulators of Osteogenesis Are Downregulated
Subsequently, we analyzed the expression of key transcriptional effectors of osteogenesis, Sox9 and Runx2, which are regulated by Wnt/Bmp crosstalk (Gaur et al. 2005; Pan et al. 2008). Immunohistochemical analyses of Sox9 and Runx2 at E13.5 revealed both proteins were expressed in overlapping domains within the nasal palatal mesenchyme (Fig. 5A, C, E). Sox9 was also highly expressed in developing craniofacial cartilages (Fig. 5A, E). The expression of both proteins was dramatically downregulated in the nasal mesenchyme while expression elsewhere was unaffected (Fig. 5B, D, F). Similarly, at E14.5, Sox9 and Runx2 were expressed in overlapping domains in the reorientated nasal mesenchyme directly beneath the midline epithelial seam (Fig. 5G, I, K) while the expression of both proteins was markedly downregulated in the nasal mesenchyme of mutant embryos (Fig. 5H, J, L). Sox9 and Runx2 are critical factors in orchestrating multiple steps of intramembranous ossification, and their expression in wild-type palatal mesenchyme defines the nasal mesenchymal contribution to palatal growth, fusion, and bone formation (Fig. 5A, C, E, G, I, K). Since Sox9 and Runx2 were downregulated and Sostdc1 is a BMP antagonist (Wu et al. 2008), we analyzed whether BMP signaling was affected in mutant embryos. Immunofluorescence for pSmad 1/5/9 revealed the effectors of BMP signaling were also downregulated in the nasal mesenchyme at E13.5 and the future palatine bone regions at E14.5 (Fig. 5M–P).
Figure 5.

Master regulators of osteogenesis are downregulated in the nasal palatal mesenchyme of Osr2-IresCre;Smo+/M2 embryos. (A–F) Immunostaining for Sox9 (A, B; red) and Runx2 (C, D; green) in adjacent sections at E13.5 indicates both markers are expressed in overlapping domains in the nasal mesenchyme of wild-type palatal shelves (A, C, E; arrows). Sox9 is also highly expressed in the developing nasal cartilage (arrowheads) and extends into the mesenchyme beneath the medial edge epithelia (A, E) while Runx2 is also expressed in the odontogenic mesenchyme (C, E). Conversely, in mutant palatal shelves, expression of both markers is absent from the nasal palatal mesenchyme while expression in the nasal cartilage (arrowheads) and odontogenic mesenchyme is unaffected (B, D, F). (G–L) At E14.5, Sox9 is expressed in the mesenchyme beneath the midline epithelial seam (G, K), while Runx2 has a characteristic expression pattern in the future bone condensations of the wild-type palate (I, K). Both markers are excluded from the mesenchyme along the oral aspect of the horizontal palate (G, I, K). In contrast, expression of both markers is absent from the palatal mesenchyme of the mutant palate while expression in the nasal cartilages (arrowheads) and associated structures is unaffected (H, J, L). pSmad 1/5/9 is expressed in the nasal mesenchyme at E13.5 and future palatine bone mesenchyme at E14.5 in wild-type embryos (M, N; red) but is absent from these regions in mutant embryos (O, P). Autofluorescence of red blood cells is identified by triple immunofluorescent images (M–P; yellow). md, mandible; ps, palate; t, tongue. Scale bars: A–P = 100 µm.
Collectively, these results demonstrate that ectopic Hh-Smo signaling in the nasal mesenchyme downregulates BMP signaling, concomitant with upregulation of Wnt/BMP antagonist Sostdc1, resulting in defective osteogenesis and cleft palate.
Foxl1 Is a Direct Target of Gli1 in the Secondary Palate
To determine if Sostdc1 and Foxl1 are direct targets of Hh signaling, we analyzed the promoters (–1 kb) of these genes for candidate Gli binding sites. No Gli sites were found near Sostdc1. However, we identified 2 highly conserved candidate binding sites in the Foxl1 promoter (−237 and −371 from the TSS), with 1 mismatch from the Gli consensus (Appendix Fig. 5A), in regions of accessible chromatin (Appendix Fig. 5B). Gli1 chromatin immunoprecipitation (ChIP)–qPCR analyses of E13.5 palatal shelves demonstrated significant enrichment of Gli1 on the Foxl1 promoter and also known direct targets, Ptch1 and Gli1 (Appendix Fig. 5C). This is the first report to demonstrate Foxl1 is a direct target of Gli1 in vivo.
Discussion
Spatiotemporal Hh-Smo signaling defines a gene regulatory network that patterns the oral axis of the secondary palate. Recent research has established that epithelial Shh expressed within rugae (Pantalacci et al. 2008) signals to the underlying mesenchyme to activate Smo (Rice et al. 2004; Lan and Jiang 2009) and direct gene expression and cell fate through Gli transcription factors. Rice and colleagues (2004) showed that Shh signaling is crucial for palate development as disruption of Fgf10-Fgfr2b-Shh mesenchymal-epithelial signaling results in cleft palate, while targeted loss of the key Shh transducer, Smo, in palatal mesenchyme also results in cleft palate (Lan and Jiang 2009). Conversely, transgenic expression of Shh in all epithelial tissues results in a severe craniofacial phenotype with cleft palate (Cobourne et al. 2009). However, the molecular mechanisms downstream of Hh-Smo signaling within the secondary palate remain poorly characterized.
Mutant mouse studies to uncover the molecular mechanisms driving secondary palate development are often confounded by early embryonic lethality or gross craniofacial abnormalities. However, the recent generation of Osr2-IresCre mice (Lan et al. 2007) allows tissue-specific manipulation of genes involved in palate development. While the use of Cre-based mouse models to interrogate gene function is an aggressive tool that can disrupt normal physiological gene expression, such an approach has been used successfully to uncover targets of Hh signaling in the early embryonic face, limb, palate, and brain (Jeong et al. 2004; Vokes et al. 2008; Heine and Rowitch 2009; Lan and Jiang 2009). In this study, we generated a palate-specific Smo gain-of-function mouse model by targeting constitutively active Smo (Xie et al. 1998) to the palatal mesenchyme. We found that mutant embryos were characterized by a fully penetrant wide cleft of the secondary palate with various skeletal defects. Taken together, this clearly illustrates that a precise level of Hh-Smo signaling is required for normal palate development.
Patterning of the secondary palate is complex, with molecular heterogeneity along both the oral-nasal and anterior-posterior axes (Hilliard et al. 2005). We and others (Rice et al. 2006; Han et al. 2009; Lan and Jiang 2009) have shown that effectors of Hh-Smo signaling are expressed on the oral side of the palate while the nasal side is reportedly characterized by transforming growth factor β (TGFβ)/BMP mediators (Iwata et al. 2011; Parada and Chai 2012). We demonstrated that elevated Hh-Smo signaling resulted in upregulation and expansion of direct and downstream targets of the Hh pathway within the palatal mesenchyme. Using transcriptional profiling and gene ontology analyses, we identified and characterized several upregulated transcriptional repressors and Wnt/BMP antagonists, particularly Foxf2, Foxl1, and Sostdc1.
Members of the Fox transcription factor family (Foxd1, Foxd2, Foxc2, Foxf1, and Foxf2) are dependent on Smo signaling in the early developing face, leading to the suggestion that Fox factors are the mediators of Hh-Smo signaling (Jeong et al. 2004). Indeed, loss of Smo in the palatal mesenchyme also results in downregulation of Foxf1 and Foxf2 (Lan and Jiang 2009). Our data demonstrate that Foxf1, Foxf2, and the novel target Foxl1 are all robustly upregulated in response to increased Hh-Smo signaling within the palate, confirming these as Smo-dependent targets. Mutations in FOXF2 have been associated with cleft palate (Jochumsen et al. 2008) while mice deficient in either Foxf1 or Foxf2 are born with cleft palate (Lan and Jiang 2009; Nik et al. 2016; Xu et al. 2016). We identified and characterized the expression of the novel Foxl1 in the secondary palate and demonstrated that Foxl1 is a direct target of Gli1. Although Foxl1–/– mice are viable, most mutant mice die before weaning, attributed to impaired development of the gastrointestinal tract (Kaestner et al. 1997). However, secondary palate formation has not been investigated in these mice and may be a contributing factor. Alternatively, other Fox family members may compensate for the loss of Foxl1. In support of this hypothesis, Foxf1 and Foxl1 have similar functions in the developing stomach and intestine (Madison et al. 2009), while partial functional redundancy between Foxf1 and Foxf2 has been demonstrated in the secondary heart field (Hoffmann et al. 2014) and palate (Xu et al. 2016). The function of Foxl1 during palate development remains unknown, but studies in other tissues have revealed Foxl1 (in addition to Foxf1 and Foxf2) can indirectly affect epithelial proliferation via modulation of Wnt–β-catenin signaling (Kaestner et al. 1997; Perreault et al. 2001; Madison et al. 2009). Therefore, we postulate that Foxl1 may play a role in coordinating palatal growth via epithelial-mesenchymal feedback.
Expansion of Hh-Smo signaling into the nasal mesenchyme resulted in a gain of oral gene expression concomitant with a loss of nasal gene expression, resulting in impaired osteogenesis of the palatine bones. We showed downregulation of extracellular matrix proteoglycans in the palatal mesenchyme, which play multiple roles in osteogenesis (Raouf et al. 2002; Waddington et al. 2003). Furthermore, we confirmed Sox9 and Runx2 were downregulated along with a failure of BMP signaling in the nasal mesenchyme of the palate, coincident with ectopic expression of the dual Wnt/BMP antagonist, Sostdc1. Ectopic expression of Sostdc1 in CNCCs directly antagonizes BMP-induced osteogenesis, resulting in cleft palate (Wu et al. 2008). Taken together, our data suggest that ectopic Sostdc1 driven by expanded Hh-Smo signaling, at least in part, underlies the failure of BMP signaling and osteogenesis defects in mutant embryos.
During tooth and lip development, Shh negatively regulates Wnt signaling (Ahn et al. 2010; Kurosaka et al. 2014), while Sostdc1 knockout mice have elevated Wnt signaling and supernumerary teeth (Zhang et al. 2009; Ahn et al. 2010; Cho et al. 2011). Our transcriptome data identified several upregulated Wnt antagonists, suggesting that Wnt signaling may be affected by ectopic Hh-Smo signaling. Furthermore, we noted that epithelial Gli1 and Ptch1 were reduced at E13.5 and Shh expression was secondarily downregulated in rugae at E15.5, which we suggest is due to epithelial-mesenchymal negative feedback mechanisms. Negative feedback via Sostdc1 has been suggested for tooth and rugae patterning (Ahn et al. 2010; Lee et al. 2011). Thus, we speculate a Wnt-Hh-Sostdc1 negative feedback loop may also be present during secondary palate development. However, it is likely that other Wnt/BMP antagonists act in concert with Sostdc1 to reinforce Wnt and BMP antagonism. In support of this hypothesis, pharmacological inhibition or genetic inactivation of Wnt antagonists rescued cleft palate in Pax9–/– embryos (Jia et al. 2017; Li et al. 2017). Further work is needed to elucidate if Sostdc1 and other Wnt/BMP antagonists are direct targets of Hh-Smo signaling during secondary palate development.
In this study, we identify Foxl1 as a direct target of Gli1 in vivo. To delineate all Hh-Smo direct from downstream targets on a genome-wide scale, ChIP-seq data sets for the Gli transcription factors (Gli1, Gli2, and Gli3) on secondary palate tissue would enable the direct regulatory networks to be uncovered and will be addressed in future studies.
Author Contributions
M.J. Dixon, contributed to conception, design, and data interpretation, drafted and critically revised the manuscript; N.L. Hammond, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; K.J. Brookes, contributed to design and data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034518785336 for Ectopic Hedgehog Signaling Causes Cleft Palate and Defective Osteogenesis by N.L. Hammond, K.J. Brookes and M.J. Dixon in Journal of Dental Research
Acknowledgments
We thank Leo Zeef and Andy Hayes of the Bioinformatics and Genomic Technologies Core Facilities at the University of Manchester for providing support with the microarray analysis.
Footnotes
A supplemental appendix to this article is available online.
This work was supported by the Medical Research Council (grant G1001601 to M.J.D.) and The Wellcome Trust Institutional Strategic Support Fund (grant 105610 to M.J.D.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
ORCID iD: N.L. Hammond  https://orcid.org/0000-0003-3086-5243
https://orcid.org/0000-0003-3086-5243
References
- Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G. 1999. Craniofacial, vestibular and bone defects in mice lacking the distal-less-related gene Dlx5. Development. 126(17):3795–3809. [DOI] [PubMed] [Google Scholar]
- Ahn Y, Sanderson BW, Klein OD, Krumlauf R. 2010. Inhibition of Wnt signaling by wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development. 137(19):3221–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. 2012. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139(2):231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kwak S, Woolley TE, Lee MJ, Kim EJ, Baker RE, Kim HJ, Shin JS, Tickle C, Maini PK, et al. 2011. Interactions between Shh, Sostdc1 and Wnt signaling and a new feedback loop for spatial patterning of the teeth. Development. 138(9):1807–1816. [DOI] [PubMed] [Google Scholar]
- Cobourne MT, Xavier GM, Depew M, Hagan L, Sealby J, Webster Z, Sharpe PT. 2009. Sonic hedgehog signaling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Dev Biol. 331(1):38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MM., Jr. 2010. Hedgehog signaling update. Am J Med Genet A. 152A(8):1875–1914. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Rossant J. 1992. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 116(2):357–368. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. 2011. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 12(3):167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS, et al. 2005. Canonical Wnt signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 280(39):33132–33140. [DOI] [PubMed] [Google Scholar]
- Han J, Mayo J, Xu X, Li J, Bringas P, Jr, Maas RL, Rubenstein JL, Chai Y. 2009. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development. 136(24):4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine VM, Rowitch DH. 2009. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J Clin Invest. 119(2):267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP. 2005. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J Anat. 207(5):655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AD, Yang XH, Burnicka-Turek O, Bosman JD, Ren X, Steimle JD, Vokes SA, McMahon AP, Kalinichenko VV, Moskowitz IP. 2014. Foxf genes integrate Tbx5 and hedgehog pathways in the second heart field for cardiac septation. PLoS Genet. 10(10):e1004604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Tung L, Urata M, Hacia JG, Pelikan R, Suzuki A, Ramenzoni L, Chaudhry O, Parada C, Sanchez-Lara PA, et al. 2011. Fibroblast growth factor 9 (FGF9)–pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor beta (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. J Biol Chem. 287(4):2353–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. 2004. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18(8):937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Zhou J, Fanelli C, Wee Y, Bonds J, Schneider P, Mues G, D’Souza RN. 2017. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development. 144(20):3819–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochumsen U, Werner R, Miura N, Richter-Unruh A, Hiort O, Holterhus PM. 2008. Mutation analysis of FOXF2 in patients with disorders of sex development (DSD) in combination with cleft palate. Sex Dev. 2(6):302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Silberg DG, Traber PG, Schutz G. 1997. The mesenchymal winged helix transcription factor Fkh6 is required for the control of gastrointestinal proliferation and differentiation. Genes Dev. 11(12):1583–1595. [DOI] [PubMed] [Google Scholar]
- Kurosaka H, Iulianella A, Williams T, Trainor PA. 2014. Disrupting hedgehog and Wnt signaling interactions promotes cleft lip pathogenesis. J Clin Invest. 124(4):1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Jiang R. 2009. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development. 136(8):1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Wang Q, Ovitt CE, Jiang R. 2007. A unique mouse strain expressing Cre recombinase for tissue-specific analysis of gene function in palate and kidney development. Genesis. 45(10):618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Miyazawa S, Shin JO, Kwon HJ, Kang DW, Choi BJ, Lee JH, Kondo S, Cho SW, et al. 2011. Shh signaling is essential for rugae morphogenesis in mice. Histochem Cell Biol. 136(6):663–675. [DOI] [PubMed] [Google Scholar]
- Li C, Lan Y, Krumlauf R, Jiang R. 2017. Modulating Wnt signaling rescues palate morphogenesis in Pax9 mutant mice. J Dent Res. 96(11):1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, McKenna LB, Dolson D, Epstein DJ, Kaestner KH. 2009. Foxf1 and Foxl1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J Biol Chem. 284(9):5936–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla MA, Cooper ME, Goldstein T, Castilla EE, Lopez Camelo JS, Marazita ML, Murray JC. 2006. Contributions of PTCH gene variants to isolated cleft lip and palate. Cleft Palate Craniofac J. 43(1):21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. 2009. Cleft lip and palate. Lancet. 374(9703):1773–1785. [DOI] [PubMed] [Google Scholar]
- Nik AM, Johansson JA, Ghiami M, Reyahi A, Carlsson P. 2016. Foxf2 is required for secondary palate development and Tgfβ signaling in palatal shelf mesenchyme. Dev Biol. 415(1):14–23. [DOI] [PubMed] [Google Scholar]
- Pan Q, Yu Y, Chen Q, Li C, Wu H, Wan Y, Ma J, Sun F. 2008. Sox9, a key transcription factor of bone morphogenetic protein-2-induced chondrogenesis, is activated through BMP pathway and a CCAAT box in the proximal promoter. J Cell Physiol. 217(1):228–241. [DOI] [PubMed] [Google Scholar]
- Pantalacci S, Prochazka J, Martin A, Rothova M, Lambert A, Bernard L, Charles C, Viriot L, Peterkova R, Laudet V. 2008. Patterning of palatal rugae through sequential addition reveals an anterior/posterior boundary in palatal development. BMC Dev Biol. 8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada C, Chai Y. 2012. Roles of BMP signaling pathway in lip and palate development. Front Oral Biol. 16:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault N, Katz JP, Sackett SD, Kaestner KH. 2001. Foxl1 controls the Wnt/beta-catenin pathway by modulating the expression of proteoglycans in the gut. J Biol Chem. 276(46):43328–43333. [DOI] [PubMed] [Google Scholar]
- Raouf A, Ganss B, McMahon C, Vary C, Roughley PJ, Seth A. 2002. Lumican is a major proteoglycan component of the bone matrix. Matrix Biol. 21(4):361–367. [DOI] [PubMed] [Google Scholar]
- Rice R, Connor E, Rice DP. 2006. Expression patterns of hedgehog signalling pathway members during mouse palate development. Gene Expr Patterns. 6(2):206–212. [DOI] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. 2004. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 113(12):1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Wong WH, McMahon AP. 2008. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 22(19):2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington RJ, Roberts HC, Sugars RV, Schonherr E. 2003. Differential roles for small leucine-rich proteoglycans in bone formation. Eur Cell Mater. 6:12–21; discussion 21. [DOI] [PubMed] [Google Scholar]
- Wallin J, Wilting J, Koseki H, Fritsch R, Christ B, Balling R. 1994. The role of Pax-1 in axial skeleton development. Development. 120(5):1109–1121. [DOI] [PubMed] [Google Scholar]
- Welsh IC, O’Brien TP. 2009. Signaling integration in the rugae growth zone directs sequential SHH signaling center formation during the rostral outgrowth of the palate. Dev Biol. 336(1):53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Li J, Engleka KA, Zhou B, Lu MM, Plotkin JB, Epstein JA. 2008. Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice. J Clin Invest. 118(6):2076–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al. 1998. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 391(6662):90–92. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu H, Lan Y, Aronow BJ, Kalinichenko VV, Jiang R. 2016. A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet. 12(1):e1005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Supowit SC, Hou X, Simmons DJ. 1999. Transforming growth factor-beta2 mRNA level in unloaded bone analyzed by quantitative in situ hybridization. Calcif Tissue Int. 64(6):522–526. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lan Y, Chai Y, Jiang R. 2009. Antagonistic actions of Msx1 and Osr2 pattern mammalian teeth into a single row. Science. 323(5918):1232–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034518785336 for Ectopic Hedgehog Signaling Causes Cleft Palate and Defective Osteogenesis by N.L. Hammond, K.J. Brookes and M.J. Dixon in Journal of Dental Research