

Abstract
Spinal muscular atrophy (SMA) is a rare, inherited neuromuscular disease caused by deletion and/or mutation of the Survival of Motor Neuron 1 (SMN1) gene. A second gene, SMN2, produces low levels of functional SMN protein that are insufficient to fully compensate for the lack of SMN1. Risdiplam (RG7916; RO7034067) is an orally administered, small‐molecule SMN2 pre‐mRNA splicing modifier that distributes into the central nervous system (CNS) and peripheral tissues. To further explore risdiplam distribution, we assessed in vitro characteristics and in vivo drug levels and effect of risdiplam on SMN protein expression in different tissues in animal models. Total drug levels were similar in plasma, muscle, and brain of mice (n = 90), rats (n = 148), and monkeys (n = 24). As expected mechanistically based on its high passive permeability and not being a human multidrug resistance protein 1 substrate, risdiplam CSF levels reflected free compound concentration in plasma in monkeys. Tissue distribution remained unchanged when monkeys received risdiplam once daily for 39 weeks. A parallel dose‐dependent increase in SMN protein levels was seen in CNS and peripheral tissues in two SMA mouse models dosed with risdiplam. These in vitro and in vivo preclinical data strongly suggest that functional SMN protein increases seen in patients’ blood following risdiplam treatment should reflect similar increases in functional SMN protein in the CNS, muscle, and other peripheral tissues.
Keywords: central nervous system (CNS), muscle, Risdiplam, SMN protein, spinal muscular atrophy (SMA), tissue distribution
Abbreviations
- BCRP
breast cancer resistance protein
- BLQ
below the limit of quantification
- BN/Crl
Brown Norway
- CK14
CytoKeratin 14
- CNS
central nervous system
- CSF
cerebrospinal fluid
- EDTA
ethylene diamine tetraacetic acid
- ER
efflux ratio
- FVB
Friend Leukemia virus B
- GI
gastro‐intestinal
- HESI
heated electrospray ionization
- HET
HETerozygous
- HPLC
high‐performance liquid chromatography
- HTRF
homogeneous time resolved fluorescence
- IP
intraperitoneal
- IV
intravenous
- K3EDTA
tri‐potassium ethylenediaminetetraacetic acid
- Ko143
(3S,6S,12aS)‐1,2,3,4,6,7,12,12a‐Octahydro‐9‐methoxy‐6‐(2‐methylpropyl)‐1,4‐dioxopyrazino[19,29:1,6]pyrido[3,4‐b]indole‐3‐propanoic acid 1,1‐dimethylethyl ester
- KO
knock out
- Kp,uu
CSF‐to‐unbound plasma partition coefficient
- LLC‐PK1
Lewis‐Lung Cancer Porcine Kidney cells
- LLOQ
lower limit of quantification
- MDR1
multidrug resistance protein 1
- PD
pharmacodynamics
- pKp,uu
predicted CSF‐to‐unbound plasma partition coefficient
- PK
pharmacoKinetics
- PND
postnatal day
- PO
Per Os (oral administration)
- QWBA
quantitative whole‐body autoradiography
- RIPA
radioimmunoprecipitation assay
- rpm
revolutions per minute
- SMA
spinal muscular atrophy
- SMN
survival of motor neuron
- SMNΔ7
SMN mRNA lacking exon 7or SMN protein translated from SMNΔ7 mRNA
- SRM
single reaction monitoring
- Δ7
deletion of exon 7
1. INTRODUCTION
Spinal muscular atrophy (SMA) is caused by the deletion and/or mutation of the Survival of Motor Neuron 1 (SMN1) gene, resulting in insufficient levels of functional SMN protein and selective vulnerability of motor neurons.1, 2 A second SMN gene, SMN2, produces only low levels of functional SMN protein which are insufficient to fully compensate for the lack of the SMN1 gene.3, 4 The orally administered small molecules risdiplam (RG7916; RO7034067, 7‐(4,7‐diazaspiro[2.5]octan‐7‐yl)‐2‐(2,8‐dimethylimidazo[1,2‐b]pyridazin‐6‐yl)pyrido[1,2‐a]pyrimidin‐4‐one)) and RG7800 (RO6885247, 2‐(4‐ethyl‐6‐methyl‐pyrazolo[1,5‐a]pyrazin‐2‐yl)‐9‐methyl‐7‐(1‐methyl‐4‐piperidyl)pyrido[1,2‐a]pyrimidin‐4‐one) were designed to modify SMN2 mRNA splicing, by promoting inclusion of exon 7, thereby increasing full‐length SMN2 mRNA production and levels of functional SMN protein in patients with SMA.
The hallmark of SMA is the progressive degeneration of α‐motor neurons in the brain stem and spinal cord that causes muscle atrophy, weakness, and disease‐related complications that can impact survival.5 Accumulating evidence challenges the idea that SMA is solely a disease of motor neurons. SMA is increasingly described as a disease affecting tissues and cell types beyond the motor neuron, suggesting that effective disease intervention may require body‐wide correction of SMN protein levels to achieve a complete reversal or amelioration of the disease state.5, 6, 7 This is consistent with the requirement of peripheral SMN restoration for long‐term rescue in a severe SMA animal model.8, 9, 10
SMN protein is ubiquitously expressed throughout the body and is implicated in several basic cellular functions.11 SMN protein may have a role in many cells and tissues, such as skeletal muscle, heart, bone, and autonomic and nervous systems; and deficiencies may contribute to the disease state.5, 7, 12
A significant clinical benefit has been observed with therapies that increase levels of functional SMN protein. Recently, an antisense oligonucleotide therapy (nusinersen, Spinraza®), which is administered intrathecally, was approved in the U.S. and European Union for the treatment of SMA in pediatric and adult patients. Both Spinraza® and risdiplam (having different molecular targets) act by promoting exon 7 inclusion in SMN2 mRNA, leading to increased production of functional SMN protein.13, 14, 15 Approval of Spinraza® as a treatment for SMA in adult and pediatric patients validated the mechanism of action and effectiveness of SMN2 mRNA‐splicing therapies.
RG7800 was the first small molecule SMN2 splicing modifier to enter human clinical trials.16 In the Phase 1 MOONFISH trial (NCT03032172) no serious adverse events were reported following RG7800 administration in patients with Type 2 and 3 SMA. However, dosing was suspended due to off‐target retinal findings of RG7800 in long‐term, nonclinical safety studies in monkeys.
Risdiplam is an orally administered, small‐molecule SMN2 pre‐mRNA splicing modifier, optimized in terms of pharmacokinetic (PK) characteristics and with enhanced specificity toward SMN2 exon 7 splicing relative to other SMN2 splicing modifiers.17 Risdiplam demonstrated a higher in vitro and in vivo efficacy in cellular and mouse models of SMA, a larger safety window in preclinical models (including to off‐target retinal findings) and an improved human PK profile compared with RG7800.17 Risdiplam increases SMN2FL mRNA production and distributes into the central nervous system (CNS) and peripheral tissues by avoiding interaction with human multidrug resistance protein 1 (MDR1); a transport protein that restricts brain exposure.
The primary objectives of these preclinical studies were to assess in vitro characteristics, as well as the extent of central and peripheral tissue distribution of risdiplam, and to evaluate biomarker levels following risdiplam dosing in animal models of SMA. These studies are of importance since the relationship between drug exposure in tissues and organs, and biomarkers evolution in preclinical models provide the basis for estimating functional SMN protein level increases in target tissues from the easily measured biomarker response in patients’ blood following risdiplam treatment. Blood sampling is minimally invasive for patients. Therefore, functional SMN protein level increases in blood can be expected to reflect similar increases in functional SMN protein levels in the CNS, muscle, and other key organs affected in SMA. Risdiplam is under investigation in the SUNFISH (Type 2/3 SMA; NCT02908685), FIREFISH (Type 1 SMA; NCT02913482), and JEWELFISH (non‐naïve Type 2/3 SMA; NCT03032172) clinical studies, with the potential to address a broad range of patients.18
2. MATERIALS AND METHODS
Risdiplam structure and method of synthesis can be found in reference.17 RG7800 structure and method of synthesis can be found in reference.16
2.1. In vitro transport assay
Parent porcine kidney epithelial LLC PK1 (Lewis Lung Cancer Porcine Kidney 1) and canine kidney epithelial MDCKII (Madin‐Darby Canine Kidney) cell lines were used. LLC‐PK1, MDCKII, L‐MDR1 (LLC‐PK1 cells transfected with human MDR1), L‐Mdr1a (LLC‐PK1 cells transfected with rodent Mdr1a), M‐BCRP (MDCKII cells transfected with human Breast Cancer Resistance Protein; BCRP), and M‐Bcrp1 (MDCKII cells transfected with rodent Bcrp1) cell lines were obtained from Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam, the Netherlands), and used under a license agreement. The rodent Mdr1a is a murine protein and shares 95% amino acid sequence identity with the rat Mdr1a,19 henceforth referred as “rodent” Mdr1a throughout the manuscript. The assays were conducted as previously described.20, 21 Briefly, cells were cultured on semipermeable 96‐well inserts (surface area 0.11 cm2, pore size 0.4 mm; Millipore), and bidirectional transport measurements were performed either at Day 3 or 4 after seeding. The medium was removed from the apical (100 mL) and basolateral (240 mL) compartments and replaced on the receiver side by culture medium without phenol red and with or without inhibitor (zosuquidar, 1 μM for L‐MDR1 and L‐Mdr1a; Ko143, 1 μM for M‐BCRP and M‐Bcrp). Transcellular transport was initiated by the addition of media to the donor compartment containing test substrate (risdiplam or RG7800, tested at 1 μM) and 10 μM Lucifer yellow (Sigma‐Aldrich). Lucifer yellow was included to confirm monolayer integrity and reference substrates were incubated as controls to MDR1/Mdr1a or BCRP/Bcrp activity. Plates were incubated for 3.5 hours at 37°C and 5% CO2 under continuous shaking (100 rpm). Samples (triplicates for each condition) were taken from the donor and receiver compartments and analyzed by scintillation counting or high‐performance liquid chromatography with tandem mass spectrometry, as previously described for liquid chromatography, 10ADvp pump system (Shimadzu, Kyoto, Japan) coupled with a PAL HTS auto‐sampler (CTC Analytics, Zwingen, Switzerland) was used, and for MS, API 4000 or QTrap4000 system equipped with a TurboIonspray source (Sciex, Framingham, MA).20
where Papp, A, C 0, and dQ/dt represent the apparent permeability, the filter surface area, the initial concentration, and the amount transported per time period, respectively. Papp values were calculated on the basis of a single time point. Transport efflux ratios (ERs) were calculated as follows:
where PappBA is the permeability value in the basolateral‐to‐apical direction, and P appAB is the permeability value in the apical‐to‐basolateral direction.
2.2. Animals
Thirteen animal studies were carried out as listed in Table 1.
Table 1.
In vivo studies design
| Study number | Species | Age (@ day1) | Animal gender and n | Compound dosed | Doses (mg/kg) | Dosing route | Number of days of dosing | Tissues sampled | Endpoints |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Mice (FVB) | PND10 | 18 Mixeda | Risdiplam | 3 | IP | 1 | Plasma, Brain | PK |
| 2 | Mice (FVB) | Adults | 15 F | Risdiplam | 10 | PO | 1 | Plasma, Brain | PK |
| 3 | Mice (C/C) | Adults | 9F 6M | Risdiplam | 1, 3, 10 | PO | 10 | Plasma (PK only), Brain | PK, SMN protein |
| 4 | Mice (Δ7) | PND3 | 24 Mixeda | Risdiplam | 0.1, 0.3, 1, 3 | IP | 7 | Plasma (PK only), Brain, Muscle | PK, SMN protein |
| 5 | Mice (Δ7) | PND3 | 18 Mixeda | Risdiplam | 1, 3, 10b | POb | 219 | Plasma/Blood, Brain | PK, SMN protein |
| 6 | Rat (Wistar) | Adults | 22F 31M | Risdiplam | 1, 3, 9 | PO | 28 | Plasma, Brain | PK |
| 7 | Rat (Wistar & partially pigmented) | Adults | 33 M | Risdiplam | 3 | PO | 4 | Plasma, Brain, Muscle, CSF | PK |
| 8 | Rat (Wistar) | PND22 | 26F 36M | Risdiplam | 1, 3, 7.5 | PO | 1 (n = 31) or 90 (n = 31) | Plasma, Brain | PK |
| 9 | Monkey (cynomolgus) | Adults | 2 M | Risdiplam | 3 | PO | 7 | Plasma, Brain, Muscle, CSF, Bone marrow, Heart, Liver, Kidney, Spleen | PK |
| 10 | Monkey (cynomolgus) | Adults | 11F 11M | Risdiplam | 1.5, 3, 5 | PO | 275 | Plasma, Brain, CSF | PK |
| 11 | Mice (C/C) | Adults | 51F 38M | RG7800 | 10 | PO | 30 | Plasma, Blood, Brain, Muscle |
PK SMN protein |
| 12 | Mice (C/C) | Adults | 5F 5M | RG7800 | 10 | PO | 10 | Plasma, Blood, Brain, Muscle, Liver, Skin, Heart, Pancreas, Spinal Cord |
PK SMN protein |
| 13 | Rat (Wistar) | Adults | 9M | 14C‐ Risdiplam |
2 (IV) 5 (PO) |
IV PO |
1 | QWBA | PK |
F, female; M, male; CSF, cerebrospinal fluid; PND, postnatal day; PO, per os; IP, intraperitoneal; PK, pharmacokinetics (measure of the compound exposure); QWBA, quantitative whole‐body autoradiography.
At PND3 or PND10 mouse gender cannot be easily determined.
In Study 5, dosing was initiated at PND3 IP at 0.3, 1, or 3 mg/kg/day and changed to PO dosing of 1, 3, or 10 mg/kg/day from PND24 onwards.
All studies were carried out in Association for Assessment and Accreditation of Laboratory Animal Care International certified facilities, and the protocols for animal experiments were approved by the Institutional Animal Care and Use Committee.
Studies 1 and 2 used Friend Leukemia virus B (FVB) heterozygous or wild‐type littermates from either the SMNΔ7 mouse colony (Study 1, age postnatal day [PND] 10) or the C/C‐allele SMA mouse colony (Study 2, adult mice). In Studies 3, 11 and 12, SMA C/C‐allele SMA mice were derived from FVB.129(B6)‐Smn1tm5(Smn1/SMN2)Mrph/J strain from The Jackson Laboratory (stock number 008604) and dosing was initiated when mice were ~7 weeks old. In Studies 4 and 5, SMNΔ7 mice were derived from FVB.Cg‐Tg(SMN2*delta7)4299Ahmb Tg(SMN2)89Ahmb strain from The Jackson Laboratory (stock number 005025) and dosing was initiated at PND3.
For Studies 6, 7, and 8, adult Wistar Hannover Crl:WI (Han) rats were obtained from Charles River Laboratories, (Margate, UK or Raleigh, NC). For Study 7, pigmented (Brown Norway BN/Crl) rats were used. For both Studies 6 and 7, dosing was initiated in adult rats and in Study 8, dosing was initiated at PND22.
In Studies 9 and 10, purpose‐bred cynomolgus monkeys (Macaca fascicularis) were between 24 and 26 months old at the start of dosing.
2.3. Study design in mice (Studies 1, 2, 3, 4, 5, 11, and 12)
Two different mouse models of SMA were utilized for these studies. For studies in adult mice, the C/C‐allele mouse model of mild SMA was utilized. C/C‐allele mice have a near‐normal life span but show decreased muscle function, reduced body weight gain, and peripheral necrosis in comparison to normal mice.22 Neonatal SMNΔ7 mice, a mouse model of severe SMA, were also used. These mice die approximately 2 weeks after birth.23
For oral dosing of adult mice, compounds were formulated as a suspension in 0.5% hydroxypropylmethyl cellulose with 0.1% Tween 80. For intraperitoneal (IP) dosing of juvenile mice, compounds were formulated in dimethyl sulfoxide and administered at a dosing volume of 2.5 mL/kg. For repeat administration, the compounds were administered once daily. For Study 5, mice were dosed IP from PND3 to PND23 and dosed by oral gavage from PND24 onward. For information on doses and length of dosing for each individual study, please see Table 1.
After dosing, blood was collected by terminal cardiac puncture at specified time points and plasma and tissues collected for compound analysis. The concentrations of test compound in plasma and tissues were quantified by liquid chromatography tandem mass spectrometry. Plasma or tissue samples were extracted by protein precipitation method and the extracts were separated by a reverse HPLC column utilizing Waters Acquity UPLC and detected using a Micromass Quattro Ultima Mass Spectrometer. Chromatograms were integrated using Masslynx 4.1 software. The lower limits of quantification (LLOQ) were 0.002 μg/mL (plasma; Studies 2, 3, 12), 0.0025 μg/mL (plasma; Study 1, 4, 5), 0.010 μg/mL (plasma; Study 11), 0.01 μg/g (brain; Studies 1, 2, 3, 11), 0.02 μg/g (brain; Study 4, 5), 0.25 μg/g (brain; Study 12), 0.01 μg/g (quadriceps; Study 3), 0.025 μg/g (quadriceps; Study 11, 12), or 0.04 μg/g (quadriceps; Study 4).
To measure increase in brain and muscle SMN protein, mice were killed with carbon dioxide as per Institutional Animal Care and Use Committee guidelines one hour after the last dose and the brain and quadriceps were collected. Tissues were not perfused before collection. Tissue samples were collected, homogenized, transferred to a 96‐well plate and were diluted in RIPA buffer. Samples were run in duplicate and averaged. SMN protein was quantified using Homogeneous Time Resolved Fluorescence (HTRF, Cisbio Bioassays) as previously published.14 Total protein content was quantified in each tissue homogenate using the BCA assay according to the manufacturer's protocol. The HTRF signal for SMN protein was normalized to the total protein concentration for each sample, generating the ΔF SMN signal/total protein. To measure SMN protein in blood, blood was obtained by cardiac puncture. SMN protein was quantified using electrochemiluminescence immunoassay utilizing a Meso Scale Discovery Sector Imager 6000 instrument as previously published. SMN protein concentrations (pg/mL) were determined by comparison to a standard curve prepared with recombinant human SMN protein.24
Changes in SMN protein levels are reported as . SMN vehicle KO is the mean SMN protein level normalized to the total protein for each control mouse and then this ratio is averaged across the vehicle‐dosed group in knock‐out (KO) mice. SMN treated KO is the SMN protein level normalized to the total protein for each compound‐dosed KO animal.
2.4. Study design in rats (Studies 6, 7, and 8)
Risdiplam was administered orally by gavage once daily as a solution in 10 mM ascorbic acid/0.01 mg/mL sodium thiosulfate pentahydrate, pH 3, and the dosing volume was 10 mL/kg (Study 6) or 4 mL/kg (Study 7, 8). For information on doses and length of dosing for each individual study, please see Table 1. Each animal was killed under isoflurane anesthesia. Animals were exsanguinated by the severing of major blood vessels. Terminal blood samples were taken from the jugular vein immediately prior to exsanguination and collected into tubes containing K3‐EDTA anticoagulant. The entire brain was collected into labeled 7 mL Precellys® homogenization tubes (CK14), snap‐frozen in liquid nitrogen and stored on dry ice. Tissues were homogenized by bead beating and/or diluted with blank tissue homogenate or blank rat plasma. The analyte was isolated from matrix (EDTA plasma or tissues homogenate) by protein precipitation with acetonitrile/ethanol containing the internal standard (13C, D2 stable isotope‐labeled risdiplam) and separated from other constituents of the sample by narrow‐bore HPLC. Detection was accomplished utilizing heated electrospray (HESI) MS/MS positive‐ion selected reaction monitoring mode (SRM). CSF and tissue samples were quantified against rat plasma calibration curves diluted with the appropriate blank tissue homogenate or blank plasma. The LLOQ for risdiplam was 0.250 ng/mL in rat plasma using 20 μL aliquots, 0.500 or 1.00 ng/mL in CSF and 2 ng/g in tissues using 20 μL of tissue homogenate.
Unbound (free) plasma concentrations (Cu_p) were calculated by multiplying the measured total concentration in plasma by the measured free fraction in plasma (16% in adult rat). The CSF‐to‐unbound plasma partition coefficient (Kp,uu) was calculated as follows: where C_CSF is the concentration in the CSF. The predicted Kp,uu (pKp,uu) was derived according to equation (5) from25: where γ is a fixed parameter (0.753) and ER the efflux ratio measured in L‐Mdr1a cells.
2.5. Study design in monkeys (Studies 9 and 10)
Risdiplam was administered orally by gavage once daily as a solution in 10 mM ascorbic acid/0.01 mg/mL sodium thiosulfate pentahydrate, pH 3, and the dosing volume was 1.5 mL/kg (Study 9) or 5 mL/kg (Study 10). For information on doses and length of dosing for each individual study, please see Table 1.
At the end of dosing, animals were killed, and terminal plasma and tissues were collected and stored frozen. In Study 10, brain stem and cortex samples (0.5 g) were separately collected. A sample of 0.5 mL of CSF was collected from all animals.
Tissues were homogenized and diluted with blank cynomolgus monkey plasma. The analyte was isolated from matrix as described for Studies 6‐8. Detection was accomplished utilizing HESI MS/MS in positive ion SRM. The LLOQ in cynomolgus monkey plasma was 0.250 ng/mL (Study 9) or 0.500 ng/mL (Study 10) using 20 μL aliquots. All other samples were quantified against cynomolgus monkey plasma calibration curves. Due to sample dilution, the resulting LLOQs were 0.500 ng/mL in CSF (Study 9) and between 0.500 and 5000 ng/g in tissues (10.0 ng/g in brain). For Study 10, a dedicated, sensitive CSF method with LLOQ 0.100 ng/mL was used. Unbound (free) plasma concentrations were calculated by multiplying the measured total concentration in plasma by the measured free fraction in plasma (15% in adult cynomolgus monkeys).
2.6. Rat Quantitative Whole‐Body Autoradiography (QWBA) study design (Study 13)
Wistar rats received either a single oral dose of 14C‐risdiplam (by gastric gavage), or a single intravenous dose of 14C‐risdiplam (by tail vein injection). Dose levels were 5 or 2 mg/kg, for oral and intravenous doses, respectively. For whole‐body autoradiography, following deep anesthesia under isoflurane, single animals were killed by cold shock (in a mixture consisting of an excess of dry‐ice in hexane) at the following times after dosing: 10 min for the IV‐dosed animals and 2, 24, 72, and 168 hours for the oral‐dosed animals. Once fully frozen, the carcasses were prepared for, and subjected to, whole‐body autoradiography procedures. Radioactivity concentration in tissues was quantified from the whole‐body autoradiograms, using a validated image analysis system. After exposure in a copper‐lined, lead exposure box for 7 days, the imaging plates were processed using a FUJI FLA 5000 or 5100 radioluminography system (Raytek Scientific Ltd). The electronic images were analyzed using a validated PC‐based image analysis package (Seescan2 software, LabLogic). While under terminal anesthesia, blood (approximately 3 mL) was collected from each of the animals by cardiac puncture into tubes precoated with lithium heparin. Blood and plasma were assayed for total radioactivity.
3. RESULTS
3.1. Transport in vitro assays
Risdiplam showed an average passive permeability in parental LLC‐PK1 cells of around 350 nm/s. Similar permeability was observed in L‐MDR1, L‐Mdra1, M‐BCRP, and M‐Bcrp cells in the presence of inhibitor. Risdiplam was found not to be a substrate of human MDR1 with an ER of 2.2, but a weak substrate of rodent Mdr1a (ER = 3.7). The predicted CSF‐to‐unbound plasma partition coefficient (pKp,uu) based on the rodent ER and model from25 is therefore 0.33. Risdiplam was a weak human BCRP substrate with an ER of 3.7 and a strong Bcrp substrate in rodents with an ER of 44.
RG7800 was also a highly permeable compound, with an average passive permeability in LLC‐PK1 cells of ≥100 nm/s and was also not a substrate of human MDR1.
3.2. Risdiplam tissue distribution
During the preclinical development of risdiplam, brain, and muscle tissues, as well as key organs affected in SMA, were collected as primary or secondary endpoints in a variety of PK, pharmacodynamics (PD), and toxicology studies. Single or repeat daily oral or IP administration of risdiplam for up to 39 weeks showed similar total drug levels in plasma, brain, and muscle of mice (n = 90), rats (n = 148), and monkeys (n = 24) (Figure 1A,B). CSF levels of risdiplam in monkeys reflected those of free, nonbound compound in plasma. A minor deviation from unity was observed in rat CSF levels compared with free plasma levels (Figure 1C) and in mice brain levels compared with total plasma (Figure 1A). The observed in vivo Kp,uu in rats was 0.28.
Figure 1.
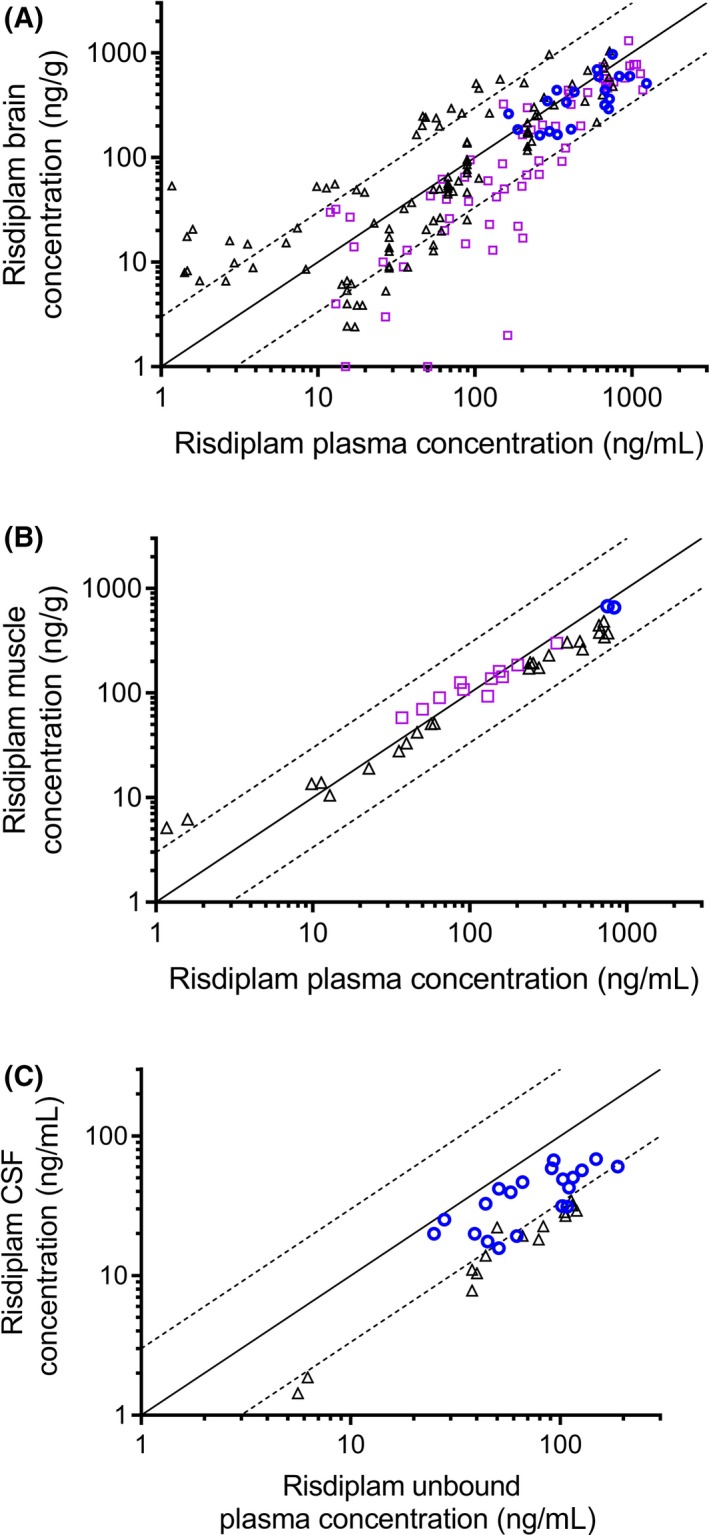
Risdiplam tissues concentration vs risdiplam plasma concentration: A, brain (n = 189) (Studies 1‐4, 6‐10). B, muscle (n = 37) (Studies 4,7,9), and C, CSF (n = 35) (Studies 7,9,10) of mice (open squares), rats (open triangles), and monkeys (open circles) following single or repeat PO or IP administration. Each symbol represents data coming from an individual animal. The solid line represents the line of unity. The dotted lines represent threefold variation around unity. CSF, cerebrospinal fluid; IP, intraperitoneal; PO, per os
A strong correlation was observed between concentrations of risdiplam in plasma and levels of risdiplam in tissues over a wide range of concentrations (Figure 1A‐C), indicating that the total plasma is representative of tissue concentrations. There was no obvious deviation from unity in one particular dose group or with increasing dose (Supplemental Data Figure 1). The largest dataset was obtained on brain tissue concentrations in 189 animals (Figure 1A). In Study 10, brain samples from the stem and cortex regions of monkeys (n = 22) demonstrated very similar risdiplam concentrations (average cortex/brain stem concentrations ratio of 1.10, range 0.71‐1.59). These two values were averaged for each individual animal shown in Figure 1A.
In Study 10, tissue drug levels were maintained after daily dosing in monkeys receiving risdiplam daily for 39 weeks. At the end of the dosing period, some monkeys remained on study but no longer received the drug (recovery animals). Drug levels were below the limit of quantification (BLQ) in plasma, CSF, and brain after 22 weeks’ treatment‐free recovery period (muscle not sampled). In nonhuman primates, the ratios of total risdiplam concentration in plasma to brain and muscle concentrations were close to 1 for all tissues measured. Specifically, in Study 9 following oral dosing of risdiplam at 3 mg/kg/day for 7 days in monkeys (n = 2), total plasma concentrations (794 ng/mL) matched those in brain (783 ng/g) and muscle (668 ng/g), whereas CSF drug level (53.5 ng/mL) were within the same range of the calculated free compound concentration in plasma (119 ng/mL) (Figures 1C and 2).
Figure 2.
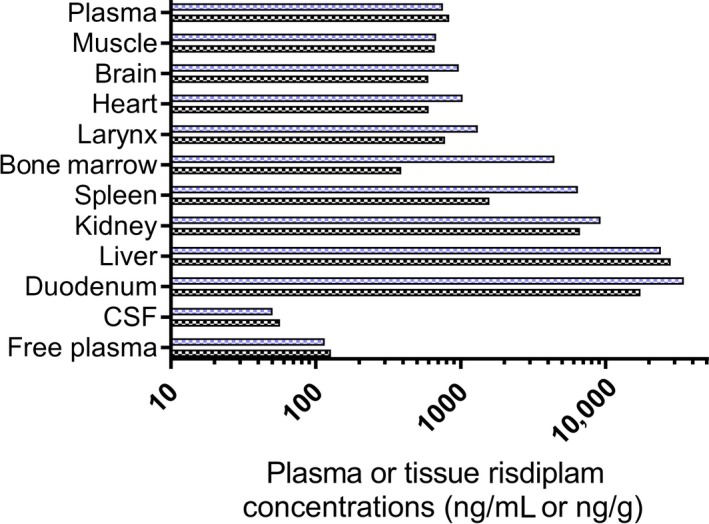
Risdiplam tissue distribution in cynomolgus monkeys. Risdiplam concentration in plasma (ng/mL), CSF (ng/mL) and tissues (ng/g) of cynomolgus monkeys (n = 2) at Day 7 following once daily PO dosing (3 mg/kg/day for 7 days, Study 9). The blue and back bars represent the individual values for each of the two individual monkeys. CSF, cerebrospinal fluid; PO, per os
The time‐course of risdiplam concentrations in tissue and plasma is shown for adult rats (Figure 3A,B) and adult FVB mice (Figure 3C,D). Parallel time courses of drug levels were measured in plasma, brain, CSF, and muscle, up to 48 hours after the last dose, confirming a parallel elimination in those tissues (Study 7, Figure 3A). A similar decreasing trend, with plasma and brain levels decreasing in parallel, was confirmed after 28 days of daily dosing in rats (Figure 3B) and after single PO or IP dose in mice (Figure 3C,D). Absolute levels of risdiplam in rat brain were in the same range as total plasma levels in both Figure 3A,B. Brain levels were slightly higher or slightly lower than plasma depending on interexperimental variability (Figure 3A,B). This is also quite apparent in Figure 1A where the symbols for rats (triangles) are on either side of the unity line. In rats dosed at 7.5 mg/kg/day PO (Study 8), brain‐to‐total‐plasma ratios after 90 days of dosing were close to 1 (0.92 average for n = 13), and drug levels in plasma and brain samples were BLQ after a further 8 weeks of recovery treatment‐free period (n = 6).
Figure 3.
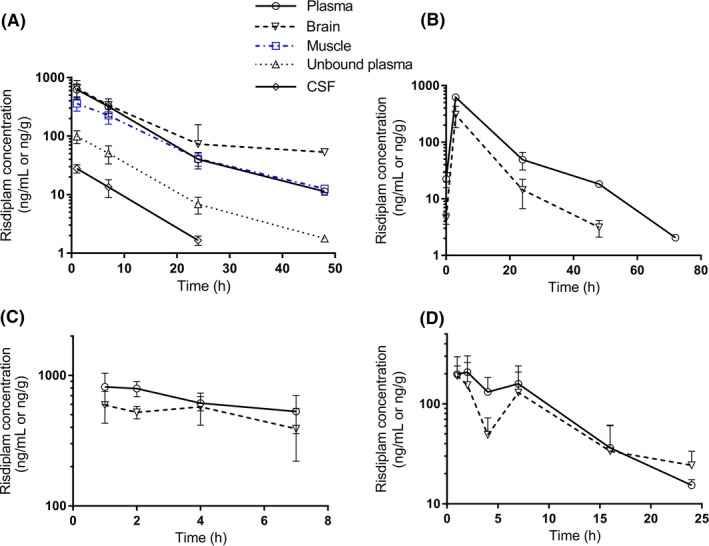
Time course of plasma and tissue risdiplam concentrations in mice or rats following PO or IP, single or repeat administration of risdiplam. A, Time course in plasma (open circles), brain (open downward triangles), muscle (open squares), unbound plasma (open upward triangles), and CSF (open lozanges) in adult rats following 4 days of 3 mg/kg daily dosing PO (Study 7, n = 6 or 7 rats per time point). B, Time course of plasma and brain in adult rats following 28 days of 3 mg/kg daily dosing PO (Study 6, n = 2 per time point). C, Time course of plasma and brain in adult FVB mice following a single dose of 10 mg/kg PO (Study 2, n = 3 per time point). D, Time course of plasma and brain in juvenile FVB mice (PND10) following a single dose of 3 mg/kg IP (Study 1, n = 3 per time point). In all graph, symbols represent mean with standard deviation. CSF, cerebrospinal fluid; FVB, friend leukemia virus B; IP, intraperitoneal; PND10, postnatal day 10; PO, per os
Radioactivity was rapidly and widely distributed following oral administration of 14C‐risdiplam in the rat QWBA, with the majority of tissues measured attaining peak concentrations at 2 hours (the first sampling point). Concentrations of radioactivity declined over the duration of the study in plasma and tissues. Tissue/plasma ratios of radioactivity were high in the bone marrow (5.5), kidney cortex (9.1), kidney medulla (4.1), liver (7.2), lung (7.1), heart (2.4), pancreas (4.7), spleen (8.0), trachea (2.7), mucosa of small intestine (3.4), large intestine (1.5), and rectum (4.1) 2 hours after oral administration. Tissue/plasma ratios 10 minutes after IV administration were similar to the oral 2‐hour ratios in all tissues including intestinal mucosa.
In a tissue distribution study in two cynomolgus monkeys, muscle, brain, heart, and larynx drug levels were all within the total plasma concentration, with less than 30% difference (Study 9, Figure 2). Bone marrow and spleen showed a high interindividual variability, with tissue/plasma ratio around 5. Tissue/plasma ratios were higher for kidney (10), liver and duodenum (both 33). In the same study, two other monkeys were killed 6 weeks after the last dose, and drug levels in plasma, CSF, muscle, brain, heart, and bone marrow were all BLQ. Risdiplam concentrations in duodenum, spleen, and larynx were below 1 ng/g and around 4 ng/g in both liver and kidney.
Similar tissue distribution studies in rat and monkeys, and rat QWBA were also performed using RG7800. Following oral administration, RG7800 widely distributed and was present in key tissues and organs affected in SMA (brain, muscle, bone, mucosa of the GI tract, pancreas, liver, lung, heart, kidney, and spleen) at concentrations equal to or higher than total plasma.
3.3. SMN protein increase in CNS and periphery
Several PD studies investigated the effect of orally administered small molecule SMN2 splicing modifiers on SMN protein levels in key target tissues of SMA mouse models. In C/C‐allele SMA mice dosed once daily for up to 30 days, RG7800 increased SMN protein levels in blood and to a similar extent in brain and muscle (Figure 4A,B). In study 11, a similar fold increase was observed in the three tissues over the 30‐day period (Figure 5). In Study 12, after 10 days of daily doses, multiple tissues were harvested from C/C‐allele SMA mice and analyzed for SMN protein. Blood, brain, spinal cord, and skin showed an SMN protein fold increase between 1.64 and 2.02, whereas in the pancreas, SMN protein increased by almost fourfold (Figure 6). In some tissues, there was little difference in baseline SMN protein levels in mutant C/C‐allele SMA mice relative to heterozygous mice reducing the window to measure an effect. Consistent with this, SMN protein fold increase was minimal in the liver (1.1 on average), whereas a 23% and 31% SMN protein increase from baseline was observed in muscle and heart, respectively (Figure 6).
Figure 4.
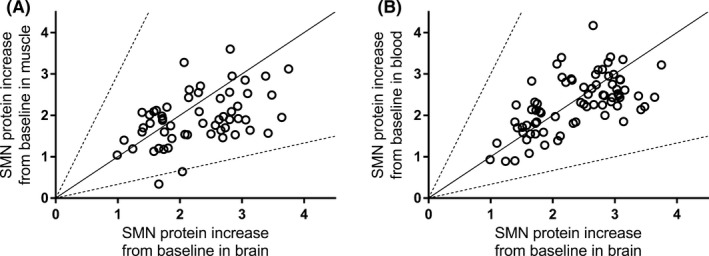
SMN protein increase in muscle and blood vs brain of SMA mice following administration of RG7800. A, SMN protein increase from baseline (reported as ratio of SMN protein levels in treated vs control) in muscle vs brain of adult C/C‐allele SMA mice following single or repeat (up to 30 days) doses of 10 mg/kg/day PO of RG7800 (studies 11 and 12, n = 59). B, SMN protein increase from baseline (reported as ratio of SMN protein levels in treated vs control) in blood vs brain of adult C/C‐allele SMA mice following single or repeat (up to 30 days) doses of 10 mg/kg/day PO of RG7800 (studies 11 and 12, n = 59) (n = 74). Each symbol represents data coming from an individual animal. The solid line represents the line of unity. PO, per os; SMN, survival motor neuron
Figure 5.
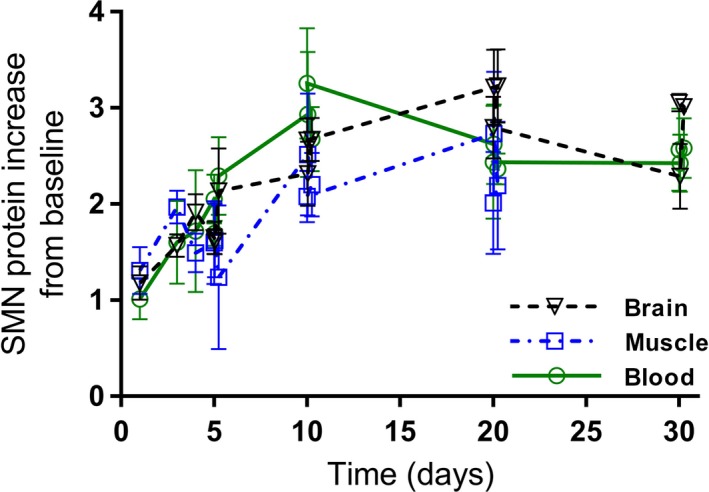
SMN protein increase in blood, brain, and muscle of SMA mice following administration of RG7800. SMN protein increase from baseline (reported as ratio of SMN protein levels in treated vs control) in brain, muscle, and blood over time (Study 11, n = 5 per time point) in adult C/C‐allele SMA mice following single or repeat once daily doses of RG7800 (10 mg/kg/day) PO for up to 30 days. Symbols represent mean with standard deviation. PO, per os; SMA, spinal muscular atrophy; SMN, survival motor neuron
Figure 6.
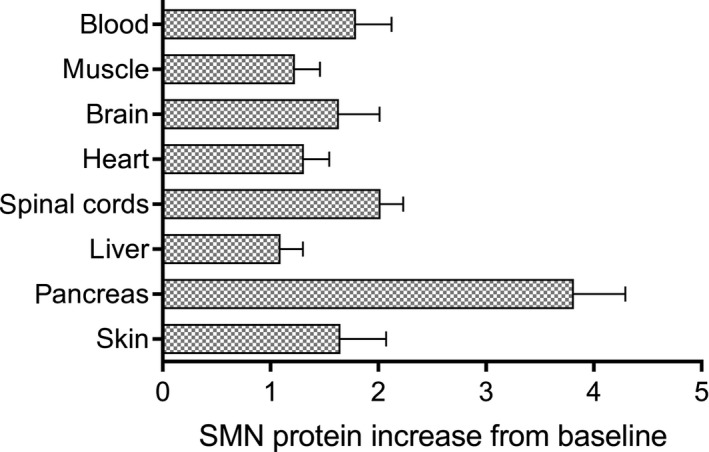
SMN protein increase in different tissues following administration of RG7800. SMN protein increase from baseline (reported as ratio of SMN protein levels in treated vs control) in different tissues in adult C/Callele SMA mice following repeat (10 days) doses of 10 mg/kg/day PO of RG7800 (Study 12, n = 5). Bars represent mean with standard deviation. PO, per os; SMA, spinal muscular atrophy; SMN, survival motor neuron
These findings were confirmed while dosing risdiplam: a similar dose‐dependent increase in SMN protein levels in brain and muscle (0.1 mg/kg/day: brain 28%, muscle 32%; 1 mg/kg/day, brain 206%, muscle 210%) was observed after 7 days of once daily dosing in SMNΔ7 mice (Study 4, n = 6 or 7 per dose) (Figure 7,17 Risdiplam also prolonged survival of SMNΔ7 mice (Study 5) when dosed once daily with risdiplam for 219 days.17 In Study 5, SMN protein level increases were measured relative to SMN levels in heterozygous control animals and a correlation was observed between increase in SMN protein in blood and in brain. In the same study, risdiplam was shown to improve phenotype and motor function, to prolong survival and to increase body weight gain in SMNΔ7 mice.17
Figure 7.
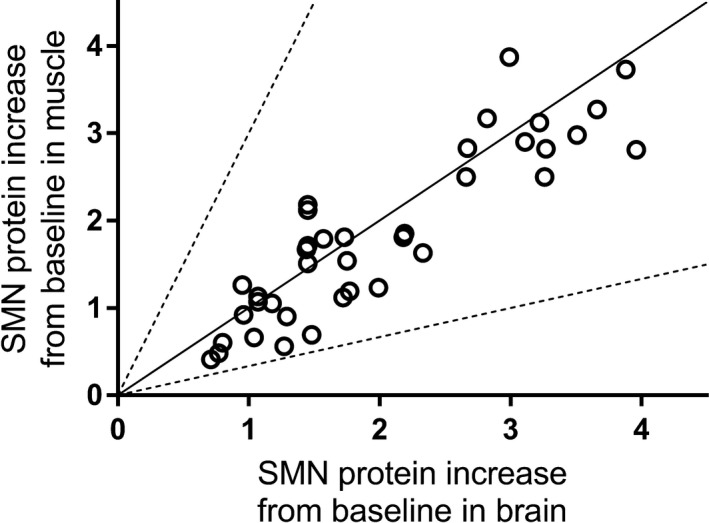
SMN protein increase in muscle vs brain in SMA mice following administration of risdiplam. SMN protein increase from baseline (reported as ratio of SMN protein levels in treated vs control) in muscle vs brain of adult C/C‐allele SMA mice (Study 3, n = 4 per dose) dosed 1, 3, or 10 mg/kg/day PO for 10 days, and postnatal day 10 SMNΔ7 mice (Study 4, n = 6 or 7 per dose) dosed 0.1, 0.3, 1, or 3 mg/kg/day PO for 7 days. The solid line represents the line of unity. Each symbol represents data coming from an individual animal. PO, per os; SMA, spinal muscular atrophy; SMN, survival motor neuron; SMNΔ7, truncated SMN protein
4. DISCUSSION
Orally administered small molecule SMN2 splicing modifiers (risdiplam and RG7800) have been developed to target SMN2 alternative splicing selectively in both the peripheral organs and CNS by penetrating the blood brain barrier. According to the free drug hypothesis, only the nonprotein‐bound molecules are available for interaction with their targets.25, 26 Hence, to achieve a meaningful prediction of pharmacological efficacy in humans, the preclinical assessment of new drug entities requires an accurate estimate of the concentration of the unbound drug at the site of action. In this study, we have shown that, as expected from in vitro data, risdiplam freely distributed from the blood into the CNS and multiple tissues in animals, and is expected to behave similarly in human based on the same mechanistic rationale.
Both risdiplam and RG7800 show high passive permeability, which is important for GI and tissue uptake, and are not MDR1 substrates in humans, which would otherwise restrict brain distribution.25 In rat pKp,uu was estimated at 0.33 based on rodent Mdr1a ER, which is in line with the observed in vivo Kp,uu of 0.28, confirming Mdr1a as the principal mechanism lowering rat CSF levels (Figure 1C) and mice brain levels (Figure 1A). Still, this property did not limit SMN protein increase in mouse brain. Risdiplam is also a strong rodent Bcrp substrate in vitro, but this interaction was not apparent in vivo, since rodent CSF levels were successfully predicted based on the weak Mdr1a interaction alone. This apparent disconnect for brain penetration of rodent Bcrp substrates has also been observed with other molecules.21, 27This is in alignment with risdiplam's high passive permeability (350 nm/s). Furthermore, while risdiplam is not a human MDR1 substrate, it is also only a weak human BCRP substrate, and CSF levels of risdiplam in cynomolgus monkeys reflected those of free plasma levels, indicating a lack of relevance of BCRP for this molecule in monkeys. Since monkeys are considered the closest species to humans regarding efflux transporters (>95% amino acid sequence homology for both BCRP and MDR1), it is reasonable to anticipate human CSF levels to be in line with what was observed in monkeys.28 Therefore, the total drug concentration in CSF or the unbound concentration in plasma in humans should be good estimates of the pharmacologically active compound available to modify SMN2 alternative splicing in the CNS.
Brain penetration in monkey brain stem and cortex areas was similar. It is expected that risdiplam will show a similar behavior in humans. In each of the three species studied (mice, rats, and monkeys) risdiplam showed elimination from the central compartment parallel to plasma, independently of species, dose or duration of dosing. Several weeks after dosing cessation, the drug was fully eliminated, with drug levels being BLQ in brain, CSF, and plasma.
Consistent with drug exposure in the CNS, SMN protein levels increased in a dose‐proportional manner in the brain of SMNΔ7 and C/C‐allele mouse models of SMA after treatment with risdiplam (Studies 3 and 4) or with RG7800 (Studies 11 and 12). Robust brain penetration and increased SMN protein levels in brain have been previously shown for several small molecules SMN2 splicing modifiers structurally related to risdiplam and administered orally.14, 16, 29 These molecules, including risdiplam, not only improved survival, body weight, phenotype, and motor behavior in different mouse models of SMA but also prevented motor neuron loss and rescued synaptic pathology in mice.14, 16, 17 These preclinical data indicate that, based on the same mechanistic rational, risdiplam should also distribute into the CNS in patients with SMA after oral administration (of an oral solution): thus, this is expected to lead to functional SMN protein level increases in the CNS and preservation of lower alpha motor neurons.
Denervation and atrophy of skeletal muscles, as well as intrinsic abnormalities in SMA skeletal muscle cells, are thought to contribute directly to SMA disease pathogenesis.6, 12 The free drug hypothesis applies to all tissues and unbound drug in plasma is also expected to be in equilibrium with free drug in the muscles. In this study, we show that risdiplam distribution to, and elimination from, muscle tissue correlates with plasma levels in mice, rats, and monkeys. Importantly, this profile is independent of dose, duration of dosing or species. We also demonstrated that SMN protein levels increased in a dose‐proportional manner in the muscle of two different mouse models of SMA treated with risdiplam. Excellent muscle penetration and increased SMN protein levels in muscle have been shown previously for several small molecules SMN2 splicing modifiers structurally related to risdiplam and administered orally. Published data showed a dose‐dependent amelioration of muscle atrophy in SMA mice, and the overall increase in muscle size reflected an increase in the myofiber size and number.14, 16, 29 Similar reductions in muscle atrophy with risdiplam were observed in mouse models of SMA.17 These preclinical results suggest that, based on the same mechanistic rational, risdiplam should also distribute into muscles of patients with SMA after oral treatment and should result in a similar increase in functional SMN protein levels in muscles than in blood.
Both the rat QWBA and the monkey tissue distribution studies confirmed a wide distribution of risdiplam with elimination parallel to plasma. Risdiplam levels equivalent to, or higher than, the total plasma levels in bone, mucosa of the GI tract, pancreas, liver, lung, heart, kidney, and spleen were therefore confirmed in rodents and nonhuman primates. Those tissues have been specifically described as affected in SMA, beyond the CNS and muscle.5, 6 We also show here that RG7800 distributed widely in C/C‐ allele mice and led to increased SMN protein levels in other key organs affected in SMA in addition to an increase in blood, CNS, and muscle (Figure 6). Data from these studies are consistent with previous reports showing systemic distribution and a similar magnitude in SMN protein level increase in blood and multiple tissues, including brain, in different mouse models of SMA treated with SMN2 splicing modifiers structurally related to risdiplam.14, 16, 29
In contrast, due to intrathecal administration and inefficient cell membrane penetration, Spinraza® effects are likely limited to the CNS.9, 10, 30 The nonclinical package for nusinersen (publicly available information [eg, from U.S. Summary Basis for Approval]) states that the antisense oligonucleotide is delivered to the CNS where SMN protein is increased and leads to improved phenotype of mouse models of SMA. Systemic administration of nusinersen to neonatal mice increased SMN protein in the spinal cord and robustly rescued severe SMA mice, much more effectively than intracerebroventricular administration. Subcutaneous injections extended the median lifespan from 16 to 108 days median survival compared with intracerebroventricular administration.9
As an orally administered product, risdiplam increases functional SMN protein levels in both the CNS and peripheral tissues in animal models and it is also expected to do so in patients based on the same mechanistic rational. This may be more beneficial than a therapeutic agent solely targeting the motor neurons of the spinal cord.
In mouse models of SMA, a 100% or greater increase in brain SMN levels above vehicle results in near‐normal phenotype.29 Here we demonstrate that, in mouse models of SMA treated with oral small molecule SMN2 splicing modifiers including risdiplam, the percentage increase in SMN protein in peripheral blood (and muscle and other tissues) correlates with that in the CNS (Figures 4, 5, 6, 7). This indicates that a change in SMN protein in blood could be used as a surrogate, or biomarker to monitor increases of SMN protein levels in the CNS. Another important advantage of risdiplam is the durability of the effect. As we have shown in this current work, protein levels follow compound exposure in plasma and tissues and will stay elevated (relative to the untreated control) as long as the drug is administered, without attenuation of the effect. As SMA is a chronic progressive disease, this persistent upregulation of functional SMN protein levels in all organs and tissues is expected to result in durable benefit to the patient.
In conclusion, this study demonstrates that risdiplam distributes well into the CNS and peripheral tissues, including muscle, blood, and brain, of mice, rats, and monkeys following single or repeated oral or IP dosing. Risdiplam increased functional SMN protein levels in the CNS and periphery of mouse models of SMA by a similar magnitude. Recent early interim data from SUNFISH Part 1, showed a dose‐dependent, up to 2.5‐fold average increase in functional SMN protein in the blood of patients with SMA treated with risdiplam.31 These in vitro and in vivo preclinical data strongly suggest that functional SMN protein increases seen in patients’ blood following risdiplam treatment should reflect similar increases in functional SMN protein in the CNS, muscle, and other peripheral tissues.
AUTHOR CONTRIBUTIONS
Participated in research design: Poirier, Weetall, Ratni, Bassett, Ullah, Senn, Naryshkin, Paushkin, and Mueller; Conducted experiments: Weetall, Heinig, Bucheli, Bassett, Alsenz, and Senn; Contributed new reagents or analytic tools: Ratni, Heinig, and Bucheli; Performed data analysis: Poirier, Weetall, Schoenlein, and Ullah; Wrote or contributed to the writing of the manuscript: Poirier, Weetall, Heinig, Ratni, Ullah, Senn, Naryshkin, Paushkin, and Mueller.
DISCLOSURES
AP, KH, FB, KS, JA, SB, MU, CS, HR, and LM are employees of F. Hoffmann‐La Roche Ltd., Switzerland. MW, NN are employees of PTC Therapeutics, NJ, USA. SP is a member of the SMA Foundation, NY, USA.
Supporting information
{kind=link}
ACKNOWLEDGMENTS
These studies were funded by F. Hoffmann‐La‐Roche Ltd. The authors acknowledge MTM for providing editorial support, which was funded by F. Hoffmann‐La‐Roche Basel Ltd, Switzerland in accordance with Good Publication Practice (GGP3) guidelines.
Poirier A, Weetall M, Heinig K, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. 2018;e447 10.1002/prp2.447
Agnès Poirier and Marla Weetall are joint first authors.
REFERENCES
- 1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120‐2133. [DOI] [PubMed] [Google Scholar]
- 2. Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AH, Pellizzoni L. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol. 2012;32:126‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell. 1995;80:155‐165. [DOI] [PubMed] [Google Scholar]
- 4. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307‐6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nash LA, Burns JK, Chardon JW, Kothary R, Parks RJ. Spinal muscular atrophy: more than a disease of motor neurons? Curr Mol Med. 2016;16:779‐792. [DOI] [PubMed] [Google Scholar]
- 6. Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med. 2013;19:40‐50. [DOI] [PubMed] [Google Scholar]
- 7. Simone C, Ramirez A, Bucchia M, Rinchetti P, Rideout H, Papadimitriou D, Re DB, Corti S. Is spinal muscular atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications? Cell Mol Life Sci. 2016;73:1003‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hua Y, Liu YH, Sahashi K, Rigo F, Bennett CF, Krainer AR. Motor neuron cell‐nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015;29:288‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long‐term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahashi K, Ling KK, Hua Y, Wilkinson JE, Nomakuchi T, Rigo F, Hung G, Xu D, Jiang YP, Lin RZ, Ko CP, Bennett CF, Krainer AR. Pathological impact of SMN2 mis‐splicing in adult SMA mice. EMBO Mol Med. 2013;5:1586‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh RN, Howell MD, Ottesen EW, Singh NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta. 2017;1860:299‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shababi M, Lorson CL, Rudnik‐Schoneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi‐organ disease? J Anat. 2014;224:15‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG, Chen G, Zhang N, Gabbeta V, Vazirani P, Bhattacharyya A, Furia B, Risher N, Sheedy J, Kong R, Ma J, Turpoff A, Lee CS, Zhang X, Moon YC, Trifillis P, Welch EM, Colacino JM, Babiak J, Almstead NG, Peltz SW, Eng LA, Chen KS, Mull JL, Lynes MS, Rubin LL, Fontoura P, Santarelli L, Haehnke D, McCarthy KD, Schmucki R, Ebeling M, Sivaramakrishnan M, Ko CP, Paushkin SV, Ratni H, Gerlach I, Ghosh A, Metzger F. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688‐693. [DOI] [PubMed] [Google Scholar]
- 15. Sivaramakrishnan M, McCarthy KD, Campagne S, Huber S, Meier S, Augustin A, Heckel T, Meistermann H, Hug MN, Birrer P, Moursy A, Khawaja S, Schmucki R, Berntenis N, Giroud N, Golling S, Tzouros M, Banfai B, Duran‐Pacheco G, Lamerz J, Hsiu Liu Y, Luebbers T, Ratni H, Ebeling M, Clery A, Paushkin S, Krainer AR, Allain FH, Metzger F. Binding to SMN2 pre‐mRNA‐protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017;8:1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ratni H, Karp GM, Weetall M, Naryshkin NA, Paushkin SV, Chen KS, McCarthy KD, Qi H, Turpoff A, Woll MG, Zhang X, Zhang N, Yang T, Dakka A, Vazirani P, Zhao X, Pinard E, Green L, David‐Pierson P, Tuerck D, Poirier A, Muster W, Kirchner S, Mueller L, Gerlach I, Metzger F. Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. J Med Chem. 2016;59:6086‐6100. [DOI] [PubMed] [Google Scholar]
- 17. Ratni H, Bendels S, Bylund J, Chen KS, Denk N, Ebeling M, Feng Z, Green L, Guerard M, Jablonski P, Jacobsen B, Kletzl H, Ko CP, Kustermann S, Metzger F, Mueller B, Naryshkin NA, Paushkin SV, Pinard E, Poirier A, Reutlinger M, Weetall M, Zeller A, Zhao X, Mueller L. Discovery of risdiplam, a selective survival of motor neuron‐2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy. J Med Chem. 2018;61:6501‐6517. [DOI] [PubMed] [Google Scholar]
- 18. Sturm S, Günther A, Jaber B, Jordan P, Kotbi NA, Parkar N, Cleary Y, Frances N, Bergauer T, Heinig K, Kletzl H, Marquet A, Ratni H, Poirier A, Müller L, Czech C, Khwaja O. A Phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol. 2018. 10.1111/bcp.1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hubbard TJP, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, Coates G, Fairley S, Fitzgerald S, Fernandez‐Banet J, Gordon L, Graf S, Haider S, Hammond M, Holland R, Howe K, Jenkinson A, Johnson N, Kahari A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Lawson D, Longden I, Megy K, Meidl P, Overduin B, Parker A, Pritchard B, Rios D, Schuster M, Slater G, Smedley D, Spooner W, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wilder S, Zadissa A, Birney E, Cunningham F, Curwen V, Durbin R, Fernandez‐Suarez XM, Herrero J, Kasprzyk A, Proctor G, Smith J, Searle S, Flicek P. Ensembl 2009. Nucleic Acids Res. 2009;37:D690‐D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poirier A, Cascais AC, Bader U, Portmann R, Brun ME, Walter I, Hillebrecht A, Ullah M, Funk C. Calibration of in vitro multidrug resistance protein 1 substrate and inhibition assays as a basis to support the prediction of clinically relevant interactions in vivo . Drug Metab Dispos. 2014a;42:1411‐1422. [DOI] [PubMed] [Google Scholar]
- 21. Poirier A, Portmann R, Cascais AC, Bader U, Walter I, Ullah M, Funk C. The need for human breast cancer resistance protein substrate and inhibition evaluation in drug discovery and development: why, when, and how? Drug Metab Dispos. 2014b;42:1466‐1477. [DOI] [PubMed] [Google Scholar]
- 22. Osborne M, Gomez D, Feng Z, McEwen C, Beltran J, Cirillo K, El‐Khodor B, Lin MY, Li Y, Knowlton WM, McKemy DD, Bogdanik L, Butts‐Dehm K, Martens K, Davis C, Doty R, Wardwell K, Ghavami A, Kobayashi D, Ko CP, Ramboz S, Lutz C. Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum Mol Genet. 2012;21:4431‐4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full‐length SMN. Hum Mol Genet. 2005;14:845‐857. [DOI] [PubMed] [Google Scholar]
- 24. Zaworski P, von Herrmann KM, Taylor S, Sunshine SS, McCarthy K, Risher N, Newcomb T, Weetall M, Prior TW, Swoboda KJ, Chen KS, Paushkin S. SMN protein can be reliably measured in whole blood with an electrochemiluminescence (ECL) immunoassay: implications for clinical trials. PLoS ONE. 2016;11:e0150640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caruso A, Alvarez‐Sanchez R, Hillebrecht A, Poirier A, Schuler F, Lave T, Funk C, Belli S. PK/PD assessment in CNS drug discovery: prediction of CSF concentration in rodents for P‐glycoprotein substrates and application to in vivo potency estimation. Biochem Pharmacol. 2013;85:1684‐1699. [DOI] [PubMed] [Google Scholar]
- 26. Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov. 2010;9:929‐939. [DOI] [PubMed] [Google Scholar]
- 27. Zhao R, Raub TJ, Sawada GA, Kasper SC, Bacon JA, Bridges AS, Pollack GM. Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood‐brain barrier. Drug Metab Dispos. 2009;37:1251‐1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng B, Doran AC, Di L, West MA, Osgood SM, Mancuso JY, Shaffer CL, Tremaine L, Liras J. Prediction of human brain penetration of P‐glycoprotein and breast cancer resistance protein substrates using in vitro transporter studies and animal models. J Pharm Sci. 2018;107:2225‐2235. [DOI] [PubMed] [Google Scholar]
- 29. Zhao X, Feng Z, Ling KK, Mollin A, Sheedy J, Yeh S, Petruska J, Narasimhan J, Dakka A, Welch EM, Karp G, Chen KS, Metzger F, Ratni H, Lotti F, Tisdale S, Naryshkin NA, Pellizzoni L, Paushkin S, Ko CP, Weetall M. Pharmacokinetics, pharmacodynamics, and efficacy of a small‐molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy. Hum Mol Genet. 2016;25:1885‐1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med. 2017;377:1723‐1732. [DOI] [PubMed] [Google Scholar]
- 31. Mercuri E, Kirschner J, Baranello G, Servais L, Goemans N, Pera M, Marquet A, Seabrook T, Sturm S, Armstrong G, Kletzl H, Czech C, Kraus D, Abdallah H, Mueller L, Gorni K, Khwaja O. Clinical studies of RG7916 in patients with spinal muscular atrophy: SUNFISH part 1 study update. Neuromuscul Disord. 2017;27:S209. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials