

Abstract
The functions of most long non-coding RNAs (lncRNAs) are unknown. In contrast to proteins, lncRNAs with similar functions often lack linear sequence homology; thus, the identification of function in one lncRNA rarely informs the identification of function in others. We developed a sequence comparison method to deconstruct linear sequence relationships in lncRNAs and evaluate similarity based on the abundance of short motifs called kmers. We found that lncRNAs of related function often had similar kmer profiles despite lacking linear homology, and that kmer profiles correlated with protein binding to lncRNAs and with their subcellular localization. Using a novel assay to quantify Xist-like regulatory potential, we directly demonstrated that evolutionarily unrelated lncRNAs can encode similar function through different spatial arrangements of related sequence motifs. Kmer-based classification is a powerful approach to detect recurrent relationships between sequence and function in lncRNAs.
The human genome expresses thousands of lncRNAs, several of which regulate fundamental cellular processes. Still, the overwhelming majority of lncRNAs lack characterized function and it is likely that physiologically important lncRNAs remain to be identified. Moreover, the mechanisms through which most lncRNAs act are not clear, limiting our understanding of the biology that they govern in cells 1–12.
A significant roadblock to progress remains the inability to detect recurrent relationships between lncRNA sequence and function. An understanding of analogous relationships in proteins has enabled the classification of protein families, functional domains, and mechanisms that, in turn, have led to discoveries that have improved the diagnosis and treatment of disease 13,14. However, with rare exceptions, the functions of lncRNAs are unrecognizable from computational analyses and must be determined empirically 10–12,15–20. As a result, classification of function in one lncRNA often provides no information about function in others. For example, the Xist and Kcnq1ot1 lncRNAs both repress gene expression in cis (meaning on the same chromosome from which they were transcribed), and both require the Polycomb Repressive Complex to do so 7. Yet, despite similarities in mechanism, the two lncRNAs share almost no sequence similarity by standard metrics. Using two common sequence alignment algorithms, nhmmer 21 and Stretcher 22, Xist and Kcnq1ot1 appear just as similar to each other as they do to randomly generated sequences (Supplementary Fig. 1). Thus, comparing the sequence of Kcnq1ot1 to a known cis-repressive lncRNA (Xist) provides no indication that Kcnq1ot1 is also a cis-repressive lncRNA. This problem extends to the thousands of lncRNAs that lack characterized functions.
Results
Kmer-based quantitation as a means to compare lncRNA sequence content
We hypothesized that lncRNAs with shared functions should harbor sequence similarities that confer the shared functions, even if conventional alignment algorithms do not detect the similarity. Our rationale follows. First, most lncRNAs likely have no catalytic activity, suggesting that the proteins they bind in cells define their function. Second, proteins often bind RNA through short motifs, or kmers, that are between 3 to 8 bases in length, where “k” specifies the length of the motif 23,24. Third, the mere presence of a set of protein binding motifs may be more important than their relative positioning within a lncRNA, meaning that functionally related lncRNAs could harbor related motif contents and still lack linear sequence similarity.
To test our hypothesis, we developed a method of sequence comparison, called SEEKR (SEquence Evaluation from Kmer Representation). In SEEKR, all kmers of a specified length “k” (i.e. k= 4, 5, or 6) are counted in one-nucleotide increments across each lncRNA in a user-defined group, such as the GENCODE annotation set 12. Kmer counts for each lncRNA are then normalized by lncRNA length and standardized across the group to derive a matrix of kmer profiles, which consist of z-scores for each kmer in each lncRNA. The relative similarity of kmer profiles between any pair of lncRNAs can then be determined via Pearson’s correlation (Fig. 1A, B; Methods).
Figure 1. Overview and initial test of kmer-based sequence comparison.

(A) LncRNAs of related function (names in black) may harbor similar sequence similarity in the form of motif content (colored bars) even if they lack linear homology. (B) In SEEKR, the abundance of all kmers of length k are counted by tiling across each lncRNA in a user-defined group in one nucleotide increments. Kmer counts are normalized for lncRNA length, and standardized across the group to derive z-scores. Similarity is evaluated by comparing lncRNA kmer profiles (lists of z-scores for each kmer in the lncRNAs) with Pearson’s correlation. (C) Number of homologous pairs detected by SEEKR vs. kmer length in a test set of conserved lncRNAs. Green and orange lines mark the homologue number detected by Stretcher and nhmmer, respectively. (D) Signal to background ratios for homologue detection via the three methods. Tukey boxplots show the lower, median, and upper quartile of values, and ±1.5x the IQR (n=161 r values for signal, n=12880 r values for background); outliers are not shown.
SEEKR offers advantages relative to existing alignment algorithms. Foremost, SEEKR does not consider positional information in similarity calculations, allowing it to quantify nonlinear sequence relationships. For reasons described above, this functionality might suit lncRNAs better than traditional alignment algorithms developed to detect linear sequence homology between evolutionarily related entities 21,22,25,26. Second, whereas traditional alignment algorithms can only quantify similarity, SEEKR can quantify similarities and differences using Pearson’s correlation. Third, SEEKR can quantify relationships in groups of lncRNAs despite differences in overall length, whereas length differences can confound traditional alignment algorithms. For example, conventional alignment of a 20kb and 4kb RNA is barely informative (80% of the 20kb RNA would not align), but their kmer contents can be compared via SEEKR. Lastly, SEEKR is algorithmically efficient; all pairwise comparisons between human GENCODE lncRNAs can be computed in under a minute.
Initially, we assessed whether SEEKR could detect previously identified sequence similarities in lncRNAs. We compared kmer profiles via SEEKR for all pairwise combinations in a set of 161 lncRNAs recently described to be conserved between human and mouse 27. We also aligned the lncRNAs to each other using two existing alignment algorithms, the hidden Markov model based nhmmer 21, and Stretcher, an implementation of the global alignment algorithm Needleman-Wunsch 22. In this test, SEEKR detected known lncRNA homologues nearly as well as or better than both algorithms (Fig. 1C). We defined signal to background in this assay as the ratio between the median similarity of homologous and non-homologous lncRNAs. By this metric, nhmmer detected homologues the most clearly, as expected (signal-to-background ratio of 0.606 : 0.000), followed by SEEKR (signal-to-background of 0.152 : −0.003 at kmer length k=6), and Stretcher (signal-to-background of 0.525 : 0.307; Fig. 1D). We conclude that kmer-based classification can detect sequence similarity between evolutionarily related lncRNAs.
We next examined if SEEKR could detect novel forms of similarity between lncRNAs with no known sequence homology. We created kmer profiles for all lncRNAs in the human and mouse GENCODE databases 12, as well as for select lncRNAs that were not included in GENCODE. Next, we compared kmer profiles between all lncRNAs in each organism using Pearson’s correlation and hierarchically clustered the resulting matrices to examine the patterns that emerged. Consistent with our hypothesis, clustering lncRNAs by SEEKR grouped many by known function in human and mouse (Fig. 2). Several known cis-repressive lncRNAs, including XIST, TSIX, KCNQ1OT1, UBE3A-ATS, ANRIL/CDKN2B-AS1, and Airn clustered together due to high abundance of AU-rich kmers, whereas several cis-activating lncRNAs, including PCAT6, HOTTIP, LINC00570, DBE-T, and HOTAIRM1, clustered separately due to high abundance of GC-rich kmers (Figs. 2A and D). These patterns were robust over differing kmer lengths (Supplementary Fig. 2). To determine if this level of clustering was significant, we curated lists of human and mouse cis-activating and cis-repressive lncRNAs from the literature (Supplementary Table 1), and compared average pairwise kmer similarities between lncRNAs in each list to pairwise similarities of 10,000 size-matched lists of randomly selected lncRNAs from the respective organism. Human and mouse cis-repressors, and human cis-activators (but not mouse cis-activators), were significantly more similar to each other than expected by random chance (Supplementary Table 2). Concordantly, SEEKR detected significant similarity between the cis-repressive Kcnq1ot1 and Xist lncRNAs where none was found by conventional alignment algorithms (Supplementary Fig. 1). We conclude that lncRNAs of related function can have related kmer profiles even if they lack linear sequence similarity.
Figure 2. LncRNAs of related function often have related kmer contents.
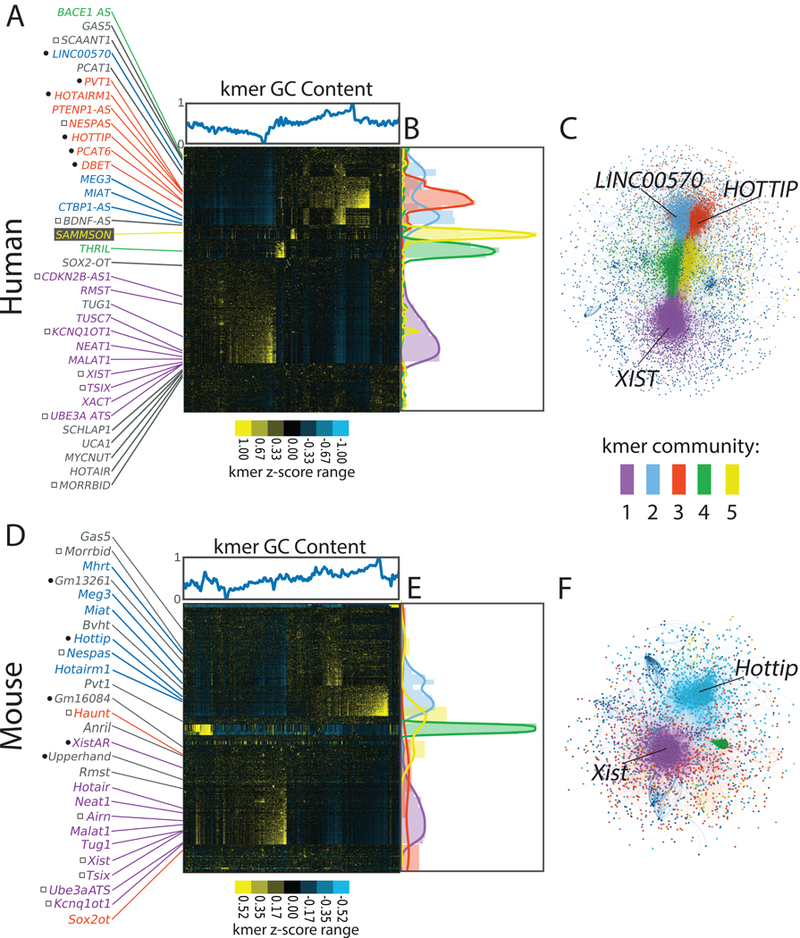
(A) Hierarchical cluster of all human GENCODE lncRNAs at kmer length 6, with lncRNAs and kmers on the x- and y-axes, respectively. Kmer z-scores (relative kmer abundance) range from blue (lowest) to yellow (highest). GC content of kmers is shown above the x-axis. Locations of select lncRNAs are marked. Left of lncRNA names, black circles indicate cis activators and squares indicate cis repressors. (B) Locations of lncRNAs assigned to communities 1 through 5 via the Louvain/network-based approach. (C)Network graph of Louvain-assigned lncRNA communities. LncRNA names in (A) are colored by their Louvain community assignment; lncRNAs in gray were assigned to the null. (C, D, E) Same as (A, B, C) but for mouse GENCODE lncRNAs.
Unexpected relationships also emerged in the hierarchical clusters of Fig 2. Most notably, the lncRNAs NEAT1 and MALAT1 showed greater than average similarity to XIST in both human and mouse. Among all human lncRNA pairwise comparisons, their Pearson’s r values fell in the 99.99th and 99.60th percentile, respectively. Likewise, in mouse, the similarities were in the 97.15th and 95.32nd percentiles. The meaning of the similarity between the three lncRNAs is unclear, but we note that all three lncRNAs seed the formation of sub-nuclear compartments and engage with actively transcribed regions of the genome 28–33. We speculate that their kmer similarity is related to these shared actions.
LncRNAs can be partitioned into communities of related kmer content
We next used a network-based approach to partition lncRNAs into communities of related kmer profiles, reasoning that such communities would provide a framework to understand the predictive value of lncRNA kmer content. We created networks of relationships between all human and mouse lncRNAs in which weighted edges connected lncRNAs in an organism if the Pearson’s correlation between their standardized kmer profiles met a threshold for similarity (Methods). We then used the Louvain method to assign lncRNAs within the largest connected component of the network representations to communities of related kmer profiles 34. Approximately half of all GENCODE lncRNAs grouped into five major communities in both human and mouse. LncRNAs not assigned to the five most populated communities were assigned to a “null” community. Our network-based approach and hierarchical clustering grouped lncRNAs in similar ways (p < 1e-324, Chi-squared; Supplementary Tables 3 and 4), signaling community robustness. LncRNA community assignments and associated summary statistics are provided in Supplementary Tables 5–12 and Supplementary Fig. 3. Differences in human and mouse community structures may be due in part to differences in completeness of lncRNA annotation. In the versions of GENCODE used for this work, there were about twice as many lncRNAs annotated in human (v22, n=15953) as there were annotated in mouse (vM5, n=8245 12).
Kmer content correlates with localization and protein binding
We next examined whether lncRNAs with related kmer profiles shared biological properties. For this analysis, we focused on human lncRNAs, where data from the ENCODE project allowed us to examine lncRNA subcellular localization and protein associations, transcriptome-wide. To determine whether kmer content provides information about lncRNA localization, we examined ENCODE subcellular fractionation RNA-Seq experiments performed in HepG2 and K562 cells 35. For each lncRNA expressed in each cell type, we computed its nuclear ratio and determined whether the distributions of nuclear ratios differed between communities. The majority of communities showed slight but significant differences in their distribution of nuclear ratios, with the largest differences found between communities #1 and #3 (Fig. 3A and Supplementary Tables 13–16). Concordantly, lncRNAs that associate with polysomes in K562 cells 36 were also non-uniformly distributed between communities (p = 3.5e-5, Chi-squared), and were the most over- and under-represented in the most cytoplasmic and nuclear lncRNA communities, respectively (communities #3 and #1 being the most cytoplasmic and nuclear, respectively; Supplementary Table 17). Lastly, we used ENCODE data to identify the most cytoplasmic and nuclear lncRNAs in HepG2 and K562 cells and determine which kmers were asymmetrically distributed between lncRNAs in the two compartments. 360 and 27 kmers were significantly enriched in cytoplasmic and nuclear lncRNAs, respectively (p-adjusted <0.05; Kolmogorov–Smirnov test; Supplementary Table 18). Consistent with our RNA-Seq and polysome analyses, 58 and 93% of the cytoplasmic- and nuclear-biased kmers were the most enriched in the most cytoplasmic and nuclear lncRNA communities, respectively (communities #3 and #1; Supplementary Table 18, last column). We conclude that kmer content provides information about the subcellular localization of a lncRNA.
Figure 3. LncRNA localization and protein binding correlate with kmer content.
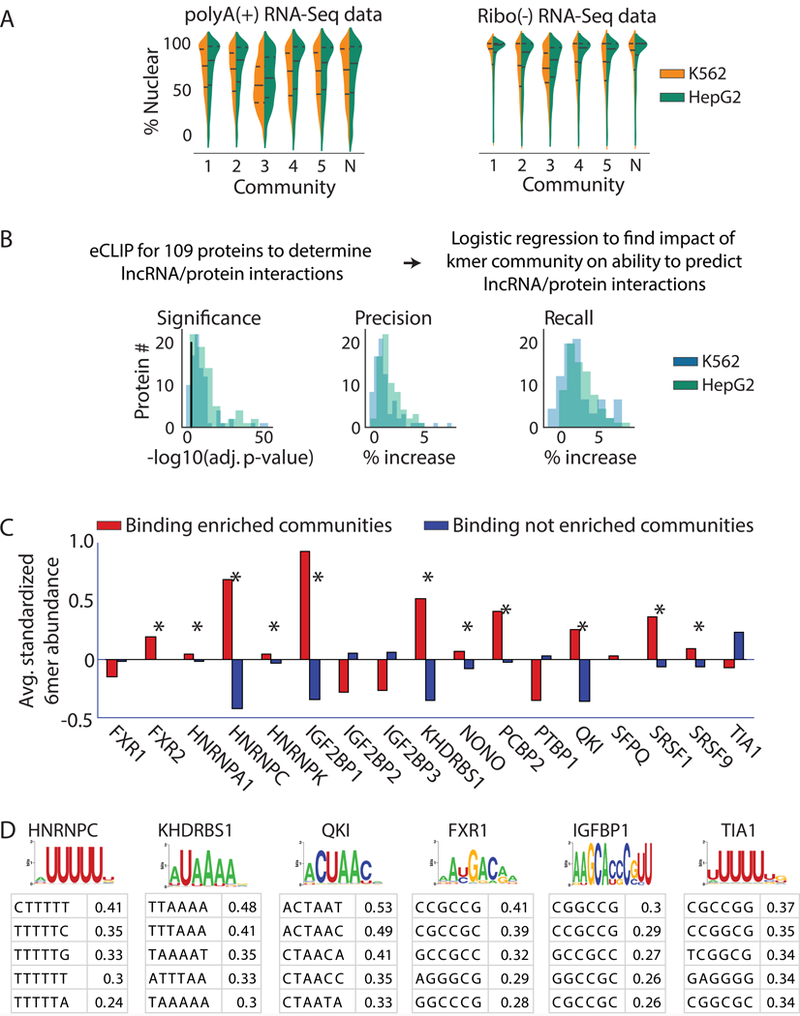
(A) Violin plots of lncRNA localization by kmer community in K562 (blue) and HepG2 (green) cells, as determined from RNA-Seq of polyA-selected and ribosome-depleted RNA. “N”, the “null” community. Lines show the lower, median, and upper quartile of values (see Supplemental Figs. 13–16 for samples sizes). (B) From left to right; Log10 significance of increase in likelihood (i), % increase in precision (ii), and % increase in recall (iii) obtained when lncRNA community information is included in a logistic regression to predict protein association. Black line in (i) corresponds to a log10(adjusted p-value) of 0.05 (n=3747 lncRNAs for HepG2, n=3278 lncRNAs for K562). (C) 11 of the 17 proteins with experimentally determined PWMs from 23 show significantly increased abundance of motif-matching kmers (n=4096) in lncRNA communities that are enriched for binding to the protein in question (p<0.01; permutation test; marked by *’s). (D) The most enriched kmers in 300 nucleotide windows surrounding motif matches in CLIP peaks do not always match the motif. PWMs from 23 are shown above average z-scores for the top 5 most enriched kmers in true positive relative to false positive binding regions for the protein in question. PWMs and top kmers are shown for all 17 proteins in Supplementary Fig. 5.
To determine if kmer content provides information about protein binding in lncRNAs, we examined ENCODE data for 156 eCLIP experiments performed for 109 proteins in HepG2 and K562 cells 37. We created binary vectors for each experiment that recorded whether the lncRNAs bound or did not bind a given protein, then built separate logistic regression models for each protein to determine if kmer community assignments could improve prediction of lncRNA/protein associations over a null model that only included lncRNA length and expression as covariates. LncRNA community assignments significantly increased the log-likelihood of detecting lncRNA/protein associations for the majority of proteins examined (p-adjusted <0.05;146/156, ~94%; Fig. 3B and Supplementary Table 19). Increases in precision and recall in community-informed models were generally modest but significant (Fig. 3B and Supplementary Table 20). In total, ~17% (25/146) of our models had an increase in precision and/or recall of 5% or more. Notably, in all cases in which recall increased, precision also increased, indicating that kmer community information increased the ability to predict true lncRNA-protein associations and simultaneously increased the fidelity of those predictions. When we used individual 6mers instead of lncRNA communities as predictive features, results were no better than the null model that used only lncRNA length and expression as predictive features. Models with more features than samples are prone to learning noise in their training set, and often lose predictive power, due to overfitting 38. Using individual 6mers brought the number of features being evaluated to 4099, more than the number of lncRNAs expressed in HepG2 and K562 cells (3745). We conclude that kmer content provides information about the protein-binding potential of a lncRNA, but that no single kmer provides an overwhelming portion of that information, and, that kmer communities provide a way to collapse high-dimensional kmer matrices down to representative variables for predictive purposes.
Protein binding to RNA is difficult to assess from motif content alone due to the degeneracy of most motifs and the challenge of predicting the effects of RNA structure 24,39–41). Supporting this notion, we found that the abundance of motif-matching kmers was consistently, but not always, higher in the communities enriched for binding of specific proteins than in the cognate communities not enriched for binding, indicating that factors in addition to motif abundance control protein/lncRNA associations (Fig. 3C). We therefore sought to determine if kmer content could distinguish between motif matches in lncRNAs that coincide with protein binding events and those that do not. We searched the lncRNAs expressed in HepG2 and K562 cells for matches to binding motifs of the 17 proteins in Fig. 3C, whose position weight matrices were determined from biochemical assays in 23. We annotated motif matches that fell inside and outside of CLIP peaks as true and false positive matches, respectively. As expected, the majority of motif matches fell outside of CLIP peaks (i.e., they were false positive matches; Supplementary Table 21). We then used SEEKR to compare regional kmer content in 300 nucleotide windows surrounding true and false positive motif matches. Remarkably, for 13 of 17 proteins examined, kmer profiles of true positive binding regions were more similar to each other than kmer profiles of randomly selected, size-matched sets of false positive regions (p-value < 0.005; Supplementary Fig. 4). These data support the notion that binding modules for the same protein in different RNAs often have sequence similarity that extends beyond the protein binding motif, and that this similarity can be quantified, in part, by local kmer content.
Moreover, SEEKR provides a simple way to visualize the density of specific kmers within CLIP enriched regions. We compared the most overrepresented kmers in true positive binding regions to protein binding motifs measured in vitro 23, and found that their relationships differed substantially from protein to protein (Fig 3D and Supplementary Fig. 5). For certain proteins, such as HNRNPC, KHDRBS1, and QKI, the most enriched kmers in true positive regions matched the PWM for the protein that was determined in vitro 23. We interpret this observation to mean that for these proteins, motif density plays a dominant role in determining RNA binding in vivo, because our kmer data show that motif-matching kmers are more abundant in true positive regions than they are in false positive regions. For other proteins, such as FXR1, IGFBP1, and TIA1, the most enriched kmers in true positive regions did not match the PWM determined in vitro 23. For these proteins, sequence beyond the binding motif may play a dominant role in dictating association with RNA, possibly due to effects from RNA structure. When PWMs were extracted from eCLIP peaks, similar relationships between kmers and in vitro defined motifs were observed (Supplementary Fig. 5). These results show how SEEKR can be used to augment traditional motif-based analyses and provide insights into mechanisms of RNA-protein interaction. SEEKR provides a way to quantify sequence similarities between any number of protein binding regions, which in turn, can provide predictive power and identify shared characteristics that are not apparent from PWM-based motif analyses.
Similarities in lncRNA communities between organisms
Given (i) that kmer content provides some indication of protein binding potential in a lncRNA, (ii) that sequence specificities of many RNA binding proteins are conserved 23,24, and (iii) that protein binding likely dictates lncRNA function, we hypothesized that kmer contents between communities of functionally related lncRNAs could be conserved even if the lncRNAs themselves lack known evolutionary relationships. In support of this idea, we identified extensive similarity between certain human and mouse lncRNA communities via SEEKR (Methods; Supplementary Fig. 6). Most notably, lncRNAs in human community #1 (the “XIST” community) had kmer profiles that were, as a group, nearly indistinguishable from lncRNAs in mouse community #1 (the “Xist” community) and were also similar to lncRNAs in mouse community #4 (p<0.0001 for both comparisons). Human community #2 and community #3 (the “HOTTIP” community) were both similar to mouse community #2 (the “Hottip” community; p<0.0001). No other major similarities between mouse and human were apparent. Extending this analysis across greater evolutionary distance, we found HOTTIP-like lncRNA communities in ten of ten vertebrates examined as well as in the sea urchin S. purpuratus, and XIST-like lncRNA communities in seven of ten vertebrates examined (Supplementary Figs. 7–9; 10). These analyses demonstrate that, at the level of kmers, subsets of human lncRNAs are more similar to lncRNAs in other genomes than they are similar to lncRNAs in their own genome, supporting the idea that groups of lncRNAs have similar function in different organisms despite lacking obvious linear sequence similarity.
SEEKR can predict Xist-like regulatory potential in lncRNAs
We next directly tested whether kmer profiles could be used to predict lncRNA regulatory potential. We focused on the ability of certain lncRNAs to repress transcription in cis. Cis-repression was one of the earliest characterized functions of lncRNAs, and is essential for normal human health and development. In the most striking example, the XIST lncRNA silences nearly all genes across an entire chromosome during X-chromosome Inactivation 7. Cis-repression is also one of most straightforward lncRNA functions to study because, by definition, cis acting lncRNAs act near their site of transcription.
We developed a reductionist assay to study lncRNA cis-repressive activity in a normalized genomic context, called TETRIS (transposable element to test RNA’s effect on transcription in cis). TETRIS enables the sequence of a lncRNA and an adjacent reporter gene to be manipulated in a plasmid, but then rapidly inserted into chromosomes via the piggyBac transposase 42,43, so that effects of the lncRNA on the reporter can be studied in genomic chromatin (Fig. 4A and Methods). Under our assay conditions, piggyBac catalyzes 4–7 insertions of each cargo per stably selected cell, and cell density estimates suggest between 100,000 to 500,000 cells receive insertions and survive selection (Fig. 4B and not shown). Thus, each TETRIS assay likely surveys 400,000 to 3.5 million insertion events. Insertion-site dependent variation in lncRNA-induced effects are averaged out in the population, bypassing the need to isolate clones of modified cells, and providing the means to quantify lncRNA regulatory potential without influence from genomic position.
Figure 4. Kmer content correlates with lncRNA repressive activity.
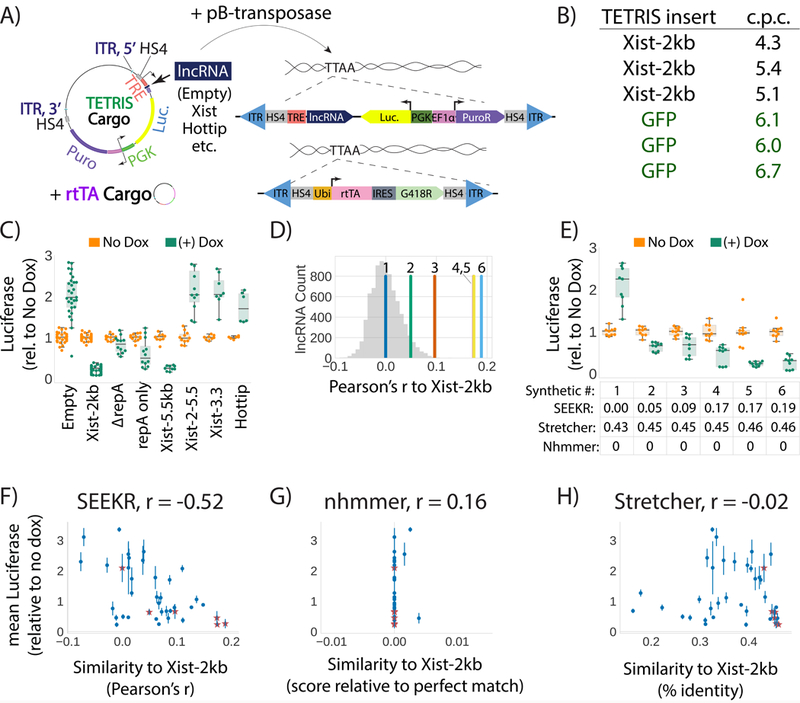
(A) Overview of vectors and concept of the TETRIS assay. (B) Number of TETRIS-lncRNA-cargo insertions per cell (“c.p.c.”) after 10-day drug selection for two separate cargos, Xist-2kb and GFP. Each row represents copy number data from independent replicates. (C) Luciferase values for different TETRIS-lncRNA constructs relative to No Dox. Tukey boxplots as in Fig. 1D. Data are from at least six independent luciferase assays from at least two biological replicate derivations of TETRIS cell lines. Exact numbers of assays and replicates performed for each TETRIS lncRNA cargo are found in Supplementary Table 22. (D) Pearson’s r similarity of kmer profiles for the six synthetic lncRNAs relative to the first 2kb of Xist. Histogram of similarity of Xist-2kb to all other GENCODE M5 lncRNAs is shown in gray. (E) Effect of synthetic lncRNA expression on luciferase activity. Tukey boxplots as in Fig. 1D. SEEKR, Stretcher, and nhmmer similarity for each synthetic lncRNA relative to the first 2kb of Xist is shown below the graph. (F, G, H) Pearson’s correlation between repressive activity and similarities to Xist-2kb as defined by SEEKR, nhmmer, and Stretcher for thirty-three endogenous lncRNAs/lncRNA fragments (dots) and six synthetic lncRNAs (stars) (mean ± standard deviation). See Supplementary Table 22 for sample sizes in panels C, E, F, G, H.
We validated TETRIS by comparing effects that expression of different lncRNAs had on luciferase activity. A cell line created from a vector that lacked a lncRNA insert (TETRIS-Empty) showed a ~2-fold increase in luciferase activity upon addition of doxycycline, representing our baseline for the assay (Fig. 4C). We attribute this mild activation to the close proximity of the dox-inducible and luciferase promoters, and to the fact that both promoters are contained within the same insulated domain 44. By contrast, expression of the first 2kb of Xist repressed luciferase 5-fold relative to uninduced control (Fig. 4C). The 2-fold activation and 5-fold repression were stable across nine and 16 independent derivations of TETRIS-Empty and TETRIS-Xist-2kb cell lines, respectively (mean ± standard deviation of 2.03 ± .50 and 0.23 ± .08), demonstrating that TETRIS assays result in reproducible effects on luciferase activity. For its repressive effect, Xist requires “Repeat A,” a 425-nucleotide long element contained within its first 2kb 45. In the context of TETRIS, deletion of Repeat A resulted in a significant, but not complete, de-repression of luciferase, whereas expression of Repeat A alone resulted in repression relative to control, but at reduced levels compared to Xist-2kb (“∆repA” and “repA only”; Fig. 4C). Similarly, expression of the first 5.5kb of Xist caused a 5-fold repression of luciferase, whereas deletion of the first 2kb from the 5.5kb construct caused complete loss of repressive activity (“Xist-5.5kb” and “Xist-2–5.5”; Fig. 4C). Expression of either the final 3.3kb of Xist or the Hottip lncRNA had no repressive effect (Fig. 4C). These experiments demonstrate (i) that TETRIS is a suitable assay to measure repression by cis-acting lncRNAs in a normalized genomic context, and (ii) in the assay, sequence elements in addition to Repeat A cooperate to encode repressive function in the 5´ end of Xist.
We next used TETRIS and SEEKR to test our hypothesis that kmer content can predict lncRNA regulatory potential. We reasoned that we could design entirely synthetic lncRNAs that lacked linear sequence similarity to any known lncRNA but nonetheless had robust Xist-like repressive activity. We generated six synthetic lncRNA sequences in silico with varying levels of kmer similarity to the first 2kb of Xist, and cloned them into TETRIS to measure their effects on luciferase activity. As measured by SEEKR, the lncRNAs had Pearson’s similarities to Xist that ranged from average (a Pearson’s r of ~0) to three standard deviations above the mean similarity for all mouse lncRNAs (a Pearson’s r of 0.19, more similar to Xist-2kb than all other lncRNAs the mouse genome; Fig. 4D). Using nhmmer or Stretcher to align the synthetic lncRNAs to the first 2kb of Xist produced either no alignments (nhmmer) or alignments that differed by only three percent across all six synthetic lncRNAs (Stretcher; Fig. 4E, grid below graph). Via BLAST, the lncRNAs had no significant similarity to the mouse genome or to each other (not shown). The lack of informative alignments was expected because the synthetic lncRNAs have no evolutionary relationship with Xist, any region in the genome, or each other. Nevertheless, as envisioned, the synthetic fragments that SEEKR classified to be most similar to Xist had the highest repressive activity (Fig. 4E). These data directly demonstrate that evolutionarily unrelated lncRNAs can encode similar function through different spatial arrangements of related sequence motifs. Thus, kmer content can be used to predict lncRNA regulatory potential.
We next examined whether SEEKR could predict Xist-like repressive activity in endogenous lncRNAs. We cloned into TETRIS thirty-three lncRNAs or lncRNA fragments that had a range of kmer similarities to the first 2kb of Xist. Included in our final set of fragments were several conserved lncRNAs and/or shorter fragments contained within them (Airn, Hottip, Kcnq1ot1, Malat1, Neat1, and Pvt1), as well as many lncRNAs with uncharacterized functions (Supplementary Table 22). Again, the more Xist-like a lncRNA fragment was at the level of kmers, the more likely it was to repress in TETRIS; the Pearson’s r value between Xist-likeness at a kmer length of 6 and luciferase activity upon dox addition was −0.41 (p=0.02). Including the six synthetic lncRNAs in the correlation brought the Pearson’s r value to −0.52 (p=0.0007; Fig. 4F). Nhmmer and Stretcher had no ability to predict repressive activity, demonstrating that these algorithms cannot detect sequence signatures correlated with repressive activity in this setting (p=0.32 and 0.91, respectively; Fig. 4G and H). LncRNA fragment length also had no ability to predict repressive activity (r=0.03, p-value=0.84).
Lastly, we examined whether kmer profiles associated with sequence elements required for repression by Xist-2kb might increase our ability to predict repressive activity in other lncRNAs. To determine the elements in Xist-2kb required for repression, we made a series of 26 deletions (Fig. 5). Surprisingly, 15 of the deletions, including ones that removed predicted stable structures, pseudoknots, and ~40% of Repeat A (“∆ΔSS1”, “ΔSS2”, “ΔPK2”, “ΔSS3”, “ΔSS4”; bottom panel in Fig. 5; 41), had no significant effect on repression. However, removal of all eight GC-rich portions of Repeat A, but not its U-rich linkers, caused a ~3-fold reduction in repression (“ΔGC repeat in rA” vs “ΔU spacer in rA”), as did removal of three predicted stable structures and their intervening sequences in the 742 nucleotides immediately downstream of Repeat A (“ΔSS2/3/4 broad”; 41). Co-deletion of Repeat A and the stable structures had an additive effect, causing a near complete loss of repression (the “ΔrAΔSS234 br.” mutant), whereas expression of Repeat A or the stable structures alone had half the repressive potency of Xist-2kb (“Only rA” and “Only SS234”). Expression of both regions together had the same repressive potency as Xist-2kb (“Minimal”). Thus, in TETRIS, the major elements required for repression are contained between nucleotides 308 and 1,476 of Xist. Based on prior structural models 41,46, we infer that the elements are comprised of protein binding sites, spacer sequences, and stable structures.
Figure 5. Mapping of elements required for repression by Xist-2kb in TETRIS.
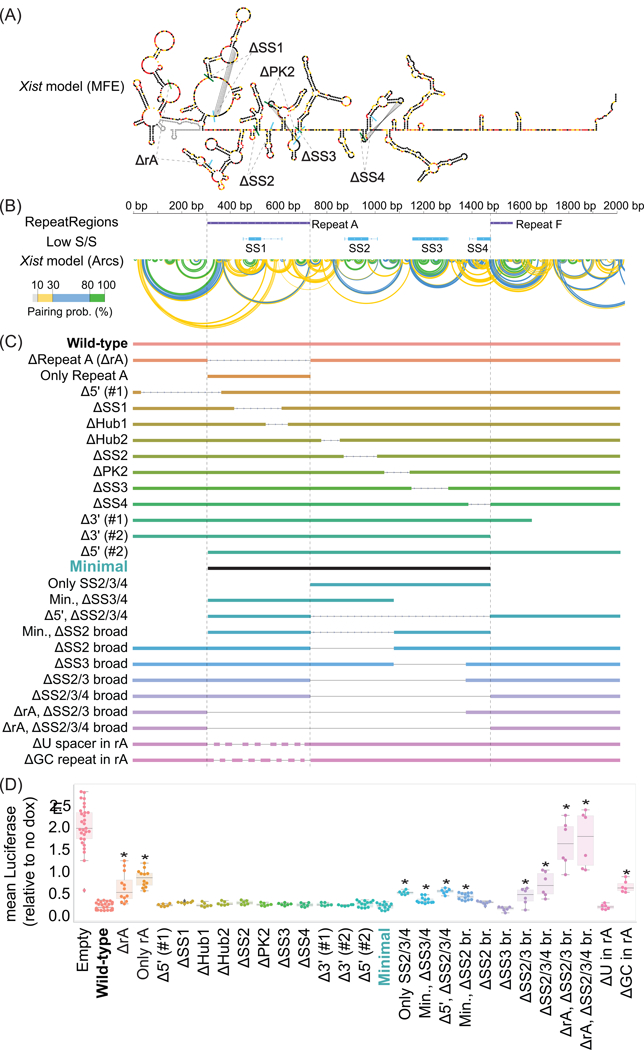
(A) Minimum Free Energy (MFE) and (B) arc-based structural models of the first 2kb of Xist from 41; green and blue bars in (i) mark starts and stops of indicated regions; locations of Xist repeats 7 and predicted stable structures (low S/S, regions of low SHAPE reactivity and Shannon entropy from 41) are also shown in (ii). (C) Deleted regions. (D) Effects on luciferase after dox addition. *, Bonferroni corrected p<0.001 relative to Wild-type/Xist-2kb via Student’s t-test. Tukey boxplots show the lower, median, and upper quartile of values, and ±1.5x the IQR (see Supplementary Table 22 for sample sizes and exact p-values).
Having mapped the elements responsible for repression in Xist-2kb, we attempted to extract subsets of 6mers from them that increased our ability to predict Xist-like repression. We also examined if kmer variance across lncRNA communities or kmer nucleotide composition could be used to extract subsets of outperforming 6mers, and if different kmer lengths had better predictive power than k=6. No rationally designed subset of 6mers could predict repression better than the full 6mer profile of Xist-2kb, nor could any other kmer length (Supplementary Fig. 10). These results support the ideas that different lncRNAs can encode similar function through related, but not necessarily identical, sequence solutions, and that the full complement of 6mers may be a broadly effective search tool to identify such similarities (not too relaxed, not too stringent).
Discussion
Collectively, our data support the notion that many lncRNAs function through recruitment of proteins that harbor degenerate RNA binding motifs, and that spatial relationships between protein binding motifs in these lncRNAs are often of secondary importance to the concentration and effectiveness of the motifs themselves. By this logic, a lncRNA may merely need to present the appropriate motifs embedded within the appropriate structural contexts to achieve a specific function. Thus, different lncRNAs likely encode similar function through vastly different sequence solutions, and nonlinear sequence comparisons can be used to discover similarities between them. By extension, because the RNA binding motifs of many proteins are conserved 23,24, it is likely that groups of lncRNAs rely on similar motifs to encode related function in different organisms even though they lack direct evolutionary relationships. This concept is supported by our observation that lncRNA communities with related kmer contents exist in human, mouse, and other organisms. We propose that nonlinear sequence homology – in which the relative abundance of a set of protein binding motifs is conserved, but the sequential relationships between them are not – is prevalent in lncRNAs. To quantify nonlinear homology, we introduce SEEKR, a method to compare sequence content between any group of lncRNAs, regardless of the size of the group, the evolutionary relationships between the lncRNAs being analyzed, or the differences in their lengths. Each lncRNA (and each functional domain within each lncRNA) has its own kmer signature, which can encode information about protein binding and RNA structure. SEEKR provides a simple way to tie this information to a biological property.
Methods
Kcnq1ot1 versus Xist comparison
Kcnq1ot1 was aligned to Xist using nhmmer and Stretcher with default parameters. To assess significance of the alignments, we generated 1,000 pseudo-Kcnq1ot1s that were the same length of real Kcnq1ot1 but composed of nucleotides randomly selected from a distribution of the mononucleotide content of Kcnq1ot1 (0.335 A: 0.205 G: 0.202 C: 0.258 T). We then aligned the pseudo-lncRNAs to Xist with nhmmer and Stretcher as well as compared their kmer contents relative to all other mouse lncRNAs at kmer length k=6 via SEEKR.
SEEKR
In SEEKR, a matrix of kmer counts for a user-defined set of lncRNAs is created by counting all occurrences of each kmer in each lncRNA in one-nucleotide increments, and then dividing those counts by the length of the corresponding lncRNA. Z-scores are then derived for each kmer in each lncRNA by subtracting the mean length-normalized abundance of each kmer in the group of lncRNAs being analyzed from the length-normalized abundance of the kmer in the lncRNA in question, and then dividing that difference by the standard deviation in abundance of that kmer in the group of lncRNAs being analyzed. We refer to the array of z-scores for each kmer in a given lncRNA as its kmer profile. Similarity between any two lncRNAs can be calculated by comparing their kmer profiles with Pearson’s correlation.
Our rationale for length normalization in SEEKR follows. Without length normalization, kmer profiles become difficult to interpret for lncRNAs of different lengths. For example, an RNA that is 10x longer than another RNA will have 10x the number of kmers. Without normalization, these lncRNAs would be considered dissimilar by SEEKR, regardless of the similarity in their relative concentrations of kmers. By length normalizing, SEEKR creates a list of relative kmer concentrations in a given lncRNA that is robust to differences in length. The idea that length normalization is important is supported by studies of known cis-repressive lncRNAs. At 18kb, the Xist lncRNA is the most potent cis-repressive lncRNA known. At least three other known cis-repressive lncRNAs are longer than Xist, but less potent: Airn, Kcnq1ot1, and Ube3a-ATS, are 90kb, 85kb, and 1.1Mb, respectively 7. Of these, the longest lncRNA, Ube3a-ATS, is the least potent, arguing that length alone does not account for lncRNA potency. In certain biological contexts, lncRNA length may not be relevant, or it may have varying influence on lncRNA function. However, what these contexts might be and to what extent length does or does not affect lncRNA function in them are not known and difficult to predict. We also note that Pearson’s correlation inherently normalizes for length. Thus, comparisons of kmer content that use Pearson’s correlation will eliminate length as a variable.
GENCODE lncRNA annotations
All GENCODE annotations used in this work were from human build v22 and mouse build vM5 12. For each lncRNA, only the major splice annotation was considered (the −001 isoform). In total, there were 15953 human and 8245 mouse transcripts. The heat maps in Fig. 2 were generated with GENCODE annotations plus the additional lncRNA sequences downloaded from the UCSC genome browser 47: SAMMSON, XACT, UBE3A-ATS, MORRBID, and NESPAS, (Human), and unspliced Airn, Anril, Bvht, Haunt, Morrbid, unspliced Tsix, Ube3a-ATS, XistAR, and Upperhand (Mouse).
Conservation analysis
Ninety-three pairs of human and mouse GENCODE lncRNAs were recently identified as putative homologues due to their high conservation at the DNA level 27. These 93 lncRNAs, plus an additional 68 lncRNA pairs that had equivalent names in mouse and human GENCODE annotations, formed the final set of 161 homologues that were used for the conservation analysis of Fig. 1C. For the Fig 1C. experiment, “signal” values were computed as the mean of the 161 homologue-to-homologue measurements in each of the three algorithms; likewise, background values were computed as the mean of the remaining 12880 non-homologous comparisons. Homologous pairs were defined as being “detected” if the signal value/average similarity (as determined via SEEKR, nhmmer, or Stretcher) was higher for homologue-to-homologue measurements than it was for all other lncRNA-to-non-homologue comparisons. For this analysis, nhmmer was downloaded as part of the HMMER package (URLs) and was run with --nonull2, --nobias, --noali, and -o flags set. Stretcher was used as part of Biopython (URLs) and was run with --gapopen=16, and –gapextend=4.
Hierarchical clustering and labeling
Hierarchical clustering was performed with the R package “amap” using Pearson’s as a distance metric and average linkage 48, and was visualized with Java Treeview 49. We used kmer length k=6 for our main analyses because it performed well in evolutionary comparisons (Fig. 1C), and it provided a feature number (4^6 = 4096 features) that is only marginally larger than the average length of a GENCODE lncRNA (1152 and 1471 nucleotides for human and mouse lncRNAs, respectively).
Clustering of known cis-activating and cis-repressive lncRNAs
We performed a literature review to curate lists of experimentally verified cis-repressive and cis-activating lncRNAs in mouse and human (Supplementary Table 1). We calculated the mean pairwise similarity between all lncRNAs in each of these groups, and compared those means the distribution of mean similarities calculated from pairwise comparisons of 10,000 randomly selected, size-matched groups of lncRNAs in their respective organism to generate p-values that describe the likelihood that the similarity observed between the functionally related cis-acting lncRNAs was greater than would have been expected from random chance (Supplementary Table 2).
Network analysis and lncRNA community definition
Networks of lncRNAs were formed from a weighted adjacency matrix in which edges between any two lncRNAs were kept only if their Pearson’s r-value was at least 0.13. We selected the lncRNAs within the largest connected component of this network representation and used the Louvain algorithm 34 at default resolution parameter to assign lncRNAs to communities of related kmer profiles (using the Python package “louvain-igraph”). This decision was supported through use of the recently developed CHAMP algorithm 50 (URLs), which found a wide domain of optimality around the default resolution parameter. We retained assignments for the lncRNAs present in the top five most populated communities, and assigned the remaining lncRNAs, including those not found in the largest connected component of the network representation, to the “null” community, which served as an important outgroup for our comparisons of kmer content and biological properties in Fig. 3. Multiple Pearson’s r value thresholds between 0.12 and 0.21 were tested for human lncRNAs and we found little to no difference in community definition, correlation with lncRNA localization, or ability to predict protein-binding patterns (not shown). Gephi was used for network visualization (URLs). Community colors were automatically assigned by Gephi according to the size of each community.
We also compared communities generated with 5mers and 7mers to those generated with 6mers. We created contingency tables that compared the distribution of lncRNAs in each of the five major 6mer communities plus the null to the distribution of lncRNAs in each of the five major 5mer and 7mer communities plus their respective nulls. P-values comparing communities between the kmer lengths were all < 1E-324 (chi-squared), indicating that community definitions are largely stable when 5mers, 6mers, or 7mers are used (Supplementary Table 9 and 10). This stability, the quality of our TETRIS predictions when using 6mers (Supplementary Fig. 10), and the computational inefficiency of performing operations on matrices of 7mers or greater provided additional support for our decision to use 6mers for the bulk of our analyses.
We applied the same r-value threshold and community assignment logic that we used for human lncRNAs to define lncRNA communities using kmer length k = 6 in all other organisms.
Comparing lncRNA groups in hierarchical clusters to lncRNA communities found by Louvain
Clusters of lncRNAs with similar kmer content in human and mouse (from Fig 2.) were created by manually making cuts in the dendrogram of the hierarchical clusters that maximized the visual similarity of kmer profiles between lncRNAs in each cluster. Five cuts were made in the hierarchical cluster from each organism to approximate the five major communities found by the Louvain algorithm. We measured the similarity of the manually made clusters to the five major Louvain-defined communities by a creating contingency table that compared lncRNA distributions between the two methods. We then tested if the distribution of lncRNAs across the two sets of communities were significantly similar via a chi-squared test. In both human and mouse, the p-value was < 1E-324 (Supplementary Table 3 and 4).
LncRNA localization analysis
Localization data were downloaded from ENCODE (URLs) as fastq files and aligned to GRCh38 with STAR using default parameters 47,51. FeatureCounts was used to tabulate the number of reads aligning to our set of lncRNAs 52. We then filtered out all lncRNAs with <0.1 RPKM from each community, and calculated the number of reads in the nuclear fraction over the total number of reads from both the nuclear and cytosolic fractions for each lncRNA.
To determine if specific kmers were enriched in cytosolic or nuclear lncRNAs, we selected cytosolic- and nuclear-enriched subgroups of lncRNAs that were expressed in HepG2 or K562 cells. Because the subcellular distribution values for HepG2 or K562 expressed lncRNAs were not normally distributed (Fig. 3A), we needed to employ different thresholds to define cytosolic and nuclear so that the two groups would include similar numbers of lncRNAs. “Cytosolic” lncRNAs were defined as any lncRNA that was more than 50% cytosolic, which resulted in 2801 transcripts, and “nuclear” lncRNAs were defined as any lncRNA that was more than 95% nuclear, which resulted in 4576 transcripts. To determine the average difference in kmer abundance between lncRNAs in the two compartments, we calculated the mean value of the z-scores for each kmer in each group, and then used the difference between the means as the metric to calculate the nuclear-enrichment score (Supplemental Table 18). To test for significant differences between the distributions of z-scores between lncRNAs in the two compartments, we used a KS-test and calculated an adjusted p-value using a Bonferroni correction. This analysis yielded 387 kmers whose distributions differed significantly between cytosolic and nuclear lncRNAs (p-value < 0.05; Supplemental Table 18).
Using only the lncRNAs from community 3, we repeated the process of applying the Louvain algorithm to define communities and measure cellular localization in order to rule out the possibility that potential sub-communities were responsible for the cytosolic nature of community 3. The Louvain algorithm found four main sub-communities and all smaller sub-communities were grouped into a fifth community. The results of ANOVA tests indicated there was no significant differences between any of the communities for either the polyA-selected or ribosome-depleted RNA RNA-Seq data. We performed this analysis again for community 1, but no sub-communities were found to be significantly different (Supplementary Fig 11). This uniformity of cellular localization among possible sub-communities provides biological support for our original community definitions.
lncRNA polysome association
A recent study found 229 lncRNAs in GENCODE v22 that were polysome associated in K562 cells 36. A chi-squared test showed these 229 lncRNAs were non-randomly distributed between the communities (p-value = 3.5E-5; Supplementary Table 17). The expected values for the chi-squared test were calculated by filtering all communities for lncRNAs expressed in K562 cells, dividing the number lncRNAs in each community by the total number of expressed lncRNAs (3277), and multiplying by the number of polysomal lncRNAs (229).
LncRNA protein association data
eCLIP data were downloaded from ENCODE 35,37. For each of the 156 eCLIP experiments “bed narrowPeak” data (representing sites of protein binding that passed a ENCODE-defined threshold for enrichment over background; 35,37) were pooled from available biological duplicates. Genomic coordinates were overlapped with lncRNA exon coordinates annotated by GENCODE. Any lncRNA which overlapped with one or more eCLIP peak was considered as having a true binding interaction with the given protein. LncRNA expression data were collected from ENCODE RNA-Seq experiments in the same cell type as that of the eCLIP experiment (HepG2 or K562).
For each protein, a vector was built for each lncRNA that encoded whether the protein-lncRNA pair did or did not interact. Next, two feature matrices (null and full) were constructed. The null matrix included the log normalized values for length and expression of each of the lncRNAs. The full matrix included log normalized length and expression, as well as an additional five columns that corresponded to each of the five lncRNA communities. Each lncRNA was assigned a value of “1” in the column representing its community.
Models of protein associations
To address if lncRNA communities contained information about lncRNA/protein associations, we used a machine learning model 53. We tested if providing the model with the community data allowed it to predict interactions better than a corresponding null model that was not given the community data but still included lncRNA length and expression values as covariates. Logistic regression models were implemented with scikit-learn, using default parameters 53. The significance of the additional community information was measured with a likelihood ratio test (LRT), where the LRT statistic, D, equaled:
A chi-squared distribution was used to determine the corresponding p-value for the LRT statistic. P-values were adjusted with a Bonferroni correction for the 156 comparisons.
To quantify the extent of the effect that community inclusion had on prediction of lncRNA/protein interactions, we used a Leave-One-Out-Cross-Validation approach to measure precision and recall metrics 53, defined as:
In our model, precision is the number of lncRNAs correctly predicted to bind a protein, divided by the total number of lncRNAs the model predicted to bind a protein. Recall is the number of lncRNAs the model correctly predicted to bind a protein, divided by the total number of lncRNAs found to bind a protein according to the eCLIP data. For each lncRNA, the logistic regression models were allowed to train on all other lncRNAs except the single “left out” lncRNA. After training, both models were asked to predict if the “left out” lncRNA did or did not bind the protein. This procedure was repeated for all lncRNAs in each eCLIP dataset to calculate precision and recall.
The methodology for training and testing the raw kmer models was exactly the same as described above except that the five community features were replaced by the 4096 relative kmer abundance features.
Calculating the abundance of motif-matching kmers in lncRNA communities
The data for the bar graph in Fig. 3C were generated by the following approach. Of the 109 proteins on which eCLIP was performed in 37, 79 showed significant association with at least one kmer community over the null (Supplementary Table 19). Of these 79 proteins, binding motifs for 17 were determined via an in vitro binding assay in 23. The PWMs for each of these 17 proteins contained relative weights for each motif matching 6mer, representing the likelihood that the kmer in question would bind the protein in question. We multiplied the weight of each motif-matching 6mer by its average standardized abundance in each of the six communities, including the null, to obtain kmer abundances that were scaled by the likelihood that the kmer in question matched the binding motif in question. For each of the 17 proteins, sums of the weighted abundance for all motif-matching kmers were created for the communities in which protein binding was enriched and not enriched over the null, respectively, then divided by the number of communities in each group to obtain the average weighted abundance of motif-matching kmers in the binding-enriched and binding-not-enriched groups. These abundances are plotted in Fig. 3C. For proteins that had more than one PWM reported in 23, the average abundance shown in Fig. 3C is comprised of the weighted abundance averaged over all reported PWMs. To calculate significance, we shuffled the communities in the binding-enriched and binding-not-enriched groups 10,000 times and determined how often the difference in kmer abundance between the randomly shuffled binding-enriched and binding-not-enriched groups was greater than the difference between the real binding-enriched and binding-not-enriched groups.
Measuring kmer similarity surrounding motif matches in lncRNAs
The lncRNAs expressed in HepG2 and K562 cells were examined for motif matches to the 17 proteins for which eCLIP data was reported in 37 and whose PWMs were determined via a high-throughput in vitro assay in 23 by using FIMO at a threshold of p<0.01 (from the MEME suite, URLs; 54; Supplementary Table 21). Each motif match was then labeled as a true positive if it overlapped an eCLIP peak, or a false positive if it did not. For each protein, the sequences surrounding the center of each true and false positive motif match (up to 150bp on either side of the center, or up to the end of the gene, whichever came first) were collected and their kmer contents were analyzed with SEEKR. Significance of the similarity between true positive regions was measured by a permutation test against randomly selected sets of false positive regions controlling for both the size of the set and the number of overlapping regions in the set (Supplementary Fig. 4).
Identifying motifs from eCLIP peaks
To find motifs in eCLIP peaks for the 17 proteins listed in Fig. 3C, we extracted the subset of sequences from eCLIP peaks whose CLIPper-defined p-value was <0.001 (peaks with the highest read densities relative to control; 37). We searched these sequences for motifs using DREME at default parameter as a part of the MEME-ChIP package 55.
Human-to-mouse and human-to-other community similarity calculations
To evaluate the similarity between human and mouse lncRNA communities, we calculated the distribution of similarities between all pairwise combinations of lncRNAs within each human kmer community (“human-to-self”), and compared this distribution to: (1) a distribution of pairwise comparisons made between all other human lncRNAs excepting lncRNAs from the community in question (“human-to-other-human”), (2) distributions of all pairwise comparisons made between all lncRNAs in each of the five mouse lncRNA communities (“human-to-mouse”), and (3) distributions of all pairwise comparisons made between all human and mouse lncRNAs that did not fall into one of the five major communities (“human-to-null”). We then performed a permutation test to determine whether a given human community was similar enough to a mouse community to overcome its intrinsic similarity to other lncRNAs in the human genome. The expectation was that, for related communities, the human-to-mouse distribution would be more similar to the human-to-self distribution than it would be to the human-to-other-human and human-to-null distributions. Bonferroni-adjusted p-values were calculated by permutation tests where we iteratively subsampled 0.1–1% of each distribution, re-measured the mean pairwise similarities, counted number trials in which the “human-to-mouse” mean subsample was closer to the “human-to-other-human” mean than it was to the “human-to-self” mean, and finally, divided by the total number of trials performed (36,000). This bootstrapping procedure provided a statistical framework to determine if the similarities uncovered between human and mouse communities were greater than what would have been expected from random chance. For example, in each of 36,000 tests, the distribution of similarities between a randomly selected subset of lncRNAs from human community #1 and size-matched subsets of lncRNAs from mouse community #1 was always more similar to the distribution of similarities between all pairwise comparisons of the human community #1 subset than it was similar to the distribution of similarities between the human community #1 subset and size-matched subsets of non-community #1 human lncRNAs (see upper left panel in Supplementary Fig 6; “H-1 vs M-1” plot; the H-1-vs-H-1 distribution in red is nearly indistinguishable from the H-1-vs-M-1 distribution in purple).
To generate the plots in Supplementary Figs. 8 and 9, identical analyses were performed that compared human lncRNA communities to lncRNA communities from Rabbit, Dog, Opossum, Chicken, Lizard, Coelacanth, Zebrafish, Stickleback, Nile Tilapia, Elephant Shark, and Sea Urchin 10. In these latter cases, the human XIST and HOTTIP lncRNAs were doped into the lncRNA annotation set from the organism in question to find the homologous communities that were the most XIST- and HOTTIP-like (Supplementary Fig. 7).
Generation of plasmids for TETRIS assays
The pTETRIS-Cargo vector was created from components of a cumate-inducible piggyBAC transposon vector (System Biosciences), pGl4.10-Luciferase (Promega), and pTRE-Tight (Clontech). Briefly, a 567bp fragment containing a minimal mouse PGK promoter was cloned into a SacI site in pGl4.10-Luciferase to generate pGI4-PGK-Luc-pA. The reverse complement of PGK-Luc-pA was cloned into a vector containing the bovine growth hormone polyA site. The entire bGHpa-[reversePGK-Luc-pA] was cloned into NotI and SalI sites of the piggyBAC vector (System Biosciences). The cumate-inducible promoter in the piggyBAC vector was then replaced with the Tetracycline Responsive Element (TRE) from pTRE-Tight (Clontech) via Gibson assembly to generate pTETRIS-Cargo in Fig. 4A, in which the lncRNA, the luciferase gene, and a gene encoding puromycin resistance are all flanked by chicken HS4 insulator elements, and inverted terminal repeats (ITRs) recognized by the piggyBAC transposase. The rtTA-cargo vector from Fig. 4A was generated by cloning the hUbiC-rtTA3-IRES-Neo cassette from pSLIK-Neo (Addgene Plasmid #25735) into SfiI and SalI sites in a piggyBAC transposon vector (System Biosciences). The piggyBAC transposase from System Biosciences was cloned into SmaI and HindIII sites into pUC19 (NEB) to allow propagation of the transposase on ampicillin plates.
Generation of TETRIS-lncRNA Cargo vectors
LncRNA fragments were PCR-amplified from genomic DNA or bacterial artificial chromosomes using Phusion DNA Polymerase (NEB), or commercially synthesized (Genewiz; IDT), and cloned via Gibson assembly into the SwaI site of pTETRIS-Cargo. Insert size was verified by restriction digestion, and the 5´ and 3´ end of each insert was verified by Sanger sequencing. To generate mutant Xist-2kb constructs, the 2kb fragment of Xist was subcloned into pGEM-T-Easy, and the regions in question were deleted using site-directed mutagenesis, or by synthesis of a mutated fragment and re-cloning back into compatible sites in pGEM-Xist-2kb (Genewiz). Deletions were verified by Sanger sequencing and then assembled into the SwaI site of pTETRIS-Cargo. The sequence of all inserted fragments, including Xist-2kb mutations, are listed in Supplementary Table 22.
Estimation of TETRIS copy number per cell
Genomic DNA was prepared from biological triplicate derivations of TETRIS-GFP and TETRIS-Xist-2kb cell lines. qPCR signal (SsoFast, Biorad) from the genomic DNA was compared to signal from a molar standard amplified from increasing amounts of the corresponding TETRIS plasmid (Supplementary Table 23).
TETRIS assays
To generate stable TETRIS-lncRNA cell lines, 8×10^5 E14 embryonic stem cells were seeded in a single well of a 6-well plate, and the next day transfected with 0.5μg TETRIS cargo, 0.5μg rtTA-cargo, and 1μg of pUC19-piggyBAC transposase. Cells were subsequently selected on puromycin [2μg/ml] and G418 [200μg/ml] for 6 to 12 days. Due to the efficiency of piggyBAC cargo integration and the rapidity of puromycin selection, all observable death from drug selection occurred within ~3 days after addition of puromycin and G418 (i.e. cells with puromycin resistance were invariably resistant to G418). For luciferase assays, 1×10^5 cells per well of 24 well plate were seeded in triplicate from each biological replicate preparation of a stable TETRIS-lncRNA cell line. 24 hours post-seeding, media was changed to include doxycycline at a final concentration of 1μg/ml. After two days of growth in dox-containing media, cells were lysed with 100 ul of passive lysis buffer (Promega), and luciferase activity was measured using Bright-Glo™ Luciferase Assay reagents (Promega) on a PHERAstar FS plate reader (BMG Labtech). Luciferase activity was normalized to protein concentration in the lysates via Bradford assay (Biorad). Each lncRNA fragment was assayed in at least in triplicate from at least two independent biological replicate preparations of stable TETRIS-lncRNA cell lines.
Synthetic lncRNA design
Synthetic lncRNAs were designed by generating 10 million, 1650 nucleotide long lncRNAs in silico that were composed of nucleotides randomly selected based on a given input ratio. To generate synthetic lncRNAs #2 through #6, the input ratio was the mononucleotide content of the 2,016-nucleotide long fragment of Xist inserted into TETRIS (0.203 A: 0.262 G: 0.204 C: 0.331 T). To generate synthetic lncRNA #1, the input ratio was an equal proportion of mononucleotides (0.250 A: 0.250 G: 0.250 C: 0.250 T). Synthetic lncRNAs with the specified kmer similarity to the 2kb fragment of Xist were then selected and synthesized as geneBlocks (Integrated DNA Technologies) and Gibson assembled into the SwaI site in TETRIS. Similarities in kmer content to the 2kb fragment of Xist are relative to all other mouse GENCODE lncRNAs.
Visualization of Xist structural models
Minimum Free Energy and probability-arc structural models of Xist-2kb were generated using SHAPE-MaP data from 41, the visualization package VARNA 56, and a modified version of the IGV browser 57. Predicted pseudoknots and regions of low SHAPE reactivity and low Shannon Entropy in Xist-2kb are from 41.
TETRIS predictions for kmer sizes and subsets
We measured SEEKR’s ability to capture the relationship between a lncRNA’s Xist-likeness and its repressive ability in the TETRIS assay using kmers from size one to eight. In each case, the correlation is measured using the means of all biological and technical replicates of each real and synthetic lncRNA, by normalizing kmer counts of Xist-2kb and the lncRNA in question in context with all mouse GENCODE lncRNAs. This process was repeated for select subsets of kmers which had the potential to increase our ability to predict repressive activity in TETRIS. Individual subsets were created by counting and normalizing kmers as normal with SEEKR then removing columns of the resulting count matrix that were not included in a given subset. Additionally, we randomly generated 100,000 kmer subsets each containing between 2 and 4095 kmers, and measured each of the subsets Pearson’s r values relative to our TETRIS data (Supplementary Fig. 10).
Statistical analyses
All statistics were performed in Python or R. Details of statistical analyses are described in the corresponding sections. All multiple comparison tests were adjusted using a Bonferroni correction. p-values are reported as exact values except in cases where the p-value was calculated using a permutation test, and no random samples were found to be more extreme than the observed value. In these cases, p-values are reported as (p <= 1/n), where n is the number of permutations performed.
Supplementary Material
Acknowledgments
We thank UNC colleagues for discussions, and JrGang Cheng for help with TETRIS cloning. This work was supported by National Institutes of Health (NIH) Grants GM121806 and GM105785, Basil O’Connor Award #5100683 from the March of Dimes Foundation, and funds from the Lineberger Comprehensive Cancer Center and UNC Department of Pharmacology (J.M.C.), the James S. McDonnell Foundation 21st Century Science Initiative - Complex Systems Scholar Award Grant #220020315 (P.J.M.), and NIH MIRA award R35 GM122532 (K.M.W.). J.M.K is an NSF Graduate Research Fellow (Grant DGE-1650116) and was supported in part by an NIH training grant in bioinformatics and computational biology (T32 GM067553). D.M.L was supported in part by an NIH training grant in genetics and molecular biology (T32 GM007092). M.J.S. was an NSF Graduate Research Fellow (Grant DGE-1144081) and was supported in part by an NIH training grant in molecular and cellular biophysics (Grant T32 GM08570).
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
The data sets generated during and/or analyzed during the current study are available within the article and its supplementary information files.
Code availability
SEEKR is open source and available on GitHub (URLs). A web instance of SEEKR is hosted at seekr.org.
URLs
SEEKR, https://github.com/CalabreseLab/seekr; nhmmer, http://hmmer.org/download.html; Biopython, http://biopython.org/; CHAMP, https://github.com/wweir827/champ; Gephi, https://gephi.org; ENCODE RNAseq http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq/; MEME, http://meme-suite.org/doc/fimo.html;
Supplementary information is included as part of this submission.
References and Notes
- 1.Iyer MK et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet, doi:10.1038/ng.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geisler S & Coller J RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 14, 699–712, doi:10.1038/nrm3679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holoch D & Moazed D RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16, 71–84, doi:10.1038/nrg3863 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, Hao L, Li D, Zhu L & Hu S Long non-coding RNAs and their biological roles in plants. Genomics Proteomics Bioinformatics 13, 137–147, doi:10.1016/j.gpb.2015.02.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinn JL & Chang HY Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166, doi:10.1146/annurev-biochem-051410-092902 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutschner T & Diederichs S The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol 9, 703–719, doi:10.4161/rna.20481 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JT & Bartolomei MS X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152, 1308–1323, doi:S0092–8674(13)00205–5 [pii]10.1016/j.cell.2013.02.016 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Wu X & Sharp PA Divergent transcription: a driving force for new gene origination? Cell 155, 990–996, doi:10.1016/j.cell.2013.10.048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cech TR & Steitz JA The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94, doi:10.1016/j.cell.2014.03.008 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Hezroni H et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 11, 1110–1122, doi:10.1016/j.celrep.2015.04.023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabili MN et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25, 1915–1927, doi:gad.17446611 [pii]10.1101/gad.17446611 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derrien T et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22, 1775–1789, doi:22/9/1775 [pii]10.1101/gr.132159.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bateman A et al. UniProt: a hub for protein information. Nucleic Acids Research 43, D204–D212, doi:10.1093/nar/gku989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman H, Henrick K & Nakamura H Announcing the worldwide Protein Data Bank. Nat Struct Biol 10, 980, doi:10.1038/nsb1203-980 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Ulitsky I & Bartel DP lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46, doi:S0092–8674(13)00759–9 [pii]10.1016/j.cell.2013.06.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kutter C et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet 8, e1002841, doi:10.1371/journal.pgen.1002841PGENETICS-D-12-00087[pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Necsulea A et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635-+, doi:10.1038/nature12943 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Eddy SR Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu Rev Biophys 43, 433–456, doi:10.1146/annurev-biophys-051013-022950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinn JJ et al. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev 30, 191–207, doi:10.1101/gad.272187.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy SR Homology searches for structural RNAs: from proof of principle to practical use. RNA 21, 605–607, doi:10.1261/rna.050484.115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler TJ & Eddy SR nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489, doi:10.1093/bioinformatics/btt403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rice P, Longden I & Bleasby A EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Ray D et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177, doi:10.1038/nature12311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefl R, Skrisovska L & Allain FH RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep 6, 33–38, doi:10.1038/sj.embor.7400325 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC & Batzoglou S Multiple sequence alignment. Curr Opin Struc Biol 16, 368–373, doi:10.1016/j.sbi.2006.04.004 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J Mol Biol 215, 403–410, doi:10.1016/S0022-2836(05)80360-2 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Pervouchine DD et al. Enhanced transcriptome maps from multiple mouse tissues reveal evolutionary constraint in gene expression. Nature communications 6, 5903, doi:10.1038/ncomms6903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chadwick BP Variation in Xi chromatin organization and correlation of the H3K27me3 chromatin territories to transcribed sequences by microarray analysis. Chromosoma 116, 147–157, doi:10.1007/s00412-006-0085-1 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Engreitz JM et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 159, 188–199, doi:10.1016/j.cell.2014.08.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mak W et al. Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr Biol 12, 1016–1020, doi:S0960982202008928 [pii] (2002). [DOI] [PubMed] [Google Scholar]
- 31.West JA et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55, 791–802, doi:10.1016/j.molcel.2014.07.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clemson CM, McNeil JA, Willard HF & Lawrence JB XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol 132, 259–275 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calabrese JM et al. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151, 951–963, doi:S0092–8674(12)01300–1 [pii]10.1016/j.cell.2012.10.037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blondel VD, Guillaume JL, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J Stat Mech-Theory E, doi:Artn P1000810.1088/1742–5468/2008/10/P10008 (2008). [Google Scholar]
- 35.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi:nature11247 [pii]10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlevaro-Fita J, Rahim A, Guigo R, Vardy LA & Johnson R Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA 22, 867–882, doi:10.1261/rna.053561.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514, doi:10.1038/nmeth.3810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawkins DM The problem of overfitting. J Chem Inf Comput Sci 44, 1–12, doi:10.1021/ci0342472 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Spitale RC et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486-+, doi:10.1038/nature14263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert N et al. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54, 887–900, doi:10.1016/j.molcel.2014.04.016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smola MJ et al. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci U S A 113, 10322–10327, doi:10.1073/pnas.1600008113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Matteo M et al. PiggyBac toolbox. Methods Mol Biol 859, 241–254, doi:10.1007/978-1-61779-603-6_14 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Ding S et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473–483, doi:10.1016/j.cell.2005.07.013 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Dowen JM et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387, doi:10.1016/j.cell.2014.09.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wutz A, Rasmussen TP & Jaenisch R Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30, 167–174, doi:10.1038/ng820ng820[pii] (2002). [DOI] [PubMed] [Google Scholar]
- 46.Liu F, Somarowthu S & Pyle AM Visualizing the secondary and tertiary architectural domains of lncRNA RepA. Nat Chem Biol 13, 282–289, doi:10.1038/nchembio.2272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tyner C et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res 45, D626–D634, doi:10.1093/nar/gkw1134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Team RCR: A language and environment for statistical computing, <https://www.R-project.org/> (2017).
- 49.Saldanha AJ Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248, doi:10.1093/bioinformatics/bth349 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Weir WH, Emmons S, Gibson R, Taylor D & Mucha PJ Post-Processing Partitions to Identify Domains of Modularity Optimization. Algorithms 10, doi:10.3390/a10030093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi:10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, doi:10.1093/bioinformatics/btt656 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Pedregosa F et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res 12, 2825–2830 (2011). [Google Scholar]
- 54.Bailey TL et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208, doi:10.1093/nar/gkp335 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Machanick P & Bailey TL MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697, doi:10.1093/bioinformatics/btr189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darty K, Denise A & Ponty Y VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975, doi:10.1093/bioinformatics/btp250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Busan S & Weeks KM Visualization of RNA structure models within the Integrative Genomics Viewer. RNA 23, 1012–1018, doi:10.1261/rna.060194.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.