

ABSTRACT
In fibroblasts, platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) stimulate the formation of actin-rich, circular dorsal ruffles (CDRs) and phosphatidylinositol 3-kinase (PI3K)-dependent phosphorylation of Akt. To test the hypothesis that CDRs increase synthesis of phosphorylated Akt1 (pAkt), we analyzed the contributions of CDRs to Akt phosphorylation in response to PDGF and EGF. CDRs appeared within several minutes of growth factor addition, coincident with a peak of pAkt. Microtubule depolymerization with nocodazole blocked CDR formation and inhibited phosphorylation of Akt in response to EGF but not PDGF. Quantitative immunofluorescence showed increased concentrations of Akt, pAkt and phosphatidylinositol (3,4,5)-trisphosphate (PIP3), the phosphoinositide product of PI3K that activates Akt, concentrated in CDRs and ruffles. EGF stimulated lower maximal levels of pAkt than did PDGF, which suggests that Akt phosphorylation requires amplification in CDRs only when PI3K activities are low. Accordingly, stimulation with low concentrations of PDGF elicited lower levels of Akt phosphorylation, which, like responses to EGF, were inhibited by nocodazole. These results indicate that when receptor signaling generates low levels of PI3K activity, CDRs facilitate local amplification of PI3K and phosphorylation of Akt.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: MTORC1, Macropinocytosis, Microtubules, PI 3-kinase, Akt
Highlighted Article: Circular dorsal ruffles in fibroblasts amplify PI3K-dependent signals leading to phosphorylation of Akt in response to EGF or low concentrations of PDGF.
INTRODUCTION
Macropinocytosis is a cellular process of solute endocytosis that occurs constitutively in macrophages (Canton et al., 2016), dendritic cells (Sallusto et al., 1995) and the soil amoeba Dictyostelium discoideum (Bloomfield and Kay, 2016). Macropinocytosis can be stimulated by growth factors, chemokines and various other stimuli (Swanson, 2008; Egami et al., 2014; Buckley and King, 2017; Yoshida et al., 2018). In macrophages, stimulation with macrophage-colony stimulating factor (M-CSF) or the chemokine CXCL12 elicits membrane ruffles, which form cup-shaped structures that close into large endocytic vesicles called macropinosomes (Yoshida et al., 2009; Yoshida et al., 2015b; Pacitto et al., 2017). Macropinosomes either recycle to the plasma membrane or fuse with lysosomes. Stimulation of murine embryonic fibroblasts (MEFs) with the growth factors platelet-derived growth factor (PDGF) or epidermal growth factor (EGF) elicits an alternative pathway to cup formation through actin-rich cell surface ruffles, which reorganize to form circular dorsal ruffles (CDRs). CDRs contract and often close to form macropinosomes (Bryant et al., 2007; Dubielecka et al., 2010; Hoon et al., 2012; Araki et al., 2007). CDRs and the circular ruffles that comprise macropinocytic cups can localize molecules associated with signal transduction, including phosphatidylinositol 3-kinase (PI3K) and its product phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (Yoshida et al., 2009, 2015b; Mercanti et al., 2006). Additionally, the formation of CDRs or closure of cups into macropinosomes requires PI3K (Wymann and Arcaro, 1994; Araki et al., 1996; Hooshmand-Rad et al., 1997; Valdivia et al., 2017), which suggests that CDRs and macropinocytic cups are self-organized structures that require PIP3 for complete morphogenesis.
Macropinocytosis provides a mechanism for activation of mTORC1 (mechanistic target of rapamycin complex-1), a protein complex that regulates metabolism and cell growth in response to signals generated by growth factors or other ligands at the plasma membrane (Saxton and Sabatini, 2017). mTORC1 is activated at lysosomal membranes by two small GTPases, Rag and Rheb (Saito et al., 2005; Sancak et al., 2010; Betz and Hall, 2013; Saxton and Sabatini, 2017). In macrophages and fibroblasts, macropinosomes induced by receptor activation deliver extracellular nutrients into lysosomes, where lysosome-associated membrane protein complexes detect the increased luminal concentrations of amino acids and trigger the activation of Rag GTPases (Yoshida et al., 2015b, 2018; Zoncu et al., 2011). Activated Rag recruits mTORC1 from cytosol to lysosomes (Sancak et al., 2008, 2010). Additionally, growth factor receptor stimulation of PI3K generates PIP3 in plasma membrane, which recruits the serine/threonine kinase Akt (Akt1; also known as protein kinase B, PKB) via its PH-domain (Manning and Toker, 2017). Akt is phosphorylated by PDK1 on threonine 308 and by mTORC2 (mTOR complex-2) on serine 473 (Ebner et al., 2017a, 2017b; Zhang et al., 2003). Phosphorylated Akt (pAkt) induces the phosphorylation of TSC2, a part of the TSC protein complex that is a GTPase-activating protein (GAP) for Rheb (Potter et al., 2002; Inoki et al., 2002, 2003; Garami et al., 2003). Phosphorylated TSC complex dissociates from lysosomes, eliminating its GAP activity towards Rheb and thereby permitting Rheb activation of mTORC1 at the lysosomal membrane (Menon et al., 2014). Thus, growth factor signaling to mTORC1 in macrophages and fibroblasts consists of a vesicular pathway, by which macropinosomes deliver extracellular amino acids to lysosomes for activation of Rag, and a cytosolic pathway in which receptor-mediated stimulation of PI3K activates the Akt–TSC1/2–Rheb pathway.
The cytosolic pathway can be initiated within macropinocytic cups and CDRs through localized amplification of PI3K. Akt–GFP is recruited to LPS-induced macropinocytic cups in macrophages (Wall et al., 2017) and to macropinosomes induced by active Ras (Porat-Shliom et al., 2008). In response to CXCL12, macrophages expressing fluorescent protein-tagged PH-domain probes show PIP3 enriched in membranes of macropinocytic cups (Pacitto et al., 2017). Immunofluorescence staining localizes Akt phosphorylated at threonine 308 to macropinocytic cups. These results suggest that PIP3 generation in CDRs or macropinocytic cups facilitates Akt phosphorylation and downstream signaling on the cytosolic pathway (Yoshida et al., 2018).
The cup structure can enhance PI3K-dependent activation of Akt. In macrophages, inhibitors of actin cytoskeleton dynamics or of macropinosome formation reduce phosphorylation of Akt in response to CXCL12 (Pacitto et al., 2017). In MDA-MB-231 cells, phosphorylation of Akt in response to the G-protein-coupled receptor (GPCR) ligand lysophosphatidic acid (LPA) requires PI3K catalytic subunit p110β and Rac-dependent macropinocytosis (Erami et al., 2017). In contrast, Akt phosphorylation in response to M-CSF and PDGF, in macrophages and MEFs, respectively, is insensitive to inhibitors of the actin cytoskeleton (Yoshida et al., 2015b). This suggests that qualitative or quantitative features of receptor signaling determine different requirements for cup-restricted PI3K activity.
Different patterns of signaling to Akt have been described for cellular responses to PDGF and EGF. Flow cytometric analysis of signaling to Akt in response to PDGF and EGF revealed differences in the kinetics and duration of cellular responses (Gross and Rotwein, 2016), which indicates different and heterogeneous cellular responses to the two growth factors. This suggests that morphological comparisons of signaling by PDGF and EGF might reveal underlying mechanistic differences.
The connections between macropinocytosis and growth factor signaling were defined using inhibitors of actin cytoskeleton dynamics, including ethylisopropyl amiloride (EIPA), jasplakinolide, blebbistatin, inhibitors of protein kinase C and genetic deletion of Rac1 (Pacitto et al., 2017; Yoshida et al., 2015a). The combination of jasplakinolide and blebbistatin (J/B), which inhibit actin filament turnover and myosin II, respectively, effectively immobilizes macrophage ruffles and inhibits macropinocytosis completely (Yoshida et al., 2015b). Earlier studies of macrophages showed that depolymerizing microtubules with nocodazole or colchicine inhibits actin cytoskeleton dynamics, cell surface ruffling and macropinocytosis (Racoosin and Swanson, 1989, 1992). Nocodazole inhibits the protrusive activities that underlie ruffling (Rosania and Swanson, 1996). However, roles for microtubules in macropinocytosis by MEFs have not been described, to our knowledge.
Here, we analyze the contributions of CDRs to Akt phosphorylation in response to PDGF and EGF in MEFs. We show that CDRs enhance the recruitment and phosphorylation of Akt at the plasma membrane, that CDRs and macropinosomes formed after stimulation with these growth factors can be inhibited by depolymerization of microtubules, and that CDRs facilitate signal amplification when receptor signaling generates lower levels of pAkt. The results indicate roles for CDRs in the amplification of PI3K and attendant signals, further supporting roles for plasma membrane domains and macropinocytosis in growth factor signaling to mTORC1.
RESULTS
Nocodazole blocks PDGF-induced macropinocytosis and mTORC1 activation
To define the time-course of the transition from CDRs to macropinosomes in PDGF-stimulated MEFs, morphological changes were monitored by time-lapse microscopy (Fig. 1A). Following overnight deprivation of MEF for growth factors, CDRs were induced within a few minutes of PDGF stimulation (t=1:00, min:s). CDRs then contracted radially, generating macropinosomes (t=6:00). To quantify CDRs, cells were fixed at various intervals after PDGF stimulation and the number of the cells with CDRs was measured. CDRs were induced to maximal levels within a minute of stimulation and decreased afterward (Fig. 1B), indicating that induction of CDRs by PDGF was an acute response. To study the transition from CDRs to macropinosomes, we measured macropinosome formation at the same time intervals (Fig. 1B). The number of macropinosomes increased over 30 min following stimulation (Fig. 1B). Thus, PDGF quickly induces CDRs, which close into macropinosomes.
Fig. 1.

Nocodazole treatment blocks PDGF-induced CDR formation, macropinocytosis and mTORC1 activation. (A) Time-lapse images of MEFs stimulated with PDGF (2 nM). CDRs appeared within 1 min (1:00, arrows), shrank (1:00–4:20) and closed into macropinosomes (arrowheads at 6:00 and 12:40). Scale bar: 10 µm. (B) The frequency of cells containing CDRs (top panel) or macropinosomes (bottom panel) at the indicated times after PDGF stimulation. More than 300 cells (top) or 25 cells (bottom) were observed at each time point; three independent experiments were carried out to calculate the mean and standard error. (C) Inhibition of CDR and macropinosome formation by nocodazole (Noco). Cells were pretreated with nocodazole (10 µM, 20 min) and stimulated by PDGF for 1 min (for CDR assay) or 15 min (macropinosome assay). (D) MEFs were incubated in DPBS containing leucine (0.4 mM) for 30 min with or without nocodazole (10 µM, 20 min pretreatment), then stimulated by PDGF for 15 min before lysis and preparation for western blotting. Nocodazole blocked PDGF-induced phosphorylation of S6K but not of Akt or ERK.
Inhibition of cytoskeletal activities by the combination of jasplakinolide and blebbistatin (J/B) blocks PDGF-induced macropinocytosis (Yoshida et al., 2015b). Additionally, we found that the microtubule polymerization inhibitor nocodazole blocked PDGF-induced CDR formation and macropinocytosis (Fig. 1C). Phase contrast and immunofluorescence staining for tubulin confirmed that nocodazole depolymerized microtubules and inhibited ruffling without otherwise altering cell morphology (Fig. S1). It was shown previously that inhibition of PDGF-induced macropinocytosis by J/B blocks mTORC1 activation, as measured by S6K phosphorylation (Yoshida et al., 2015b), which indicates a role for macropinocytosis in PDGF signaling to mTORC1. Like J/B, nocodazole blocked CDR formation, macropinocytosis and S6K phosphorylation in response to PDGF (Fig. 1D), but did not alter phosphorylation of either Akt or ERK1/2. These results are consistent with earlier studies showing that PDGF-induced macropinocytosis provides a vesicular pathway for extracellular amino acids to activate mTORC1 (Yoshida et al., 2015b).
Nocodazole attenuates EGF-induced Akt phosphorylation
To test whether EGF induces CDR formation and macropinocytosis in MEFs, we observed fixed cells by phase contrast microscopy following stimulation for different times (Fig. 2A,B, Fig. S2A). Maximal CDR formation was observed after 5 min of stimulation and decreased over the next 25 min (Fig. 2A,B). Macropinosome formation increased at 15 min (Fig. S2A). Nocodazole blocked CDR formation (Fig. 2C) and macropinocytosis (Fig. S2B) in response to EGF. Activation of mTORC1 (pS6K) in response to PDGF requires extracellular leucine and is blocked by inhibition of macropinocytosis, indicating that the amount of leucine delivered by macropinosomes to lysosomes determines the magnitude of S6K phosphorylation (Yoshida et al., 2015b). Similarly, EGF-induced phosphorylation of S6K was similarly dependent on extracellular leucine and macropinocytosis (Fig. 2D). Although Akt and ERK phosphorylation levels under these conditions were constant after stimulation, the phosphorylation of S6K increased with increasing leucine concentration (Fig. 2D), indicating that EGF-induced mTORC1 activation follows delivery of extracellular leucine into lysosomes. Consistent with a macropinocytosis-mediated delivery of leucine into lysosomes, we observed that nocodazole blocked EGF-induced S6K phosphorylation (Fig. 2E). EGF-induced phosphorylation on threonine 308 of Akt was diminished by nocodazole, whereas ERK phosphorylation was unaffected (Fig. 2E), which suggested that EGF signaling to Akt was enhanced by CDR formation, unlike PDGF signaling. Quantitative analysis of western blots showed that phosphorylation of Akt at threonine 308 [forming pAkt(308)] or serine 473 [forming pAkt(473)] in response to EGF was inhibited by nocodazole (Fig. S3A), but PDGF-stimulated phosphorylation of Akt was not (Fig. S4A). The Akt-specific inhibitor MK2206 blocked synthesis of both pAkt(308) and pAkt(473), but did not affect CDR formation, macropinocytosis or ERK phosphorylation (Fig. S5 and data not shown), indicating that Akt functions downstream of CDRs. Thus, for both EGF and PDGF, activation of mTORC1 requires macropinocytosis of leucine (vesicular pathway) and activation of Akt (cytosolic pathway). However, inhibition of CDR formation reduced Akt phosphorylation in response to EGF but not PDGF.
Fig. 2.
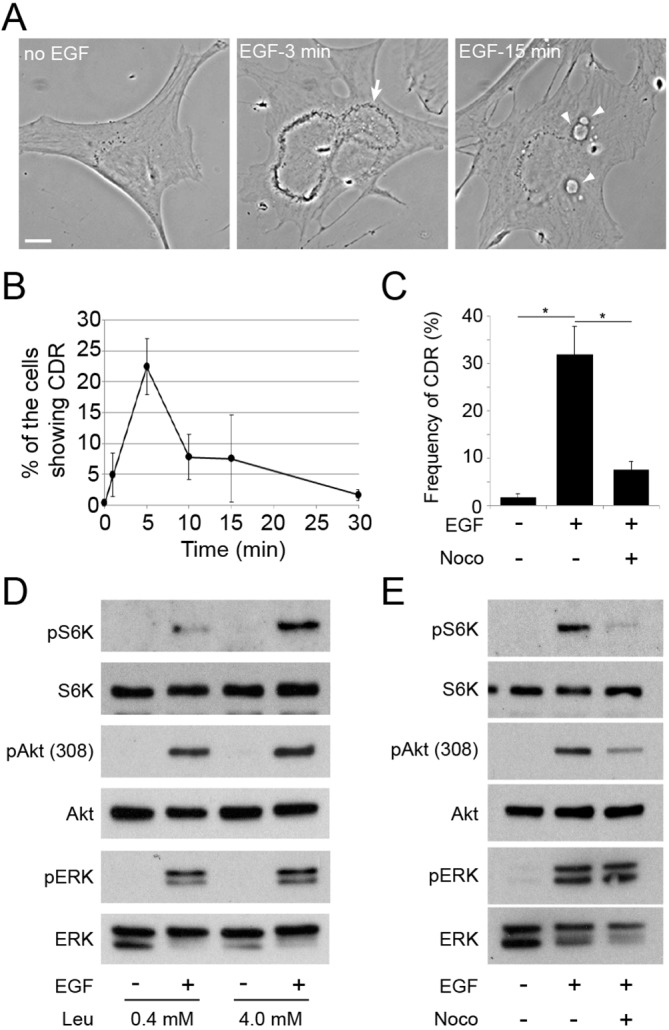
EGF-induced CDR formation, mTORC1 activity and Akt phosphorylation are inhibited by nocodazole. (A) CDR (arrow, 3 min) and macropinosome formation (arrowheads, 15 min) in response to EGF (16 nM) in MEFs. Scale bar: 10 µm. (B) The frequency of cells inducing CDRs at the indicated times after EGF stimulation. (C) Nocodazole (Noco) treatment blocked EGF-induced CDR formation. (D) Effects of leucine concentration on EGF-induced S6K phosphorylation (mTORC1). Cells were incubated in DPBS containing 0.4 mM or 4.0 mM leucine for 30 min and then stimulated by EGF for 15 min. Phosphorylation of S6K but not Akt or ERK required leucine. (E) Effects of nocodazole treatment on EGF-induced signal pathways. Cells were incubated in DPBS containing 0.4 mM leucine for 30 min with or without nocodazole and then stimulated by EGF for 15 min. Nocodazole treatment inhibited S6K phosphorylation and attenuated Akt phosphorylation without inhibiting EGF-stimulated ERK phosphorylation.
Inhibition of the cytoskeleton attenuates Akt phosphorylation in response to EGF
We reported earlier that formation of pAkt(308) induced by CXCL12 in macrophages was blocked by inhibition of macropinocytic cup formation, and that the maximal level of pAkt(308) generated in response to CXCL12 was less than that elicited by M-CSF (Pacitto et al., 2017). Based on that study, we hypothesized that the different inhibitory effects of nocodazole or J/B on levels of pAkt in response to PDGF and EGF are related to the different magnitudes of Akt phosphorylation elicited by the two growth factors. To test this, we measured the phosphorylation of Akt [synthesis of pAkt(308) and pAkt(473)] induced by different doses of EGF or PDGF. When cells were stimulated with concentrations of EGF and PDGF that saturate receptors (Ozcan et al., 2006; Fretto et al., 1993), the level of pAkt induced by EGF was less than that induced by PDGF (Fig. 3A). Notably, the magnitude of ERK phosphorylation under each experimental condition was the same. Similar to the effects of nocodazole, inhibition of the actin cytoskeleton by J/B decreased Akt phosphorylation in response to EGF, without affecting ERK phosphorylation (Fig. 3B). Quantitative analysis of western blots showed that synthesis of pAkt(308) or pAkt(473) in response to EGF was inhibited by J/B (Fig. S3B), but PDGF-stimulated phosphorylation of Akt was not (Fig. S4B). Akt phosphorylation induced by higher concentrations of EGF (160 nM) was attenuated by nocodazole treatment (Fig. 3C). Thus, EGF induced lower maximal levels of Akt phosphorylation than did PDGF, and those low levels of phosphorylation were sensitive to cytoskeletal inhibition. These measurements were obtained from cells lysed 15 min after stimulation, which is the optimal time for measuring signaling to mTORC1 (see Figs 1 and 2), but might not be the time of peak Akt phosphorylation. To address the possibility that cytoskeleton-dependent phosphorylation of Akt is time dependent, we compared the kinetics of signaling induced by PDGF and EGF. The kinetics of ERK phosphorylation were similar for both PDGF and EGF, but differed for Akt phosphorylation. Akt phosphorylation peaked at 5 min for both growth factors, but persisted after stimulation with PDGF (Fig. 3D,E). Nocodazole inhibited Akt phosphorylation measured at 5 min in response to EGF (Fig. 3F and Fig. S3C). Therefore, the differential sensitivities of EGF and PDGF to nocodazole or J/B were present at early time points.
Fig. 3.

Inhibition of cytoskeletal activity attenuates EGF-induced Akt phosphorylation. (A) Comparison of the magnitude of Akt phosphorylation [pAkt(308) and pAkt(473)] induced after 15 min stimulation by PDGF (0.4–4 nM) or EGF (16 and 160 nM). Intensities of pAkt(308) and pAkt(473), but not pERK, were stronger in response to PDGF than to EGF. Comparison of Akt phosphorylation induced by PDGF and EGF required using two different exposure times for the western blots (short versus long). (B) J/B attenuated pAkt(308) and pAkt(473) responses induced by EGF (16 nM), but not by PDGF (2 nM). J/B did not attenuate phosphorylation of ERK. (C) Nocodazole treatment (Noco) attenuated pAkt(308) and pAkt(473) formation induced by EGF. pERK levels were unaffected. (D) Time course of signaling in response to EGF (16 nM); increases in pAkt(308), pAkt(473) and pERK were transient. (E) Time course of signaling in response to PDGF (2 nM). Both pAkt(308) and pAkt(473) increases were prolonged, whereas that of pERK was transient. (F) Nocodazole inhibition of both pAkt(308) and pAkt(473) formation was evident as early as 5 min after stimulation with 16 nM EGF. pERK formation was not affected.
CDRs correlate with increased Akt phosphorylation
We developed quantitative immunofluorescence methods to determine the magnitudes of Akt phosphorylation in individual cells (Pacitto et al., 2017). To determine the applicability of this method for MEFs, we measured and compared the intensities of pAkt(308) and Akt by microscopy and western blot. Cells were stimulated with PDGF for 15 min, with or without inhibitors for PI3K catalytic subunits p110α (A66) (Jamieson et al., 2011) or p110β (TGX-221) (Jackson et al., 2005). Samples were then fixed and pAkt(308) and Akt labeled for immunofluorescence microscopy (Fig. S6). The ratio images (pAkt/Akt) were calculated and the intensities quantified relative to unstimulated cells. The ratio values of the PDGF-stimulated samples were higher than those of controls, and were reduced by the PI3K inhibitors (Fig. S6A,B). Cell lysates were prepared under the same experimental conditions and pAkt(308) was measured by western blot analysis. The results indicate that quantitative ratiometric immunofluorescence of pAkt and Akt in individual cells reflected the results of western blot analysis of cell lysates (Fig. S6C).
Using this method, we investigated the relationship between CDRs and Akt phosphorylation in single cells. After stimulation with growth factors, cells were labelled for immunofluorescence microscopy, and the relative intensities of pAkt(308) to Akt were measured ratiometrically. Three different conditions were used: no growth factor treatment, with EGF and with PDGF. After imaging, the growth factor-stimulated cells were subdivided based on phase contrast morphology into those with or without CDRs. Analysis of phase contrast and ratio images showed that, after EGF treatment, ratio values were higher in cells with CDRs than in cells lacking CDRs (Fig. 4A,B). The ratio values of EGF/no CDRs were significantly higher than those with no growth factor treatment, indicating that CDR formation was not required for increased pAkt(308) (Fig. 4A,B). The ratio values of PDGF-stimulated cells with CDRs were slightly higher than in PDGF-stimulated cells lacking CDRs (Fig. 4A); however, the differences were not statistically significant (Fig. 4B). Similar relationships were observed for pAkt(473) (Fig. S7A,B). The ratiometric images showed elevated pAkt/Akt ratios extending throughout the cell or broadly across the cell margins, rather than localized within CDRs (Fig. 4A and Fig. S7A). However, the immunofluorescence images of pAkt showed both antigens enriched in CDRs (Fig. S7C,D), which suggests that Akt recruitment to CDRs was followed by redistribution of pAkt elsewhere within the cell. Similar to the results of western blot analysis (Fig. 3F), quantitative immunofluorescence analysis also determined that nocodazole attenuated pAkt(308)/Akt ratios in EGF-stimulated cells (Fig. 4C). Thus, the Akt phosphorylation responses were greater in response to PDGF than to EGF, and were significantly higher in EGF-stimulated cells containing CDRs.
Fig. 4.

Correlation between the formation of CDRs and the magnitude of Akt phosphorylation. (A) Phase contrast and ratio [pAkt(308)/Akt] images for MEFs under different conditions. Cells were treated with or without EGF (16 nM) or PDGF (2 nM) for 3 min. After fixation, samples were stained with anti-pAkt(308) and anti-Akt antibodies. Phase contrast, pAkt(308) and Akt images were taken and then the ratio images were obtained by dividing the pAkt(308) image by the Akt image. Arrows indicate CDRs. Scale bar: 10 µm. Color bar indicates relative values of ratio intensities. (B) Quantification of ratiometric imaging in A. The relative ratio value of EGF-stimulated cells with CDRs (EGF, CDR+) was significantly higher than that of EGF-stimulated cells without CDRs (EGF, CDR−). More than 10 cells were observed for each condition. (C) Quantification of ratiometric imaging of pAkt(308) and Akt with or without nocodazole (Noco). MEFs were stimulated by EGF (16 nM) for 3 min with or without nocodazole pretreatment. After fixation, samples were stained with anti-pAkt(308) and anti-Akt antibodies. Phase, pAkt(308) and Akt images were taken. More than 15 cells were observed for each condition. One-way ANOVA was applied for the statistics; *P<0.05.
PIP3, Akt and pAkt are enriched in CDRs and cell ruffles
Akt activation requires it be recruited to the plasma membrane via the interaction of the Akt PH domain with PIP3 or PI(3,4)P2 (Ebner et al., 2017a). To observe distributions of PIP3 during CDR formation, we performed ratiometric imaging of yellow fluorescent protein (YFP)-tagged Btk-PH, a probe for PIP3 (Varnai et al., 1999) and free cyan fluorescence protein (CFP). In the absence of stimulation, intracellular ratios of YFP–Btk-PH to CFP were relatively even, except for a preferential accumulation of YFP–Btk-PH in the nucleus (Fig. S8). After stimulation with EGF, YFP–Btk-PH recruitment to CDRs coincided with CDR formation and was observed in all cells examined (Fig. 5A,B). Quantitative analysis confirmed that the ratio value inside CDRs was higher than that of the whole cell area (Fig. 5C). The results suggested that Akt preferentially binds plasma membrane inside CDRs. To localize Akt, MEFs expressing free CFP, which corrects for variations in cell thickness (Swanson, 2002), were stimulated for 3 min with EGF, then were fixed and labeled for immunolocalization of Akt. Ratiometric imaging of Akt–Alexa Fluor 594 and CFP indicated higher concentrations of Akt within CDRs (Fig. 5D). This was confirmed by quantitative analysis (Fig. 5E). These results indicate that Akt was recruited to CDRs by the local generation of PIP3 inside the structure. Next, we observed the distributions of pAkt(308) and pAkt(473) in EGF-stimulated MEFs. Cells expressing CFP were stimulated by EGF for 3 min, then fixed and prepared for ratiometric immunofluorescence microscopy of pAkt(308) or pAkt(473). Inside CDRs, the ratio values of pAkt(308)/CFP and pAkt(473)/CFP were higher than the cell average, although high values were also observed in ruffle-rich regions outside CDRs (Fig. 5F,G). Together, the quantitative imaging supports a model in which CDRs create domains of plasma membrane that support PI3K-dependent phosphorylation of Akt.
Fig. 5.

Characterization of CDRs as platforms for EGF-induced Akt signaling. (A) Live-cell imaging of MEFs expressing YFP–Btk-PH and CFP after stimulation by EGF. Time after addition of growth factor is indicated at bottom of phase contrast images (minutes:seconds). Comparison of phase contrast and ratio (YFP–BtkPH/CFP) images shows high ratio values inside CDRs (arrows). (B) MEFs expressing YFP–Btk-PH and CFP were fixed after 3 min stimulation with EGF. High ratio values were observed inside CDRs (arrow). (C) Quantitation of images in B. In 11 MEFs with CDRs, ratio intensities for the entire cell (Cell) and CDR area (CDR) were measured then compared. (D) Fluorescence microscopy of Akt recruitment to CDRs in MEFs. Cells expressing CFP were stimulated with EGF for 3 min and fixed for immunofluorescence staining for Akt (Alexa Fluor 594-tagged secondary antibody). Comparison of phase contrast and ratio images (Akt–Alexa Fluor 594/CFP) shows higher ratio values in CDRs (arrows). (E) Quantitation of images in D. In 12 MEFs with CDRs, the intensities of the ratio over the entire cell and CDR area were measured and compared. (F,G) Fluorescence microscopy was used to observe the distribution pattern of EGF-induced pAkt(308) (F) or pAkt(473) (G) in MEFs. Strong signals were identified in CDRs (cropped images) and peripheral lamellipodia (arrows). All color bars indicate relative values of ratio intensities. Student's t-test was applied for the statistics; *P<0.05. Scale bars: 10 µm.
Similar patterns of localization were observed in MEFs stimulated with PDGF. Live cell ratiometric imaging of YFP–Btk-PH and CFP indicated increased concentrations of PIP3 in CDRs (Fig. 6A). Aggregate measurements of ratios in fixed cells showed similar patterns (Fig. 6B) and quantitative analysis of ratios in CDRs versus the entire cell indicated that ratios were significantly increased (Fig. 6C). Similarly, PDGF induced CDRs in which the relative value of Akt–Alexa Fluor 594/CFP was higher than the average for the entire cell (Fig. 6D,E).
Fig. 6.

Akt signaling at PDGF-induced CDRs. (A) Live-cell imaging of MEFs expressing YFP–Btk-PH and CFP after stimulation by PDGF (2 nM). Time after addition of growth factor is indicated at bottom of phase contrast images (minutes:seconds). Comparison of phase contrast and ratio (YFP–BtkPH/CFP) images shows sustained high ratio values inside CDRs (arrows). (B) MEFs expressing YFP–Btk-PH and CFP were fixed after 3 min stimulation with PDGF (2 nM). High ratio values were observed inside CDRs (arrow). (C) Quantitation of images in B. In 9 MEFs with CDRs, the intensities of the ratio over the cell (Cell) and CDR area (CDR) were measured and compared. (D) Fluorescence microscopy of Akt recruitment to CDRs in MEFs. MEFs expressing CFP were stimulated with PDGF (2 nM) for 3 min and fixed for immunofluorescence staining for Akt (Alexa Fluor 594-tagged secondary antibody). Comparison of phase contrast and ratio images (Akt–Alexa Fluor 594/CFP) shows higher ratio values in CDRs (arrow). (E) Quantitation of images in D. In 9 MEFs with CDRs, the intensities of the ratio over the cell and CDR area were measured then compared. Student's t-test was applied for the statistics; *P<0.05. All color bars indicate relative values of ratio intensities. Scale bars: 10 µm.
The differential sensitivities of Akt phosphorylation to cytoskeletal inhibition in response to PDGF and EGF could be a result of qualitative differences in signaling between the two growth factor receptors, which render EGF receptor signaling to Akt sensitive to cytoskeletal inhibition. The different kinetics of Akt signaling (Fig. 3D,E) are consistent with different mechanisms. Alternatively, the differential sensitivities to these inhibitors could reflect a shared mechanism of Akt signal amplification, which is only apparent when signals are low, as observed previously in macrophages (Pacitto et al., 2017). To address this possibility, we determined the concentration of PDGF that elicited levels of Akt phosphorylation comparable to those obtained with EGF (10 pM PDGF; Fig. 7A), and measured the sensitivity of that low level of Akt phosphorylation to cytoskeletal inhibition. The kinetics of the responses were similar (Fig. 7B). Moreover, nocodozole or J/B attenuated pAkt(308) and pAkt(473) responses to 10 nM PDGF but not to higher concentrations of PDGF (Fig. 7C,D and Fig. S4C,D). Quantification of immunofluorescence imaging of pAkt and Akt supported the western blot analysis (Fig. 7E). Together, these results indicate that cytoskeletal activities facilitate Akt phosphorylation in response to weaker stimuli.
Fig. 7.

Low concentrations of PDGF induce cytoskeleton-dependent Akt phosphorylation. (A) Dose-dependent assay of PDGF stimulation. At 15 min following stimulation with PDGF (0.01–2.0 nM), MEFs were lysed for biochemical analysis. The intensity of pAkt induced by PDGF decreased at 0.01 nM (10 pM). (B) Time course of signaling in response to 10 pM PDGF. Akt phosphorylation was prolonged but ERK phosphorylation was transient. (C) Nocodazole (Noco) treatment attenuated pAkt(308) and pAkt(473) induced by 10 pM PDGF, but not by 2 nM PDGF. (D) J/B treatment attenuated pAkt(308) and pAkt(473) induced by 10 pM PDGF, but not by 2 nM PDGF. (E) Quantification of immunofluorescence images of pAkt(308) and Akt. MEFs were stimulated by PDGF (2 nM or 10 pM) for 3 min with or without nocodazole. Ratio images of pAkt(308) and Akt were calculated to quantify the intensity of pAkt in each condition. The relative ratio value of 10 pM PDGF-stimulated cells was significantly higher than for untreated cells and was attenuated by nocodazole. The values for cells stimulated with 2 nM PDGF were not attenuated by nocodazole. More than 15 cells were observed for each condition. One-way ANOVA was applied for the statistics; *P<0.05.
DISCUSSION
This study adds EGF to the growing number of extracellular ligands that can signal to mTORC1 by delivering amino acids into lysosomes via macropinocytosis and advances the concept that growth factor-mediated activation of Akt can be enhanced by localized stimulation of PI3K in domains of plasma membrane. Live-cell imaging and immunofluorescence localized PIP3 inside CDRs in MEFs. Western blot analysis showed that inhibition of cytoskeletal activities limited CDR formation and Akt phosphorylation in response to EGF or to low concentrations of PDGF, but not to higher concentrations of PDGF. Quantitative analysis of immunofluorescence staining demonstrated greater levels of Akt phosphorylation in cells with CDRs than in cells without. Thus, as restricted areas of PIP3 accumulation, CDRs provide platforms for Akt phosphorylation. We propose that CDRs amplify Akt phosphorylation when receptor signaling generates low levels of PI3K activity, either by low concentrations of ligand or by innate differences in receptor signaling to PI3K (Fig. 8).
Fig. 8.
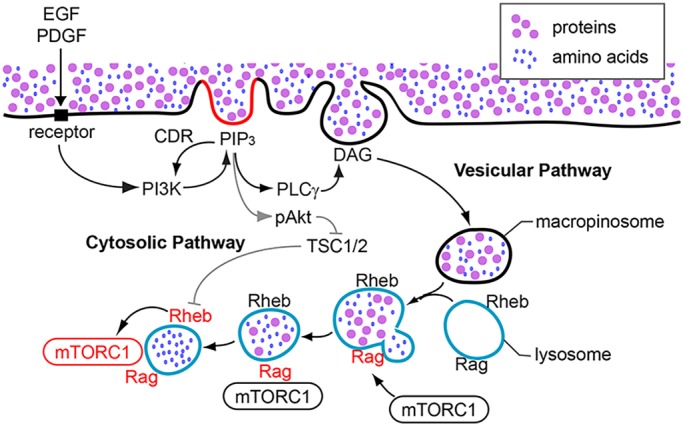
A model for CDR-dependent Akt phosphorylation and activation of mTORC1. EGF and low concentrations of PDGF induce mTORC1 activation via cytosolic and vesicular pathways, both of which are regulated by CDRs. After stimulation, CDRs are formed by cytoskeletal activities and activated PI3K generates PIP3 at the plasma membrane. The molecular mechanism of PIP3 restriction to CDRs is still unknown, but could be caused by positive feedback amplification of PI3K by PIP3, supported by diffusion barriers in CDRs. Elevated concentrations of PIP3 inside CDRs recruit Akt to the plasma membrane (cytosolic pathway). Akt localized to the plasma membrane is phosphorylated on threonine 308 and serine 473 by PDK1 and mTORC2, respectively. pAkt then phosphorylates TSC2, resulting in the loss of GAP function of the TSC1/2 complex towards Rheb. Rheb is then activated at the lysosomes. Meanwhile, PIP3 at CDRs also activates PLCγ, which generates diacylglycerol (DAG) in CDRs, leading to the formation of macropinosomes (vesicular pathway). Macropinosomes convey extracellular nutrients into lysosomes, which induce the activation of Rag. Rag recruits mTORC1 to the lysosomes, where mTORC1 is activated by Rheb. This study supports a role for CDRs in amplification of PI3K and pAkt.
We also identified a role for microtubules in CDR formation, macropinocytosis and the amplification of growth factor receptor signaling to Akt. The mechanism by which nocodazole inhibits CDR formation and macropinocytosis is unknown, but is probably related to microtubule-associated signals that regulate Rac1. In macrophages, microtubule depolymerization by nocodazole or colchicine inhibits macropinocytosis (Racoosin and Swanson, 1989, 1992). Ruffling is inhibited by microtubule-depolymerizing drugs and by taxol, a microtubule-stabilizing agent, which indicates that microtubule dynamics influence actin dynamics in ruffle formation (Rosania and Swanson, 1996). This might be caused by Rac1 GEF activities, which are linked to dynamic microtubules (Waterman-Storer et al., 1999).
How does Akt signal amplification localize to CDRs or macropinocytic cups? Structural barriers in cup margins limit lateral diffusion of membrane-tethered proteins within the inner leaflet of cup membranes (Welliver et al., 2011). Such barriers should confine phospholipids or the enzymes that modify them within CDRs and cups, consequently localizing signal cascades to these regions of plasma membrane (Welliver and Swanson, 2012). PIP3 synthesized by PI3K within CDRs might be retained there by the diffusion barrier, allowing positive feedback stimulation of PI3K by PIP3, which could be mediated by Ras, Rac or the adapter protein Gab1 (Rodrigues et al., 2000; Castellano and Downward, 2011; Fritsch et al., 2013; Ding et al., 2015). The elevated concentrations of PIP3 within CDRs could also enhance barrier formation through activation of Rac1 or Ras (Kay et al., 2018; Delos Santos et al., 2015; Hoon et al., 2012; Lien et al., 2017). If PIP3 in membranes must exceed some concentration threshold to allow binding of Akt, PDK1 or mTORC2, or if ruffles or cups somehow enhance coupling between PDK1 and Akt, then activation of PI3K in CDRs could provide a mechanism for amplification of Akt phosphorylation. We should note, however, that localized increases in concentrations of PIP3 or pAkt were not always confined to CDRs (Fig. 5A,F,G and Fig. 6B), or to macropinocytic cups in macrophages. Often, the first response to acute stimulation of receptors was a more generally distributed increase in signals at lamellipodia and peripheral regions of ruffling, followed by a more punctuated response localized to cups (Yoshida et al., 2009; Pacitto et al., 2017). Thus, diffusion barriers created by cup margins do not circumscribe the earliest PI3K responses, which suggests that other features of ruffle or lamellipodia structure might localize the early amplification of PI3K.
Macropinocytosis in Dictyostelium provides a mechanism for nutrient uptake leading to cell growth. Dictyostelium macropinosomes do not form in response to receptor stimulation, but their mechanisms of formation are very similar to macropinosomes in metazoan cells. Most notably, the membrane of macropinocytic cups is enriched in PIP3 and Ras, and the cup margin is enriched in proteins that regulate actin polymerization through Rac. Thus, like growth factor or chemokine signaling in macrophages and fibroblasts, the macropinocytic cups of Dictyostelium define a sharply delineated region of localized PI3K activity. Kay et al. proposed that growth factor signaling is an evolutionary adaptation of an ancient ‘macropinocytic amplifier’ regulating Akt and cell growth (Kay et al., 2018). This study and other recent studies of signaling in metazoan cells support this proposal (Pacitto et al., 2017; Erami et al., 2017).
Unlike EGF and low concentrations of PDGF (10 pM), higher concentrations of PDGF (2–4 nM) induced Akt phosphorylation that was independent of CDRs. Western blotting (Fig. 3) and microscopy assays (Fig. 4) showed that, at receptor-saturating concentrations, PDGF induced stronger Akt phosphorylation than did EGF. Although EGF elicited lower maximal levels of Akt phosphorylation compared with PDGF, levels of ERK phosphorylation stimulated by the two growth factors were comparable. This suggests that receptors for EGF and PDGF initiate different overall levels of PI3K activity. A similar relationship appears in macrophages, where CXCL12 generates low levels of pAkt(308), which can be reduced by inhibition of macropinocytic cup formation, and M-CSF generates higher levels of pAkt(308), which are not reduced by such inhibition (Pacitto et al., 2017). These results indicate that both CDRs in MEFs and macropinocytic cups in macrophages provide a mechanism to boost Akt phosphorylation when PI3K generates relatively little PIP3 (Fig. 8).
The kinetics of Akt phosphorylation differ in response to different ligands and to different concentrations of ligands (Gross and Rotwein, 2016). The timing of CDR formation corresponds to the transient activation of Akt by EGF and PDGF and may reflect a transient amplification of PI3K within CDRs. Sustained activation of Akt observed in response to PDGF (Gross and Rotwein, 2016) could reflect an additional cytoskeleton-independent mechanism of PIP3 generation. The timing of Akt phosphorylation responses to EGF (early and transient) and the absence of significant staining of pAkt on macropinosomes suggests that CDRs are the principal location of Akt phosphorylation, rather than fully formed macropinosomes. However, the persistent Akt phosphorylation after PDGF treatment suggests that later stages of macropinocytosis carry Akt signals into the cell. Future experiments should address how pAkt reaches TSC1/2 on lysosomes to influence mTORC1 signaling.
This and previous studies of signaling to Akt (Yoshida et al., 2015b; Pacitto et al., 2017) support a concept in which cells amplify weak PI3K signals using confined subregions of plasma membrane that limit lateral diffusion of PIP3 in the membrane (Kay et al., 2018). Cytoskeleton-dependent amplification of Akt signaling may reflect the normal behavior of growing cells, in which constant concentrations of growth factors trigger stochastic, localized signal cascades (Yoshida et al., 2009, 2018). Cytoskeleton-independent signaling could be an innate feature of some receptor signaling pathways, such as those for M-CSF and PDGF, but might also appear as an aberrant consequence of receptor overexpression (e.g., EGF receptor) or dysregulation of PI3K signaling pathways.
MATERIALS AND METHODS
Reagents, plasmids and antibodies
Recombinant murine PDGF-BB and recombinant murine EGF were from Peprotech. Fluorescein isothiocyanate-labeled dextran, average molecular weight 70,000 (FDx70), was from Molecular Probes. Nocodazole and blebbistatin were from Abcam. Jasplakinolide was from Enzo Life Science. MK 2206 was from ApexBio. A66 and TGX 221 were from Symansis. Anti-phosphorylated S6K(Thr389) (#9234), anti-S6K (#2708), anti-pAkt(308) (#4056), anti-pAkt(473) (#4060), anti-Akt (#9272), anti-pERK1/2(Thr202/Tyr204) (#4376), and anti-ERK1/2 (#4695) antibodies for western blot analysis were from Cell Signaling Technology. Anti-pAkt(308) (#2695), anti-pAkt(473) (#4060), and anti-Akt (#2920) for immunofluorescence staining were from Cell Signaling Technology. Anti-α-tubulin was from Abcam (DM1A). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG was from GE Healthcare. Anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 594 antibodies were from Invitrogen. Plasmids pECFP-N1 for free CFP and pmCitrine-BtkPH-N1 for YFP–Btk-PH were described previously (Yoshida et al., 2009). FuGENE HD was from Promega.
Cell culture, inhibitor treatment and transfection
Mouse embryonic fibroblasts (MEFs) were cultured as described previously (Yoshida et al., 2015b; Gupta et al., 2007). Briefly, cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies 11995) with 10% fetal calf serum (FCS) and penicillin/streptomycin. For inhibitor treatments, cells were pretreated with nocodazole (10 µM, 20 min), MK2206 (2 µM, 30 min), A66 (3 µM, 30 min) o TGX 221 (0.5 µM, 30 min) in low-glucose DMEM (Life Technologies 11885). A combination of blebbistatin (75 µM for 35 min) and jasplakinolide (1 µM for 15 min) was also used. FuGENE HD was used for transfection according to the manufacturer's protocol.
Microscopy and live-cell imaging
A Nikon Eclipse TE-300 inverted microscope with a 60× numerical aperture 1.4, oil-immersion PlanApo objective lens (Nikon, Tokyo, Japan) and a Lambda LS xenon arc lamp for epifluorescence illumination (Sutter Instruments, Novato, CA, USA) were used to collect phase-contrast and fluorescence images. Fluorescence excitation and emission wavelengths were selected using a 69008 set (Chroma Technology, Rockingham, VT, USA) and a Lambda 10-2 filter wheel controller (Shutter Instruments) equipped with a shutter for epifluorescence illumination control. A Photometrics CoolSnap HQ cooled CCD camera (Roper Scientific, Tucson, AZ, USA) was used for recording. For live-cell imaging, cells plated onto glass-bottom dishes of 35 mm diameter (MatTek Corp.) were cultured in low-glucose DMEM overnight. Cells were stimulated with 2 nM PDGF or 16 nM EGF, and imaged at 20-s intervals. Time-lapse images were processed using MetaMorph v6.3 software (Molecular Devices).
Circular dorsal ruffle assay
MEFs were cultured on coverslips in low-glucose DMEM without FBS overnight. Cells were stimulated with 2 nM PDGF or 16 nM EGF (1–3 min), then fixed with fixation buffer 1 (20 mM HEPES pH 7.4, 4% paraformaldehyde, 4.5% sucrose, 70 mM NaCl, 10 mM KCl, 10 mM MgCl2, 2 mM EGTA, 70 mM lysine-HCl, 10 mM sodium periodate) at room temperature for 15 min. The fixed cells were washed three times with washing buffer (Tris-buffered saline, TBS, consisting of 20 mM Tris-HCl pH 7.4, 150 mm NaCl, 4.5% sucrose) at room temperature for 10 min each time and mounted for microscopy. The frequency of cells inducing CDRs was determined from images of more than 300 cells per condition. Cells were randomly selected and the number of cells with CDRs was counted. The frequency was calculated as (number of the cells with CDR)/(total number of cells observed). The average and standard error of the frequencies were calculated from three independent experiments. One-way ANOVA was applied for the statistics.
Macropinosome assay
Macropinosome assays were performed as described previously (Yoshida et al., 2015a). Cells were cultured on coverslips in low-glucose DMEM without FBS overnight. PDGF (2 nM) or EGF (16 nM) were added together with fluorescein dextran (FDx70; 1 mg/ml) and the cells incubated for 15 min at 37°C. Cells were fixed with fixation buffer 2 (20 mM HEPES pH 7.4, 2% paraformaldehyde, 4.5% sucrose, 70 mM NaCl, 10 mM KCl, 10 mM MgCl2, 2 mM EGTA, 70 mM lysine-HCl, 10 mM sodium periodate) at 37 C for 30 min following three washes with warmed DPBS to remove extracellular FDx70. The fixed cells were washed with washing buffer three times for 10 min at room temperature and mounted. Phase-contrast and FDx70 images of each sample were taken and merged after subtracting the background signal using MetaMorph. More than 25 cells were observed, and the number of induced macropinosomes per cell was determined by counting FDx70-positive vesicles on the merged images. If the signal of the FDx70-positive structure on the merged images was not clearly a macropinosome, then both FDx70 and phase contrast images were observed separately to confirm macropinosome identity. The average number of induced macropinosomes per cell was calculated using the results from three independent experiments. One-way ANOVA was applied for the statistics.
Immunofluorescence staining
Immunofluorescence staining of Akt and pAkt was carried out as described previously (Pacitto et al., 2017). MEFs were treated for 3 or 15 min with or without PDGF or EGF, then fixed at room temperature for 10 min with fixation buffer 1. Cells were washed with TBST buffer (50 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.6), permeabilized in freshly prepared 0.2% saponin in TBS (w/v) for 15 min at room temperature and then incubated in 1% BSA in TBST as the blocking buffer (30 min at room temperature). Anti-Akt, anti-pAkt(308) and anti-pAkt(473) antibodies were diluted 1:50 in blocking buffer and incubated with the samples for 2 h at room temperature as primary antibody treatment. Samples were washed with TBST (three times for 10 min). Anti-rabbit IgG Alexa Fluor 488 and anti-mouse IgG Alexa Fluor 594 antibodies were diluted 1:200 in blocking buffer and incubated with the samples for 1.5 h at room temperature as secondary antibody treatment. Samples were washed three times with TBST for 10 min and mounted. For microtubule staining, stimulated cells were fixed and samples incubated in cold ethanol for 20 s to permeabilize cells. Then samples were washed with TBST (three times for 10 min), labeled with mouse anti-α-tubulin (Abcam) primary antibody (1: 200) and anti-mouse IgG Alexa Fluor 488 secondary antibody (1:100) and mounted for microscopic observation.
Quantification of pAkt/Akt ratios of immunofluorescence images
Quantification of pAkt/Akt ratio images was carried out as described previously (Pacitto et al., 2017). pAkt and Akt images were corrected for shade, bias and background (Pacitto et al., 2017; Hoppe, 2012). A binary image map was created from the corrected Akt (denominator) image. The corrected Akt images and binary images were combined using the ‘Logical AND’ command of MetaMorph and the resulting image saved as ‘AND image.’ The corrected pAkt image was divided by the AND image, multiplied by 100 and saved as ‘Ratio image’. These ratio images were used to quantify the average pAkt/Akt ratio value in single cells. Ratio images were thresholded to exclude background areas, and cellular subregions were selected manually. Using the ‘region measurements’ command of MetaMorph, the integrated intensity of the pAkt/Akt ratio and threshold area for a target cell were logged in Excel. The integrated intensities were divided by the threshold areas to yield relative intensities as the average pAkt/Akt ratio. More than 10 cells (Fig. 4 and Figs S2 and 3) or 20 cells (Fig. 5) were observed for each condition. One-way ANOVA was applied for the statistics.
Ratio imaging and quantitative analysis of fixed CFP-expressing cells
MEFs expressing CFP were treated with EGF or PDGF for 3 min. Cells were fixed and stained with Akt or pAkt antibody with Alexa Fluor 594-tagged secondary antibody. Phase contrast, CFP, Akt or pAkt images were taken. Alexa Fluor 594/CFP ratio images were generated after shade–bias–background correction. Cell area and CDR area(s) were determined on the phase image using the drawing tool of MetaMorph, and the confined regions transferred to the ratio image by MetaMorph command. The average ratio value inside the cell areas and CDR areas were measured by applying the region measurement tool. The results were logged to the Excel sheet and the (average ratio value at CDR)/(average ratio value of entire cell) calculated. For the quantification (Fig. 7B), nine or more cell images of EGF- or PDGF-stimulated MEFs were observed. Student's t-test was applied for the statistics.
Ratiometric live-cell imaging
Plasmids pECFP-N1 and pmCitrine-BtkPH-N1 were used for free CFP and YFP–Btk-PH, respectively. Plasmids were transfected into MEFs by FuGENE HD according to the manufacturer's protocol. After PDGF or EGF stimulation, phase contrast, YFP and CFP images of live MEFs were captured every 20 s for 10 min. Ratio images of YFP–Btk-PH relative to CFP were generated as described previously (Yoshida et al., 2015b; Pacitto et al., 2017).
Cell lysates and western blotting
Cells were pretreated with the inhibitors and stimulated with PDGF for 15 min, then lysed for 10 min in cold lysis buffer (40 mM HEPES pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 1.5 mM Na3Vo4, 0.3% CHAPS, and a mixture of protease inhibitors from Roche Applied Science) (Yoshida et al., 2011). Lysates were centrifuged at 13,000×g for 15 min at 4°C. The supernatant was mixed with 4× SDS sample buffer and boiled for 5 min. The samples were subjected to SDS-PAGE and western blotting with the indicated antibodies as described previously (Yoshida et al., 2011, 2015b). For the quantification of western blots, the results from at least three independent experiments were scanned and saved as jpeg images. Intensities of bands were measured using Metamorph. The jpeg images were inverted and binary images generated. Binary images were applied as masks for the measurement of band intensity. Each band on the masked image was cropped and the intensities of Akt bands were used as reference to calculate the ratios of pAkt(308)/Akt and pAkt(473)/Akt. Ratio values from samples with and without inhibitor treatments were compared using the values of untreated control samples as the reference. Student's t-test was applied for the statistics.
Supplementary Material
Acknowledgement
The authors thank Drs David Friedman and Jonathan Backer for suggestions. S.Y. thanks Drs Peggy Pachke and Ling Qi for their support.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.Y., J.A.S.; Methodology: S.Y., R.P., C.S.; Resources: L.K.; Data curation: S.Y.; Writing - original draft: S.Y., J.A.S.; Writing - review & editing: S.Y., R.P., C.S., J.A.S.; Visualization: S.Y.; Supervision: S.Y., J.A.S.; Funding acquisition: J.A.S.
Funding
This work was supported by the National Institutes of Health (grant GM-110215 to J.A.S.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.220517.supplemental
References
- Araki N., Johnson M. T. and Swanson J. A. (1996). A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135, 1249-1260. 10.1083/jcb.135.5.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki N., Egami Y., Watanabe Y. and Hatae T. (2007). Phosphoinositide metabolism during membrane ruffling and macropinosome formation in EGF-stimulated A431 cells. Exp. Cell Res. 313, 1496-1507. 10.1016/j.yexcr.2007.02.012 [DOI] [PubMed] [Google Scholar]
- Betz C. and Hall M. N. (2013). Where is mTOR and what is it doing there? J. Cell Biol. 203, 563-574. 10.1083/jcb.201306041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield G. and Kay R. R. (2016). Uses and abuses of macropinocytosis. J. Cell Sci. 129, 2697-2705. 10.1242/jcs.176149 [DOI] [PubMed] [Google Scholar]
- Bryant D. M., Kerr M. C., Hammond L. A., Joseph S. R., Mostov K. E., Teasdale R. D. and Stow J. L. (2007). EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J. Cell Sci. 120, 1818-1828. 10.1242/jcs.000653 [DOI] [PubMed] [Google Scholar]
- Buckley C. M. and King J. S. (2017). Drinking problems: mechanisms of macropinosome formation and maturation. FEBS J. 284, 3778-3790. 10.1111/febs.14115 [DOI] [PubMed] [Google Scholar]
- Canton J., Schlam D., Breuer C., Gütschow M., Glogauer M. and Grinstein S. (2016). Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat. Commun. 7, 11284 10.1038/ncomms11284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E. and Downward J. (2011). RAS interaction with PI3K: more than just another effector pathway. Genes Cancer 2, 261-274. 10.1177/1947601911408079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos Santos R. C., Garay C. and Antonescu C. N. (2015). Charming neighborhoods on the cell surface: plasma membrane microdomains regulate receptor tyrosine kinase signaling. Cell. Signal. 27, 1963-1976. 10.1016/j.cellsig.2015.07.004 [DOI] [PubMed] [Google Scholar]
- Ding C.-B., Yu W.-N., Feng J.-H. and Luo J.-M. (2015). Structure and function of Gab2 and its role in cancer (Review). Mol. Med. Rep. 12, 4007-4014. 10.3892/mmr.2015.3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubielecka P. M., Cui P., Xiong X., Hossain S., Heck S., Angelov L. and Kotula L. (2010). Differential regulation of macropinocytosis by Abi1/Hssh3bp1 isoforms. PLoS ONE 5, e10430 10.1371/journal.pone.0010430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner M., Lucic I., Leonard T. A. and Yudushkin I. (2017a). PI(3,4,5)P3 engagement restricts Akt activity to cellular membranes. Mol. Cell 65, 416-431 e6. 10.1016/j.molcel.2016.12.028 [DOI] [PubMed] [Google Scholar]
- Ebner M., Sinkovics B., Szczygiel M., Ribeiro D. W. and Yudushkin I. (2017b). Localization of mTORC2 activity inside cells. J. Cell Biol. 216, 343-353. 10.1083/jcb.201610060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egami Y., Taguchi T., Maekawa M., Arai H. and Araki N. (2014). Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front. Physiol. 5, 374 10.3389/fphys.2014.00374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erami Z., Khalil B. D., Salloum G., Yao Y., Lopiccolo J., Shymanets A., Nurnberg B., Bresnick A. R. and Backer J. M. (2017). Rac1-stimulated macropinocytosis enhances GbetaUpsilon activation of PI3Kbeta. Biochem. J. 474, 3903-3914. 10.1042/BCJ20170279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretto L. J., Snape A. J., Tomlinson J. E., Seroogy J. J., Wolf D. L., Larochelle W. J. and Giese N. A. (1993). Mechanism of platelet-derived growth factor (PDGF) AA, AB, and BB binding to alpha and beta PDGF receptor. J. Biol. Chem. 268, 3625-3631. [PubMed] [Google Scholar]
- Fritsch R., De Krijger I., Fritsch K., George R., Reason B., Kumar M. S., Diefenbacher M., Stamp G. and Downward J. (2013). RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050-1063. 10.1016/j.cell.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A., Zwartkruis F. J., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L. and Thomas G. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457-1466. 10.1016/S1097-2765(03)00220-X [DOI] [PubMed] [Google Scholar]
- Gross S. M. and Rotwein P. (2016). Mapping growth-factor-modulated Akt signaling dynamics. J. Cell Sci. 129, 2052-2063. 10.1242/jcs.183764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Ramjaun A. R., Haiko P., Wang Y., Warne P. H., Nicke B., Nye E., Stamp G., Alitalo K. and Downward J. (2007). Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 129, 957-968. 10.1016/j.cell.2007.03.051 [DOI] [PubMed] [Google Scholar]
- Hoon J.-L., Wong W.-K. and Koh C.-G. (2012). Functions and regulation of circular dorsal ruffles. Mol. Cell. Biol. 32, 4246-4257. 10.1128/MCB.00551-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshmand-Rad R., Claesson-Welsh L., Wennstrom S., Yokote K., Siegbahn A. and Heldin C.-H. (1997). Involvement of phosphatidylinositide 3'-kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp. Cell Res. 234, 434-441. 10.1006/excr.1997.3636 [DOI] [PubMed] [Google Scholar]
- Hoppe A. D. (2012). FRET-based imaging of Rac and Cdc42 activation during Fc-receptor-mediated phagocytosis in macrophages. Methods Mol. Biol. 827, 235-251. 10.1007/978-1-61779-442-1_16 [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Zhu T., Wu J. and Guan K.-L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648-657. 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Xu T. and Guan K. L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829-1834. 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. P., Schoenwaelder S. M., Goncalves I., Nesbitt W. S., Yap C. L., Wright C. E., Kenche V., Anderson K. E., Dopheide S. M., Yuan Y. et al. (2005). PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat. Med. 11, 507-514. 10.1038/nm1232 [DOI] [PubMed] [Google Scholar]
- Jamieson S., Flanagan J. U., Kolekar S., Buchanan C., Kendall J. D., Lee W.-J., Rewcastle G. W., Denny W. A., Singh R., Dickson J. et al. (2011). A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem. J. 438, 53-62. 10.1042/BJ20110502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay R. R., Williams T. D. and Paschke P. (2018). Amplification of PIP3 signalling by macropinocytic cups. Biochem. J. 475, 643-648. 10.1042/BCJ20170785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E. C., Dibble C. C. and Toker A. (2017). PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62-71. 10.1016/j.ceb.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B. D. and Toker A. (2017). AKT/PKB signaling: navigating the network. Cell 169, 381-405. 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S., Dibble C. C., Talbott G., Hoxhaj G., Valvezan A. J., Takahashi H., Cantley L. C. and Manning B. D. (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771-785. 10.1016/j.cell.2013.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercanti V., Charette S. J., Bennett N., Ryckewaert J. J., Letourneur F. and Cosson P. (2006). Selective membrane exclusion in phagocytic and macropinocytic cups. J. Cell Sci. 119, 4079-4087. 10.1242/jcs.03190 [DOI] [PubMed] [Google Scholar]
- Ozcan F., Klein P., Lemmon M. A., Lax I. and Schlessinger J. (2006). On the nature of low- and high-affinity EGF receptors on living cells. Proc. Natl. Acad. Sci. USA 103, 5735-5740. 10.1073/pnas.0601469103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacitto R., Gaeta I., Swanson J. A. and Yoshida S. (2017). CXCL12-induced macropinocytosis modulates two distinct pathways to activate mTORC1 in macrophages. J. Leukoc. Biol. 101, 683-692. 10.1189/jlb.2A0316-141RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat-Shliom N., Kloog Y. and Donaldson J. G. (2008). A unique platform for H-Ras signaling involving clathrin-independent endocytosis. Mol. Biol. Cell 19, 765-775. 10.1091/mbc.e07-08-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter C. J., Pedraza L. G. and Xu T. (2002). Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4, 658-665. 10.1038/ncb840 [DOI] [PubMed] [Google Scholar]
- Racoosin E. L. and Swanson J. A. (1989). Macrophage colony stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J. Exp. Med. 170, 1635-1648. 10.1084/jem.170.5.1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racoosin E. L. and Swanson J. A. (1992). M-CSF-induced macropinocytosis increases solute endocytosis but not receptor-mediated endocytosis in mouse macrophages. J. Cell Sci. 102, 867-880. [DOI] [PubMed] [Google Scholar]
- Rodrigues G. A., Falasca M., Zhang Z., Ong S. H. and Schlessinger J. (2000). A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 20, 1448-1459. 10.1128/MCB.20.4.1448-1459.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosania G. R. and Swanson J. A. (1996). Microtubules can modulate pseudopod activity from a distance inside macrophages. Cell Motil. Cytoskel. 34, 230-245. [DOI] [PubMed] [Google Scholar]
- Saito K., Araki Y., Kontani K., Nishina H. and Katada T. (2005). Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 137, 423-430. 10.1093/jb/mvi046 [DOI] [PubMed] [Google Scholar]
- Sallusto F., Cella M., Danieli C. and Lanzavecchia A. (1995). Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 182, 389-400. 10.1084/jem.182.2.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L. and Sabatini D. M. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496-1501. 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S. and Sabatini D. M. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290-303. 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R. A. and Sabatini D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960-976. 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J. A. (2002). Ratiometric fluorescence microscopy. In Molecular Cellular Biology, Vol. 31 (ed. Sansonetti P. and Zychlinsky A.). pp. 11-18. New York: Academic Press; 10.1016/S0580-9517(02)31002-X [DOI] [Google Scholar]
- Swanson J. A. (2008). Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 9, 639-649. 10.1038/nrm2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia A., Goicoechea S. M., Awadia S., Zinn A. and Garcia-Mata R. (2017). Regulation of circular dorsal ruffles, macropinocytosis, and cell migration by RhoG and its exchange factor, Trio. Mol. Biol. Cell 28, 1768-1781. 10.1091/mbc.e16-06-0412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P., Rother K. I. and Balla T. (1999). Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in living cells. J. Biol. Chem. 274, 10983-10989. 10.1074/jbc.274.16.10983 [DOI] [PubMed] [Google Scholar]
- Wall A. A., Luo L., Hung Y., Tong S. J., Condon N. D., Blumenthal A., Sweet M. J. and Stow J. L. (2017). Small GTPase Rab8a-recruited phosphatidylinositol 3-kinase gamma regulates signaling and cytokine outputs from endosomal toll-like receptors. J. Biol. Chem. 292, 4411-4422. 10.1074/jbc.M116.766337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer C. M., Worthylake R. A., Liu B. P., Burridge K. and Salmon E. D. (1999). Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1, 45-50. 10.1038/9018 [DOI] [PubMed] [Google Scholar]
- Welliver T. P. and Swanson J. A. (2012). A growth factor signaling cascade confined to circular ruffles in macrophages. Biol. Open 1, 754-760. 10.1242/bio.20121784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welliver T. P., Chang S. L., Linderman J. J. and Swanson J. A. (2011). Ruffles limit diffusion in the plasma membrane during macropinosome formation. J. Cell Sci. 124, 4106-4114. 10.1242/jcs.091538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann M. and Arcaro A. (1994). Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem. J. 298, 517-520. 10.1042/bj2980517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Hoppe A. D., Araki N. and Swanson J. A. (2009). Sequential signaling in plasma-membrane domains during macropinosome formation in macrophages. J. Cell Sci. 122, 3250-3261. 10.1242/jcs.053207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Hong S., Suzuki T., Nada S., Mannan A. M., Wang J., Okada M., Guan K. L. and Inoki K. (2011). Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 286, 32651-32660. 10.1074/jbc.M111.238014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Gaeta I., Pacitto R., Krienke L., Alge O., Gregorka B. and Swanson J. A. (2015a). Differential signaling during macropinocytosis in response to M-CSF and PMA in macrophages. Front. Physiol. 6, 8 10.3389/fphys.2015.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Pacitto R., Yao Y., Inoki K. and Swanson J. A. (2015b). Growth factor signaling to mTORC1 by amino acid-laden macropinosomes. J. Cell Biol. 211, 159-172. 10.1083/jcb.201504097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Pacitto R., Inoki K. and Swanson J. (2018). Macropinocytosis, mTORC1 and cellular growth control. Cell. Mol. Life Sci. 75, 1227-1239. 10.1007/s00018-017-2710-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Gao X., Saucedo L. J., Ru B., Edgar B. A. and Pan D. (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578-581. 10.1038/ncb999 [DOI] [PubMed] [Google Scholar]
- Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y. and Sabatini D. M. (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678-683. 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.