

ABSTRACT
Terminal differentiation generates the specialized features and functions that allow postmitotic cells to acquire their distinguishing characteristics. This process is thought to be controlled by transcription factors called ‘terminal selectors’ that directly activate a set of downstream effector genes. In Caenorhabditis elegans, the differentiation of both the mechanosensory touch receptor neurons (TRNs) and the multidendritic nociceptor FLP neurons uses the terminal selectors UNC-86 and MEC-3. The FLP neurons fail to activate TRN genes, however, because a complex of two transcriptional repressors (EGL-44/EGL-46) prevents their expression. Here, we show that the ZEB family transcriptional factor ZAG-1 promotes TRN differentiation not by activating TRN genes but by preventing the expression of EGL-44/EGL-46. As EGL-44/EGL-46 also inhibits the production of ZAG-1, these proteins form a bistable, negative-feedback loop that regulates the choice between the two neuronal fates.
KEY WORDS: Cell fate, Neuronal differentiation, Binary decision, Repressors, Touch receptor neurons, ZEB
Summary: ZAG-1, a ZEB family transcription factor, safeguards fate specification of C. elegans touch receptor neurons by inhibiting a TEA domain-containing repressor, revealing its role in neuronal fate choice.
INTRODUCTION
The terminally differentiated state or cell fate of neurons distinguishes them from other neurons through specialized features and functions, and the expression of effector genes, termed as ‘terminal differentiation genes’ (Hobert, 2011). Although individual effector genes may not be expressed in a cell type-specific manner, the collective expression of a battery of terminal differentiation genes serves as a signature for the cell fate. For example, the cell fate of mammalian photoreceptors is defined by the expression of over 600 genes, including those encoding proteins directly involved in light detection (Blackshaw et al., 2001; Hsiau et al., 2007). The expression of the terminal differentiation genes is often activated by transcription factors called ‘terminal selectors’ (Garcia-Bellido, 1975; Hobert, 2011) that act alone or in combination and bind to common cis-regulatory elements in the terminal differentiation gene.
Terminal selectors or transcriptional activators are not, however, sufficient to specify neuronal fates. Neuron type-specific transcriptional repressors sculpt the expression profiles of terminal differentiation genes by repressing effector genes that do not contribute to a given fate (Kerk et al., 2017; Mitani et al., 1993; Pflugrad et al., 1997; Vallstedt et al., 2001; William et al., 2003). Such repressors appear to be particularly important, as the same terminal selectors are often expressed in several distinct types of neuron and promote their differentiation into distinct fates (Hobert, 2016). For example, although UNC-3 is the terminal selector for all cholinergic motor neurons in the C. elegans ventral nerve cord, combinations of class-specific repressors control the subtype identity of those motor neurons by antagonizing the activity of UNC-3 on selective effector genes (Kerk et al., 2017). Because repressors restrict selector activity, their expression needs to be tightly regulated to ensure the proper differentiation of a particular cell fate.
To understand the mechanism that controls the expression of repressors, we have studied the choice of cell fate that generates either the touch receptor neurons (TRN) or the FLP neurons in C. elegans. The six mechanosensory TRNs detect gentle mechanical stimuli along the body (Chalfie and Sulston, 1981), whereas the two FLP neurons are multidendritic nociceptors that sense harsh touch, noxious temperature and humidity (Chatzigeorgiou and Schafer, 2011; Chatzigeorgiou et al., 2010; Kaplan and Horvitz, 1993; Russell et al., 2014).
The heterodimer of the POU homeodomain transcription factor UNC-86 and the LIM homeodomain transcription factor MEC-3 acts as a terminal selector that promotes TRN cell fate (Xue et al., 1993). Specifically, the heterodimer activates a set of TRN terminal differentiation genes (the mechanosensory channel genes mec-4 and mec-10, the tubulin genes mec-7 and mec-12, the tubulin acetyltransferase gene mec-17, and others) by binding to conserved regulatory elements in their proximal promoters (Duggan et al., 1998; Zhang et al., 2002).
MEC-3 and UNC-86 are expressed in and are needed for the differentiation of the FLP neurons and the two postembryonic multidendritic PVD neurons. The expression of unc-86 and mec-3 in these cells does not lead to the normal expression of many TRN terminal differentiation genes, resulting in cells whose fate is very different from the TRNs (Finney and Ruvkun, 1990; Way and Chalfie, 1988). Our previous work suggested that the TEA domain transcription factor EGL-44 and the zinc-finger protein EGL-46, which form a complex abbreviated as EGL-44/EGL-46, in FLP neurons prevent these cells from acquiring the TRN fate (Mitani et al., 1993; Wu et al., 2001). Loss of these genes in the FLP cells turns them into TRN-like cells; and misexpression of these genes in the TRNs prevent them from taking on TRN characteristics. Thus, repression by EGL-44 and EGL-46 in FLP neurons distinguishes them from the TRNs.
In this paper, we extend this model of TRN and FLP neuronal determination by identifying another cell fate regulator, the zinc-finger homeodomain transcription factor ZAG-1, which is expressed in TRNs but not FLP (or PVD) neurons. ZAG-1 promotes the TRN fate not by directly activating the TRN terminal differentiation genes but by preventing egl-44 and egl-46 expression in the TRNs. In FLP neurons, the EGL-44/EGL-46 complex simultaneously represses the TRN genes, activates FLP genes and represses zag-1. The mutual inhibition by EGL-44/EGL-46 and ZAG-1 establishes a bistable switch between TRN and FLP fates. Our work suggests that UNC-86/MEC-3 serves as a ground-state selector, resulting in a common state in both TRNs and FLP neurons, and that individual fates are subsequently controlled by this bistable switch. We hypothesize that ZAG-1, particularly, regulates the choice of cell fate by inhibiting fate repressors to safeguard the activation of terminal differentiation genes.
RESULTS
An RNAi screen for transcription factors affecting TRN cell fate
The six C. elegans TRNs include two pairs of embryonically derived and bilaterally symmetric neurons (the anterior ALML and ALMR, and posterior PLML and PLMR), and the unpaired, postembryonically derived AVM and PVM neurons (Chalfie and Sulston, 1981; Sulston and Horvitz, 1977). To search systematically for transcription factors specifying TRN fate, we knocked down the expression of transcription factor genes using RNAi and looked for animals that no longer expressed mec-17p::RFP, a TRN marker (see Materials and Methods for details). Using RNAi against unc-86 as a positive control, we tested various genetic backgrounds that were previously found to enhance the effects of RNA interference and found that eri-1; lin-15B mutants had the highest penetrance for the loss of RFP expression. About 80% of these animals did not express RFP in either of the two ALM neurons when treated with unc-86 RNAi (Fig. 1A,B). The posterior TRNs (PLM neurons) were less affected by the RNAi treatment (Fig. 1B), as seen previously (Calixto et al., 2010). Therefore, we focused on the disappearance of mec-17p::RFP expression in the ALM neurons in the screen.
Fig. 1.

A RNAi screen identifies positive regulators of TRN fate. (A) TU4429, eri-1(mg366); lin-15B(n744); uIs134[mec-17p::RFP] animals treated with RNAi against unc-86 or GFP. Arrows indicate the absence of RFP expression in ALM neurons. (B) Percentage of animals that showed RFP or GFP expression in at least one ALM and one PLM (ALM+PLM+), in no ALM but at least one PLM (ALM−PLM+), and in no ALM and no PLM (ALM−PLM−) neurons. Strains tested for the efficiency of RNAi are TU4429 (wild type), TU3595 [sid-1(pk3321) him-5(e1490); lin-15B(n744); uIs72[unc-119p::sid-1; mec-18::GFP; myo-2p::mCherry]], TU4301 [lin-15B(n744); uIs115[mec-17p::RFP]], TU4396 [lin-15B(hd126) nre-1(hd20); uIs134[mec-17p::RFP]] and TU4429. (C) The positive RNAi clones identified from the screen. P>0.05 for all the positives using the data from all five rounds of screen. Scale bars: 20 µm.
Among the 443 bacterial clones expressing dsRNA against 392 transcription factors and associated proteins (Table S1), we identified 14 genes that were required for the expression of TRN markers (Fig. 1). Four of these genes (unc-86, mec-3, ldb-1 and ceh-20) were known to affect the expression of TRN terminal differentiation genes (Cassata et al., 2000; Way and Chalfie, 1988; Zheng et al., 2015b). We examined null mutants for all the remaining ten genes but could only confirm the loss of mec-17p::RFP expression in zag-1 mutants.
We were surprised that RNAi against nine genes affected TRN expression through six rounds of testing, but loss-of-function mutations in these genes did not. These false-positive results are unlikely to result from the specific genetic background of the RNAi strain, as mutation of several of the genes (zip-4, hmbx-1 and nhr-119) in eri-1; lin-15B animals did not affect TRN fate. Activating the RNAi pathway non-specifically by RNAi against GFP in those triple mutants (e.g. zip-1; eri-1; lin-15B mutants) did not cause the loss of mec-17p::RFP expression either. The discrepancy between the RNAi and mutant phenotypes has been seen in other systems (Kok et al., 2015; Poole et al., 2011) and may be due to mistargeting of the dsRNAs or genetic compensation in mutants (see Discussion for details).
ZAG-1 is required for the expression of TRN fate markers but acts independently of UNC-86/MEC-3
zag-1 encodes the sole C. elegans homolog of ZEB transcription factors, all of which contain a homeodomain flanked by clusters of C2H2-type zinc fingers. Human ZEB transcription factors induce the epithelial-to-mesenchymal transition (EMT) and are essential for normal embryonic development; mutations in ZEB genes cause defects in neural crest development and are linked to malignant tumor progression (Vandewalle et al., 2009). C. elegans zag-1 regulates the differentiation and axonal guidance of several types of neurons, including the command interneurons, GABAergic motor neurons, dopaminergic sensory neurons ADE and PDE, the posterior interneuron PVQ, and the pharyngeal neuron M4 (Clark and Chiu, 2003; Ramakrishnan and Okkema, 2014; Wacker et al., 2003).
Previous research on the TRNs has shown that a hypomorphic zag-1 allele (zd86) caused defects in ALM migration and axonal growth (Clark and Chiu, 2003) and made PVM adopt the shape of the multidendritic nociceptor neuron PVD (Smith et al., 2013). Using a zag-1 null mutation (hd16), we found that the complete loss of zag-1 led to larval arrest in the L1 stage (before AVM and PVM arise) and failure to express several TRN fate markers in the ALM and PLM neurons (Fig. 2A,B, Table 1). The zag-1 null allele hd16 deletes part of the first exon and part of the first intron (Wacker et al., 2003), whereas zd86 and two other viable alleles (zd85 and rh315) all cause premature stops in the fifth exon, leaving the first two zinc fingers and the homeodomain intact (Fig. 2B). Thus, ZAG-1 is important for general TRN fate specification.
Fig. 2.

zag-1 is required for the expression of TRN markers independently of mec-3. (A) The expression of mec-17p::RFP and mec-3p::GFP reporters in zag-1(hd16) L1 larvae. (B) The structure of zag-1 and the positions of zag-1 mutations. (C) The number of mec-3 transcripts in TRNs from wild-type and zag-1 animals from smFISH experiments. (D) The expression of the fosmid-based reporter zag-1::EGFP in TRNs but not FLPs. Outline indicates the absence of GFP expression in FLP neurons. (E) zag-1:EGFP expression in mec-3 mutants. Arrows indicate expression in PLM neurons. Scale bars: 20 µm.
Table 1.
The penetrance for TRN marker expression in zag-1 null mutants
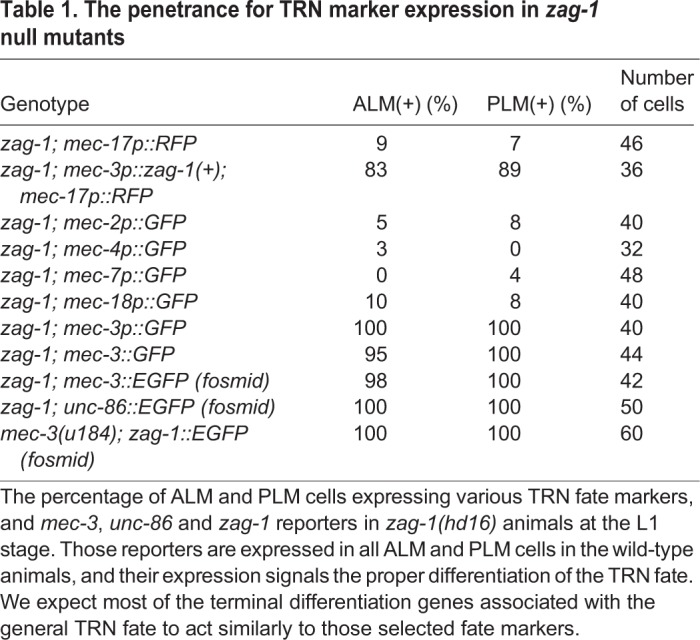
We next tested the genetic interaction between zag-1 and mec-3, and found that the zag-1 (hd16) null allele did not affect the expression of the mec-3 using a transcriptional reporter (Fig. 2A), quantitative mRNA measurements with single molecule fluorescent in situ hybridization (smFISH; Fig. 2C) or MEC-3::GFP translational fusions (Table 1). In addition, expression of zag-1(+) from the mec-3 promoter restored the expression of the TRN fate marker mec-17p::RFP in zag-1 mutants, suggesting that ZAG-1 induces the expression of TRN terminal differentiation genes cell-autonomously. As mec-3 expression is completely dependent on unc-86, we expected and found that the expression of unc-86 was not changed in zag-1 mutants.
Using a fosmid-based GFP translational fusion, we found that zag-1 was expressed in the six TRNs but not in the FLP and PVD neurons (Fig. 2D), and the expression of zag-1 in TRNs was not affected by mutations in mec-3 (Fig. 2E). Therefore, zag-1 and mec-3 are transcriptionally independent of each other. We also failed to find any physical interaction between ZAG-1 and MEC-3 in yeast two-hybrid assays (Fig. S1). Together, our results suggest that ZAG-1 promotes TRN fate independently of UNC-86 and MEC-3; the expression of the three transcription factors only overlaps in the TRNs and thus forms a unique combinatorial code for TRN fate.
Smith et al. (2013) found that another conserved transcription factor, AHR-1 (aryl hydrocarbon receptor), controls the differentiation of AVM; in ahr-1 null mutants, AVM cells adopted a PVD-like multidendritic shape. They hypothesized that AHR-1 and ZAG-1 function in parallel to specify TRN morphology in the AVM and PVM, respectively. We found that ahr-1 is expressed in all six TRNs but is only required for the expression of TRN markers in AVM neurons, suggesting that AHR-1 plays a subtype-specific role in TRN fate specification (Fig. S2A-C). In contrast, ZAG-1 is required for TRN fate adoption in general.
ZAG-1 promotes TRN fate by suppressing TRN fate inhibitors EGL-44 and EGL-46
The TEA domain transcription factor EGL-44 and the zinc-finger protein EGL-46 repress TRN fate in the FLP neurons (Wu et al., 2001). These proteins appeared to work together to regulate gene expression and physically interacted in yeast two-hybrid assays (Fig. S3). Both genes were normally expressed in the FLP neurons but not the TRNs, and the expression of egl-46 was dependent on egl-44 (Wu et al., 2001) (Fig. 3A, Table 2). Furthermore, egl-44 and egl-46 mutations caused the ectopic expression of mec-17p::GFP and other TRN reporters in FLP neurons (Wu et al., 2001) (Fig. 3B, Table 2; Table S2).
Fig. 3.

ZAG-1 promotes TRN fate by repressing egl-44 and egl-46. (A) The expression of the TRN marker mec-17p::GFP in FLP neurons of egl-44 animals and in TRNs of egl-44; zag-1 and zag-1; egl-46 animals. Penetrance of the expression of various reporters. Arrows indicate GFP expression and outlines indicate no expression. (B,C) The expression of egl-44::GFP and egl-46::GFP reporters in FLPs, but not TRNs, of wild-type animals and in TRNs of zag-1 animals. (D) The number of egl-44 mRNA molecules in TRNs of wild-type and zag-1 animals. Scale bars: 20 µm.
Table 2.
The penetrance of TRN marker expression in egl-44 and egl-46 single mutants, and in egl-44; zag-1 and egl-46; zag-1 double mutants
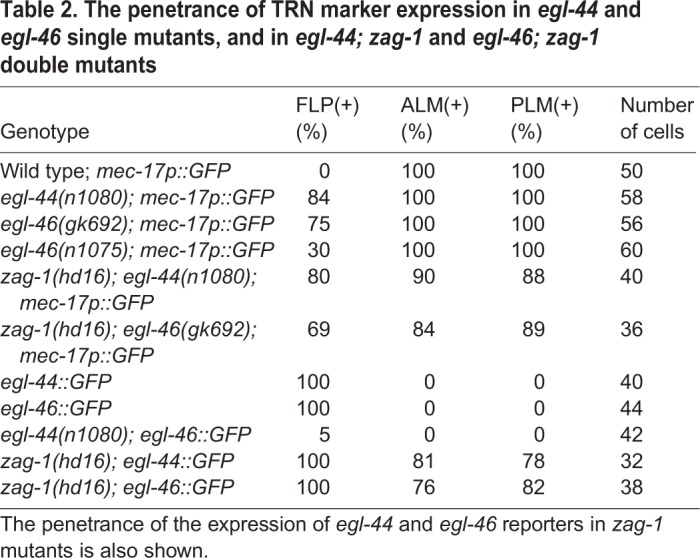
As zag-1 was selectively expressed in TRNs but not FLP neurons, we tested whether ZAG-1 promoted the TRN fate by preventing the activation of EGL-44/EGL-46 in these cells. Consistent with this hypothesis, GFP reporters for both egl-44 and egl-46 were ectopically expressed in the TRNs in zag-1 mutants (Fig. 3C). In addition, egl-44 mRNA, as measured by smFISH, was increased in zag-1 TRNs, an indication that ZAG-1 transcriptionally repressed egl-44 (Fig. 3D). Importantly, egl-44 and egl-46 are epistatic to zag-1, as mutations in them restored the expression of TRN fate markers in zag-1-deficient animals (Fig. 3B; Table S2). Thus, the TRN cell fate did not require ZAG-1 in the absence of EGL-44 and EGL-46. Instead, ZAG-1 promoted the TRN fate through a double inhibition mechanism by preventing the expression of the TRN fate repressors egl-44 and egl-46.
We also found that loss of ZAG-1 affected general neurite growth and guidance in TRNs, as egl-44; zag-1 and zag-1; egl-46 mutants showed shortened and misguided TRN neurites. For example, ALM neurites and the PLM anterior neurites were absent or severely shortened in about 50% and 70% (n=40) of those double mutants, respectively (Fig. S4). Our results using the zag-1 null allele revealed its essential role in regulating neurite development, which extends previous observation that hypomorphic zag-1 alleles caused axonal guidance defects in multiple neurons (Clark and Chiu, 2003; Wacker et al., 2003). Because these outgrowth defects are not seen in mec-3 mutants, ZAG-1 has a second, independent function in TRNs in addition to inhibiting egl-44 and egl-46.
Misexpression of ZAG-1 converts FLP neurons into TRN-like cells
To address whether zag-1 could affect FLP differentiation, we misexpressed zag-1 in FLP neurons using the mec-3 promoter. This misexpression led to the expression of mec-17p::GFP and other TRN markers (Fig. 4A; Table S2) and diminished expression of egl-44 and egl-46 (Fig. 4A, Table 3) in FLP neurons. These data suggest that misexpressed ZAG-1 converts FLP neurons to a TRN-like fate by inhibiting egl-44 and egl-46. The expression of mec-3p::zag-1 transgene also activated TRN markers in PVD neurons (Fig. 4A). Thus, ZAG-1 not only prevents PVM neurons from taking on the PVD fate (Smith et al., 2013) but can also turn PVD neurons into TRN-like cells. As egl-44 was not expressed in PVD neurons, the misexpressed ZAG-1 presumably directs this conversion of cell fate by inhibiting some unidentified factor(s) that normally prevents the acquisition of the TRN fate by PVD neurons.
Fig. 4.

Mutual inhibition between zag-1 and egl-44 regulates TRN fate decision. (A) The activation of the TRN marker mec-17p::GFP and the loss of the expression of egl-44 and egl-46 reporters in FLP neurons of animals carrying the mec-3p::zag-1 transgene. Arrows indicate GFP expression and outlines indicate no expression. (B) The expression of zag-1::EGFP in FLP neurons of egl-44 and egl-46 mutants, and weak expression of zag-1 reporter in animals misexpressing egl-44 from the mec-3 promoter. (C-E) The expression of mec-17p::GFP in animals carrying various transgenes. Scale bars: 20 µm in A,C; 100 µm in D-F.
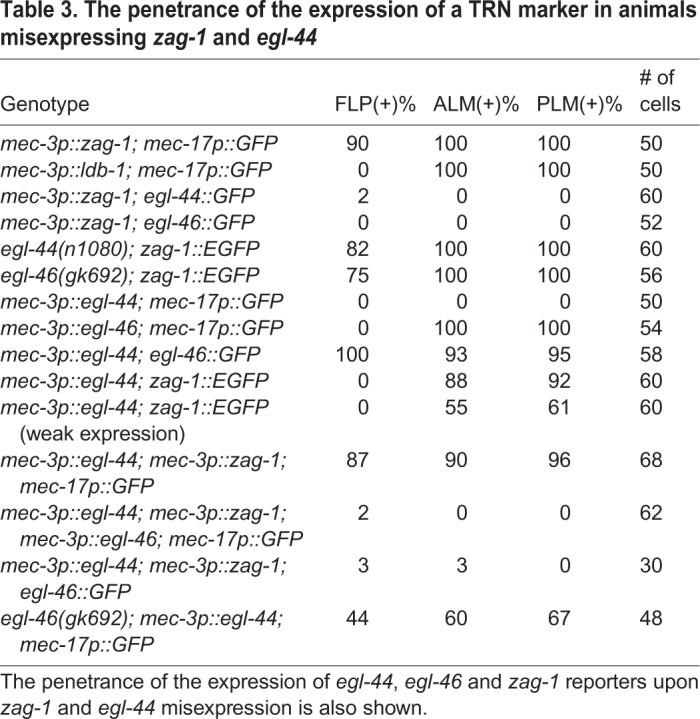
Table 3.
The penetrance of the expression of a TRN marker in animals misexpressing zag-1 and egl-44
We also expressed ahr-1 from the mec-3 promoter and found that misexpressed AHR-1 could activate TRN fate markers in PVD but not FLP neurons (Fig. S2D,E). Overexpression of AHR-1 in TRNs also caused morphological defects, such as the growth of an ectopic ALM posterior neurite (Fig. S2D), which does not occur when ZAG-1 was overexpressed. These results further support the hypothesis that ZAG-1 and AHR-1 have different functions.
EGL-44/EGL-46 prevents zag-1 expression in FLP neurons
Given the mutually exclusive patterns of zag-1 and egl-44/egl-46 expression in FLP neurons and TRNs, we next tested whether EGL-44/EGL-46 regulated zag-1 expression. Mutations in egl-44 and egl-46 resulted in ectopic expression of a zag-1::EGFP reporter in FLP neurons (Fig. 4B, Table 3). Misexpression of egl-44 from the mec-3 promoter is sufficient to suppress the TRN fate, because it activates the endogenous egl-46, a co-factor that is required for EGL-44 functions (Wu et al., 2001) (Fig. 4C; Table 3). We found that EGL-44 misexpression reduced, but did not completely eliminate, the expression of zag-1 in TRNs (Fig. 4B; Table 3). Therefore, the positive TRN fate regulator ZAG-1 and the negative regulator EGL-44/EGL-46 reciprocally inhibit each other's expression.
ZAG-1 also inhibited endogenous egl-46 expression. When EGL-44 and ZAG-1 were simultaneously misexpressed from the heterologous mec-3 promoter, the presence of both proteins led to the activation of TRN markers in all mec-3-expressing neurons (TRNs, FLP and PVD; Fig. 4D), because ZAG-1-mediated inhibition of endogenous egl-46 blocked the effects of EGL-44. Expression of an egl-46::GFP reporter was reduced by ZAG-1 despite the presence of misexpressed EGL-44 (Table 3), suggesting that ZAG-1 can repress egl-46 transcription both through egl-44 and independently of egl-44. To rule out the possibility of direct protein-protein interaction and interference, we misexpressed EGL-44, EGL-46 and ZAG-1 from the heterologous mec-3 promoter and found that the expression of TRN markers was turned off, suggesting that EGL-44/EGL-46 can repress TRN fate in the presence of ZAG-1 (Fig. 4E). These data suggest that the mutual inhibition between ZAG-1 and EGL-44/EGL-46 occurs only at the transcription level and not likely through direct interaction. Consistent with this idea, ZAG-1 failed to interact with either EGL-44 or EGL-46 in yeast two-hybrid assays (MEC-3 also failed to interact with EGL-44, EGL-46 and ZAG-1; Fig. S3).
We also tested the touch sensitivity of the above transgenic animals and the functional results are consistent with the marker expression results. Animals carrying the uIs211[mec-3p::egl-44] transgene were completely insensitive to gentle touch; co-expression of zag-1 from the mec-3 promoter restored the sensitivity in animals where the TRN markers were reactivated but failed to do so when mec-3p::egl-46 was also co-expressed (Fig. S5).
EGL-44/EGL-46 and ZAG-1 regulate a switch between FLP and TRN fates
We next investigated how EGL-44/EGL-46 and ZAG-1 regulated FLP fate using several genes (the stomatin gene sto-5, the glycoprotein gene dma-1, the dynein regulator bicd-1 and the FMRFamide-like peptide gene flp-4) that were expressed in FLP neurons (Aguirre-Chen et al., 2011; Kim and Li, 2004; Liu and Shen, 2011) but not the TRNs (Fig. 5A). FLP expression of all four genes depended on EGL-44 and EGL-46 (Fig. 5B,C). Moreover, the expression of the FLP and TRN markers was mutually exclusive; FLP neurons in egl-44 and egl-46 mutants or animals carrying mec-3p::zag-1 transgene never showed mixed expression of the FLP- and TRN-specific genes (Table 4). Mutation in zag-1 or the misexpression of egl-44 in TRNs activates the FLP markers in addition to turning off the TRN markers (Fig. 5B; Fig. S6), and we never observed mixed expression of both types of markers (Table 4). As FLP markers were not expressed in egl-44; zag-1 double mutants, our results suggest that EGL-44/EGL-46 not only repressed the TRN fate but also promoted the FLP fate. Thus, the action of EGL-44/EGL-46 and ZAG-1 results in two mutually exclusive fates.
Fig. 5.

EGL-44/EGL-46 simultaneously induces FLP genes and suppresses TRN genes. (A,B) Various FLP fate reporters were expressed in wild-type FLP neurons (A) but not in egl-44 mutants (B). (C) The expression of FLP and TRN fate markers was mutually exclusive in FLP neurons of egl-44 mutants. (D) The expression of mec-19 was not affected by mutations in egl-44. Scale bars: 20 µm in A-C; 100 µm in D.
Table 4.
Percentages of FLP and ALM neurons expressing the green (G) and red (R) markers in various genetic backgrounds

The EGL-44/EGL-46 ZAG-1 bistable switch affected neuronal morphology as well as transcriptional reporters. For example, egl-44 misexpression in ALM neurons resulted in an ectopic posterior neurite that was characteristic of FLP neurons (Fig. S7), and zag-1 expression in FLP and PVD neurons drastically reduced the number of dendritic branches (Fig. S7). This latter result is consistent with a role for ZAG-1 in preventing PVM from adopting a PVD-like morphology (Smith et al., 2013).
dma-1, bicd-1 and flp-4 were also expressed in PVD neurons, suggesting that FLP and PVD share a common genetic program. The expression of these PVD markers was blocked by the misexpression of ZAG-1 in PVD neurons, suggesting that ZAG-1 inhibited the unknown PVD fate regulator (repressor). On the other hand, the sto-5 reporter was expressed in FLP but not in PVD neurons, indicating differences in the transcriptome of the two cell types (Fig. S8).
The activation of the FLP and PVD markers, like the TRN markers, required UNC-86 and MEC-3, which are the common terminal selectors for the three fates (Table 4; Fig. S6B). We identified one common terminal differentiation gene, mec-19, which was expressed in TRN, FLP and PVD neurons at similar levels (and no other cells); its expression depended on UNC-86 and MEC-3 but was not affected by the loss of EGL-44/EGL-46 and ZAG-1 (Fig. 5D; Table 4). Thus, unc-86 and mec-3 serve as the ‘ground-state selectors’ that promote a common ground state shared by the three types of neurons, and mec-19 is a marker for this ground state. Subsequently, zag-1 and egl-44 act as ‘modulators’ that shift the ground state towards specific fates. Therefore, the development of a particular fate requires the activities of both the ground-state selectors and the fate-restricting modulators. Our results are consistent with the findings in C. elegans cholinergic motor neurons, where ground-state markers, such as cho-1 (choline transporter), cha-1 (choline acetyltransferase) and unc-17 (vesicle acetylcholine transporter), are activated by selectors and not regulated by subtype-specific repressors (Kerk et al., 2017).
Among the six TRNs, ALM maintains the default TRN state, whereas PLM undergoes further differentiation controlled by the posterior Hox protein EGL-5, which represses UNC-86/MEC-3-dependent ALM genes and activates UNC-86/MEC-3-independent PLM genes (Zheng et al., 2015a). We found that the ALM gene mir-84, but not the PLM gene rfip-1, was expressed in the FLP neurons of egl-44 mutants (FLP neurons do not express EGL-5; Zheng et al., 2015b; Fig. S9). These results suggest that that the bistable switch controls the general TRN fate not TRN subtype identity.
EGL-44/EGL-46 inhibits the expression of TRN genes by binding to UNC-86/MEC-3-binding sites and by suppressing ALR-1 in FLP neurons
We next investigated the mechanism by which EGL-44/EGL-46 prevented the expression of TRN terminal differentiation genes. Although we found two UNC-86/MEC-3-binding sites required for the expression of a minimal TRN-specific promoter (a 184 bp mec-18 promoter) in TRNs, mutational analysis of this minimal promoter failed to identify any additional, discrete cis-regulatory element that mediated its repression in FLP neurons (Fig. S10). Thus, EGL-44/EGL-46 seems unlikely to suppress TRN markers via repressive elements that are distinct from the UNC-86/MEC-3-binding sites. We then tested the possibility that EGL-44/EGL-46 acts through the UNC-86/MEC-3-binding site, as EGL-44 belongs to the TEA domain class transcription factors, which recognize DNA sequences similar to the UNC-86/MEC-3-binding site (Fig. 6A) (Jiang et al., 2000; Zhang et al., 2002). EGL-44 bound to previously identified UNC-86/MEC-3 motifs in the mec-4, mec-7, mec-17 and mec-18 promoters in electrophoretic mobility shift assays (Fig. 6B), suggesting that EGL-44 directly contacts the TRN promoters. The EGL-44/EGL-46 complex also bound to the same EGL-44-binding motif in the mec-4 promoter, suggesting EGL-46 may act as a co-repressor (Fig. 6C). The association of EGL-44/EGL-46 with the cis-regulatory element bound by UNC-86/MEC-3 suggests that EGL-44/EGL-46 in FLP neurons may prevent the activation of TRN genes by occluding the UNC-86/MEC-3-binding sites essential for the expression of TRN fate.
Fig. 6.

EGL-44/EGL-46 inhibits the expression of TRN genes by binding to the same cis-regulatory elements bound by UNC-86/MEC-3. (A) The alignment of various cis-regulatory motifs tested in electrophoretic mobility shift (EMSA) assays. Positions 1-4 were assigned to the four nucleotides following the consensus UNC-86/MEC-3-binding site (Zhang et al., 2002). Nucleotides in red were considered important for EGL-44 binding and nucleotides in blue were responsible for the lack of EGL-44 binding. (B) The binding of recombinant EGL-44 proteins to various probes in EMSA assays. Arrows indicate the band of EGL-44::DNA complexes. (C) The binding of EGL-44/EGL-46 to the mec-4 probes. (D) The expression of TRN fate markers in egl-44; alr-1 double mutants and in animals misexpressing ALR-1 from the mec-3 promoter, and the expression of a fosmid-based alr-1 reporter wgIs200[alr-1::EGFP] in wild-type animals and in egl-44 mutants. Arrows and outlines indicate the position of the nonexpressing FLP neuron cell bodies.
However, how EGL-44/EGL-46 avoids repressing UNC-86/MEC-3 targets that are commonly expressed in FLP and TRNs is unclear. One such target is mec-3 itself, the activation and maintenance of which depend on two UNC-86/MEC-3-binding sites (Xue et al., 1992). We found that the two sites were not bound by EGL-44 (Fig. 6B), which explains why mec-3 expression is not affected by the presence of EGL-44/EGL-46 in FLP neurons. Comparing the cis-regulatory motif sequences on the mec-3 promoter with those on TRN-specific promoters, we found that the four nucleotides (positions 1 to 4 in Fig. 6A) following the UNC-86/MEC-3-binding sequence showed significant divergence between EGL-44-binding and nonbinding sites. In particular, the EGL-44-binding sites all contain adenine at the fourth position and the nonbinding sites do not (Fig. 6A). Changing this adenine to thymine eliminated the binding of EGL-44 to the site on the mec-4 promoter (Fig. 6B). However, converting other nucleotides to adenine at the fourth position was not sufficient to enable the binding of EGL-44 to mec-3 promoter motifs and coordinated change of the nucleotides on the first and second positions are needed to evoke EGL-44 binding (Fig. 6B). Similar results were also obtained for the UNC-86/MEC-3 target gene mec-10, which is expressed in both TRN and FLP cells (Huang and Chalfie, 1994). The UNC-86/MEC-3 site on mec-10 promoter was not bound by EGL-44 and changing the nucleotides on the first and fourth position enabled EGL-44 binding. Thus, our results suggest that EGL-44/EGL-46 can differentiate TRN/FLP common genes from TRN-specific genes via the cis-regulatory sequences in their promoters.
We next tested whether converting the EGL-44-binding site to a nonbinding site in a TRN promoter could prevent the suppression by EGL-44/EGL-46 in FLP neurons in vivo. Contrary to our expectation, a mec-4 promoter reporter harboring the adenine-to-thymine change at the fourth position was not activated in FLP neurons, although its TRN expression was preserved. In the 184 bp mec-18 promoter, changing the four nucleotides following the UNC-86/MEC-3-binding sequence from ACCA to CTAT completely abolished EGL-44 binding in vitro; however, the mutant reporter was only weakly expressed in ∼30% of FLP neurons and remained silenced in the rest ∼70% (Fig. S11). In addition, creating an EGL-44-binding site in the mec-3 promoter by mutating the regulatory motif did not suppress mec-3 expression in FLP neurons (Fig. S11A). The discrepancy between the in vitro and in vivo results suggests that the lack of EGL-44 binding to TRN differentiation genes per se is not sufficient to distinguish TRN fate from the TRN/FLP ground state. In addition to directly binding to the cis-regulatory elements of TRN promoters, EGL-44/EGL-46 may also inhibit TRN genes by activating or repressing the expression of other trans-acting factors.
One such factor may be ALR-1, which is an ortholog of human Arx and Drosophila aristaless, and is needed for the robust differentiation of TRN fate (Topalidou et al., 2011). Loss of alr-1 variably reduced but did not eliminate the expression of TRN markers in the TRNs in wild type and strongly eliminated the ectopic expression of TRN markers in FLP neurons in egl-44 and egl-46 mutants (Fig. 6D; Fig. S12A). This difference between reduction and elimination may result from the fact that FLP neurons have lower mec-3 expression than the TRNs (Topalidou et al., 2011) and thus a stronger need for ALR-1 to activate TRN markers. The observation that alr-1 is epistatic to egl-44 and egl-46 suggested that ALR-1 is a downstream effector suppressed by EGL-44/EGL-46. Indeed, a fosmid-based alr-1 translational reporter, which was normally expressed in TRNs but not FLP neurons, became de-repressed in FLP cells in egl-44 and egl-46 mutants (Fig. 6D; Fig. S12A).
The lack of ALR-1 and the lower level of mec-3 in wild-type FLP neurons may explain the inactivation of the mutated TRN fate reporters that cannot be bound by EGL-44/EGL-46. Supporting this hypothesis, forced expression of ALR-1 in FLP neurons ectopically activated even the wild-type TRN fate reporters (Topalidou et al., 2011) (Fig. 6D), suggesting that ALR-1 not only promotes TRN fate but also overcomes the direct suppression from EGL-44/EGL-46 on the TRN genes. This ability of ALR-1 may result from ALR-1 upregulating mec-3 (Topalidou et al., 2011) and directly interacting with TRN promoters (ChIP-seq data; Fig. S12B). Therefore, EGL-44/EGL-46 inhibits TRN-specific genes by both occupying the UNC-86/MEC-3 site in the TRN promoters and suppressing TRN fate-promoting transcription factor ALR-1 (Fig. 7). Consistent with this model, overexpression of MEC-3 in FLP neurons could overcome EGL-44/EGL-46-mediated inhibition and activate the TRN program (Topalidou and Chalfie, 2011) presumably by both retaking the UNC-86/MEC-3 sites and by activating alr-1, which is a mec-3-dependent gene (Topalidou et al., 2011).
Fig. 7.

A model for the regulatory mechanisms that control cell fate specification among FLP, PVD and TRN fates. Gene names in green, red and blue indicate genes expressed in FLP, TRN and PVD neurons, respectively. Black text indicates genes that were commonly expressed in all three types of neuron; gray text indicates repressed genes and proteins.
Spatial and temporal expression of zag-1 and egl-44 are mutually exclusive
Given the mutual inhibition of zag-1 and egl-44 in the FLP neurons and the TRNs, we asked whether the two genes were generally expressed in different cells in the nervous system. Using the neurotransmitter maps for glutamatergic, cholinergic and GABAergic neurons (Gendrel et al., 2016; Pereira et al., 2015; Serrano-Saiz et al., 2013), we found that, in addition to TRNs, zag-1 was expressed in the AIB, AIM, AIN, AIZ, AVA, AVB, AVD, AVE, AVG, AVK, AVL, M4, M5, RIA, RIB, RIF, RIG, RIM, RIV, RMD, RME, RMF, RMH, SIA and SMD neurons in the head; in all the DD, VD and VC neurons in the ventral cord; and in the DVA, DVB, LUA, PDA, PVC, PVP, PVQ, PVR and PVT neurons in the tail. zag-1 is also expressed in the serotonergic HSN neurons. In comparison, egl-44 expression was much more restricted, being found only in the FLP, ADL and SAB neurons, as well as a few VA and VB motor neurons. Moreover, egl-44 was widely expressed in hypodermis, pharynx and intestine, whereas zag-1 expression was absent in these tissues. We also constructed an egl-44::RFP reporter and crossed it with zag-1:EGFP but did not observe overlapping expression in any cell (Fig. S13A). The above data support the hypothesis that zag-1 and egl-44 expression are mutually exclusive in the nervous system and throughout all tissues of the animal. Given the mutual inhibition of EGL-44 and ZAG-1 in the TRNs/FLPs, we expected that the loss of zag-1 might affect egl-44 expression more broadly. We did not, however, observe a systematic upregulation of egl-44::GFP expression in the nervous system in either the zag-1(zd86) hypomorphic mutants or the arrested L1 animals of zag-1(hd16) null mutants (Fig. S13B,C). Similarly, tissues like hypodermis, pharynx and intestine did not gain zag-1 expression upon the loss of egl-44 either, suggesting that, in addition to the reciprocal inhibition, other activating signals are needed to create the expression pattern of the two transcription factors.
A few neurons expressed both zag-1 and egl-44 but did so at different times. egl-44 and egl-46 were expressed in the precursors of the postembryonic TRNs (the AVM and PVM neurons) and persisted in these cells for a few hours after their generation (Feng et al., 2013; Wu et al., 2001), but they were not expressed in terminally differentiated AVM and PVM. These differentiated cells expressed zag-1, which promoted the TRN fate. The transient expression of egl-44 and egl-46 may be to temporarily block selector functions and to ensure the correct timing of differentiation. Similarly, egl-44 and egl-46 were transiently expressed in the early embryos in HSN neurons before they migrated and differentiated (Wu et al., 2001) (Fig. S13D), whereas zag-1 was expressed in the terminally differentiated HSN neurons in adults and was required for the activation of the HSN fate marker tph-1p::GFP (Fig. S13E,F).
DISCUSSION
Using RNAi to identify genes involved in cell fate determination
We demonstrate here that a systematic RNAi screen using a library of transcription factors can identify neuronal cell fate regulators, particularly those genes whose mutations lead to lethality or sterility. Our previous forward genetic screens, which searched for viable mutants with touch-sensing defects, despite reaching saturation, only identified unc-86 and mec-3 as the TRN fate determinants (Chalfie and Au, 1989; Chalfie and Sulston, 1981). Using the RNAi screen, we not only recovered unc-86 and mec-3 blindly, but also identified ceh-20, ldb-1, and zag-1 as genes required for the expression of TRN fate. These latter genes would not have been identified in our previous screens, because null mutations in them lead to early larval arrest. In particular, our RNAi screen yielded ZAG-1 as a new TRN fate determinant and the third piece in a combinatorial code (UNC-86, MEC-3, and ZAG-1) that defines TRN fate.
Our experiments raise two concerns about the reliability of such RNAi screens. First, only 5 of the 14 genes identified from the screen were confirmed with mutants, and the remaining 9 genes (65%) appeared to be false positives. A similar problem was encountered in a genome-wide RNAi screen for genes involved in the specification of ASE neuron in C. elegans (Poole et al., 2011). More recently, Kok et al. (2015) found that approximately 80% of morpholino-induced phenotypes in zebrafish embryos were not observed in mutants. Similar discrepancies between RNAi and mutant phenotypes were also observed in mice (Daude et al., 2012) and Arabidopsis (Gao et al., 2015). Such widespread differences may be attributed to the mistargeting of dsRNAs or to genetic compensation (the upregulation of a network of genes that compensate for genetic loss but not knockdown; Rossi et al., 2015). Second, our screen failed to recover ceh-13, which is known to affect the TRN fate in ALM neurons (Zheng et al., 2015b), suggesting the existence of false negatives. In addition, because the PLM cell body is located in the tail region (posterior to the intestine), the bias of feeding RNAi towards more anterior cells led to the failure of recovering egl-5, whose loss only affects PLM differentiation (Zheng et al., 2015b).
ZAG-1 prevents the inappropriate expression of repressors and safeguards cell fate determination
In this study, we found that neurons develop protective mechanisms to prevent the improper activation of repressors and to ensure the differentiation of the correct cell fate. Specifically, ZAG-1 prevents the expression of repressors EGL-44 and EGL-46 in the TRNs. Because the EGL-44/EGL-46 complex is a powerful inhibitor of TRN fate (its derepression in zag-1 mutants or misexpression in TRNs can completely shut off the expression of TRN genes), the function of ZAG-1 is essential for ensuring the specification of TRN fate.
As a differentiation regulator, ZAG-1 is required for the adoption of TRN fate but does not directly bind to the cis-regulatory elements within TRN promoters and so does not act as a selector. Moreover, unlike the highly confined expression of terminal selectors and repressors (e.g. both mec-3 and egl-44 are expressed in few neurons), zag-1 is expressed in many neurons in the head and tail ganglia, and ventral cord motor neurons, as well as in various muscles (this study; Wacker et al., 2003). Thus, ZAG-1, as a transcriptional repressor, may serve as a cell fate protector for many different neurons. Already, ZAG-1 has been found to be required for the fate specification of at least TRN, HSN, PVQ and M4 neurons (this study; Clark and Chiu, 2003; Ramakrishnan and Okkema, 2014), although whether ZAG-1 also regulates HSN, PVQ and M4 fates by preventing repressor expression is unclear.
The widespread expression of zag-1 in neurons is also consistent with its function in controlling neurite outgrowth (this study; Clark and Chiu, 2003; Wacker et al., 2003). This separate function, observed in many different neurons, appears to be independent of its role in inhibiting the expression of repressors in cell-fate specification.
Diverse mechanisms of repressor function
Repressors affect effector gene expression in several ways. Repressors can act through discrete cis-regulatory elements that are close to but separate from the selector binding site on the promoter of effector genes (Kerk et al., 2017), and the mechanism of repression may involve histone modification and the recruitment of histone deacetylase (Winnier et al., 1999). Our study suggests that repressors (EGL-44/EGL-46) may directly occupy the selector binding site (for UNC-86/MEC-3) and, thus, could prevent selector binding.
Repressors that inhibit effector genes directly can also activate effector genes indirectly by inhibiting other repressors (Kerk et al., 2017). EGL-44/EGL-46 does activate FLP genes in addition to inhibiting TRN genes, but whether it does so by repressing other repressors is unclear.
All of these methods shape the final collection of effector genes that define a particular cell fate. ZAG-1, however, appears to act differently because it does not interact with effector genes directly (we did not find any UNC-86/MEC-3-induced effector gene that was directly repressed by ZAG-1). Instead, a major function for ZAG-1 in TRNs is to prevent the expression of repressors, including EGL-44/EGL-46 and possibly the unknown repressor that inhibits TRN fate in PVD neurons. Thus, ZAG-1 may be devoted to inhibiting the alternative FLP and PVD fates, whereas UNC-86/MEC-3 induces TRN fate. The uncoupling between the two modules suggests that they may have evolved independently. We distinguish the action of EGL-44/EGL-46, which prevents downstream gene expression, from that of ZAG-1, which regulates egl-44 and egl-46 expression, by calling the former a repressor and the latter an inhibitor.
Negative-feedback loop controls binary fate choice and neuronal diversification
The repressors reported by Kerk et al. (2017) formed a hierarchy that regulated effector gene expression in motor neurons. In this study, however, we showed that by downregulating each other's gene expression, a repressor and an inhibitor can form a bistable switch that ensures proper cell differentiation. The diversification of three types of sensory neurons (TRNs, FLP and PVD neurons) that share the same selectors (UNC-86 and MEC-3) use a set of binary fate choices. The TRN versus FLP fate choice is controlled by the negative-feedback loop between ZAG-1 and EGL-44, whereas the TRN versus PVD choice might be similarly controlled by mutual inhibition between ZAG-1 and an unknown repressor that helps specify PVD fate and represses TRN fate (Fig. 7).
In the absence of both components of the switch (egl-44; zag-1 mutants), both TRNs and FLP neurons expressed the TRN genetic program, suggesting that the ground state is a TRN-like fate and the selectors UNC-86/MEC-3 activate TRN genes by default. EGL-44/EGL-46-induced modification of the default genetic program gives rise to the FLP fate. Our results are different from the observation in motor neurons, where the loss of repressors leads to a ‘mixed ground state’ that is not similar to any of the five motor neuron types (Kerk et al., 2017). This distinction may result because ZAG-1 only inhibits EGL-44 and does not directly regulate effector genes, whereas in motor neurons all repressors interact with effector genes.
The choosing of one of two alternative fates appears to be a common way to diversify neuronal types and subtypes. Notable examples include the fate choices between Drosophila R7 and R8 photoreceptors (Mikeladze-Dvali et al., 2005), and between the left and right ASE gustatory neurons in C. elegans (Sarin et al., 2007); both decisions are mediated by bistable feedback loops. In vertebrates, mutual repression between Hox proteins directs motor neuron diversification in mouse spinal cord (Dasen et al., 2003, 2005), and the neurotransmitter identity of cortical neurons is also subject to binary regulation (Lodato et al., 2014; Nakatani et al., 2007).
The function of ZEB family transcription factors in regulating such binary cell fate choices appears to be evolutionarily conserved. The Drosophila homolog of ZAG-1, Zfh1, promotes a GW motor neuron fate over an EW interneuron fate in the 7-3 neuroblast lineage, and its expression is suppressed by the steroid receptor family transcription factor Eagle in the EW interneuron (Lee and Lundell, 2007); whether Zfh1 also suppressed Eagle in GW motor neuron is unclear. In another example, the mutual antagonism between Zfh1 and another zinc-finger transcription factor, Lame duck, regulates the decision between pericardial cell and fusion competent myoblast fates in the mesoderm (Sellin et al., 2009). Although a genetic interaction between Zfh1 and the Drosophila EGL-44, Scalloped, has not been reported, these results suggest that Zfh1 can form regulatory switches with other transcription factors. Mouse homologs of ZAG-1, ZEB1 and ZEB2, repress tissue differentiation during early embryogenesis, induce epithelial mesenchymal transition (EMT) and are essential for neural tube development (Vandewalle et al., 2009). Their roles in specifying terminal cell fates, however, are unclear, because their knockout leads to embryonic lethality (Miyoshi et al., 2006).
Binary fate switches may enable the generation of neuronal diversity
Because the introduction of self-reinforcing, bistable switches can generate diversity within pre-existing neuronal fates, we envision that the extraordinary variety of neuronal types in the nervous system evolved from a few primitive neuronal fates through stepwise addition of binary switches. For example, as TRN, FLP and PVD fates all require the same selectors, UNC-86 and MEC-3, they may be derived from a common ancestral fate. In fact, the existence of UNC-86/MEC-3 target genes such as mec-19, which is expressed in all the FLP, PVD and TRNs but not any other neuron, suggests such an ancestral fate.
One possible evolutionary derivation of these cells is that some ancestral TRN-like cells acquired the ability to express egl-44 and egl-46. This expression led to the suppression of TRN genes, the activation of FLP genes and the emergence of the FLP neuron type. The fact that the ground state is a TRN-like state seems to support this hypothesis. Moreover, as egl-44 is primarily expressed in non-neuronal tissues, mutations in the regulatory elements of egl-44 might have allowed expression in some neurons. Thus, EGL-44 may have been co-opted to induce divergence among neurons that share a common fate, and this cell fate divergence was subsequently stabilized by the establishment of a negative-feedback loop between EGL-44 and ZAG-1. Alternatively, a regulatory element in the zag-1 gene, which was already present in the ancestral TRNs to repress egl-44 and egl-46, may have been mutated to prevent its expression in some ancestral TRN cells. This loss led to the de-repression of egl-44 and egl-46, and the subsequent acquisition of the FLP fate.
Overall, we imagine that a limited number of ground-state selectors may first define a handful of shared states, and subsequently a series of binary fate switches carry out further differentiation that modifies the ground state to generate a diverse array of terminal neuronal fates. The broad expression of ZAG-1 in the nervous system and the lack of increased EGL-44 expression in zag-1 mutants suggests that ZAG-1 may form bistable regulatory loops with many different inhibitors. Moreover, the fact that ZAG-1 expression is found only in a subset of neurons that express the same selector supports the hypothesis that ZAG-1 may ensure diversification from a shared ground state by inhibiting repressors; e.g. zag-1 is expressed in 7/17, 6/15 and 3/11 classes of neurons that use UNC-86, UNC-3 and CEH-14 as a terminal selector, respectively (classification according to Hobert, 2016). Selective loss of zag-1 expression in some cells may prevent these cells from committing to particular fates and allowing them to adopt alternative ones. Finally, the broad expression of ZEB family transcription factors in the nervous system of both Drosophila (Lai et al., 1991) and mice (Vandewalle et al., 2009) possibly suggests a conserved role for them in regulating binary fate choices and the generation of neuronal diversity.
MATERIALS AND METHODS
Strains, constructs and transgenes
C. elegans wild-type (N2) and mutant strains were maintained as previously described (Brenner, 1974). Most strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440) or the National BioResource Project of Japan. VH514, zag-1(hd16)/unc-17(e113) dpy-13(e184) was used as the balanced null allele for zag-1 and zag-1(zd86) was used as a hypomorphic allele. For other genes identified from the RNAi screen (see below), we tested zip-4(tm1359), nhr-119(gk136908), nhr-166(gk613), nhr-159(tm2323), egl-38(ok3510)/nT1[qIs51], hmbx-1(ok3467), fkh-2(ok683), lin-40(ku285), lin-40(s1506) unc-46(e177)/eT1, elt-6(gk723) and elt-6(gk754). Other mutant alleles used in this study include ahr-1(ju145), egl-44(n1080), egl-46(n1127), mec-3(u184) and unc-86(u5).
A 2.4 kb mec-3 promoter, a 2.2 kb zag-1 promoter, a 2.2 kb bicd-1 promoter, a4.9 kb dma-1 promoter, a 3.1 kb flp-4 promoter, a 2.3 kb sto-5 promoter and a 1.3 kb mec-19 promoter were cloned from wild-type (N2) genomic DNA into the Gateway pDONR221 P4-P1r vector. The genomic coding regions of GFP, zag-1, egl-44, egl-46, alr-1 and ahr-1 were cloned into Gateway pDONR221. The resultant entry vectors, together with pENTR-unc-54-3′ UTR and the destination vector pDEST-R4-R3, were used in the LR reaction to create the final expression vectors. Gateway cloning was performed according to the manual provided by Life Technologies. To generate transgenic animals, we injected DNA constructs (5 ng/µl for each expression vector) into the animals to establish stable lines carrying an extrachromosomal array; at least three independent lines were tested. In some cases, the transgene was integrated into the genome using γ-irradiation (Mello et al., 1991), and at least three integrant lines were outcrossed and examined. For the mec-3p::zag-1; mec-3p::egl-44; mec-3p::egl-46 triple transgenic animals, the injection was repeated twice, and three stable lines were obtained from each injection. Similar results were obtained from the replicates.
DNA constructs TU#625 and TU#626 (Wu et al., 2001) contain translational GFP fusion of egl-44 and egl-46, respectively, and were injected into animals to form reporters for the two genes: uIs215[egl-44::GFP] and uEx927[egl-46::GFP]. TU#924 contains a 400 bp mec-18 promoter inserted into pPD95.75 between HindIII and BamHI sites, and this mec-18p::GFP construct was used as a template to create a series of promoter variants shown in Fig. S10 using the Q5 site-directed mutagenesis kit from New England Biolabs.
Transgenes zdIs5[mec-4p::GFP] I, muIs32[mec-7p::GFP] II, uIs31[mec-17p::GFP] III, uIs115[mec-17p::RFP] IV, uIs134[mec-17p::RFP] V and uIs72[mec-18p::mec-18::GFP] were used as fluorescent markers for the TRN cell fate. uEx1104[Pbicd-1::GFP], uEx1105[dma-1p::GFP], uEx1106[flp-4p::GFP] and uIs232[sto-5p::GFP] served as FLP fate markers. zdIs13[tph-1p::GFP] and vsIs97[tph-1p::DsRed2] were used as HSN fate markers. uIs22[mec-3p::GFP] and uIs152[mec-3p::RFP] were used as mec-3 transcriptional reporters; uEx1007[mec-3p::mec-3::GFP] and wgIs55[mec-3::TY1::EGFP::3xFLAG] were used as mec-3 translational reporters. wgIs83[zag-1::TY1::EGFP::3xFLAG], wgIs476[unc-86::TY1::EGFP::3xFLAG], wgIs200[alr-1::TY1::EGFP::3xFLAG], leEx1709[ahr-1::GFP] and uEx1107[mec-19p::GFP] served as the reporters for zag-1, unc-86, alr-1, ahr-1 and mec-19 respectively. uIs211[mec-3p::egl-44], uEx926[mec-3p::zag-1] and uEx1027[mec-3p::alr-1] were used for misexpression.
To perform cell identification, we crossed wgIs83[zag-1::TY1::EGFP::3xFLAG] and uIs215[egl-44::GFP] into otIs518[eat-4::SL2::mCherry::H2B], otIs544[cho-1::SL2::mCherry::H2B] and otIs564 [unc-47::SL2::H2B::mChopti], labeling glutamatergic, cholinergic and GABAergic neurons (Gendrel et al., 2016; Pereira et al., 2015; Serrano-Saiz et al., 2013), respectively. We identified zag-1- and egl-44-expressing neurons, based on the position and neurotransmitter identity of the cells expressing GFP.
RNAi screen
RNAi screen was performed using a modified bacteria-feeding protocol previously reported (Kamath et al., 2003; Poole et al., 2011). We used the Ahringer RNAi library from Source Bioscience (www.lifesciences.sourcebioscience.com/) and the list of 392 RNAi clones targeting transcription factors were generated by searching WormBase WS238 using Gene Ontology terms related to ‘DNA binding’ and ‘transcription factor activity’ (see the complete list in Table S1). To perform the RNAi experiments, we seeded bacteria expressing dsRNA on NGM agar plates containing 6 mM IPTG and 100 µg/ml ampicillin. One day later, eggs from TU4429, eri-1 (mg366); lin-15B (n744); uIs134[mec-17p::RFP] animals were placed onto these plates; the eggs hatched and grew to adults at 20°C. The F1 progeny of these worms were scored for the expression of TRN markers at the second larval stage. RNAi clones were considered to be positive if more than 15% (n>20) of the treated animals failed to show RFP expression in the ALM neurons in at least two of the three replicate plates. Three initial rounds of screens were performed on all the 392 clones, and 14 clones were found to be positive in all the three rounds. Two more screening rounds were then conducted on these 14 RNAi clones, which were all confirmed to be positive. We sequenced the inserts of all positive clones to confirm the identity of the target genes.
Yeast two-hybrid assay
Yeast media and plates were prepared according to recipes from Clontech and yeasts were grown at 30°C. The yeast strain PJ69-4a (provided by Songtao Jia at Columbia University, NY, USA) used for the two-hybrid assays contains GAL1-HIS3, GAL2-ADE2 and GAL7-lacZ reporters. Vectors pGAD424 and pGBT9 (Clontech) were used to express proteins fused to the yeast-activating domain (AD) and -binding domain (BD), respectively. cDNA fragments of mec-3, zag-1, ldb-1, egl-44 and egl-46 were cloned into the two-hybrid vectors using either restriction enzymes or with Gibson Assembly (NEB).
Combinations of the AD or BD vectors were co-transformed into yeast using the Frozen-EZ II kit from Zymo Research and using empty vectors as negative controls. Growth assays were performed by growing individual colonies overnight in selective media lacking tryptophan and leucine. Cultures were then diluted to let OD600 become 0.5, and 10 µl of a further 1:10 diluted culture were spotted onto plates lacking histidine to test the expression of the HIS3 reporter. Plates were imaged after 2 days of growth. Liquid β-galactosidase assays were performed using the Yeast β-Galactosidase Assay Kit (Thermo Scientific).
Electrophoretic mobility shift assay
Recombinant GST::EGL-44 proteins were produced in E. coli BL21 (DE3) using the expression vector pGEX-6p-1 (Amersham Pharmacia Biotech) and purified using affinity chromatography columns filled with glutathione sepharose 4B beads (Amersham). EGL-44 was cleaved off the column using PreScission Protease (Amersham). EGL-46 was expressed using the pET32a vector (Novagen) and purified using the S-Tag rEK purification kit (Novagen).
Gel mobility shift assays were performed using a modified protocol previously reported (Xue et al., 1993). DNA probes were labeled with Biotin using a Biotin 3′ End labeling kit (Pierce) and then annealed into double strands. 100 ng of proteins were incubated with 20 fmol (0.5∼0.7 ng) of probe at room temperature for 30 min, and the mixture was loaded onto a 10% TBE polyacrylamide mini gel (Bio-Rad). 2 pmol unlabeled probe (100×) was added for the cold probe competition. The gel was transferred to a 0.2 mm nylon membrane, which was then treated with UV light to crosslink DNA and proteins. Biotin-labeled DNA was detected using LightShift chemiluminescence EMSA kit (Pierce). The sequences of the probes are listed in Table S3.
smFISH, phenotypic scoring and statistical analysis
Single-molecule fluorescence in situ hybridization (smFISH) was performed as described previously (Topalidou et al., 2011). Imaging was conducted on a Zeiss Axio Observer Z1 inverted microscope with a CoolSNAP HQ2-FW camera (Photometrics).
To examine the expression pattern of TRN markers, we grew animals at 20°C, examined them using the same microscope and recorded the percentages of TRN cells that express the fluorescent reporter in three independent experiments. The results are presented as aggregates; no significant differences were seen between replicates. For transgenic animals, at least three independent lines were examined.
Statistical significance was determined using the Student's t-test for the majority of comparisons of two sets of data. For multiple comparisons, the Holm-Bonferroni method was used to correct the P values.
Supplementary Material
Acknowledgements
We thank Alex Bounoutas for contributing to the initial studies on DNA binding of EGL-44, and Songtao Jia and Elizabeth Miller for sharing materials and reagents. We also thank Oliver Hobert, Richard Mann and the members of our laboratory for helpful discussions and comments.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.Z., M.C.; Methodology: C.Z., F.Q.J., B.L.T., J.W., M.C.; Validation: C.Z., F.Q.J., B.L.T.; Formal analysis: C.Z., F.Q.J., B.L.T., J.W., M.C.; Investigation: C.Z., F.Q.J., B.L.T., J.W.; Resources: C.Z., J.W.; Data curation: C.Z.; Writing - original draft: C.Z.; Writing - review & editing: C.Z., M.C.; Visualization: C.Z., F.Q.J.; Supervision: C.Z., M.C.; Project administration: M.C.; Funding acquisition: M.C.
Funding
This work was supported by National Institutes of Health [GM30997 and GM122522 to M.C.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.168096.supplemental
Reference
- Aguirre-Chen C., Bulow H. E. and Kaprielian Z. (2011). C. elegans bicd-1, homolog of the Drosophila dynein accessory factor Bicaudal D, regulates the branching of PVD sensory neuron dendrites. Development 138, 507-518. 10.1242/dev.060939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshaw S., Fraioli R. E., Furukawa T. and Cepko C. L. (2001). Comprehensive analysis of photoreceptor gene expression and the identification of candidate retinal disease genes. Cell 107, 579-589. 10.1016/S0092-8674(01)00574-8 [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto A., Chelur D., Topalidou I., Chen X. and Chalfie M. (2010). Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554-559. 10.1038/nmeth.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassata G., Röhrig S., Kuhn F., Hauri H.-P., Baumeister R. and Bürglin T. R. (2000). The Caenorhabditis elegans Ldb/NLI/Clim orthologue ldb-1 is required for neuronal function. Dev. Biol. 226, 45-56. 10.1006/dbio.2000.9846 [DOI] [PubMed] [Google Scholar]
- Chalfie M. and Au M. (1989). Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science 243, 1027-1033. 10.1126/science.2646709 [DOI] [PubMed] [Google Scholar]
- Chalfie M. and Sulston J. (1981). Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev. Biol. 82, 358-370. 10.1016/0012-1606(81)90459-0 [DOI] [PubMed] [Google Scholar]
- Chatzigeorgiou M. and Schafer W. R. (2011). Lateral facilitation between primary mechanosensory neurons controls nose touch perception in C. elegans. Neuron 70, 299-309. 10.1016/j.neuron.2011.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou M., Yoo S., Watson J. D., Lee W.-H., Spencer W. C., Kindt K. S., Hwang S. W., Miller D. M. III, Treinin M., Driscoll M. et al. (2010). Specific roles for DEG/ENaC and TRP channels in touch and thermosensation in C. elegans nociceptors. Nat. Neurosci. 13, 861-868. 10.1038/nn.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. G. and Chiu C. (2003). C. elegans ZAG-1, a Zn-finger-homeodomain protein, regulates axonal development and neuronal differentiation. Development 130, 3781-3794. 10.1242/dev.00571 [DOI] [PubMed] [Google Scholar]
- Dasen J. S., Liu J.-P. and Jessell T. M. (2003). Motor neuron columnar fate imposed by sequential phases of Hox-c activity. Nature 425, 926-933. 10.1038/nature02051 [DOI] [PubMed] [Google Scholar]
- Dasen J. S., Tice B. C., Brenner-Morton S. and Jessell T. M. (2005). A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell 123, 477-491. 10.1016/j.cell.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Daude N., Wohlgemuth S., Brown R., Pitstick R., Gapeshina H., Yang J., Carlson G. A. and Westaway D. (2012). Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrP(C)-deficiency. Proc. Natl. Acad. Sci. USA 109, 9035-9040. 10.1073/pnas.1202130109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan A., Ma C. and Chalfie M. (1998). Regulation of touch receptor differentiation by the Caenorhabditis elegans mec-3 and unc-86 genes. Development 125, 4107-4119. [DOI] [PubMed] [Google Scholar]
- Feng G., Yi P., Yang Y., Chai Y., Tian D., Zhu Z., Liu J., Zhou F., Cheng Z., Wang X. et al. (2013). Developmental stage-dependent transcriptional regulatory pathways control neuroblast lineage progression. Development 140, 3838-3847. 10.1242/dev.098723 [DOI] [PubMed] [Google Scholar]
- Finney M. and Ruvkun G. (1990). The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63, 895-905. 10.1016/0092-8674(90)90493-X [DOI] [PubMed] [Google Scholar]
- Gao Y., Zhang Y., Zhang D., Dai X., Estelle M. and Zhao Y. (2015). Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc. Natl. Acad. Sci. USA 112, 2275-2280. 10.1073/pnas.1500365112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bellido A. (1975). Genetic control of wing disc development in Drosophila. Ciba Found Symp 0, 161-182. [DOI] [PubMed] [Google Scholar]
- Gendrel M., Atlas E. G. and Hobert O. (2016). A cellular and regulatory map of the GABAergic nervous system of C. elegans. Elife 5, e17686 10.7554/eLife.17686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. (2011). Regulation of terminal differentiation programs in the nervous system. Annu. Rev. Cell Dev. Biol. 27, 681-696. 10.1146/annurev-cellbio-092910-154226 [DOI] [PubMed] [Google Scholar]
- Hobert O. (2016). A map of terminal regulators of neuronal identity in Caenorhabditis elegans. Wiley Interdiscip Rev. Dev. Biol. 5, 474-498. 10.1002/wdev.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiau T. H.-C., Diaconu C., Myers C. A., Lee J., Cepko C. L. and Corbo J. C. (2007). The cis-regulatory logic of the mammalian photoreceptor transcriptional network. PLoS ONE 2, e643 10.1371/journal.pone.0000643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M. and Chalfie M. (1994). Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature 367, 467-470. 10.1038/367467a0 [DOI] [PubMed] [Google Scholar]
- Jiang S.-W., Trujillo M. A., Sakagashira M., Wilke R. A. and Eberhardt N. L. (2000). Novel human TEF-1 isoforms exhibit altered DNA binding and functional properties. Biochemistry 39, 3505-3513. 10.1021/bi991048w [DOI] [PubMed] [Google Scholar]
- Kamath R. S., Fraser A. G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M. et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231-237. 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kaplan J. M. and Horvitz H. R. (1993). A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 90, 2227-2231. 10.1073/pnas.90.6.2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerk S. Y., Kratsios P., Hart M., Mourao R. and Hobert O. (2017). Diversification of C. elegans motor neuron identity via selective effector gene repression. Neuron 93, 80-98. 10.1016/j.neuron.2016.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. and Li C. (2004). Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. J. Comp. Neurol. 475, 540-550. 10.1002/cne.20189 [DOI] [PubMed] [Google Scholar]
- Kok F. O., Shin M., Ni C.-W., Gupta A., Grosse A. S., van Impel A., Kirchmaier B. C., Peterson-Maduro J., Kourkoulis G., Male I. et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97-108. 10.1016/j.devcel.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z. C., Fortini M. E. and Rubin G. M. (1991). The embryonic expression patterns of zfh-1 and zfh-2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech. Dev. 34, 123-134. 10.1016/0925-4773(91)90049-C [DOI] [PubMed] [Google Scholar]
- Lee H.-K. and Lundell M. J. (2007). Differentiation of the Drosophila serotonergic lineage depends on the regulation of Zfh-1 by Notch and Eagle. Mol. Cell. Neurosci. 36, 47-58. 10.1016/j.mcn.2007.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu O. W. and Shen K. (2011). The transmembrane LRR protein DMA-1 promotes dendrite branching and growth in C. elegans. Nat. Neurosci. 15, 57-63. 10.1038/nn.2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S., Molyneaux B. J., Zuccaro E., Goff L. A., Chen H. H., Yuan W., Meleski A., Takahashi E., Mahony S., Rinn J. L. et al. (2014). Gene co-regulation by Fezf2 selects neurotransmitter identity and connectivity of corticospinal neurons. Nat. Neurosci. 17, 1046-1054. 10.1038/nn.3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C. C., Kramer J. M., Stinchcomb D. and Ambros V. (1991). Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. 10.1002/j.1460-2075.1991.tb04966.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikeladze-Dvali T., Wernet M. F., Pistillo D., Mazzoni E. O., Teleman A. A., Chen Y.-W., Cohen S. and Desplan C. (2005). The growth regulators warts/lats and melted interact in a bistable loop to specify opposite fates in Drosophila R8 photoreceptors. Cell 122, 775-787. 10.1016/j.cell.2005.07.026 [DOI] [PubMed] [Google Scholar]
- Mitani S., Du H., Hall D. H., Driscoll M. and Chalfie M. (1993). Combinatorial control of touch receptor neuron expression in Caenorhabditis elegans. Development 119, 773-783. [DOI] [PubMed] [Google Scholar]
- Miyoshi T., Maruhashi M., Van De Putte T., Kondoh H., Huylebroeck D. and Higashi Y. (2006). Complementary expression pattern of Zfhx1 genes Sip1 and deltaEF1 in the mouse embryo and their genetic interaction revealed by compound mutants. Dev. Dyn. 235, 1941-1952. 10.1002/dvdy.20799 [DOI] [PubMed] [Google Scholar]
- Nakatani T., Minaki Y., Kumai M. and Ono Y. (2007). Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development 134, 2783-2793. 10.1242/dev.02870 [DOI] [PubMed] [Google Scholar]
- Pereira L., Kratsios P., Serrano-Saiz E., Sheftel H., Mayo A. E., Hall D. H., White J. G., LeBoeuf B., Garcia L. R., Alon U. et al. (2015). A cellular and regulatory map of the cholinergic nervous system of C. elegans. Elife 4, e12432 10.7554/eLife.12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrad A., Meir J. Y., Barnes T. M. and Miller D. M. III (1997). The Groucho-like transcription factor UNC-37 functions with the neural specificity gene unc-4 to govern motor neuron identity in C. elegans. Development 124, 1699-1709. [DOI] [PubMed] [Google Scholar]
- Poole R. J., Bashllari E., Cochella L., Flowers E. B. and Hobert O. (2011). A genome-Wide RNAi screen for factors involved in neuronal specification in Caenorhabditis elegans. PLoS Genet. 7, e1002109 10.1371/journal.pgen.1002109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan K. and Okkema P. G. (2014). Regulation of C. elegans neuronal differentiation by the ZEB-family factor ZAG-1 and the NK-2 homeodomain factor CEH-28. PLoS ONE 9, e113893 10.1371/journal.pone.0113893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A., Kontarakis Z., Gerri C., Nolte H., Hölper S., Krüger M. and Stainier D. Y. R. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230-233. 10.1038/nature14580 [DOI] [PubMed] [Google Scholar]
- Russell J., Vidal-Gadea A. G., Makay A., Lanam C. and Pierce-Shimomura J. T. (2014). Humidity sensation requires both mechanosensory and thermosensory pathways in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 111, 8269-8274. 10.1073/pnas.1322512111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S., O'Meara M. M., Flowers E. B., Antonio C., Poole R. J., Didiano D., Johnston R. J. Jr, Chang S., Narula S. and Hobert O. (2007). Genetic screens for Caenorhabditis elegans mutants defective in left/right asymmetric neuronal fate specification. Genetics 176, 2109-2130. 10.1534/genetics.107.075648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin J., Drechsler M., Nguyen H. T. and Paululat A. (2009). Antagonistic function of Lmd and Zfh1 fine tunes cell fate decisions in the Twi and Tin positive mesoderm of Drosophila melanogaster. Dev. Biol. 326, 444-455. 10.1016/j.ydbio.2008.10.041 [DOI] [PubMed] [Google Scholar]
- Serrano-Saiz E., Poole R. J., Felton T., Zhang F., De La Cruz E. D. and Hobert O. (2013). Modular control of glutamatergic neuronal identity in C. elegans by distinct homeodomain proteins. Cell 155, 659-673. 10.1016/j.cell.2013.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. J., O'Brien T., Chatzigeorgiou M., Spencer W. C., Feingold-Link E., Husson S. J., Hori S., Mitani S., Gottschalk A., Schafer W. R. et al. (2013). Sensory neuron fates are distinguished by a transcriptional switch that regulates dendrite branch stabilization. Neuron 79, 266-280. 10.1016/j.neuron.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J. E. and Horvitz H. R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110-156. 10.1016/0012-1606(77)90158-0 [DOI] [PubMed] [Google Scholar]
- Topalidou I. and Chalfie M. (2011). Shared gene expression in distinct neurons expressing common selector genes. Proc. Natl. Acad. Sci. USA 108, 19258-19263. 10.1073/pnas.1111684108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalidou I., van Oudenaarden A. and Chalfie M. (2011). Caenorhabditis elegans aristaless/Arx gene alr-1 restricts variable gene expression. Proc. Natl. Acad. Sci. USA 108, 4063-4068. 10.1073/pnas.1101329108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallstedt A., Muhr J., Pattyn A., Pierani A., Mendelsohn M., Sander M., Jessell T. M. and Ericson J. (2001). Different levels of repressor activity assign redundant and specific roles to Nkx6 genes in motor neuron and interneuron specification. Neuron 31, 743-755. 10.1016/S0896-6273(01)00412-3 [DOI] [PubMed] [Google Scholar]
- Vandewalle C., Van Roy F. and Berx G. (2009). The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 66, 773-787. 10.1007/s00018-008-8465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker I., Schwarz V., Hedgecock E. M. and Hutter H. (2003). zag-1, a Zn-finger homeodomain transcription factor controlling neuronal differentiation and axon outgrowth in C. elegans. Development 130, 3795-3805. 10.1242/dev.00570 [DOI] [PubMed] [Google Scholar]
- Way J. C. and Chalfie M. (1988). mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell 54, 5-16. 10.1016/0092-8674(88)90174-2 [DOI] [PubMed] [Google Scholar]
- William C. M., Tanabe Y. and Jessell T. M. (2003). Regulation of motor neuron subtype identity by repressor activity of Mnx class homeodomain proteins. Development 130, 1523-1536. 10.1242/dev.00358 [DOI] [PubMed] [Google Scholar]
- Winnier A. R., Meir J. Y.-J., Ross J. M., Tavernarakis N., Driscoll M., Ishihara T., Katsura I. and Miller D. M. III (1999). UNC-4/UNC-37-dependent repression of motor neuron-specific genes controls synaptic choice in Caenorhabditis elegans. Genes Dev. 13, 2774-2786. 10.1101/gad.13.21.2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Duggan A. and Chalfie M. (2001). Inhibition of touch cell fate by egl-44 and egl-46 in C. elegans. Genes Dev. 15, 789-802. 10.1101/gad.857401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D., Finney M., Ruvkun G. and Chalfie M. (1992). Regulation of the mec-3 gene by the C.elegans homeoproteins UNC-86 and MEC-3. EMBO J. 11, 4969-4979. 10.1002/j.1460-2075.1992.tb05604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D., Tu Y. and Chalfie M. (1993). Cooperative interactions between the Caenorhabditis elegans homeoproteins UNC-86 and MEC-3. Science 261, 1324-1328. 10.1126/science.8103239 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Ma C., Delohery T., Nasipak B., Foat B. C., Bounoutas A., Bussemaker H. J., Kim S. K. and Chalfie M. (2002). Identification of genes expressed in C. elegans touch receptor neurons. Nature 418, 331-335. 10.1038/nature00891 [DOI] [PubMed] [Google Scholar]
- Zheng C., Diaz-Cuadros M. and Chalfie M. (2015a). Hox genes promote neuronal subtype diversification through posterior induction in caenorhabditis elegans. Neuron 88, 514-527. 10.1016/j.neuron.2015.09.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C., Jin F. Q. and Chalfie M. (2015b). Hox Proteins Act as Transcriptional Guarantors to Ensure Terminal Differentiation. Cell Rep 13, 1343-1352. 10.1016/j.celrep.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.