

ABSTRACT
Although it is known that inflammation plays a critical role in prostate tumorigenesis, the underlying processes are not well understood. Based on analysis of genetically engineered mouse models combined with correlative analysis of expression profiling data from human prostate tumors, we demonstrate a reciprocal relationship between inflammation and the status of the NKX3.1 homeobox gene associated with prostate cancer initiation. We find that cancer initiation in aged Nkx3.1 mutant mice correlates with enrichment of specific immune populations and increased expression of immunoregulatory genes. Furthermore, expression of these immunoregulatory genes is similarly increased in human prostate tumors having low levels of NKX3.1 expression. We further show that induction of prostatitis in Nkx3.1 mutant mice accelerates prostate cancer initiation, which is coincident with aberrant cellular plasticity and differentiation. Correspondingly, human prostate tumors having low levels of NKX3.1 have de-regulated expression of genes associated with these cellular processes. We propose that loss of function of NKX3.1 accelerates inflammation-driven prostate cancer initiation potentially via aberrant cellular plasticity and impairment of cellular differentiation.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Prostate cancer, Cancer initiation, NKX3.1, Inflammation, Differentiation
Summary: Chronic inflammation collaborates with loss of function of the prostate-specific tumor-suppressor NKX3.1 to promote prostate cancer initiation, increase cellular plasticity and impair cellular differentiation.
INTRODUCTION
Located at the base of the bladder and upstream of the male reproductive system, the prostate provides the first line of defense against foreign agents and pathogens originating from these organs. Consequently, many adult men display signs of inflammation of the prostate, albeit with varying degrees of severity (Sfanos et al., 2017). Such inflammation is often accompanied by infiltration of specific immune cells into the prostate, whose presence has been associated with increased cancer risk and poor prognosis (McArdle et al., 2004; Nonomura et al., 2011, Strasner and Karin, 2015). Indeed, epidemiological and experimental evidence highlight the importance of inflammation for prostate tumorigenesis, and suggest that an inflammatory microenvironment promotes prostate cancer (Sfanos et al., 2014; De Marzo et al., 2007b), while the histological appearance of proliferative inflammatory atrophy (PIA) has been associated with prostate cancer initiation (De Marzo et al., 2007a). Direct evidence that inflammation plays a causal role in prostate cancer includes its association with the promotion of gene fusions that are prevalent in prostate cancer (Mani et al., 2016). In other cancer types, inflammation has been shown to promote carcinogenesis by influencing cellular differentiation, with consequential effects on lineage plasticity (Le Magnen et al., 2018; Coussens and Werb, 2002). Accordingly, in the prostate, inflammation has been associated with impaired differentiation of prostate epithelial cells, including expansion of the pool of progenitor cells (Liu et al., 2016) and aberrant basal-to-luminal differentiation (Kwon et al., 2014), which are accompanied by the promotion of prostate cancer initiation.
A hallmark of prostate cancer initiation is loss of the NKX3.1 homeobox gene, whose functions have been associated with promotion of lineage plasticity, cellular differentiation and response to inflammation (Abate-Shen et al., 2008). NKX3.1 is a prostate-specific tumor suppressor located on chromosome 8p, whose loss or reduction represents a key initiating event in prostate cancer (Cancer Genome Atlas Research, 2015; Baca et al., 2013). Germline loss-of-function of Nkx3.1 in mutant mice results in pre-malignant lesions resembling prostatic intraepithelial neoplasia (PIN), a known precursor of prostate cancer, and shares molecular features conserved with indolent prostate cancer in humans (Bhatia-Gaur et al., 1999; Irshad et al., 2013; Kim et al., 2002a). Moreover, NKX3.1 is essential for prostatic epithelial specification and proper differentiation, and is required for maintenance of luminal stem cells (Dutta et al., 2016; Wang et al., 2009; Bhatia-Gaur et al., 1999; Talos et al., 2017). Notably, inflammation has been shown to be associated with loss of NKX3.1 expression in mouse and human prostate (Khalili et al., 2010; Markowski et al., 2008; Bethel et al., 2006).
In the current study, we have investigated the bidirectional relationship between inflammation and NKX3.1 loss of function, and its relevance for prostate cancer initiation, by comparative analyses of the consequences of its loss of function in mutant mice and its expression status in human prostate cancer. We demonstrate that loss of function of Nkx3.1 in the mouse prostate is associated with enrichment of specific immune cell populations, and additionally that its loss or reduction in mouse prostate and human prostate tumors is associated with increased expression of immunoregulatory genes. Furthermore, induction of chronic inflammation using a relevant prostatitis model accelerates prostate cancer initiation in Nkx3.1 mutant mice, which is associated with aberrant cellular plasticity and impaired differentiation. Thus, we propose that loss of function of NKX3.1 augments inflammation-induced cancer initiation potentially via affecting aberrant cellular plasticity and impaired cellular differentiation.
RESULTS
NKX3.1 loss correlates with an increase in specific immune cells and expression of immunoregulatory genes in prostate
To investigate the relationship between loss of function of Nkx3.1 and inflammation in the mouse prostate in vivo, we studied the germline Nkx3.1 mutant mouse model, which develops PIN as a consequence of aging and cooperates with loss of function of other tumor-suppressor genes in prostate tumorigenesis (Kim et al., 2002a,b; Bhatia-Gaur et al., 1999). In particular, we examined the abundance and nature of immune cells that infiltrate the prostate epithelium (hereafter referred to as ‘prostate-infiltrating immune cells’) in Nkx3.1 wild-type (Nkx3.1+/+) and mutant (Nkx3.1−/−) mice at specific time points, ranging from 3 to 15 months, during which time the prostate phenotype of the Nkx3.1 mutant mice progresses from hyperplasia to dysplasia to PIN (Fig. 1A) (Bhatia-Gaur et al., 1999). We examined the expression of CD45, a pan-leukocyte marker, and found that the percentage of CD45-positive (CD45+) prostate-infiltrating immune cells was increased in aged Nkx3.1−/− prostates compared with Nkx3.1+/+ prostates (3-fold increase at 12 months, P=0.02; n=3 per group; Fig. 1B,C and Table S1), which coincides with the occurrence of PIN in these mice.
Fig. 1.

Loss of NKX3.1 correlates with an increase in specific immune cells and expression of immunoregulatory genes in prostate. (A) Experimental design and timeline of analyses. Shown is the approximate time-range at which the specific phenotypes occur. Note that Nkx3.1 mutant mice develop prostate intraepithelial neoplasia (PIN) as a consequence of aging. (B) Representative images of immunohistochemical staining for CD45 in the anterior prostates of Nkx3.1+/+ and Nkx3.1−/− mice at the indicated ages. Scale bars: 50 µm. Insets show higher magnification of a representative region. (C) Quantification of CD45-positive cells infiltrating the prostatic epithelium as assessed by immunohistochemical staining for CD45 expression in Nkx3.1+/+ and Nkx3.1−/− mice. Analyses were done using a 2-tailed Welch's t-test. Data are represented as means and the error bars represent standard deviation (s.d.) for each genotype (n=3 mice/group). (D) Representative images of immunohistochemical staining for CD45, CD3, F4/80, B220 and Ly6G in the anterior prostate of Nkx3.1−/− mice (9 months); additional time points and quantification of cell types is provided in Fig. S1. Scale bar: 25 µm. Insets show higher magnification of a representative region. (F) Heat-map representations of a subset of genes involved in immunoregulatory and inflammatory processes that are differentially expressed in Nkx3.1−/− versus Nkx3.1+/+ mouse prostate (15 months; n=3 mice/group). Differential gene expression was estimated with a 2-sample 2-tailed Welch's t-test. Additional pathway and differential gene expression data are provided in Fig. S2. (E,G) Whisker plots showing the relative expression of the indicated human genes in TCGA Gleason 6 and 7 human prostate tumors stratified based on having high or low levels of NKX3.1 expression. n=145 prostate tumors/group. Statistical analyses were performed using a 2-tailed Welch's t-test.
To further investigate the phenotype of prostate-infiltrating immune cells, we examined cell-specific markers, namely the T-cell marker CD3, the macrophage marker F4/80, the B-cell-specific marker B220 and the granulocyte marker Ly6G, by immunohistochemistry as well as expression profiling (Fig. 1D; Fig. S1A). We found that the prostate-infiltrating immune cells in the Nkx3.1−/− prostate were mainly represented by CD3+ T cells and F4/80+ macrophages, which are the main mediators of chronic inflammation (Coussens and Werb, 2002) and in human prostate are present in both normal and inflamed contexts (Sfanos et al., 2017). A rare population of B220+ B cells was also occasionally observed in Nkx3.1−/− prostates, whereas Ly6G+ neutrophils were not detected (Fig. 1D; Fig. S1A). This was confirmed by quantification of prostate-infiltrating immune populations by flow cytometry, where we found that macrophages and T cells, which include CD4+ and CD8+ cells, were the most prominent populations in Nkx3.1−/− prostates, whereas B cells and granulocytic myeloid-derived suppressor cells (G-MDSCs), which include neutrophils, were less abundant (Fig. S1B).
To examine the relevance of these findings for human prostate cancer, we analyzed the expression levels of human genes encoding for markers of the corresponding immune cells by querying expression profiles of prostate tumors with low/intermediate Gleason score (i.e. Gleason 6 and 7) from The Cancer Genome Atlas (TCGA; Cancer Genome Atlas Research, 2015). In particular, following from our previous study in which we showed that NKX3.1 mRNA expression levels provide a read-out of NKX3.1 function in human prostate cancer patients (Dutta et al., 2017) and considering the relevance of NKX3.1 for early-stage prostate cancer (Abate-Shen et al., 2008), we segregated the low/intermediate Gleason score (Gleason 6 and 7) TCGA samples into two groups based on having ‘low’ (i.e. below the median) or ‘high’ (i.e. above the median) levels of NKX3.1 mRNA expression (n=145 per group). We found that the tumors having ‘low’ NKX3.1 expression had relatively increased expression of PTPRC (coding for CD45), CD3G, CD68 and CD19, which mark leukocytes, T cells, macrophages and B cells, respectively, but not CEACAM8 (CD66b) a marker of granulocyte cells, which was generally expressed at very low levels or not detected in the human prostate tumors (Fig. 1E).
Furthermore, analyses of gene expression profiles from aged Nkx3.1+/+ and Nkx3.1−/− mouse prostates (Ouyang et al., 2005) revealed that 14 of the top 20 most significantly upregulated biological pathways (P<0.005) in Nkx3.1−/− prostates were related to immunoregulatory or inflammatory processes, including ‘chemokine signaling’, ‘signaling in immune system’ and ‘immunoregulatory interactions between a lymphoid and a non-lymphoid cell’ (Fig. S2A; n=3 per group; Table S5). Among the positively upregulated leading genes in these pathways were those associated with inflammation-related functions, including the T-cell chemokine receptors Cxcr4 and Cxcr3, the inflammatory chemokine Ccl5 (RANTES), the adhesion proteins Sell and Vcam1, and a member of the danger receptor complex, Ly86 (Fig. 1F; Fig. S2B; n=3 per group). Notably, the human homologs of these genes were each significantly upregulated in Gleason 6 and 7 prostate tumors having ‘low’ levels of NKX3.1 (Fig. 1G). Taken together, these findings in mouse and human prostate show that loss or reduction of NKX3.1 is associated with enrichment of specific immune populations and increased expression of immunoregulatory genes.
Chronic inflammation accelerates the phenotypic consequences of Nkx3.1 loss in vivo
Considering that previous studies have shown that inflammation results in reduced expression of NKX3.1 in a mouse model of prostatitis (Khalili et al., 2010), we sought to investigate the consequences of inducing inflammation in the context of Nkx3.1 loss-of-function for prostate cancer initiation. Toward this end, we induced prostatitis in Nkx3.1 wild-type or mutant mice using CP1 bacteria, which had been isolated from the prostate of a patient with prostatitis and therefore likely to represent a clinically relevant model of prostatic inflammation (Rudick et al., 2011; Simons et al., 2015). Infection of mice with CP1 has been shown to induce a short phase of intense inflammation (i.e. ‘acute’ inflammation), followed by a less intense but persistent inflammation (i.e. ‘chronic’ inflammation), similar to the chronic inflammation that characterizes human prostates (Sfanos et al., 2017). Therefore, we infected adult Nkx3.1+/+ and Nkx3.1−/− mice with CP1, or PBS as a control, via transurethral inoculation, and investigated the phenotypic consequences from 2 weeks to 1 year post-infection (Fig. 2A; Table S2).
Fig. 2.
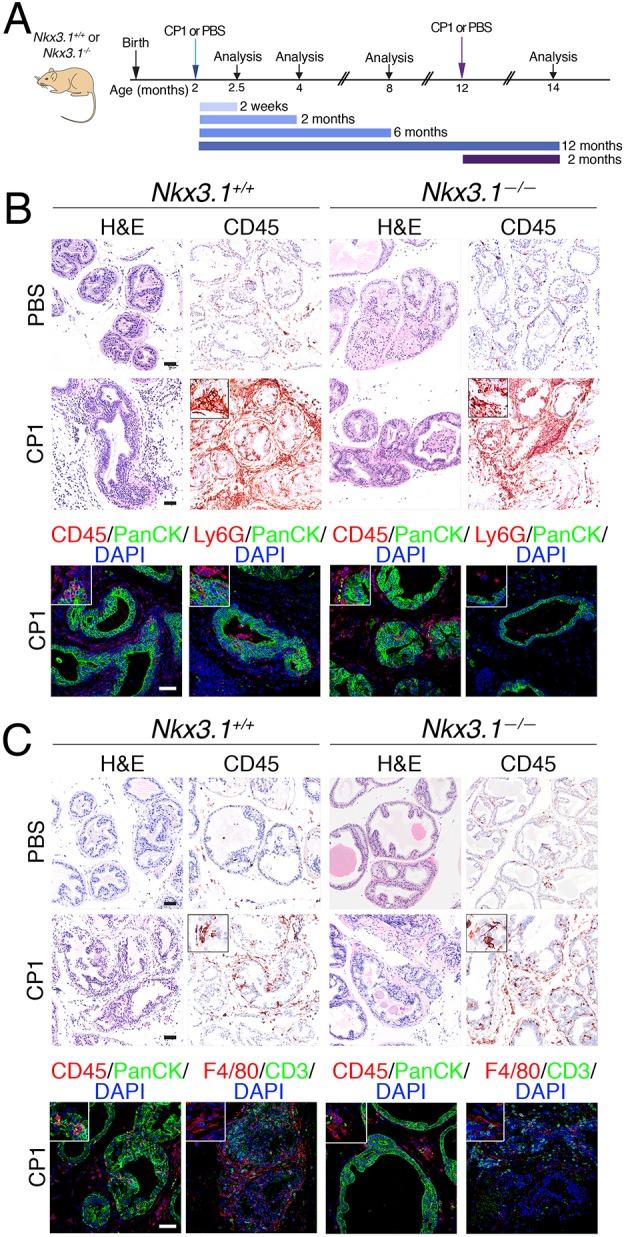
Modeling prostatitis in Nkx3.1 mutant mice. (A) Experimental design for the prostatitis model. Nkx3.1+/+ and Nkx3.1−/− mice were inoculated with the CP1 bacteria or PBS (as a control) at 2 months of age and analyzed at 2 weeks, 2 months, 6 months and 12 months post-infection. In the ‘aged’ experimental setting (dark purple), mice were inoculated at 12 months of age and analyzed at 2 months post-infection. (B) Representative images of H&E or immunohistochemical and immunofluorescence staining with the indicated antibodies at 2 weeks post-inoculation. Insets show higher magnification of a representative region. (C) Representative images of H&E or immunohistochemical and immunofluorescence staining with the indicated antibodies at 2 months post-inoculation. Insets show higher magnification of a representative region. Scale bars: 50 µm. The total number of mice analyzed in each group is provided in Table S2.
At 2 weeks post-infection, we observed acute inflammation in both the Nkx3.1+/+ and Nkx3.1−/− prostate, which was evident from the profound histological phenotype and massive infiltration of CD45-stained immune cells in the CP1-infected but not the control mice (Fig. 2B; n=4-5 mice per group; Table S2). Inflammation was observed in all three prostatic lobes, namely the anterior prostate (AP), the dorsolateral prostate (DLP) and the ventral prostate (VP), although to different degrees (Fig. S3A). At this early time point, the inflammatory response was still mainly acute as evident by the prominent presence of Ly6G+ neutrophils (Fig. 2B), which are primary mediators of acute inflammation (Kolaczkowska and Kubes, 2013).
At later time points post-infection, the Nkx3.1+/+ and Nkx3.1−/− prostates displayed evidence of chronic inflammation (Fig. 2C; Fig. S3B). In particular, at 2 months post-infection, both Nkx3.1+/+ and Nkx3.1−/− mice had sustained presence of immune cells in the CP1-infected but not the control prostates, evident by immunostaining for CD45 (Fig. 2C; n=5-7 mice per group; Table S2). As expected, the immune infiltration at this later time point was less prominent than at 2 weeks and characterized by the presence of other immune subsets, including CD3+ T cells and F4/80+ macrophages (Fig. 2C). This inflammatory reaction persisted over time since sustained increased immune infiltration continued to be observed at 6- and 12-months post-infection (Fig. S3B; n=4-5 mice per group; Table S2).
Although both the Nkx3.1+/+ and Nkx3.1−/− prostates displayed evidence of chronic inflammation, the latter were prone to acceleration of the PIN phenotype. This was evident by histological inspection by the increased cellularity, more pronounced dysplasia, and areas of microinvasion in the Nkx3.1−/− but not Nkx3.1+/+ prostate (Fig. 3; n=4-5 mice/group; Table S3). Notably, this phenomenon was not correlated with significant increased proliferation, as suggested by comparable levels of Ki67 expression in CP1- and PBS-infected Nkx3.1−/− prostates (Fig. S3C). In some cases, the phenotype was accompanied by some areas with disrupted alpha-smooth muscle actin (SMA) expression and focal positivity for the cell-death marker activated-Caspase 3 (Fig. S4). Although the tendency toward more aggressive PIN phenotypes was evident as early as 2 months after induction of prostatitis, the most striking examples were observed at 6 months and 12 months post-infection (Fig. 3A,B; Fig. S4; Table S3), highlighting the importance of aging for the pre-cancerous phenotype of the Nkx3.1 mutant mice, as we have observed previously (Bhatia-Gaur et al., 1999; Irshad et al., 2013). To further investigate the relationship of aging for consequences of inflammation, we induced prostatitis in ‘aged’ (i.e. at 12 months) Nkx3.1+/+ and Nkx3.1−/− mice and analyzed the prostate phenotype 2 months after infection (i.e. at 14 months). We found that the aged Nkx3.1−/− mice displayed a similar accelerated PIN phenotype specifically in the CP1-infected mice (Fig. 3C; Table S3). Taken together, these findings suggest that loss of function of Nkx3.1 promotes inflammation-mediated acceleration of prostate cancer initiation in vivo.
Fig. 3.
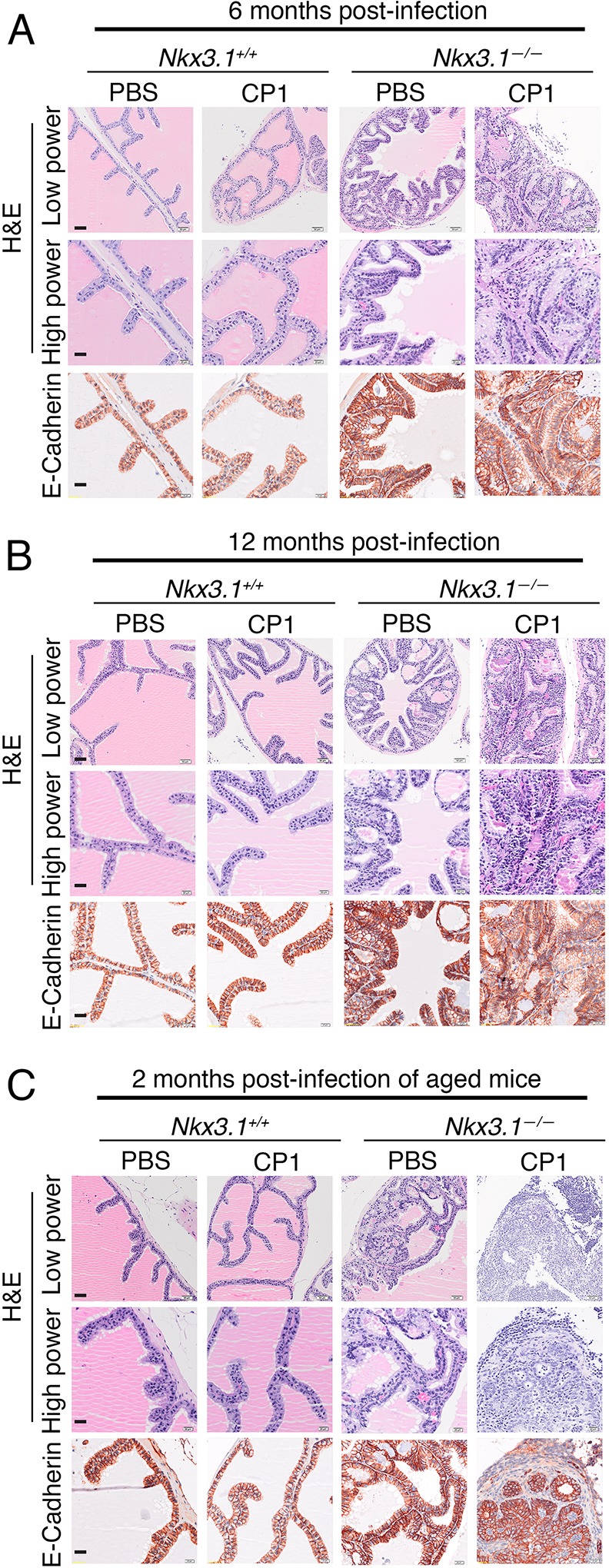
Chronic inflammation accelerates prostate cancer initiation in Nkx3.1 mutant prostate. Representative images of Nkx3.1+/+ and Nkx3.1−/− anterior prostate showing H&E or immunohistochemical staining at the indicated time points following infection with CP1 (as in Fig. 2). Scale bars: 50 µm in low-power images and 20 µm in high-power images. Additional analyses of the phenotype are provided in Figs S3 and S4. The total number of mice analyzed in each group is provided in Table S2, and a summary of the histological phenotype is provided in Table S3.
Inflammation promotes basal-to-luminal differentiation in Nkx3.1 mutant mice
Previous studies have shown that acute prostatic inflammation is associated with basal-to-luminal differentiation in vivo (Kwon et al., 2014). Therefore, we asked whether chronic inflammation induced by CP1 promotes changes in the differentiation potential of prostatic epithelial cells consistent with prostatic epithelial plasticity and, if so, whether this is influenced by the status of Nkx3.1. Toward this end, we performed lineage tracing using a tamoxifen-inducible Cre (CreERT2) under the control of cytokeratin 5 (CK5-CreERT2) or cytokeratin 8 (CK8-CreERT2) promoters to target gene recombination specifically to basal or luminal cells, respectively (Indra et al., 1999; Van Keymeulen et al., 2011). Notably, the CK5-CreERT2 allele is distinct from the CK14-CreERT2 allele used in a previous study (Kwon et al., 2014). These Cre drivers were crossed with a conditionally-activatable fluorescent reporter allele (R26CAG-EYFP; Madisen et al., 2010) as well as with the Nkx3.1 wild-type or mutant mice to generate mice having the following genotypes: CK5-CreERT2 or CK8-CreERT2; Nkx3.1+/+ or Nkx3.1−/−; R26CAG-EYFP/+ (Wang et al., 2013). Accordingly, tamoxifen induction resulted in lineage tracing specifically of basal (Ck5-expressing) or luminal (Ck8-expressing) cells, respectively, in the context of wild-type or null Nkx3.1. One month after tamoxifen induction, mice were infected with CP1 (or PBS as a control) and analyzed at 2-3 months post-infection (n=3 or 4 mice/group; Fig. 4A; Table S2B), during the phase of chronic inflammation (as above). We quantified the relative percentage of lineage-marked basal or luminal cells based on co-staining for YFP and Ck5 and/or Ck8, respectively, in prostates of mice infected with CP1 or the control (PBS) (Table 1).
Fig. 4.

Inflammation is associated with increased basal-cell plasticity in the Nkx3.1 mutant prostate. (A) Experimental design and timeline. CK5-CreERT2 or CK8-CreERT2; Nkx3.1+/+ or Nkx3.1−/−; YFP mice were induced with tamoxifen at 2 months of age and inoculated with a single dose of CP1 (or PBS as a control) 1 month after tamoxifen induction. Expression of specific markers was analyzed and quantified 3 months post-inoculation. Whole-mount images are provided in Fig. S5. (B) Quantification of YFP-positive cells that co-express CK5 (red), CK8 (gray) or both CK5 and CK8 (black) using the CK5-CreERT2 (left) or the CK8-CreERT2 (right) driver. For each group, at least three animals were analyzed (n numbers in Table S2), and the quantification of the lineage-marked cells is provided in Table 1. A two-tailed Welch's t-test was performed to compare the percentage of YFP-positive cells that co-express Ck5 or Ck8 in all groups. Significant differences are indicated (***P<0.001). Data are represented as means and the error bars represent standard error (s.e.m.) for each group. (C,D) Lineage tracing with CK5-CreERT2 (C) and CK8-CreERT2 (D). Representative images of immunofluorescence staining with the indicated antibodies. Insets show higher magnification of a representative region. Scale bars: 25 µm.
Table 1.
Quantification of lineage-marked cells in prostatitis-infected Nkx3.1 mutant mice

Consistent with previous studies (Wang et al., 2013; Choi et al., 2012), lineage tracing of control (i.e. PBS-treated) Nkx3.1+/+ prostates with CK5-CreERT2 marked predominantly (>98%) Ck5-expressing basal cells, a small percentage (<2%) of cells co-expressing Ck5 and Ck8 (presumptive intermediate cells; Wang et al., 2013) and no Ck8-expressing luminal cells (n=3; Fig. 4B,C; Table 1). Conversely, lineage tracing with CK8-CreERT2 marked primarily luminal cells (>99%), very few intermediate cells (<1%) and no Ck5-expressing basal cells (n=3; Fig. 4B,D; Table 1). Although these lineage relationships were largely maintained in CP1-infected Nkx3.1+/+ prostates, lineage tracing with CK5-CreERT2 revealed a small percentage (3.6%) of Ck8-expressing cells that had a luminal-like shape, while the converse was not observed for lineage-tracing with CK8-CreERT2 (n=4 or 3, respectively; Fig. 4B-D; Table 1).
In contrast to these findings for wild-type prostate, chronic inflammation was associated with profound epithelial plasticity in Nkx3.1−/− prostate. In particular, CK5-CreERT2 lineage tracing of control-treated Nkx3.1−/− prostates primarily marked Ck5-expressing basal cells (95.6%, n=3) and some intermediate cells (2.6%); although, unlike the Nkx3.1+/+ prostate, a small percentage of Ck5 lineage marked cells (1.8%) were Ck8-expressing luminal cells (n=3; Fig. 4B,C; Fig. S5; Table 1). In striking contrast, in the CK5-CreERT2 lineage-traced CP1-infected Nkx3.1−/− prostates, more than half of the cells (52.3%, n=4) were Ck8-expressing luminal cells, representing a 29-fold increase compared to the PBS-treated Nkx3.1−/− prostates (P<0.001, t-test; n=4; Fig. 4B,C; Fig. S5; Table 1).
Such inflammation-enhanced plasticity was not observed for luminal cells since lineage tracing of CK8-CreERT2 mice predominantly marked Ck8-expressing luminal cells (>98%) in both the PBS- and CP1-treated Nkx3.1−/− prostates (n=3 or 4, respectively; Fig. 4B,D; Table 1). Taken together, these findings suggest that inflammation greatly augments an inherent potential for basal-to-luminal differentiation in the Nkx3.1 mutant prostate.
Nkx3.1-driven gain of basal features and loss of luminal features is enhanced upon inflammation
Since NKX3.1 is required for proper prostatic epithelial differentiation and specification of luminal prostatic epithelial cells (Dutta et al., 2016; Bhatia-Gaur et al., 1999; Talos et al., 2017), we considered whether the enhanced plasticity of basal cells in the chronically inflamed Nkx3.1−/− prostate was due to impaired differentiation. In particular, we examined markers of luminal or basal cell differentiation by immunohistochemistry in CP1-infected (or control) Nkx3.1+/+ and Nkx3.1−/− prostate during acute or chronic inflammation (Fig. 5A,B). In the acute-inflammation stage (i.e. 2 weeks post-infection), both Nkx3.1+/+ and Nkx3.1−/− prostates exhibited features of altered differentiation as evident by an increased number of Ck5-expressing cells (Fig. 5A). Furthermore, this altered differentiation was maintained in the chronic-inflammation stage in Nkx3.1−/− prostates. In particular, the aged cohort (i.e. infected at 12 months and analyzed at 14 months) displayed reduced expression of cells expressing luminal markers (Nkx3.1, AR, probasin) and increased prevalence of Ck5-expressing cells (Fig. 5B; Fig. S6).
Fig. 5.

Inflammation promotes loss of luminal features in Nkx3.1 mutant prostate. (A) Representative images of immunohistochemical and immunofluorescence staining with the indicated antibodies at 2 weeks post-inoculation. Insets show higher magnification of a representative region. Scale bars: 50 µm. (B) Representative images of staining with the indicated antibodies at 2 months post-infection in the aged mice (see Fig. 2). Insets show higher magnification of a representative region. Scale bars: 25 µm for immunohistochemical analyses and 50 µm for immunofluorescence analyses. (C) Representative images of immunohistochemical staining of human prostate cancer (PCa) samples with the indicated antibodies. Scale bars: 50 µm. Black arrow shows inflammation; red arrows show loss of NKX3.1 in proximity to inflamed areas. Additional examples are shown in Fig. S7. (D) Whisker plots showing the relative expression of the indicated human genes in TCGA Gleason 6 and 7 human prostate tumors stratified based on having high or low levels of NKX3.1 expression. n=145 prostate tumors/group. Statistical analyses were performed using 2-tailed Welch's t-test.
Notably, at the acute-inflammation stage, Nkx3.1+/+ prostates exhibited a loss of Nkx3.1 protein expression (Fig. S3D), consistent with a previous study (Khalili et al., 2010). This loss of Nkx3.1 protein expression occurred specifically in the inflamed prostatic ducts, which were highly proliferative as evident by Ki67 staining, while non-inflamed ducts had normal levels of Nkx3.1 protein and normal proliferation (Fig. S3D). To determine whether this relationship between NKX3.1 loss and inflammation occurs in human prostate cancer, we performed immunohistochemistry on human prostatectomy samples (n=11) to detect expression of NKX3.1 or E-cadherin, an epithelial cell marker. Strikingly, in these human prostate cancers, NKX3.1 protein expression was reduced specifically in areas of inflammation (Fig. 5C; Fig. S7).
Considering these findings, we sought to investigate whether NKX3.1 loss is associated with impaired differentiation in human prostate cancer. In particular, we analyzed the expression levels of genes associated with luminal or basal differentiation in low/intermediate Gleason TCGA primary tumors (Cancer Genome Atlas Research, 2015) segregated by ‘low’ or ‘high’ levels of NKX3.1 expression, as described above. Notably, genes associated with luminal differentiation (i.e. FOXA1, TMPRSS2, HOXB13, KLK3, KLK2) had significantly reduced expression in the ‘low’ NKX3.1 prostate tumors (Fig. 5D). In contrast, expression of basal markers (TP63, KRT5) was significantly increased in the ‘low’ NKX3.1 prostate tumors (Fig. 5D). Furthermore, correlation of the expression levels of NKX3.1 with both immunoregulatory and differentiation-related genes in the TCGA cohort revealed a significant positive correlation between NKX3.1 and genes encoding luminal markers, and a significant negative correlation with genes encoding both basal markers and specific immune- and inflammation-related markers, as evident by Pearson correlation and heat-map analyses (Fig. S8). Together, these findings establish a link between loss of NKX3.1, inflammation and aberrant differentiation in both mouse and human prostate cancer.
DISCUSSION
Chronic inflammation has long been linked to prostate cancer, although the underlying cellular processes are not well understood. In the current study, we demonstrate that loss of a key tumor-suppressor gene associated with prostate cancer initiation, namely NKX3.1, exacerbates inflammation-induced prostate cancer initiation, which is coincident with enhanced epithelial plasticity and defects in cellular differentiation. In particular, we have modeled prostatitis in vivo using the CP1 bacterial strain (Simons et al., 2015), which induces long-term chronic inflammation similar to that observed in aged men (Sfanos and De Marzo, 2012). This enabled us to study the consequences of chronic inflammation for both cancer-related and differentiation-related phenotypes in the context of Nkx3.1 loss-of-function in vivo. Importantly, we observed a striking correlation between the status of NKX3.1 and response to inflammation and cellular differentiation, which is exacerbated upon aging. Furthermore, by complementing analyses of this mouse model with correlative analyses of expression profiling data from human patients, we have demonstrated the relevance of our findings in mice for human prostate cancer. We propose that such an inverse relationship between NKX3.1 status and response to inflammation has consequences for maintaining the differentiation status of the prostatic epithelium, which impacts prostate cancer initiation particularly in the context of aging.
While previous studies have shown reduced expression of NKX3.1 following inflammation in mouse and human prostate (Khalili et al., 2010; Markowski et al., 2008; Bethel et al., 2006), our current studies establish that this relationship is bidirectional and link these findings to cancer initiation as well as to altered cellular differentiation. In particular, we find that loss of NKX3.1 is associated with enrichment of specific immune populations and immune/inflammation markers, and alteration of differentiation markers in both mouse and human prostate. Conversely, in contexts of Nkx3.1 deficiency as occurs in tissues in which chromosomal region 8p21 is lost, chronic inflammation accelerates prostate cancer initiation, which is coincident with impaired cellular differentiation and aberrant cellular plasticity. Notably, NKX3.1 is known to control prostate specification (Dutta et al., 2016) and to be required for normal functioning of luminal stem cells (Wang et al., 2009). Therefore, our current findings suggest that impairment of these functions in the context of NKX3.1 loss impedes the response of the prostate to inflammatory assaults, thereby making it more susceptible to cancer initiation. Moreover, our data showing the increased expression of immunoregulatory genes in NKX3.1-deficient contexts in mouse and human prostate warrants further investigation of the roles of specific immune-cell populations in these processes.
Our study also suggests an important link between NKX3.1 status and aging, as we have observed previously (Bhatia-Gaur et al., 1999; Irshad et al., 2013), since the phenotypic changes associated with inflammation are more profound in aged mice. Considering that aging and inflammation are two well-known etiological factors of cancer (Ahmad et al., 2009), it is not surprising that they may synergize in promoting cancer initiation. In particular, it is widely accepted that aging is associated with important compromises of the immune system and in inflammatory changes, which may affect responses to inflammatory and oncogenic stimuli (Ahmad et al., 2009; Licastro et al., 2005). In addition, experimental evidence suggests that tissue-specific reserves of stem cells decline and their normal activity (e.g. differentiation and tissue repair) is impaired with advancing age (Rossi et al., 2008). Thus, aging, inflammation and impaired differentiation are biological processes that are all linked to cancer initiation and progression, and NKX3.1 may serve as an important mediator for regulating these processes.
Curiously, NKX3.1 is primarily expressed in luminal cells, whereas enhanced plasticity following inflammation mainly occurs in basal cells in NKX3.1-deficient contexts. In particular, chronic inflammation promotes basal-to-luminal differentiation in Nkx3.1 mutant mice, a phenomenon that rarely occurs in normal conditions (Lee and Shen, 2015). However, other conditions that perturb the prostate epithelium, such as acute inflammation or anoikis, have been shown to result in increased basal-to-luminal differentiation (Toivanen et al., 2016; Kwon et al., 2014). Interestingly, in the current study, in which we are investigating a chronic inflammatory context, we find that lineage plasticity is rarely seen in wild-type basal cells, although a previous study showed it to be more frequent in an acute inflammatory context (Kwon et al., 2014). This difference may be attributed to the specific features of the CP1 model or to chronic versus acute inflammation, which may impact the cellular phenotype or differentiation status. Notably, markers of chronic inflammation, such as CD3 receptors or CD68, are strongly correlated with NKX3.1 status in human prostate tumors, which is not the case for markers of acute inflammation such as CEACAM8 or CD177. The current study suggests that, following chronic inflammatory assault, NKX3.1-deficient luminal cells may ultimately be replaced by basal cells, which may have important implications for prostate cancer initiation.
In summary, our study highlights the importance of NKX3.1 status for cellular plasticity, inflammation and aging during prostate cancer initiation. The implication of our findings is that analysis of the status of NKX3.1 combined with markers of inflammation and differentiation may provide insight regarding prognosis of men during initial stages of prostate cancer. Indeed, given that NKX3.1 maps to a key region of the chromosome, 8p21, that undergoes allelic loss in up to 80% of early-stage prostate cancer cases, incorporating analyses of inflammation status combined with NKX3.1 status may enhance models for risk assessment of prostate cancer in a precision cancer-prevention context.
MATERIALS AND METHODS
Description of mouse models
All experiments using animals were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Columbia University Medical Center, NY, USA. The Nkx3.1 germline mutant mice have been described previously (Bhatia-Gaur et al., 1999); the studies performed herein were done using Nkx3.1 wild-type mice (Nkx3.1+/+) and homozygous-null mice (Nkx3.1−/−). These mice have been maintained in our laboratory on a predominantly C57BL/6 background. For lineage-tracing studies, the following mouse alleles were obtained from the Jackson Laboratory (https://www.jax.org) and maintained in a mixed C57BL/6/129S strain background: R26CAG-EYFP (stock number 007903; Madisen et al., 2010), CK5-CreERT2 (stock number 018394/K5-Cre-ERT2; Indra et al., 1999) and CK8-CreERT2 (stock number 017947/K8-CreER; Van Keymeulen et al., 2011). These Cre drivers were crossed for several generations with the reporter allele (R26CAG-EYFP) as well as with the Nkx3.1+/+ and Nkx3.1−/− alleles to generate a series of mice with the following genotypes: CK5-CreERT2 or CK8-CreERT2; Nkx3.1+/+ or Nkx3.1−/−; R26CAG-EYFP/+. To induce gene recombination for lineage-tracing studies, mice were administered tamoxifen (Sigma; 9 mg/40 g body weight) suspended in corn oil by oral gavage once daily for 4 consecutive days as reported previously (Wang et al., 2009, 2013). All studies were done using littermates that were genotyped prior to enrollment; only male mice were used because of the focus on prostate.
Molecular and phenotypic analyses of Nkx3.1 wild-type and mutant mice
Expression profiling of anterior prostate from 15-month-old Nkx3.1+/+ and Nkx3.1−/− mutant mice on an Affymetrix platform (Mu74AV2) has been described (Ouyang et al., 2005; Irshad et al., 2013). Analyses of differentially expressed genes and biological pathways were reported (Dutta et al., 2016). Briefly, a differential gene expression signature was defined between Nkx3.1−/− and Nkx3.1+/+ using a 2-sample 2-tailed Welch’s t-test. This signature was subjected to pathway enrichment analysis using the C2 database, which includes pathways from REACTOME (Fabregat et al., 2016), KEGG (Ogata et al., 1999) and BioCarta (https://cgap.nci.nih.gov/Pathways/BioCarta_Pathways). Pathway analysis was performed using gene set enrichment analysis (GSEA) (Subramanian et al., 2005), with the significance of enrichment estimated using 1000 gene permutations.
For phenotypic analysis, mice were sacrificed at the indicated time points, and prostate tissues were collected and fixed in 10% formalin or zinc (BD Pharmingen). Fixed tissues were embedded in paraffin, and 3-µm sections were used for all analyses. Histological and immunohistochemical analyses were performed as described (Dutta et al., 2016). For immunohistochemistry on formalin-fixed tissues, sections underwent antigen retrieval by boiling in citrate-acid-based antigen unmasking solution (Vector Labs) for 16 min, while this step was omitted for zinc-fixed tissues. Detection of mouse Nkx3.1 in immunofluorescence assays was enhanced using tyramide amplification (Perkin Elmer) with a horseradish peroxidase (HRP)-conjugated secondary antibody (Invitrogen), followed by incubation with tyramide 488 as described (Dutta et al., 2016). All other antibodies were detected using fluorochrome-coupled secondary antibodies. Details of all antibodies and fixation used for immunohistochemical analyses are provided in Table S4.
Histopathological scoring was performed according to the classification of Park and colleagues (Park et al., 2002). Images were captured using an Olympus VS120 whole-slide scanning microscope. For lineage-tracing experiments, tissues were stained by immunofluorescence and images captured using a Leica TCS SP5 confocal microscope. Quantification of lineage tracing was performed blinded (i.e. without knowledge of the genotype or treatment) by manual counting of cells using 5-10 images from 3-4 independent mice as described (Dutta et al., 2016).
Flow cytometry analyses of immune cells
Single-cell suspensions from prostate tissue were prepared using the Mouse Tumor Dissociation Kit according to the manufacturer's recommendations (Miltenyi). Cells were Fc-blocked with purified rat anti-mouse CD16/CD32 (clone: 2.4 G2, Becton Dickinson BD) for 30 min at 4°C. Dead cells were discriminated using the LIVE/DEAD (LD) Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) and samples were stained for the extracellular and intracellular markers as detailed in Table S4. Staining was visualized by fluorescence-activated cell sorting (FACS) analysis using an LSRFortessa™ (BD Biosciences) and analyzed using FlowJo® (FlowJo LLC). Macrophages are referred to as CD45+/B220−/CD11b+/F4/80+, T lymphocytes as CD45+/CD11b−/CD3+, CD4+ T cells as CD45+/CD11b−/CD3+/CD4+, CD8+ T cells as CD45+/CD11b−/CD3+/CD8+, B cells as CD45+/B220+/CD11b−, and G-MDSCs represent granulocytic myeloid-derived suppressor cells that include neutrophils and are defined as CD45+/B220−/CD11b+/Gr1high.
Bacteria-induced prostatitis model
The CP1 E. coli bacterial strain was kindly provided by A. Schaeffer and colleagues (Johns Hopkins University) and was initially isolated from the prostatic secretions of a patient with chronic prostatitis (Rudick et al., 2011). Bacterial cultures and transurethral inoculation were performed as previously described (Simons et al., 2015). Mice were inoculated with a single dose of CP1, or PBS, by transurethral infection.
Human patient samples
All studies using human patient tissues were performed following protocols approved by the institutional review board of Columbia University Medical Center. Anonymized patient specimens (n=11) were obtained from the Molecular Pathology Core, representing patients with clinically-localized prostate cancer (Gleason grade ranging from 6 to 9) who had undergone prostatectomy in the Department of Urology at Columbia University Medical Center. Immunohistochemistry was performed on paraffin-embedded tissues, following standard procedures using a rabbit polyclonal anti-human NKX3.1 antibody and a mouse monoclonal anti E-cadherin antibody (see Table S4 for antibody details). Analyses of expression of specific genes in human prostate cancer was performed using a cohort of primary tumors from TCGA (Cancer Genome Atlas Research, 2015), which includes RNA sequencing data from 290 patients with Gleason 6 and 7 primary prostate tumors. Median of expression was used as a threshold to separate patients in two groups with either a low or high level of NKX3.1 gene expression (n=145 patients per group).
Statistical analyses
Statistical analyses were conducted using a 2-sample 2-tailed Welch's t-test, χ2 test or Fisher's exact test as appropriate. Differential gene expression was estimated with a 2-sample 2-tailed Welch’s t-test. A P-value <0.05 was considered significant. GraphPad Prism software (version 5.0) was used for statistical analyses and data representation.
Supplementary Material
Acknowledgements
The authors thank Dr Alex Papachristodoulou for assistance with the mouse dissections, Drs Anthony Schaeffer, Karen Sfanos and colleagues (Johns Hopkins) for the kind gift of the CP1 bacteria and technical advice, and Michael Shen and Edward Gelmann for advice on the manuscript.
Footnotes
Competing interests
A.C. is founder, equity holder, consultant and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. C.G.D. has ownership interests in Compugen, Harpoon, Kleo, Potenza and Tizona; has served in a consulting or advisory role for Agenus, Dendreon, Janssen, Lilly, Merck, Medimmune, Pierre Fabre and Roche/Genentech; has received research funding from Aduro Biotech, Bristol Myers Squibb and Janssen; and holds patents, receives royalties or has other intellectual property interests with Bristol Myers Squibb, AstraZeneca Medimmune and Janssen. The other authors declare no competing or financial interests
Author contributions
Conceptualization: C.L.M., A.M., C.A.-S.; Methodology: C.L.M., J.Y.K., R.K.V., S.P., Z.A.L.-B., A.M.; Validation: C.L.M.; Formal analysis: C.L.M., R.K.V., A.D., J.Y.K., S.P., Z.A.L.-B., A.M.; Investigation: C.L.M.; Data curation: R.K.V., A.D.; Writing - original draft: C.L.M., C.A.-S.; Writing - review & editing: C.L.M., A.D., Z.A.L.-B., C.A.-S.; Supervision: A.C., C.G.D., A.M., C.A.-S.; Project administration: C.A.-S.; Funding acquisition: C.A.-S.
Funding
These studies used the resources of the Herbert Irving Comprehensive Cancer Center Flow Core Facility and the Molecular Pathology Core (Columbia University) funded in part through National Cancer Institute Grant P30 CA013696. The research in the C.A.-S. laboratory is supported, in part, by funding by National Institutes of Health grants CA196662, CA193442, CA183929 and CA173481, and American Cancer Society grant RP-16-237-06-COUN. C.L.M. was supported by Swiss National Science Foundation grants PBBSP3_146959 and P300P3_151158. A.D. was supported in part by the US National Center for Advancing Translational Sciences, National Institutes of Health, Grant number UL1 TR000040. A.C. is supported by US National Institutes of Health grants R35 CA197745 (Outstanding Investigator Award) and U54 CA209997 (Centers for Cancer Systems Therapeutics). A.M. is supported by the Rutgers School of Health Professions (Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey) Dean's Research Grant and start-up funds. C.G.D. is supported by the Melanoma Research Alliance, the Patrick C. Walsh Prostate Cancer Research Fund (Johns Hopkins University), the One-in-Six Foundation (Akron Community Foundation) and the Prostate Cancer Foundation. C.A.-S. is an American Cancer Society Research Professor supported in part by a generous gift from the F. M. Kirby Foundation.
Data availability
The raw and normalized data files are deposited in Gene Expression Omnibus (GEO) (GSE81440).
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.035139.supplemental
References
- Abate-Shen C., Shen M. M. and Gelmann E. (2008). Integrating differentiation and cancer: the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Differentiation 76, 717-727. 10.1111/j.1432-0436.2008.00292.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A., Banerjee S., Wang Z., Kong D., Majumdar A. P. and Sarkar F. H. (2009). Aging and inflammation: etiological culprits of cancer. Curr. Aging. Sci. 2, 174-186. 10.2174/1874609810902030174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca S. C., Prandi D., Lawrence M. S., Mosquera J. M., Romanel A., Drier Y., Park K., Kitabayashi N., Macdonald T. Y., Ghandi M. et al. (2013). Punctuated evolution of prostate cancer genomes. Cell 153, 666-677. 10.1016/j.cell.2013.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethel C. R., Faith D., Li X., Guan B., Hicks J. L., Lan F., Jenkins R. B., Bieberich C. J. and De Marzo A. M. (2006). Decreased NKX3.1 protein expression in focal prostatic atrophy, prostatic intraepithelial neoplasia, and adenocarcinoma: association with gleason score and chromosome 8p deletion. Cancer Res. 66, 10683-10690. 10.1158/0008-5472.CAN-06-0963 [DOI] [PubMed] [Google Scholar]
- Bhatia-Gaur R., Donjacour A. A., Sciavolino P. J., Kim M., Desai N., Young P., Norton C. R., Gridley T., Cardiff R. D., Cunha G. R. et al. (1999). Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 13, 966-977. 10.1101/gad.13.8.966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2015). The molecular taxonomy of primary prostate cancer. Cell 163, 1011-1025. 10.1016/j.cell.2015.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N., Zhang B., Zhang L., Ittmann M. and Xin L. (2012). Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 21, 253-265. 10.1016/j.ccr.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens L. M. and Werb Z. (2002). Inflammation and cancer. Nature 420, 860-867. 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marzo A. M., Nakai Y. and Nelson W. G. (2007a). Inflammation, atrophy, and prostate carcinogenesis. Urol. Oncol. 25, 398-400. 10.1016/j.urolonc.2007.05.007 [DOI] [PubMed] [Google Scholar]
- De Marzo A. M., Platz E. A., Sutcliffe S., Xu J., Grönberg H., Drake C. G., Nakai Y., Isaacs W. B. and Nelson W. G. (2007b). Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 7, 256-269. 10.1038/nrc2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A., Le Magnen C., Mitrofanova A., Ouyang X., Califano A. and Abate-Shen C. (2016). Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 352, 1576-1580. 10.1126/science.aad9512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A., Panja S., Virk R. K., Kim J. Y., Zott R., Cremers S., Golombos D. M., Liu D., Mosquera J. M., Mostaghel E. A. et al. (2017). Co-clinical analysis of a genetically engineered mouse model and human prostate cancer reveals significance of NKX3.1 expression for response to 5alpha-reductase inhibition. Eur. Urol. 72, 499-506. 10.1016/j.eururo.2017.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S. et al. (2016). The Reactome pathway Knowledgebase. Nucleic Acids Res. 44, D481-D487. 10.1093/nar/gkv1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra A. K., Warot X., Brocard J., Bornert J. M., Xiao J.-H., Chambon P. and Metzger D. (1999). Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 27, 4324-4327. 10.1093/nar/27.22.4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irshad S., Bansal M., Castillo-Martin M., Zheng T., Aytes A., Wenske S., Le Magnen C., Guarnieri P., Sumazin P., Benson M. C. et al. (2013). A molecular signature predictive of indolent prostate cancer. Sci. Transl. Med. 5, 202ra122 10.1126/scitranslmed.3006408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili M., Mutton L. N., Gurel B., Hicks J. L., De Marzo A. M. and Bieberich C. J. (2010). Loss of Nkx3.1 expression in bacterial prostatitis: a potential link between inflammation and neoplasia. Am. J. Pathol. 176, 2259-2268. 10.2353/ajpath.2010.080747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. J., Bhatia-Gaur R., Banach-Petrosky W. A., Desai N., Wang Y., Hayward S. W., Cunha G. R., Cardiff R. D., Shen M. M. and Abate-Shen C. (2002a). Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 62, 2999-3004. [PubMed] [Google Scholar]
- Kim M. J., Cardiff R. D., Desai N., Banach-Petrosky W. A., Parsons R., Shen M. M. and Abate-Shen C. (2002b). Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. USA 99, 2884-2889. 10.1073/pnas.042688999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E. and Kubes P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159-175. 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- Kwon O.-J., Zhang L., Ittmann M. M. and Xin L. (2014). Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc. Natl. Acad. Sci. USA 111, E592-E600. 10.1073/pnas.1318157111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H. and Shen M. M. (2015). Cell types of origin for prostate cancer. Curr. Opin. Cell Biol. 37, 35-41. 10.1016/j.ceb.2015.10.002 [DOI] [PubMed] [Google Scholar]
- Le Magnen C., Shen M. M. and Abate-Shen C. (2018). Lineage plasticity in cancer progression and treatment. Annu. Rev. Cancer Biol. 2, 271-289. 10.1146/annurev-cancerbio-030617-050224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licastro F., Candore G., Lio D., Porcellini E., Colonna-Romano G., Franceschi C. and Caruso C. (2005). Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun. Ageing 2, 8 10.1186/1742-4933-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Grogan T. R., Hieronymus H., Hashimoto T., Mottahedeh J., Cheng D., Zhang L., Huang K., Stoyanova T., Park J. W. et al. (2016). Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep 17, 2596-2606. 10.1016/j.celrep.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R. et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133-140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R. S., Amin M. A., Li X., Kalyana-Sundaram S., Veeneman B. A., Wang L., Ghosh A., Aslam A., Ramanand S. G., Rabquer B. J. et al. (2016). Inflammation-induced oxidative stress mediates gene fusion formation in prostate Cancer. Cell Rep 17, 2620-2631. 10.1016/j.celrep.2016.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowski M. C., Bowen C. and Gelmann E. P. (2008). Inflammatory cytokines induce phosphorylation and ubiquitination of prostate suppressor protein NKX3.1. Cancer Res. 68, 6896-6901. 10.1158/0008-5472.CAN-08-0578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle P. A., Canna K., McMillan D. C., McNicol A.-M., Campbell R. and Underwood M. A. (2004). The relationship between T-lymphocyte subset infiltration and survival in patients with prostate cancer. Br. J. Cancer 91, 541-543. 10.1038/sj.bjc.6601943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura N., Takayama H., Nakayama M., Nakai Y., Kawashima A., Mukai M., Nagahara A., Aozasa K. and Tsujimura A. (2011). Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 107, 1918-1922. 10.1111/j.1464-410X.2010.09804.x [DOI] [PubMed] [Google Scholar]
- Ogata H., Goto S., Sato K., Fujibuchi W., Bono H. and Kanehisa M. (1999). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29-34. 10.1093/nar/27.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X., Deweese T. L., Nelson W. G. and Abate-Shen C. (2005). Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 65, 6773-6779. 10.1158/0008-5472.CAN-05-1948 [DOI] [PubMed] [Google Scholar]
- Park J.-H., Walls J. E., Galvez J. J., Kim M., Abate-Shen C., Shen M. M. and Cardiff R. D. (2002). Prostatic intraepithelial neoplasia in genetically engineered mice. Am. J. Pathol. 161, 727-735. 10.1016/S0002-9440(10)64228-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D. J., Jamieson C. H. M. and Weissman I. L. (2008). Stems cells and the pathways to aging and cancer. Cell 132, 681-696. 10.1016/j.cell.2008.01.036 [DOI] [PubMed] [Google Scholar]
- Rudick C. N., Berry R. E., Johnson J. R., Johnston B., Klumpp D. J., Schaeffer A. J. and Thumbikat P. (2011). Uropathogenic Escherichia coli induces chronic pelvic pain. Infect. Immun. 79, 628-635. 10.1128/IAI.00910-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfanos K. S. and De Marzo A. M. (2012). Prostate cancer and inflammation: the evidence. Histopathology 60, 199-215. 10.1111/j.1365-2559.2011.04033.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfanos K. S., Hempel H. A. and De Marzo A. M. (2014). The role of inflammation in prostate cancer. Adv. Exp. Med. Biol. 816, 153-181. 10.1007/978-3-0348-0837-8_7 [DOI] [PubMed] [Google Scholar]
- Sfanos K. S., Yegnasubramanian S., Nelson W. G. and De Marzo A. M. (2017). The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 15, 11-24. 10.1038/nrurol.2017.167 [DOI] [PubMed] [Google Scholar]
- Simons B. W., Durham N. M., Bruno T. C., Grosso J. F., Schaeffer A. J., Ross A. E., Hurley P. J., Berman D. M., Drake C. G., Thumbikat P. et al. (2015). A human prostatic bacterial isolate alters the prostatic microenvironment and accelerates prostate cancer progression. J. Pathol. 235, 478-489. 10.1002/path.4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasner A. and Karin M. (2015). Immune infiltration and prostate cancer. Front. Oncol. 5, 128 10.3389/fonc.2015.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S. et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545-15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos F., Mitrofanova A., Bergren S. K., Califano A. and Shen M. M. (2017). A computational systems approach identifies synergistic specification genes that facilitate lineage conversion to prostate tissue. Nat. Commun. 8, 14662 10.1038/ncomms14662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivanen R., Mohan A. and Shen M. M. (2016). Basal progenitors contribute to repair of the prostate epithelium following induced luminal anoikis. Stem Cell Reports 6, 660-667. 10.1016/j.stemcr.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Keymeulen A., Rocha A. S., Ousset M., Beck B., Bouvencourt G., Rock J., Sharma N., Dekoninck S. and Blanpain C. (2011). Distinct stem cells contribute to mammary gland development and maintenance. Nature 479, 189-193. 10.1038/nature10573 [DOI] [PubMed] [Google Scholar]
- Wang X., Kruithof-De Julio M., Economides K. D., Walker D., Yu H., Halili M. V., Hu Y.-P., Price S. M., Abate-Shen C. and Shen M. M. (2009). A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 461, 495-500. 10.1038/nature08361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. A., Mitrofanova A., Bergren S. K., Abate-Shen C., Cardiff R. D., Califano A. and Shen M. M. (2013). Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 15, 274-283. 10.1038/ncb2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.