

Abstract
Five syndromes share predominantly hyperplastic glands with primary excess of hormone. 1) NEONATAL SEVERE PRIMARY HYPERPARATHYROIDISM, from homozygous mutated CASR, begins severely in utero. 2) CONGENITAL NON-AUTOIMMUNE THYROTOXICOSIS, from mutated TSHR, varies from severe with fetal onset to mild with adult onset. 3) FAMILIAL MALE-LIMITED PRECOCIOUS PUBERTY, from mutated LHR, expresses testosterone over-secretion in young boys. 4) HEREDITARY OVARIAN HYPERSTIMULATION SYNDROME, from mutated FSHR, expresses symptomatic vascular permeabilities during pregnancy. 5) FAMILIAL HYPERALDOSTERONISM TYPE IIIA, from mutated KCNJ5, presents in young children with hypertension and hypokalemia.
Grouping of these 5 syndromes highlights predominant hyperplasia as a stable tissue endpoint, and as their tissue stage for all the hormone excess. Comparisons were made among this and 2 other groups of syndromes, forming a continuum of tissue staging [A-B-C]. A) Predominant oversecretions express little or no hyperplasia. B) Predominant hyperplasias express little or no neoplasia. C) Predominant neoplasias express nodules, adenomas, or cancers. Hyperplasias may progress (5 of 5) to neoplastic stages while predominant oversecrertions rarely do (1 of 6; frequencies differ P<0.02). Hyperplasias do not show tumor multiplicity (0 of 5) unlike neoplasias that do (14 of 20; P<.02). Hyperplasias express mutation of a plasma membrane-bound sensor (5 of 5) while neoplasias rarely do (RET) (1 of 18; P<.0002).
In conclusion, the multiple distinguishing themes within the hyperplasias establish a robust pathophysiology. It has the shared and novel feature of mutant sensors in the plasma membrane, suggesting that these are a major contributor to hyperplasia.
Keywords: neonate, proliferation, GPCR, cyclic AMP, signal transduction, parathyroid, thyroid, gonad, adrenal cortex
INTRODUCTION
Hyperplasia was identified long ago in pathologic tissues, including the thyroid and parathyroid, and long before the secreted hormone of each tissue had become identified (Hirsch A 1885; De Crecchio, L 1865; Erdheim J 1907). Hyperplasia is frequently a secondary state, a response to extracellular stimuli, such as in a goiter caused by thyroid stimulating antibodies or by TSH.
Alternately, and whether accompanied by oversecretion, hyperplasia can be a primary state, originating from an intrinsic process of the same cells (Derwahl M, Studer H 2002; Arnold A 2011; Snow AL, Xiao W, et al 2012). Primary hyperplasia usually accompanies a pathologic over-secretion of a hormone from the same cells. For this, it can be divided into two broad formats. The first is subtle hyperplasia as a precursor or an accompaniment to predominating adenomas or cancers (Mete O, Asa SL 2013; Marx SJ 2013). The second format is predominating hyperplasia but with little or no progression to adenomas or cancer. This second format is the main subject here.
METHODS
The main foci for review were histologic staging in hormone-secretory glands, secretion processes in the glands, germline mutation, and molecular functions around the mutant protein. I analyzed reports and reviews about: (a) primary over-secretion of hormone(s), (b) descriptions of light microscopy for the over-secreting glands, (c) predominating hyperplasia in the hormone-secretory gland, and (d) origin of the syndrome from a known mutation.
The main criterion for hyperplasia of a tissue is an increase in the number of relatively normal-appearing cells (Kumar V, Abbas AK, 2010). Hyperplasia is distinct from hypertrophy; hypertyrophy is an increase in cell size, and it can accompany hyperplasia. Hyperplastic cells may be of uniform or of mixed types. When of mixed types, there is a variable decrease in the fraction with adipocytes and other stroma. The parenchymal pattern is diffuse throughout a hyperplastic tissue, in contrast to neoplasia which is focal or multifocal. Differential immunostaining for reticulin has been used only occasionally to distinguish hyperplasia from neoplasia, but mainly for hepatic or pituitary tissues (Hong H, Patonay B, et al 2011; Mete O, Asa SL 2013).
Exclusion criteria (Supplementary Table S-1) included insufficient information about gland histology, predominantly normal or near-normal tissue grade (Marx 2014), or higher grades of neoplasia (small nodules, large nodules, adenoma, or cancer). Some of the excluded syndromes were later grouped for comparisons to the reviewed group.
We used Fisher’s exact test to compare frequencies of features among syndrome groups.
RESULTS
Neonatal severe primary hyperparathyroidism
Clinical.
Neonatal severe primary hyperparathyroidism (NSHPT) is very rare (Thompson NW, Carpenter LC, et al 1978; Cooper L, Wertheimer J, et al 1986; Key L, Thorne M, et al 1990; Arnold A, Marx SJ 2013). It includes an under-mineralized skeleton, sub-periosteal resorption, bell-shaped thorax, and extremely high blood levels of calcium (20–30 mg/dl) and of PTH (500% or more of the upper normal limit). Biallelic inactivation of the CaS-R in bone cells may contribute to the severe skeletal features (Goltzman D, Hendy GN 2015). These severely ill neonates suffer respiratory distress, worsened by multiple rib fractures and hypotonia. They need complex support. After subtotal parathyroidectomy, hyperparathyroidism in severe form recurred rapidly, at 2 and 5 months for two cases (Thompson NW, Carpenter LC, et al 1978; Key L, Thorne M, et al 1990) (Supplement). The preferred treatment includes urgent total parathyroidectomy.
Parathyroids in NSHPT.
The severe skeletal expressions at birth in NSHPT indicate that harmful over-secretion of PTH had begun from the parathyroid glands of the fetus. In fact, the murine fetus with homozygous CASR knockout, has serum PTH about 20-fold the maternal level at day 18 of gestation (Simmonds CS, Karsenty G, et al 2010). Surgery for NSHPT at 2–12 weeks postpartum has shown parathyroid size 4–10 times normal (Supplementary Table S-2). The histologic pattern is diffuse chief cell hyperplasia, mainly large clear cells and some small chief cells. Parathyroid nodularity or cancer have not been reported, and monoclonality has not been analyzed. However, double adenoma was identified in a variant, i.e. one case with germline biallelic inactivation of the CASR and much milder onset in adulthood (Hannan FM, Nesbit MA, et al 2010).
Molecules and genes.
The normal CASR encodes the extracellular calcium sensing receptor (CaS-R). It is expressed mainly on the parathyroid cell surface, but also on the renal tubular cell, the thyroidal C-cell, and elsewhere. It is central in sensing levels of extracellular calcium and in regulating PTH secretion (Brown EM 2013). A role in proliferation of the parathyroid has been supported indirectly by CaS-R deficiencies in the large parathyroids of primary or secondary hyperparathyroidism and also by calcimimetic drug actions against hyperparathyroidism (Kifor O, Moore FD, et al 1996; Farnebo P, Enberg U, et al 1997; Miller G, Davis J, et al 2011).
Cases of NSHPT have biallelic inactivation of the CASR (Marx SJ, Fraser D, et al 1985; Pollak MR, Brown EM, et al 1993; Hannan FM, Nesbit MA, et al 2012). The same mutation in heterozygous form expresses as familial hypocalciuric hypercalcemia (FHH) (Arnold A, Marx SJ 2013). GNA11 and AP2S1, two other genes less frequently mutated in FHH heterozygotes (Nesbit MA, Hannan F, et al 2013a; Nesbit MA, Hannan FM, et al 2013b), have not yet been implicated in NSHPT. 30% of the CASR mutations causing NSHPT predict truncation of the CaS-R vs 3% from the CASR mutations causing FHH; most of the missense CASR mutations are clustered in a limited extracellular domain (cleft of its “Venus fly-trap” domain) (Hannan FM, Nesbit MA, et al 2012). In vitro, most shift the suppression curve between extracellular Ca++ and PTH secretion towards higher Ca++ values (Pearce SHS, Bai M, et al 1996).
Sporadic tumor from somatic mutation of CASR.
The parathyroids from common primary hyperparathyroidism show decreased sensitivity to extracellular calcium (Brown EM, Gardner DG, et al 1979). Both primary and secondary tumors of the parathyroids show lowered concentrations of CaS-R protein (Kifor O, Moore FD, et al 1996; Farnebo P, Enberg U, et al 1997); however, somatic mutation of the CASR in the parathyroids has not been found in either of these common states (Hosokawa Y, Pollak MR, et al 1995; Arnold A, Brown ME, et al J Clin Invest 1995; Cetani F, Pinchera A, et al 1999). Rare cases of FHH present with a clinical diagnosis of parathyroid adenoma (Burski K, Torjussen B, et al 2002; Yamauchi M, Sugimoto T, et al 2002; Brachet C, Boros E, et al 2009; Yabuta T, Miyauchi A, et al 2009); these have not been evaluated for a second hit at the CASR or at another gene.
Variants related to NSHPT.
Rare homozygous CASR mutations are expressed as a milder variant, presenting as hypocalciuric hypercalcemia in adulthood (Leitman SA, Tenenbaum-Rakover Y, et al 2009; Hannan FM, Nesbit MA, et al 2010).
A different variant of intermediate severity (serum calcium 11–15 mg/dl and PTH 300% or more of upper normal) may occur in a neonate with heterozygous mutated CASR, but born to a mother without the mutation. In these cases, secondary hyperparathyroidism starting in the fetus in utero may worsen temporarily the otherwise mild expressions in the heterozygous neonate (Marx SJ, Attie MF, et al 1982; Bai M, Pearce SHS, et al 1997).
Congenital non-autoimmune thyrotoxicosis
Clinical.
Congenital non-autoimmune thyrotoxicosis (CNT) is rare and is generally caused by heterozygous activating mutation of the TSH receptor (TSH-R) (Kopp P, van Sande J, et al 1995; Vassart G 2010; Hebrant A, van Staveren WCG, et al 2011). It must be distinguished from the more frequent neonatal Graves disease, caused by antibodies that activate the TSH-R. Severe expressions can sometimes be recognized at birth; cases with earliest expressions show hyperthyroidism (50%), prematurity (70%), low birth weight (85%), mental retardation (60%), advanced bone age (50%), and cranial synostosis (50%) (Vassart G 2010; Hebrant A, van Staveren WCG, et al 2011). Most severely affected neonates present sporadically with a new mutation (Kopp P, van Sande J, et al; 1995; Polak M, et al 1996; Gozu HI, Lublinghoff J, et al 2010; Vassart G 2010; Hebrant A, van Staveren WCG, et al 2011). Alternately and rarely, an affected neonate was the offspring of an affected father or mother, who had been adequately managed as a severely affected child (Supornsilchai V, Sahakitrungruang T, et al 2009). In some families with TSH-R mutation, all carriers show onset of thyrotoxicosis after age 10 years (Arturi F, Chiefari E, et al 2002; Nishihara E, Chen C-R, et al 2010). Recurrence of thyrotoxicosis with CNT is likely after subtotal treatments, including after withdrawal of anti-thyroid drugs (Hebrant A, van Staveren WCG, et al 2011; Paschke R, Niedziela M, et al 2012). The usual treatment is uninterrupted antithyroid drugs and/or thyroid ablation, total or near-total.
Thyroid gland in CNT.
The severe expressions in some cases with CNT indicate that there had been over-secretion of thyroid hormones by the fetus. Thyroid histology near parturition in CNT has not been reported, but over 50% show goiter at birth. At all older ages, there is diffuse thyroid follicular hyperplasia, with or without goiter. Average thyroid size may be increased 3–5 fold or more. There are clusters of small or large follicles, similar to toxic thyroid adenoma. At later stages, small or large nodules may occur (Hebrant A, van Staveren WCG, et al 2011; Gozu HI, Lublinghoff J, et al 2010). The frequency of thyroid cancer is not increased.
Molecules and genes.
The normal TSH-R, LH receptor (LH-R or LH/CG-R), and FSH receptor (FSH-R) are closely related. Similarly the gonadotropin hormones, TSH, LH, FSH, and CG, are closely related (Themmen APN 2005; Kleinau G, Neumann S, et al 2013; Jiang X, Dias JA, et al 2014). All recently reported hereditary cases of CNT have had a heterozygous activating TSHR mutation. Most of the mutations are modeled along the transmembrane loops of the TSH-R, with roughly similar distribution of severe and less severe mutations (Gozu HI, Lublinghoff J, et al 2010). Mutation sequences from the severest cases can also be identical to mutations in sporadic adenoma. In contrast, less severe cases are from other private germline mutations. The mutated, activated TSH-R causes in vitro a 2–7 fold higher basal cyclic AMP than controls (Gozu HI, Lublinghoff J, et al 2010). Responsivity to TSH in vitro is conserved, and apparent affinity for TSH is sometimes increased (Vassart G 2010). Activating mutation of the TSH-R also stimulates thyrocyte proliferation in vitro (Ludgate M, Gire V, et al 1999).
Sporadic tumors from somatic mutation of the TSHR.
50% of autonomous solitary thyroid adenomas have a somatic activating mutation of the TSHR (Vassart G 2010; Hebrant A, van Staveren WCG, et al 2011). TSHR activating mutations have rarely been identified as an initiator in sporadic follicular thyroid cancer (Spambalg D, Sharifi N, et al 1996).
Variant from TSHR mutation, expressed as gestational thyrotoxicosis.
Thyrotoxicosis beginning in pregnancy is usually caused by Graves disease or by CG activation of the normal TSH-Rs. In one family, a mother and daughter showed severe hyperthyroidism, occurring and recurring during six pregnancies of one or the other (Rodien P, Bremont C, et al 1998). TSH was low and CG was normal for stage of gestation. A small diffuse goiter was recognized during a recurrence. The same germline change (mutation) of the TSHR was found in both cases. In vitro, this increased markedly the TSH-R sensitivity to CG but not to TSH.
Familial male-limited precocious puberty
Clinical.
Familial male-limited precocious puberty (FMPP) or testotoxicosis is initially recognized as male iso-sexual precocious puberty. Female carriers have no disease phenotype. Expression usually begins between ages 1–3 years (Beas FO, Zurbrugg RP, et al 1962; Egli CA, SM, Grumbach MM, et al 1985); the occasional expression as increased genital size at birth indicates onset in the fetus of such a case (Beas FO, Zurbrugg RP, et al 1962; Rosenthal SM, Grumbach MM, et al 1983; Müller J, Gondos B, et al 1998). Testosterone levels in blood are increased and gonadotrophins are low. Drugs against androgen synthesis or action have accomplished partial success (Reiter EO, Mauras N, et al 2010; Fuqua JS T 2013).
Testis in FMPP.
In FMPP, there is bilateral enlargement of the genitals, including modest enlargement of the testes. The testis in FMPP shows hyperplasia of Leydig cells, precocious spermatogenesis, and rarely bilateral nodularity of Leydig cells, (Gondos B, Egli CA, et al 1985; Leschek EW, Chan W-Y, et al 2001; McGee SR, Narayan P. 2013).
Molecules and genes.
FMPP is usually attributable to germline activating mutation of the LH-R (Themmen APN 2005; Shenker A, Laue L, et al 1993). The germline and somatic activating LHR mutations 2011). In vitro, these mutations elevate basal cyclic AMP but decrease the maximal cyclic AMP response to CG (Leschek EW, Chan W-Y, et al 2001).
Sporadic tumor arising from somatic mutation of LHR.
Most sporadic Leydig cell tumors have a somatically mutated LHR. Most adenomas show LHR D578H. The abnormalities of cyclic AMP regulation in vitro are more severe (higher basal cyclic AMP and absent response to CG) from D578H than from germline mutations; furthermore, D578H has not been identified in the germline and thus may be lethal in the very early embryo (Boot AM, Lumbroso S, et al 2011).
Hereditary ovarian hyperstimulation syndrome
Clinical.
When severe, hereditary ovarian hyperstimulation syndrome (OHSS) can be a life-threatening complex of ovarian enlargement and diffuse vascular permeability (ascites, pleural effusion, hemo-concentration, thromboembolism). It reflects over-secretion from the ovarian corpus luteum of pregnancy for several factors, including estrogens, progestins, and cytokines (Fiedler K, Ezcurra D 2012). Most frequently, ovarian hyperstimulation syndrome occurs sporadically during administration of exogenous FSH or CG for in vitro fertilization; in this setting, FSH may have been given to compensate for a subtle deficiency of FSH. Rarely, during an unassisted pregnancy, OHSS occurs and can recur spontaneously in several pregnancies of the same woman, and it may arise in several women within a family (Vasseur C, Rodien P, et al 2003; Smits G, Olatunbosun O, et al 2003). OHSS remits after delivery. The management is general support until and after delivery.
Corpus luteum of pregnancy in OHSS.
In OHSS, the ovaries are larger than in normal pregnancy and multicystic; there are layers of hyperplasia of luteinized granulosa and theca cells (Stocco C, Telleria C, et al 2007; Meduri G, Bachelot A, et al 2008). The ovaries in OHSS return to normal size by 8 weeks after delivery (Smits G, Olatunbosun O, et al 2003).
Molecules and genes.
Most gain of function changes or mutations in the FSHR were in the transmembrane domains (Desai SS, Roy BS, et al 2013). In vitro these mutations do not alter basal cyclic AMP; however, they broaden or shift the increase of cyclic AMP to lower concentrations of CG and sometimes also to TSH.
Sporadic tumor arising from somatic mutation of FSHR.
Mutation of the FSHR has not been reported in sporadic gonadal tumors.
Variant with autonomous spermatogenesis in males.
Normal spermatogenesis despite undetectable FSH was reported in two unrelated males with germline activating mutation of the FSHR. Undetectable FSH in one was attributed to prior surgery for a pituitary tumor and was from unknown cause in the other (Gromoll J, Simoni M, et al 1996: Casas-González P, Scaglia HE, et al 2012). It is likely that FSH release was also inhibited by oversecretion of inhibin or other factors from the testis.
Familial hyperaldosteronism type IIIA
Clinical.
Familial hyperaldosteronism type IIIA (HAIIIA) (Footnote 1) is rare (Geller DS, Zhang J, et al 2008; Choi M, Scholl UI, et al 2011; Scholl UI, Nelson-Williams C, et al 2012; Scholl UI, Lifton RP 2013; Mulatero P, Monticone S, et al 2013). It is generally recognized at ages 1–7 years as hypokalemia, mild hypertension, and very high aldosterone levels. Treatment with blockade of the aldosterone receptors is unsuccessful; and subtotal adrenalectomy generally is followed by persistence or rapid recurrence. Total adrenalectomy is the preferred treatment.
Adrenal cortex in HAIIIA.
Normal secretion of aldosterone is stimulated by increases of extracellular K+ or by angiotensin II (Spat A, Hunyady L 2004; Bollag WP 2014; Romero CA, Orias M, et al 2015). The renin/angiotensin system is not otherwise covered here. In HAIIIA, the adrenal cortex shows massive hyperplasia that is occasionally micronodular and thst is mainly in the fasciculata, with atrophy in the glomerulosa ((Geller DS, Zhang J, et al 2008; Scholl UI, Lifton RP 2013). This distribution contrasts to the glomerulosa predominant location of normal secretion of aldosterone The cause of this distribution of steroid synthesis is not known, but it might relate to stronger KCNJ5 expression in the normal glomerulosa.
The adrenal enlargement is age-dependent (Scholl UI, Nelson-Williams C, et al 2012); from extrapolation, the hyperplasia might have begun only after birth.
Molecules and genes.
There are more than 80 mammalian genes in the family of potassium channel subunits. Some of the inwardly rectifying K+ channels (thus “Kir”) also function as K+ sensors (Spat A, Hunyady L 2004; Hibino H, Inanobe A, et al 2010). They allow a small outflow or “leak” of K+ from cytoplasm to the exterior, while they restrict external Na+ from traversing inward through its K+-selective pore. Recent studies in aldosteronomas first identified Kir3.4 (encoded by the KCNJ5 gene) as a major K+ channel subunit and a major regulator of aldosterone secretion (Choi M, Scholl UI, et al 2011). Kir3.4 is normally expressed in adrenal glomerulosa, nerve, and muscle (Kokunai Y, Nakata T, et al 2014).
Patients with HAIIIA have germline heterozygous missense mutation of KCNJ5. Most of its inactivating mutations in HAIII or in sporadic aldosteronoma model to within the pore of the K+ selectivity filter (Scholl UI, Nelson-Williams C, et al 2012; Murthy M, Xu S, et al 2014). Most mutations are G151R, T158A, or I157S. A milder familial phenotype has also been recognized recently from 3 different mutations of KCNJ5 that model outside of the K+ selectivity pore (Murthy M, Xu S, et al 2014). All the evaluated germline and somatic KCNJ5 mutations in HAIIIA, HAIIIB, or sporadic aldosteronoma cause a loss of function of Kir3.4 (Scholl UI, Lifton RP 2013). They cause loss of K+ selectivity, and thus increased influx of Na+ through Kir3.4. Some mutations also cause decreased surface expression of Kir3.4 (Cheng CJ, Sung CC, et al 2014).
Another loss of function mutation, restricted to one sequence, KCNJ5 G151E, causes HAIIIB, a different syndrome of hereditary primary hyperaldosteronism, with normal adrenocortical size and normal morphology (Scholl UI, Nelson-Williams C, et al 2012; Mulatero P, Tauber P, et al 2012; Marx SJ 2014) (Footnote 1).
Sporadic tumor arising from somatic mutation of KCNJ5.
KCNJ5 is mutated in 40% of sporadic aldosteronomas, moreso in aldosteronoma of women than men, and not in adrenocortical cancers (Scholl UI, Nelson-Williams C, et al 2012; Scholl UI, Lifton RP 2013; Mulatero P, Monticone S, et al 2013). KCNJ5 mutation also has been found selectively in the dominant nodule of sporadic multinodular adrenal glands (Dekkers T, Meer T, et al 2014). Though considered as loss of function mutations, the missense mutations of KCNJ5 have been heterozygous in aldosteronomas, i.e. haploinsufficient or without inactivation of the normal allele (Choi M, Scholl UI, et al 2011).
DISCUSSION:
Broad Themes Among Many Syndromes
Broad theme: Wide range of severity of a clinical feature within a syndrome
I reviewed major features within 5 selected syndromes (Table 1, Table 2). Some of the themes were shared and important but were also shared among many other syndromes. For example, overall clinical severity can cover a broad spectrum. The main determinant of severity of a syndrome is often the sequence of the germline mutation (Vassart G 2010; Hebrant A, van Staveren WCG, et al 2011; Christensen SE, Nissen PH, et al 2011; Murthy M, Xu S, et al 2014).
Table 1.
Some clinical features of hereditary syndromes of primary hyperplasia with hormone excess (See also Table 2).
| Syndrome | Normal serum stimulus of mutant sensor | Hormone over-secreted | Typical early age of onset of hormone excess | Selected comments about expressions |
|---|---|---|---|---|
| NSHPT# | Low Ca++ | PTH | In fetus | Severe defects at birth reflect onset by fetal over-secretion of PTH |
| CNT | TSH | T4, T3 | In fetus | Severe defects at birth reflect onset by fetal over-secretion of iodo-thyronines |
| FMPP | LH | Testosterone | 1-3 yr | Not expressed in female carriers of the mutation |
| OHSS | CG | Estrogens, progestins, cytokines | Pregnant female | CG from the normal placenta stimulates the mutant FSH receptors in the corpus luteum of pregnancy |
| HAIIIA | K+ | Aldosterone | 1 yr | Hyperplasia is in the adrenal fasciculata with atrophy in the adrenal glomerulosa |
Abbreviations: Congenital neonatal thyrotoxicosis CNT; Neonatal severe primary hyperparathyroidism NSHPT; hyperaldosteronism type III HAIII; Familial male-limited precocious puberty FMPP; Ovarian hyperstimulation syndrome OHSS
Table 2.
Features of mutated tissue in hereditary syndromes of primary hyperplasia with hormone excess (See also Table 1).
| Syndrome nodules | Genes and germline mutations @ | Mutated sensor moleule | Tissue over-functioning | Predominat hyperplasia | Progress to or adenomas – – – – – – – – – |
|
|---|---|---|---|---|---|---|
| Sporadic | Germline | |||||
| origin | origin | |||||
| NSHPT# | CASR= | CaS-R | Parathyroid | Yes | Rare | No |
| CNT | TSHR+ | TSH-R | Thyroid follicle | Yes | Yes | Yes |
| FMPP | LHR+ | LH-R | Leydig cell of testis | Yes | Rare | Yes |
| OHSS | FSHR+ | FSH-R | Corpus luteum of pregnancy | Yes | Yes | No |
| HAIIIA | KCNJ5− | Kir3.4 | Adrenal cortex | Yes | Yes | Yes |
Abbreviations: Congenital neonatal thyrotoxicosis CNT; Neonatal severe primary hyperparathyroidism NSHPT; hyperaldosteronism type III HAIII; Familial male-limited precocious puberty FMPP; Ovarian hyperstimulation syndrome OHSS. CaS-R extracellular calcium sensing receptor; TSH-R TSH receptor; LH-R LH receptor; FSH-R FSH receptor; Kir3.4 inward rectifying potassium channel subunit 3.4;.
Mutation types are: − heterozygous loss of function (inactivation); = homozygous loss of function (inactivation); + heterozygous gain of function (activation).
A less frequent determinant of severity is change of gene dosage in the germline. In particular, a double dose of the mutated CASR causes severe expressions in the form of NSHPT, whereas a single dose of the same CASR mutation is expressed far more mildly as FHH (Pollak MR, Brown EM, et al 1993; Pollak MR, Chou Y-H W, et al 1994; Arnold A, Marx SJ 2013).
Broad theme: Wide range of ages at onset
Earlier age of onset and greater severity of expression often go together; the earliest onsets may even be lethal to the embryo. Embryonic lethality was speculated for certain mutations of the TSHR or of the LHR, mainly because those mutations had been found in sporadic tumors but not in a germline (Hebrant A, van Staveren WCG, et al 2011; Boot AM, Lumbroso S, et al 2011).
For some severely affected neonates with either CNT or NSHPT, the syndrome must have started in the fetus with gland hyperplasia and toxicity from a hormone over-secreted. Furthermore, some less severe forms of expression are also likely to have begun in utero with or without recognition of their prenatal onset in utero (Beas FO, Zurbrugg RP, et al 1962; Rosenthal SM, Grumbach MM, et al 1983; Müller J, Gondos B, et al 1998). Still milder forms of expression show later onsets that are usually consistent within a family - during infancy, childhood, or adulthood. Lastly, some of the mildest carrier states have remained occult even during genetic evaluation in an adult (Nishihara E, Chen C-R, et al 2010). The latest onsets probably reflect the mildest forms of expression and the slowest gland enlargement over years.
An exception can be the requirement for late onset. In particular, either of the two syndromes, of OHSS or gestational thyrotoxicosis, can be expressed only in a female and only selectively during the unique window of her pregnancy.
Broad theme: Wide time interval until post-operative recurrence
Post-operative recurrence generally reflects dysfunctioin in residual tissues after surgery (Marx SJ 2013). Each cell in the remnant secretory tissue carries the germline defect; furthermore, remnant cells might have already become over-active (such as predominantly mono-clonal) before the time of surgery.
The average time interval until recurrence after subtotal surgery is another feature that might help to characterize a syndrome. The interval until recurrence of hyperparathyroidism (severe) was 2 and 5 months in two cases of NSHPT (Thompson NW, Carpenter LC, et al 1978; Key L, Thorne M, et al 1990;) (Supplement). This differs from the much longer interval of 12 years until recurrence (mild) for hyperparathyroidism in MEN1 (Rizzoli R, Green J III, et al 1985). And this also differs importantly from the even shorter average recurrence interval of 5 days (for mild hyperparathyroidism) after subtotal parathyroidectomy in FHH; the latter reflects immediate post-operative over-secretion independent of recurrent gland growth (Law WM jr Heath H III 1985; Marx SJ 2014).
Duration of the interval until postoperative recurrence was not documented in detail for the other 4 syndromes herein. It seems likely that hyperplasia from severe mutations expressed early (as in NSHPT or CNT) would recur more rapidly, and that mild mutations would have slower growth of the gland and would recur more slowly or, for some cases, not at all during prolonged followup (Nishihara E, Chen C-R, et al 2010).
Broad theme: Wide relevance of the genes to neoplasia
Any primary or secondary hyperplasia is typically regarded as a polyclonal process (Derwahl M, Studer H 2002; Arnold A 2011; Mete O, Asa SL 2013). However, mono-clonal components may also be inherent parts within hyperplasia. (Arnold A, Brown ME, et al 1995; Diaz-Cano SJ, de Miguel M, et al 2001; Korpershoek E, Petri BJ, et al 2014; Hartmann LC, Degnim AC, et al 2015).
Mutations can also contribute to sporadic cancers in diverse tissues, with any being a likely driver mutation in 0.1–5.0 % of most or all common cancer types (Bamford S, Dawson E, et al 2004; O’Hayre M, JV, Kufeva I, et al 2013). For example, the large intestine from over 1000 cancers tested per gene shows mutations in the following frequencies: CASR 4.5%, TSHR 5.7%, LHR 3.6%, FSHR 2.6%, KCNJ5 1.9% (Bamford S, Dawson E, et al 2004).
Distinguishing Themes Within 5 Hyperplasia Syndromes
Distinguishing theme: Predominant hyperplasia as an endpoint of expression in a gland
Hyperplasia results from an increased rate of cell birth and/or decreased rate of cell death in the gland. The quantitative disturbances of either process have not been analyzed herein, excepting indirectly as contributions by neoplasia-related processes (see below). I assume that predominantly hyperplasia herein is mainly from an increased rate of cell birth.
Beyond being an inclusion criterion herein, this histologic feature of predominant hyperplasia is a self-supporting status and a robust endpoint in a gland. It is not simply a small or brief step towards progression to adenoma or cancer.
Of course, hyperplasia in a hormone-secreting gland is not uncommon in pathology. Predominant hyperplasia is also a fundamental process in many examples of excess. Furthermore, primary or secondary hyperplasia may progress to nodules, adenomas, and occasionally cancers (Arnold A 2011; Qureshi IA, Khabaz MN, et al 2015).
Distinguishing theme: Predominant hyperplasia as the cause of hormone over-secretion
I focus upon the process of hyperplasia and increased gland size, but hyperplasia also has a close relation or coupling with hormone over-secretion. The hyperplastic gland tissue must be the source of over-secreted hormones in these 5 syndromes, because hyperplasia is the predominant and stable tissue type in the gland. Some other hereditary syndromes of primary hormone excess differ from these five insofar as either normal-appearing tissue, or small or large nodules, adenoma, or cancer can predominate in the gland and can be the main source of over-secreted hormone (Supplementary Table S3 and Supplementary Table S4).
The 5 over-secreted hormones in these 5 hyperplasia syndromes represent 3 distinct categories of chemical: steroid, iodo-thyronine, and polypeptide. Their biosynthetic pathways are specific to their chemical structure and to the differentiated cell of secretion for each. Even the mechanisms of final “secretion” or exit from the cell differ among some of the 3 (Spat A, Hunyady L 2004; Rizo J, Sudhof TC 2012; Miot F, Dupuy C 2013).
The partial contributions to total over-secretion of hormone may be divided among broad functions within the mutated and over-secreting cell. First, a high basal release rate may be attributable to increased number of secreting cells. The enlarged gland may be over-secreting even despite a lower than normal secretion rate per cell (Assie G, Libé R, et al 2013). Among the 5 syndromes reviewed here, I estimate that the hyperplastic mass is typically increased over normal by 3–10 fold and is one major if not the principal determinant of the total amount of hormone secretion (Supplementary Table S-2).
Second, there may be increased basal secretion rate per cell (Brown EM, LeBoff MS, et al 1987); this is supported by high basal cyclic AMP level per cell in vitro with activating mutations of the TSH-R or the LH-R.
Third, some part of the over-secretion may be dependent on a mutant protein’s responsivity to its normal extracellular regulator ((Pearce SHS, Bai M, et al 1996). However, among the syndromes here, most of the extracellular ligands of the mutated molecules are down-regulated in serum by the feedbacks in their syndrome (expressed as high Ca++, low TSH, low LH, low FSH, and low K+, respectively; excepting CG, which is not down-regulated).
Distinguishing theme: The causative mutated molecule is a sensor in the plasma membrane
Four of five syndromes examined here are from germline mutation of a gene (CASR, TSHR, LHR, FSHR) that encodes a GPCR in the plasma membrane (Vassart G, Costagliola SG 2011; Lefkowitz RJ 2013), and mediates response to an extracellular ligand. The activating mutations of the GPCR subfamily of receptors for gonadotropins (TSR, LH, FSH) are modeled mainly within their 7 transmembrane loops. The CASR mutations in NSHPT cause nactivation of the CaS-R protein and model mainly to an extracellular domain of the CaS-R. The focus of germline mutations among these four GPCRs reflects that members of this largest of all gene families can sense highly diverse extracellular ligands, that they may transduce to diverse differentiated functions, and that their response may include hyperplasia (Lefkowitz RJ 2013; Katrich V, Cherezov V, et al 2013).
KCNJ5 encodes Kir3.4, a membrane-bound protein that functions as a direct sensor for extracellular K+ (Spat A, Hunyady L 2004; Choi M, Scholl UI, et al 2011); its structure as a membrane channel is not related to the GPCRs (Hibino H, Inanobe A, et al 2010).
Thus, each of the 5 mutated genes in these 5 syndromes encodes a plasma membrane protein that senses an extracellular regulator (O’Hayre M, JV, Kufeva I, et al 2013; Vogelstein B, Papadopoulos N, et al 2013).
Distinguishing theme: Each of 5 mutated sensors regulates a downstream pathway
This review included three mutation-directed pathways, immediately downstream of sensing for a serum factor (Figure 1). These pathways can be grouped narrowly as transducing information from the plasma membrane to an adjacent intracellular messenger. They transduce from plasma membrane G-protein coupled receptor (GPCR) to cyclic AMP, or from GPCR to inositol phosphates, or from plasma membrane K+ channel to cytoplasmic Ca++.
Figure 1.
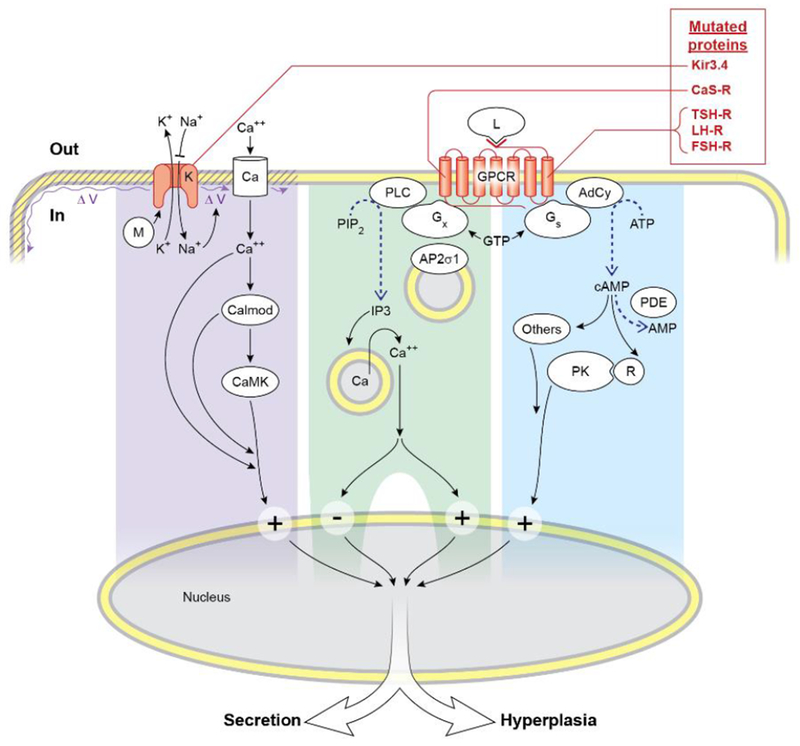
Overview of parts of the signaling pathways of the five mutated proteins in the 5 syndromes of this review. The mutated K+ channel (or Kir3.4) is shown with abnormally decreased ion selectivity of its mutation, so that fluxes of K+ and Na+ are in directions reversed from normal. Mutated proteins causing syndromes of this review are shown in red. Early downstream parts of each of three pathways are highlighted by a different background color. The pathway from Gx divides and includes a large plus or minus sign to illustrate that the CaS-R has opposite downstream effects in the parathyroid cell versus in the C-cell. By mostly unknown mechanisms, the four pathways likely converge downstream to regulate secretion and hyperplasia.
Abbreviations: G-protein coupled receptor GPCR; Calmodulin Calmod; Calcium-calmodulin dependent protein kinase CaMK; Ion channels K or Ca; Phospholipase C PLC; Sigma subunit of AP2S1 AP2σ1; Adenylyl cyclase Ad Cy; Ligand L; Heterotrimeric stimulatory G-protein Gs; Heterotrimeric G-protein other than Gs Gx; GTP-binding protein modulator of Kir3.4 M; Phosphatidyl inositol diphosphate PIP2; Inositol triphosphate IP3; Phospho-diesterase PDE; Protein kinase A catalytic subunit A PK; Regulatory inhibitory subunit 1A of PKA R; Voltage of plasma membrane V.
The CaS-R on the parathyroid cell transduces hypercalcemia through Gq and/or Ga11, to activate phospholipase C, and thereby to raise inositol phosphates and to mobilize Ca++ from stores in the cytoplasm (Wettschureck N, Lee E, et al 2007; Conigrave AD, Ward DT 2013; Brown EM 2013; Hillenbrand M, Schori C, et al 2015; Cocco L, Follo MY, et al 2015; Nesbit MA, Hannan FM, et al. 2013B ). Lowering of extracellular Ca++ or loss of function mutations of the CaS-R as in NSHPT stimulate secretion and hyperplasia in the parathyroid cells.
Unlike the effect of rising Ca++ to inhibit secretion of PTH, a rise of extracellular Ca++ acts through the CaS-R of thyroidal parafollicular C-cells to stimulate secretion of calcitonin (Garrett JE, Tamir H, et al 1995; McGehee DS, Aldersberg M, et al 1997). It may not, however, cause hyperplasia of C-cells (Conte-Devolx B, Morlet-Barla N, et al 2010). The C-cell may also respond to Ca++ in part through a plasma membrane Ca++ channel (Kantham L, Quinn SJ, et al 2009). Overall, parathyroid cells and C-cells have contrasting hormone-secretory responses to serum calcium, with contrasts that are transduced at unknown steps.
The normal TSH-R transduces both secretion and growth in the thyrocyte mainly through Gsa and rise of cyclic AMP (Vassart G 2010; Kleinau G, Neumann S, et al 2013). However, TSH-R transduction of secretion and hyperplasia has also been reported through Gq/G11 (New DC, Wong YH 2007: Kero J, Ahmed K, et al. 2007). Similarly, the normal LH-R transduces a rise of serum LH mainly through Gs and cyclic AMP. D578H, the most severe activating human mutation of LHR, caused in vitro not only much higher basal cyclic AMP than other mutations but also much higher inositol phosphates, suggesting transduction also through a G-protein other than Gs (Boot AM, Lumbroso S, et al 2011). Lastly, the normal FSH-R transduces mainly through Gs and cyclic AMP. However, it also can transduce through different G-protein(s), causing rises of inositol phosphates and Ca++ in cytoplasm (Thomas RM, Nechamen CA, et al 2011). Normally cyclic AMP has potential to transduce to any of three major signaling pathways that start with one among the following molecules: protein kinase A, the guanine nucleotide exchange factor EPAC, and ion channels (Sassone-Corsi P 2012).
Kir3.4 is one of 4 G-protein-coupled Inward Rectifying K+ channels; therefore, it is also termed GIRK4. It can bind directly to a cytoplasmic modulator, such as the beta-gamma portion of a heterotrimeric G-protein or RGS (regulator of G-protein signaling) (Wickman KD, Iñiguez-Lluhl JA, et al 1994; Zhou H, Chisari M, et al 2012; Luscher C, Slesinger PA 2010; Velarde-Miranda C, Gomez-Sanchez EP, et al 2013; Bollag WP 2014). On the aldosterone cell, rise of extracellular K+ or influx of Na+ depolarizes the plasma membrane; this opens a plasma membrane Ca++ channel as a major transduction step towards aldosterone secretion (Velarde-Miranda C, Gomez-Sanchez EP, et al 2013; Bollag WP 2014). Effectors downstream from the rises of cytoplasmic Ca++ in the aldosterone-secreting cell may include calmodulin and several calcium calmodulin-dependent kinases (Spat A, Hunyady L 2004; Bollag WP 2014).
The mechanisms for sharing predominating hyperplasia are not known and represent an important topic for future studies. For e xample, hyperplasia can have unique features in diverse other settings, such as normal expansion of cartilage in the embryo or reversible development of the breast for lactation, (Hassiotou F, Geddes D 2013; Kozhemyakina E, Lasser AB, et al 2015).
Distinguishing themes: features within the hyperplasia group versus in 2 other groups with lower or higher histologic grade
Defining three groups for comparisons:
These 5 syndromes (the “hyperplasia” group) have important shared features that suggest both universal and also distinguishing aspects in their pathophysiology. Insights about their distinctinguishing aspecs can derive from comparison to different groups with hereditary primary excess of hormones (Supplementary Table S-1; and Tables S-3 – S-6). We compare hyperplasias to another group with primary and predominant over-secretion of hormones but little or no hyperplasia (abbreviated as the “over-secretion” group) (Marx SJ 2014) (Supplementary Table S-4 & S-6; an example is FHH. A second comparison group is dominated by adenomas or cancers (the “neoplasia” group) (Supplementary Table S-1, S-3, S-5 & S-6); an example is MEN1. The three groups form a continuum among three distinct histologic grades. Furthermore, the variables for comparison are organized for a yes or no entry, with the result that all comparisons are from simple integers.
Progression to neoplasia:
Hyperplasia sometimes (5 of 5 syndromes) progresses to nodules or other neoplastic features (Table 2) but oversecretion rarely does (1 of 6 syndromes; P<0.02; Table S-6B). This supports the observations many other primary hyperplastic tissues (whether or not they over-secrete a hormone) have an increased likelihood of progression to neoplasic stages (Gorgoulis VG, Vassiliou LV, et al Nature. 2005; Barcellos-Hoff ME, Lyden D, et al 2013).
Expression as neoplasia in sporadic tissue:
Capability to cause sporadic neoplasia is expressed by some hyperplasias (3 of 5) (Supplementary Table S-7) and by some oversecretions (1 of 5; P=0.53).. Neoplasias do this more consistently than hyperplasias (18 of 19; P<0.05).
Expression as tumor multiplicity:
In the hyperplasia grouip, an underlying germline mutation is rarely expressed as tumor in multiple tissues (0 of 5); in the neoplasia group, multiplicity is expressed in 14 of 20 syndromes (the two frequencies differ; P<0.02; Table S-6C).
Focus of mutant functions is among plasma membrane sensors:
Another difference for the hyperplasia group is the focus of all mutant gene functions among plasma membrane sensors (5 of 5 functions) versus one such gene function (RET) in the neoplasia group (1 of 18; P<0.0002; Table S-6E)). The functions of most causative genes in the neoplasia group are incompletely understood. It seems that functions of the mutated genes in the neoplasia group are mainly outside of the sensor-related cluster, and they probably contribute to hyperplasia and cancer through other pathways (Agarwal SK, Mateo C, et al 2009; Huang J, Gurung B, et al 2012; Vogelstein B, Papadopoulos N, et al 2013; Mulligan LM 2014; Dahia PM 2014).
Major clinical and molecular themes in the hyperplasia group help to distinguish it from the two other comparison groups and have strengthenedits robust identity.
The consistent sensor focus of the five mutant proteins on the background of this robust identity suggests that this abnormal biochemical focus is the cause of predominant hyperplasia. Since this mutated sensor might enhance cell numbers exponentialy, there might be limits to the capacity of cell numbers to expand. Alternately I speculate that one or more members of a downregulating network is expressed as a counterbalance. The arrestins are but one well-identified downregulating system for GPCRs, but others could be operational herein (Luttrell LM, Gesty-Palmer D 2010). It will be interesting to explore further the relation between these membrane sensors and predominant hyperplasia.
Supplementary Material
ACKNOWLEDGMENTS
I thank members of the NIH Inter-institute Endocrine Training Program for stimulating discussions. I thank Constantine Stratakis for comments about this manuscript. I thank Bin Guan for advice about mutation databases. I thank Elizabeth Wright (Biostatistics Program, NIDDK) for advice about statistics. I thank Ethan Tyler (Medical Arts Department, NIH) for work on the figure.
FUNDING
Support was from the Intramural Research Programs of the NIH, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Institute of Child Health and Human Development. No support was from grants.
Abbreviations:
- CaS-R
calcium sensing receptor
- GPCR
G-protein coupled receptor
- CNT
congenital neonatal thyrotoxicosis
- FHH
familial hypocalciuric hypercalcemia
- FMPP
familial male-limited precocious puberty
- HAIII
hyperaldosteronism type III
- MAH
micronodular adrenocortical hyperplasia
- MEN
multiple endocrine neoplasia
- NSHPT
neonatal severe primary hyperparathyroidism
- OHSS
hereditaryovarian hyperstimulation syndrome
- PPNAD
primary pigmented micro-nodular adrenocortical disease
Footnotes
Footnote 1.: Hyperaldosteronism type IIIA (HAIIIA) is HAIII with severe adrenocortical hyperplasia from germline mutations of KCNJ5, such as frequently G151R but notably excluding G151E. HAIIIB is HAIII with little or no adrenocortical hyperplasia, only from germline KCNJ5 G151E (Scholl UI, Nelson-Williams C, et al 2012). Several mutations of KCNJ5 cause 50% of sporadic aldosteronomas, but G151E never does this (Scholl UI, Lifton RP 2013; Mulatero P, Monticone S, et al 2013).
DECLARATION OF INTEREST
The author declares that he has no conflict of interest.
REFERENCES
- Agarwal SK, Mateo C, Marx SJ 2009. Rare germline mutations in cyclin-dependent kinase inhibitor genes in MEN1 and related states. J Clin Endocrinol Metab/94 1826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albores-Saavedra J, Krueger JE 2001. C-cell hyperplasia and medullary thyroid microcarcinoma. Endocr Pathol/12 365–377. [DOI] [PubMed] [Google Scholar]
- Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M Lyssikatos C, Horvath A, Carney JA, Stratakis CA 2012. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab/97 351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A 2011. Pathogenesis of endocrine tumors In Williams Textbook of Endocrinology, edn 12, pp 1719–1727, Eds Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Philadelphia: Elsevier/Saunders. [Google Scholar]
- Arnold A, Brown ME, Urena P, Gaz RD, Sarfati E, Drueke TB 1995. Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest/95 2047–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A, Marx SJ 2013. Familial hyperparathyroidism (Including MEN, FHH, and HPT-JT) Primer on the Metabolic Bone Diseases and Mineral Metabolism, edn 8, pp 553–561, Eds Rosen C, Bouillon R, Compston JE, Rosen V. John Wiley & Sons, Inc. [Google Scholar]
- Arturi F, Chiefari E, Tumino S, Russo D, Squatrito S, Chazenbalk G, Persani L, Rapoport B, Filetti S 2002. Similarities and differences in the phenotype of members of an Italian family with hereditary non-autoimmune hyperthyroidism associated with an activating TSH receptor germline mutation. J Endocrinol Invest/25 696–701. [DOI] [PubMed] [Google Scholar]
- Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, Mannelli M, Mantero F, Papathomas T, De Krijger R, Tabarin A, Kerlan V, Baudin E, Tissier F, Dousset B, Groussin L, Amar L, Clauser E, Bertagna X, Ragazzon B, Beuschlein F, Libé R, de Reyniès A, Bertherat J 2014. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet /46 607–12. [DOI] [PubMed] [Google Scholar]
- Assie G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, Barreau O, Lefèvre L, Sibony M, Guignat L, Rodriguez S, Perlemoine K, René-Corail F, Letourneur F, Trabulsi B, Poussier A, Chabbert-Buffet N, Borson-Chazot F, Groussin L, Bertagna X, Stratakis CA, Ragazzon B, Bertherat J 2013. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med /369 2105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai M, Pearce SHS, Kifor O, Trivedi S, Stauffer UG, Thakker RV, Brown EM, Steinmann B 1997. In vivo and invitro characterization of neonatal hyperparathyroidism resulting from a de novo heterozygous mutation of the Ca2+-sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest/99 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R 2004. The COSIC (Catalog of Somatic Mutations in Cancer) database and website. In the Sanger Institute Catalogue of Somatic Mutations in Cancer web site, http://www.sanger.ac.uk/cosmic Brit J Cancer/91 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff ME, Lyden D., Wang TC 2013. .The evolution of the cancer niche during multistage carcinogenesis. Nature Rev Cancer/13 511–518. [DOI] [PubMed] [Google Scholar]
- Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML10, Lidov HG11, Taha H12, Martinez-Lage M13, Santi M14, Storm PB15, Lee JY16, Palmer JN17, Adappa ND18, Scott RM19, Dunn IF20, Laws ER Jr20, Stewart C6, Ligon KL21, Hoang MP5, Van Hummelen P4, Hahn WC22, Louis DN5, Resnick AC15, Kieran MW23, Getz G24, Santagata S25 2014. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet /46 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beas FO, Zurbrugg RP, Liebow SG, Patton RG, Gardner LI 1962. Familial male sexual precocity: Report of the eleventh kindred found, with observations on blood group linkage and urinary C19-steroid excretion. J Clin Endocrinol Metab/22 1095–1102. [DOI] [PubMed] [Google Scholar]
- Beckers A, Aaltonen LA, Daly AF, Karhu A 2013. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev/34 239–7711.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM 2014. Allolio B Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med/370 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertherat J 2006. Carney complex. (CNC) Orphanet J Rare Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA 2003. Molecular and functional analysis of PRKAR1A and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res./63 5308–19. [PubMed] [Google Scholar]
- Blair JW, Carachi R 1991. Neonatal primary hyperparathyroidism – A case report and review of the literature. Eur J Pediatr Surg/1 110–114. [DOI] [PubMed] [Google Scholar]
- Bollag WP 2014. Regulation of aldosterone synthesis and secretion. Compr Physiol/4 1017–1055. [DOI] [PubMed] [Google Scholar]
- Boot AM, Lumbroso S, Verhoef-Post M, Richter-Unruh A, Loojenga LHJ, Funaro A, Beishuizen A, van Maarle A, Drop SLS, Themmen APN 2011. Mutation analysis of the LH receptor gene in Leydig cell adenoma and hyperplasia and functional and biochemical studies of activating mutations of the LH receptor gene. J Clin Endocrinol Metab/96 E1197–E1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachet C, Boros E, Lissens W, Andry G, Martin P, Heinrichs C 2009. Association of parathyroid adenoma and familial hypocalciuric hypercalcemia in a teenager. Eur J Endocrinol/161 207–210. [DOI] [PubMed] [Google Scholar]
- Brown EM 2013. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract & Res Clin Endocrinol Metab/27 333–343. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gardner DG, Brennan MF, Marx SJ, Spiegel AM, Attie MF, Downs RW Jr, Doppman JL, and Aurbach GD 1979. Calcium-regulated PTH release in primary hyperparathyroidism. Studies in vitro with dispersed parathyroid cells. Am J Med/66 923–931. [DOI] [PubMed] [Google Scholar]
- Brown EM, LeBoff MS, Oetting M, Posillico JT, Chen C 1987. Secretory control in normal and abnormal parathyroid tissue. Rec Prog Horm Res/43 337–374. [DOI] [PubMed] [Google Scholar]
- Burski K, Torjussen B, Paulsen Q, Boman H, Bollersley J 2002. Parathyroid adenoma in a subject with familial hypocalciuric hypercalcemia: coincidence or causality? J Clin Endocrinol Metab/87 1015–1016. [DOI] [PubMed] [Google Scholar]
- Ch’ng JL, Kaiser A, Lynn J, Joplin JF 1984. Post-parathyroidectoy restoration of normal calcium homeostasis in neonatal primary hyperparathyroidism. Acta Endocr/105 350–353. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E 2001. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest./107 277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JA, Gaillard RC, Bertherat J, Stratakis CA 2010. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol/34 547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VLW 1985. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine/67 270–283. [DOI] [PubMed] [Google Scholar]
- Casas-González P, Scaglia HE, Pérez-Solís MA, Durand G, Scaglia J, Zariñán T, Dias JA, Reiter E, Ulloa-Aguirre A 2012. Normal testicular function without detectable follicle-stimulating hormone. A novel mutation in the follicle-stimulating hormone receptor gene leading to apparent constitutive activity and impaired agonist-induced desensitization and internalization. Mol Cell Endocrinol/364 71–82. [DOI] [PubMed] [Google Scholar]
- Cetani F, Pinchera A, Pardi E, Cianferotti L, Vignali E, Picone A, Miccoli P, Viacava P, Marcocci C 1999. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Min Res/14 878–882. [DOI] [PubMed] [Google Scholar]
- Cheng CJ, Sung CC, Wu ST, Lin YC, Sytwu HK, Huang CL, Lin SH 2014. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J Clin Endocrinol Metab/EPub ahead of print. [DOI] [PubMed] [Google Scholar]
- Cheung J, Ginter C, Cassidy M, Franklin MC, Rudolph MJ, Robine N, Darnell RB, Hendrickson WA 2015. Structural insights into mis-regulation of protein kinase A in human tumors. Proc Nat Acad Sci (USA)/112 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP 2011. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science/331 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SE, Nissen PH, Vestergaard P, Moselilde L. 2011. Familial hypocalciuric hypercalcemia: a review. Curr Opinion Endocrinol, Diab, Obes/18 359–370. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Ward DT 2013. Calcium-sensing receptor (CASR): pharmacological properties and signaling pathways. Best Pract & Res Cliin Endocrinol Metab/27 315–331. [DOI] [PubMed] [Google Scholar]
- Conte-Devolx B, Morlet-Barla N, Roux F, Sebag F, Henry JF, Niccoli P 2010. Could primary hyperparathyroidism-related hypercalcemia induce hypercalcitoninemia? Horm Res Paediatr/73 372–5. [DOI] [PubMed] [Google Scholar]
- Cocco L, Follo MY, Suh P-G 2015. Phosphoinositide-specific phospholipase C (PI-PLC) in health and disease. J Lipid Res/First published March 27, 2015 Doi:10.1194/jlr.R05794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper L, Wertheimer J, Levey R, Brown E, Leboff M, Wilkinson R, Anast CS 1986. Severe primary hyperparathyroidism in a neonate with two hypercalcemic parents: management with parathyroidectomy and heterotopic autotransplantation. Pediatrics/78 263–268. [PubMed] [Google Scholar]
- Dahia PM 2014. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nature Reviews Cancer /14 108–119. [DOI] [PubMed] [Google Scholar]
- De Crecchio L 1865. Sopra un caso di apprenzi virile in una donna Morgagni/7 154–158. [Google Scholar]
- Dekkers T, Meer T, Lenders JWM, Hermus ARM, Schultze Kool HL, Langerhuijsen JF, Nishimoto K, Ogishima T, Mukai K, Azizan EAB, tops B, Deinum J, Kusters B 2014. Adrenal nodularity and somatic mutatioins in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab/99 E1341–E1351. [DOI] [PubMed] [Google Scholar]
- de Roux N, Polak M, Couet J, Leger J, Czernichow P, Milgrom E, Misrahi M 1996. A neomutation of the thyroid-stimulating hormone receptor in a severe neonatal hyperthyroidism J Clin Endocrinol Metab/81 2023–6. [DOI] [PubMed] [Google Scholar]
- Derwahl M, Studer H 2002. Hyperplasia versus adenoma in endocrine tissue: are they different? Trends Endocrinol Metab/13 23–28. [DOI] [PubMed] [Google Scholar]
- Desai SS, Roy BS, Mahale SD 2013. Mutations and polymorphisms in the FSH receptor: functional implications and human reproduction Reproduction/146 R235–R248. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullié M, Mantero F, Rubin B, Waldmann J, Bartsch DK, Pasquali R, Lohse M, Allolio B, Fassnacht M, Beuschlein F, Reincke M 2014. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study J Clin Endocrinol Metab/99 E2093–100. [DOI] [PubMed] [Google Scholar]
- Diaz-Cano SJ, de Miguel M, Blanes A, Tashjian R, Wolfe HJ 2001. Germline RET 634 mutation positive MEN 2A-related C-cell hyperplasias have genetic features consistent with intraepithelial neoplasia J Clin Endocrinol Metab 3948–57. [DOI] [PubMed] [Google Scholar]
- Egli CA, S M, Grumbach MM, Montalvo JM, Gondos B 1985. Pituitary gonadotropin-independent male-limited autosomal dominant sexual precocity in nine generations: familial testotoxicosis J Pediatr/106 33–40. [DOI] [PubMed] [Google Scholar]
- Erdheim J Abt 1907. III Über Epithelkörperbefundebei Osteomalacie Sitzungsber d kais Akad d Wissensch In Wien, math naturw, edn 116, pp 311–370 Eds Klasse. [Google Scholar]
- Farnebo P, Enberg U, Grimelius L, Backdahl M, Schalling M, Larsson C, Farnebo L-O 1997. Tumor-specific decreased expression of calcium sensing rerceptor messenger ribonucleic acid in sporadic primary hyperparathyroidism J Clin Endocrinol Metab/82 3481–3486. [DOI] [PubMed] [Google Scholar]
- Fiedler K, Ezcurra D 2012. Predicting and preventing ovarian hyperstimulation syndrome (OHSS): the need for individualized not standardized treatment Reprod Biol Endocrinol/10 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, Zuffardi O 2014. PRKACB and Carney complex N Eng J Med /370 1065–1067. [DOI] [PubMed] [Google Scholar]
- Fuqua JS 2013. Treatment and outcome of precocious puberty: an update. J Clin Endocrinol Metab/98 2198–2207. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Tamir H, Kifor O, Simin RT, Rogers KV, Mithal A, Gagel RF, Brown EM 1995. Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology/136 5202–11. [DOI] [PubMed] [Google Scholar]
- Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP 2008. A novel form of human Mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab/93 3117–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour JR, Martin WJ 1937. The weight of the parathyroid glands. J Pathol Bacteriol/34 431–462. [Google Scholar]
- Goldbloom RB, Gillis DDA, Prasad M 1972. Hereditary parathyroid hyperplasia: A surgical emergency of early infancy. Pediatr/49 514–523. [PubMed] [Google Scholar]
- Goltzman D and Hendy GN 2015. The calcium-sensing receptor in bone – mechanistic and therapeutic insights. Nat Rev Endocrinol /advance online publication 10 March 2015 doi:10.1038/nrendo.2015.30. [DOI] [PubMed] [Google Scholar]
- Gondos B, Egli CA, Rosenthal SM, Grumbach MM 1985. Testicular changes in gonadotropin-independent familial male sexual precocity. Familial testotoxicosis Arch Pathol Lab Med /109 990–995. [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature/434 907–13. [DOI] [PubMed] [Google Scholar]
- Gossage L, Eisen T & Maher ER 2015. VHL, the story of a tumour suppressor gene. Nat Rev Cancer/15; 55–64. [DOI] [PubMed] [Google Scholar]
- Gozu HI, Lublinghoff J, Bircan R, Paschke R 2010. Genetics and phenomics of inherited and sporadic non-autoimmune hyperthyroidism. Mol Cell Endocrinol/322 125–134. [DOI] [PubMed] [Google Scholar]
- Grantmyre EB 1973. Roentgenographic features of “primary” nyperparathyroidism in infancy. J de L’Associat Canad des Radiol/24 257–260. [PubMed] [Google Scholar]
- Grogan RH, Pacak K, Pasche L, Huynh TT, Greco RS 2011. Bilateral adrenal medullary hyperplasia associated with an SDHB mutation. J Clin Oncol/29 e200–2. [DOI] [PubMed] [Google Scholar]
- Gromoll J, Simoni M, Nieschlag E 1996. An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab/81 1367–1370. [DOI] [PubMed] [Google Scholar]
- Hannan FM, Nesbit MA, Christie PT, Lissens W, Van der Schueren B, Bex M, Bouillon R, Thakker RV 2010. A homozygous inactivating calcium-sensing receptor mutation, Pro339Thr, is associated with isolated primary hyperparathyroidism: correlation between location of mutations and severity of hypercalcaemia. Clin Endocrinol (Oxf)/73 715–22. [DOI] [PubMed] [Google Scholar]
- Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, Harding B, Fratter C, Rust N, Christie PT, Turner JJO, Lemos MC, Bowl MR, Bouillon R, Brain C, Bridges N, Burren C, Connell JM, Jung H, Marks E, McCredie DM, Mughal Z, Rodda C, Tollefsen S, Brown EM, Yang JY, Thakker RV 2012. Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence of clustering of extracellular domain mutations of calcium-binding sites. Hum Molec Genet/21 2767–2778. [DOI] [PubMed] [Google Scholar]
- Hartmann LC, Degnim AC, Santen RJ, Dupont WD, Ghosh K 2015. Atypical hyperplasia of the breast — Risk assessment and management options. N Engl J Med/372 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassiotou F, Geddes D 2013. Anatomy of the human mammary gland. Clin anatom /26 29–48. [DOI] [PubMed] [Google Scholar]
- Hebrant A, van Staveren WCG, Maenhaut C, Dumont JE,, Leclere J 2011. Genetic hyperthyroidism: hyperthyroidism due to activating TSHR mutations. Eur J Endocrinol/164 1–9. [DOI] [PubMed] [Google Scholar]
- Henry RK, Keil MF, Stratakis CA, Fechner PY 2010. June Cushing’s syndrome secondary to isolated micronodular adrenocortical disease (iMAD) associated with rapid onset weight gain and negative abdominal MRI findings in a 3 year old male. J Pediatr Endocrinol Metab / 23(6) 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y 2010. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev./90 291–366. [DOI] [PubMed] [Google Scholar]
- Hillenbrand M, Schori C, Schöppe J, Plückthun A 2015. Comprehensive analysis of heterotrimeric G-protein complex diversity and their interactions with GPCRs in solution. Proc Natl Acad Sci U S A/112 E1181–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman DA, Scriver CR, Pedvis S, Shragovitch I 1964. Neonatal familial primary hyperparathyroidism. N Eng J Med/270 483–488. [DOI] [PubMed] [Google Scholar]
- Hirsch A 1885. Handbook of geographical and historical pathology: Volume II. London: New Sydenham Society; 121–202. (Translated from the Second German Edition of 1883) [Google Scholar]
- Hong H, Patonay B, Finley J 2011. Unusual reticulin staining pattern in a well-differentiated hepatocellular carcinoma. Diagnostic Pathol /6 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, Bell RS, Jüppner H, Andrulis IL, Wunder JS, Alman BA 2002. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet/30 306–10. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E 2015. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res / 43(Database issue) D512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libè R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA 2006. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet/38 794–800. [DOI] [PubMed] [Google Scholar]
- Horvath A, Mericq V, Stratakis CA 2008. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med/358 750–2. [DOI] [PubMed] [Google Scholar]
- Hosokawa Y, Pollak MR, Brown EM, Arnold A.1995. Mutational analysis of the extracellular Ca2+-sensing receptor gene in human parathyroid tumors. J Clin Endocrinol Metab/80 3107–3110. [DOI] [PubMed] [Google Scholar]
- Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M 2012. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature/482 542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Dias JA, He X 2014. Structural biology of glycoprotein hormones and their receptors: insights to signaling. Mol Cell Endocrinol/382 424–51. [DOI] [PubMed] [Google Scholar]
- Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, Pollak MR, Goltzman D, Brown EM 2009. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab/297 E915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrich V, Cherezov V, Stevens RC 1990. Structure-function of the G protein-coupled receptor superfamily. Ann Rev Pharmacol Toxicol/53 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kero J, Ahmed K, Wettschureck N, Tunaru S, Wintermantel T, Greiner E, Schütz G, Offermanns S 2007. Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J Clin Invest/117 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key L, Thorne M, Pitzer B, Volberg F, Turner C 1990. Management of neonatal hyperparathyroidism with parathyroidectomy and autotransplantation. J Pediatr/116 923–926. [DOI] [PubMed] [Google Scholar]
- Kifor O, Moore FD, Wang P, Goldstein M, Vassilev P, Kifor I, Hebert SC, Brown EM 1996. Reduced immunostaining for the extracellular Ca2+ sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab/81 1598–1600. [DOI] [PubMed] [Google Scholar]
- Kleinau G, Neumann S, Gruters A, Krude H, Beiberman H 2013. Novel insights on thyroid stimulating receptor signal transduction. Endocr Rev/34 691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokunai Y, Nakata T, Furuta M, Sakata S, Kimura H, Aiba T, Yoshinaga M, Osaki Y, Nakamori M, Itoh H, Sato T, Kubota T, Kadota K, Shindo K, Mochizuki H, Shimizu W, Horie M, Okamura Y, Ohno K, Takahashi MP 2014. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology/82 1058–64. [DOI] [PubMed] [Google Scholar]
- Kopp P, van Sande J, Parma J, Duprez L, Gerber H, Joss E,. Jameson JL, Rodd C 1995. Congenital hyperthyroidism caused by a mutation in the thyrotropin-receptor gene. N Eng J Med/332 150–154. [DOI] [PubMed] [Google Scholar]
- Korpershoek E, Petri BJ, Post E, van Eijck CHJ, Oldenburg RA, Belt EJT, de Herder WW, de Krijger RR, Dinjens WNM 2014. Adrenal medullary hyperplasia is a precursor lesion for pheochromocytoma in MEN2 syndrome. Neoplasia/16 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozhemyakina E, Lasser AB, Zelzer E 2015. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development/142 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Abbas AK, Fausto N, Aster JC 2010. Cellular responses to stress, toxins, and insults: Adaptation, injury, and death In Robbins and Cotran Pathologic Basis of Disease, edn 8, pp 3–42, Eds Kumar V, Abbas AK, Fausto N, Aster JC. Philadelphia: Saunders/Elsevier. [Google Scholar]
- Law WM jr 1985. Heath H III Familial benign hypercalcemia (hypocalciuric hypercalcemia): clinical and pathologenetic studies in 21 families. Ann Int Med/102 511–519. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ 2013. A brief history of the G-protein coupled receptors (Nobel Lecture). Angew Chem Int /52 6367–637. [DOI] [PubMed] [Google Scholar]
- Leitman SA, Tenenbaum-Rakover Y, Jap TS, Yi-Chi W, De-Ming Y, Ding C, Kussiny N, Levine MA 2009. A novel loss –of-function mutation, Gln459Arg, of the calcium-sensing receptor gene associated with apparent autosomal recessive inheritance of familial hypocalciuric hypercalcemia. J Clin Endocrinol Metab/94 4372–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschek EW, Chan W-Y, Diasmond DA, Kaefer M, Jones J, Barnes KM, Cutler GB 2001. Nodular Leydig cell hyperplasia in a boy with familial male-limited precocious puberty. J Pediatr/138 949–951. [DOI] [PubMed] [Google Scholar]
- Lodish MB, Yuan B, Levy I, Braunstein GD, Lyssikatos C, Salpea P, Szarek E, Karageorgiadis AS, Belyavskaya E, Raygada M, Faucz FR, Izatt L, Brain C, Gardner J, Quezado M, Carney JA, Lupski JR, Stratakis CA 2015. Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: molecular mechanisms and clinical presentations. Eur J Endocrinol/172 803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, Libé R, Bram Z, Groussin L, Caron P, Tabarin A, Grunenberger F, Christin-Maitre S, Bertagna X, Kuhn JM, Anouar Y, Bertherat J, Lefebvre H 2013. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. New Eng J Med/369 2115–25. [DOI] [PubMed] [Google Scholar]
- Ludgate M, Gire V, Ajjan R, Weetman A, Ivan M, Wynford-Thomas D 1998. Contrasting effects of activating mutations of GaS and the thyrotropin receptor on proliferation and differentiation of thyroid follicular cells. Oncogene/18 4798–4807. [DOI] [PubMed] [Google Scholar]
- Luscher C, Slesinger PA 2010. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci/11 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Gesty-Palmer D. 2010. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharm Rev 62:305–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz P, Kane O, Pfrsdorff A, Seiller F, Sauvage P, Levy JM 1986. Neonatal primary hyperparathyroidism: Total parathyroidectomy with autotransplantation of cryopreserved parathyroid tissue. Acta Paed Scand/75 179–182. [DOI] [PubMed] [Google Scholar]
- See comment in PubMed Commons belowMachens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, van Vroonhoven TJ, Roeher HD, Wahl RA, Lamesch P, Raue F, Conte-Devolx B, Dralle H 2003. European Multiple Endocrine Neoplasia (EUROMEN) Study Group. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med/349 1517–25. [DOI] [PubMed] [Google Scholar]
- Marx SJ 2013. Multiplicity of hormone-secreting tumors: common themes about development, expression, and management. J Clin Endocrinol Metab/98 3139–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SJ 2014. Uncoupling of secretion from size in some hormone secretory tissues. J Clin Endocrinol Metab/99 4051–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SJ, Attie MF, Spiegel AM, Levine MA, Lasker RD, and Fox M 1982. An association between neonatal severe primary hyperparathyroidism and familial hypocalciuric hypercalcemia in three kindreds. N Engl J Med/306 257–264. [DOI] [PubMed] [Google Scholar]
- Marx SJ, Fraser D, and Rapoport A 1985. Familial hypocalciuric hypercalcemia: Mild expression of the gene in heterozygotes and severe expression in homozygotes. Am J Medicine/78 15–22. [DOI] [PubMed] [Google Scholar]
- Marx SJ and Wells SA Jr 2011. Multiple Endocrine Neoplasia, In Williams Textbook of Endocrinology, edn 13, pp 1728–67. Eds Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Philadelphia: Elsevier Saunders. [Google Scholar]
- McGehee DS, Aldersberg M, Liu KP, Hsuing S, Heath MJ, Tamir H 1997. Mechanism of extracellular Ca2+ receptor-stimulated hormone release from sheep thyroid parafollicular cells. J Physiol/502 ( Pt 1) 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SR, Narayan P 2013. Precocious puberty and Leydig cell hyperplasia in male mice with a gain of function mutation in the LH receptor gene. Endocrinology/154 3900–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meduri G, Bachelot A, Cocca MP. Vasseur C, Rodien P, Kuttenn F, Touraine P, Misrahi M 2008. Molecular pathology of the FSH receptor: New insights into FSH physiology. Moll Cell Endocrinol /282 130–142. [DOI] [PubMed] [Google Scholar]
- Mete O, Asa SL 2013. Precursor lesions of endocrine system neoplasms. Pathology/45 316–330. [DOI] [PubMed] [Google Scholar]
- Mete O, Tischler AS, de Krijger R, McNicol AM, Eisenhofer G, Pacak K, Ezzat S, Asa SL 2014. Protocol for the examination of specimens from patients with pheochromocytomas and extra-adrenal paragangliomas. Arch Pathol Lab Med/138 182–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G, Davis J, Shatzen E, Colloton M, Martin D, Henley CM 2011. Cinacalcet HCl prevents development of parathyroid gland hyperplasia and reverses established parathyroid gland hyperp;asia in a rodent model of CKD. Nephrol Dial Transplant /0 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miot F, Dupuy C, Dumont JE, rousset BA 2013. Thyroid hormone synthesis and secretion In Endocrine Education, Chapter 2, www.thyroidmanager.org South Darmouth. [Google Scholar]
- Mulatero P, Monticone S, Rainey WE, Veglio F, Williams TA 2013. Role of KCNJ5 in familial and sporadic primary aldosteronism. Nat Rev Endocrinol/9 104–112. [DOI] [PubMed] [Google Scholar]
- Mulligan LM 2014. RET revisited: expanding the oncogenic portfolio. Nature Rev Cancer/14 173–186. [DOI] [PubMed] [Google Scholar]
- Müller J, Gondos B, Kosugi S, Mori T, A 1998. Severe testotoxicosis phenotype associated with Asp578-->Tyr mutation of the lutrophin/choriogonadotrophin receptor gene. J Med Genet/35 340–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, Fischer E, Tizzani D, Pallauf A, Viola A, Amar L, Williams TA, Strom TM, Graf E, Bandulik S, Penton D, Plouin PF, Warth R, Allolio B, Jeunemaitre X, Veglio F, Reincke M 2012. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension/59 235–40. [DOI] [PubMed] [Google Scholar]
- Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M, O’Shaughnessy KM 2014. Role for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronism. Hypertension/63 783–789. [DOI] [PubMed] [Google Scholar]
- Nesbit MA, Hannan F, Howles SA, Reed AAC, Cranston T, Thakker CE, Gregory L, Rimmer AJ, Rust N, Graham U, Morrison PJ, Hunter SJ, Whyte M, McVean G, Buck D, Thakker R 2013A. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet /45 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Babinski VN, Head RA, Cranston T, Rust N, Hobbs MR, Heath H III, Thakker R 2013B. Mutations affecting G-protein subunit a11 in hypercalcemia and hypocalcemia. N Eng J Med/368 2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DC, Wong YH 2007. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J Mol Signaling/2 http://dx.doc.org/10.1186/1750-2187-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara E, Chen C-R, Higashiyama T, Mizutori-Sasai Y, Ito M, Kubota s, Amino N, Miyauchi A, Rapoport B 2010. Subclinical nonautoimmune hyperthyroidism in a family segregates with thyrotropin receptor mutation with weakly increased constitutive activity. Thyroid/20 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, J V, Kufeva I, Sawiski EW, T M, Seshagiri Somasekar, I, J S 2013. The Emerging Mutational Landscape of G-proteins and G-protein Coupled Receptors in Cancer. Nat Rev Cancer/13 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack SD, Kirschner LS, Pak E, Zhuang Z, Carney JA, Stratakis CA 2000. Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Clin Endocrinol Metab/85 3860–5. [DOI] [PubMed] [Google Scholar]
- Pappioannou VE, Behringer RR 2012. Early embryonic lethality in genetically engineered mice: diagnosis and phenotypic analysis. Vet Pathol/49 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschke R, Niedziela M, Vaidya B, Persani L, Rappoport B, Leclere J 2012. 2012 European Thyroid Association guideline for the management of familial and persistent sporadic non-autoimmune hyperthyroidism caused by thyroid-stimulating hormone receptor germline mutations. Eur Thyroid J/1 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DE 2010. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol/11 9–22. [DOI] [PubMed] [Google Scholar]
- Pearce SHS, Bai M, Kifor O, Brown EM, Thakker RV 1996. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest/98 1860–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak MR, Brown EM, Chou Y-H W, Hebert SC, Marx SJ, Steinman B, Levi T, Seidman CE, Seidman JG 1993. Mutations in the human Ca2+-sensing recptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell/75 1297–1303. [DOI] [PubMed] [Google Scholar]
- Pollak MR, Chou Y-H W, Marx SJ, Steinmann B, Cole DEC, Brandi ML Papapoulos SE, Menko F, Hendy GN, Brown EM, Seidman CE, Seidman JG 1994. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest/93 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Khabaz MN, Baig M, Begum B, Abdelrehaman AS, Hussain MB 2015. Histopathological findings in goiter: A review of 624 thyroidectomies. Neuro Endocrinol Lett /19 36(1). [Epub ahead of print] [PubMed] [Google Scholar]
- Reiter EO, Mauras N, McCormick K, Kulshreshtha B, Amrhein J, de Luca F, O’Brien S, Armstrong J, Melezinkova H 2010. Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testotoxicosis: a phase II, open-label pilot study (BATT). J Pediatr Endocrinol Meta /23 999–1009. [DOI] [PubMed] [Google Scholar]
- Rhone DP 1975. Primary neonatal hyperparathyroidism: Report of a case and review of the literature. Am J Clin Pathol/64 488–499. [DOI] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC 2012. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices- guilty as charged. Ann Rev Cell Dev Biol/28 279–309. [DOI] [PubMed] [Google Scholar]
- Rizzoli R, Green J III, Marx SJ 1985. Primary hyperparathyroidism in familial multiple endocrine neoplasia type I: Long-term followup of serum calcium after parathyroidectomy. Amer J Med/78 467–474. [DOI] [PubMed] [Google Scholar]
- Rodien P, Bremont C, Sanson M-L R, Parma J, van Sande J, Costagliola S, Luton J-P, Vassart G, Duprez L 1998. Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. N Eng J Med/339 1823–1826. [DOI] [PubMed] [Google Scholar]
- Romero CA, Orias M, Weir MR 2015. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol/Published online February 2015 Doi: 10.1038/nrendo.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal SM, Grumbach MM, Kaplan SL 1983. Gonadotropin-independent familial sexual precocity with premature leydig cell and germinal cell maturation (familial testotoxicosis: effect of a potent luteinizing hormone-releasing factor agonist and medroxyprogesterone acetate therapy in four cases. J Clin Endocrinol Metab/57 571–579. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P 2012. The cyclic AMP pathway. Cold Spring Harb Perspect Biol/4 a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Nelson-Williams C, Yue P 2012. et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Nat Acad Sci (USA)/109 2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Lifton RP 2013. New insights into aldosterone-producing adenomas and hereditary aldosteronism: mutations in the K+ channel KCNJ5. Curr Opin Nephrol Hypertens/22 141–147. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, Couch R, Hammer LK, Harley FL, Farhi A, Wang WH, Lifton RP 2012. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Nat Acad Sci (USA) /109 2533–2538 (and Supplement). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker A, Laue L, Kosugi S, Merendino JJ Jr, Minegishi T, Cutler GB Jr. 1993. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature/14 365(6447):652–4. [DOI] [PubMed] [Google Scholar]
- Simmonds CS, Karsenty G, Karaplis A, Kovacs CS 2010. Parathyroid hormone regulates fetal-placental mineral homeostasis. J Bon Min Res/25 594–605. [DOI] [PubMed] [Google Scholar]
- Singhal A, Ostermaier MK, Vishnivetskiy SA, Panneels V, Homan KT, Tesmer JJ, Veprintsev D, Deupi X, Gurevich VV, Schertler GF, Standfuss J 2013. Insights into congenital stationary night blindness based on the structure of G90D rhodopsin. EMBO Rep. 2013/14 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos B, Sperveslage J, Anlauf M, Hoffmeister M, Henopp T, Buch S, Hampe J, Weber A, Hammel P, Couvelard A, Höbling W, Lieb W, Böhm BO, Klöppel G 2015. Glucagon cell hyperplasia and neoplasia with and without glucagon receptor mutations. J Clin Endocrinol Metab/jc20144405 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S 2003. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. N Engl J Med/349 760–6. [DOI] [PubMed] [Google Scholar]
- Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delande B, Zheng L, Pittalugu S et al. 2012. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med/209 2247–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spambalg D, Sharifi N, Elisei R, Gross JL, Medeiros-Neto G, Fagin JA 1996. Structural studies of the thyrotropin receptor and Gs alpha in human thyroid cancers: low prevalence of mutations predicts infrequent involvement in malignant transformation. J Clin Endocrinol Metab/81 3898–901. [DOI] [PubMed] [Google Scholar]
- Spat A, Hunyady L 2004. Control of aldosterone secretion: a model for convergence in cellular signaling paathways. Physiol Rev/84 489–539. [DOI] [PubMed] [Google Scholar]
- Stanley CA and De Leon DD 2012. Eds. Monogenic Hyperinsulinemic Hypoglycemia Disorders. Karger Basel. [Google Scholar]
- Steinmann B, Gnehm HE, Rao VH, Kind HP, Prader A 1984. Neonatal severe primary hyperparathyroidism and alkaptonuria in a boy born to related parents with familial hypocalciuric hypercalcemia. Helv Paediatr Acta/39 171–86. [PubMed] [Google Scholar]
- Stocco C, Telleria C, Gibori G 2007. The molecular control of corpus luteum formation, function, and regression. Endoc/28 117–149. [DOI] [PubMed] [Google Scholar]
- Stratakis CA 2008. Cushing syndrome caused by adrenocortical tumors and hyperplasia (corticotrophin-independent Cushing syndrome). Endocr Dev/13 117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA 2014. E pluribus unum? The main protein kinase A subunit (PRKACA), a likely oncogene, and cortisol-producing tumors. J Clin Endocrinol Metab/99 3629–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA, Kirschner LS, Carney JA 2001. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab/86 4041–6. [DOI] [PubMed] [Google Scholar]
- Supornsilchai V, Sahakitrungruang T, Wongjitrat N, Wacharasindhru S, Suphapeetiporn K, Shotelersul V 2009. Expanding clinical spectrum of non-autoimmune hyperthyroidism due to an activating germline mutation, p.M435T, in the thyrotropin receptor gene. Clin Endocrinol/70 623–628. [DOI] [PubMed] [Google Scholar]
- Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, Seminara SB, Mendonca BB, Kaiser UB, Latronico AC 2008. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med/358 709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themmen APN 2005. An update of the pathophysiology of gonadotrophin subunit and receptor gene mutations and polymorphisms. Reproduction/130 263–274. [DOI] [PubMed] [Google Scholar]
- Thomas RM, Nechamen CA, Mazurkiewicz JE, Ulloa-Aguirre A, Dias JA 2011. The adapter protein APPL1 links FSH receptor to inositol 1,4,5-trisphosphate production and is implicated in intracellular Ca(2+) mobilization. Endocrinology/152 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NW, Carpenter LC, Nishiyama RH 1978. Hereditary neonatal hyperparathyroidism. Arch Surg/113 100–103. [DOI] [PubMed] [Google Scholar]
- Toledo SP, Lourenço DM Jr, Sekiya T, Lucon AM, Baena ME, Castro CC, Bortolotto LA, Zerbini MC, Siqueira SA, Toledo RA, Dahia PL 2014. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J Clin Endocrinol Metab/100 E308–18. [DOI] [PubMed] [Google Scholar]
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, Castermans E, Villa C, Dimopoulos A, Chittiboina P, Xekouki P, Shah N, Metzger D, Lysy PA, Ferrante E, Strebkova N, Mazerkina N, Zatelli MC, Lodish M, Horvath A, de Alexandre RB, Manning AD, Levy I, Keil MF, Sierra Mde L, Palmeira L, Coppieters W, Georges M, Naves LA, Jamar M, Bours V, Wu TJ, Choong CS, Bertherat J, Chanson P, Kamenický P, Farrell WE, Barlier A, Quezado M, Bjelobaba I, Stojilkovic SS, Wess J, Costanzi S, Liu P, Lupski JR, Beckers A, Stratakis CA 2014. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med/371 2363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassart G 2010. TSH receptor mutations and diseases. In De Groot LJ, ed www.thyroidmanager.org, Endocrine Education Inc, South Darmouth, MA: 02748. [Google Scholar]
- Vassart G, Costagliola S 2011. G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol/7 362–37. [DOI] [PubMed] [Google Scholar]
- Vasseur C, Rodien P, Beau I, Desroches A, Gérard C, de Poncheville L, Chaplot S, Savagner F, Croué A, Mathieu E, Lahlou N, Descamps P, Misrahi M 2003. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. N Engl J Med/349 753–9. [DOI] [PubMed] [Google Scholar]
- Velarde-Miranda C, Gomez-Sanchez EP, Gomez-Sanchez CE 2013. Regulation of aldosterone biosynthesis by the Kir3.4 (KCNJ5) potassium channel. Clin Exp Pharmacol Physiol/40 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzosi D, Libé R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, René-Corail F, Ragazzon B, Stratakis CA, Vandecasteele G, Bertherat J 2012. Phosphodiesterase 11A (PDE11A) gene defects in patients with ACTH-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab/97 E2063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See comment in PubMed Commons belowVilla C, Lagonigro MS, Magri F, Koziak M, Jaffrain-Rea ML, Brauner R, Bouligand J, Junier MP, Di Rocco F, Sainte-Rose C, Beckers A, Roux FX, Daly AF, Chiovato L 2011. Hyperplasia-adenoma sequence in pituitary tumorigenesis related to aryl hydrocarbon receptor interacting protein gene mutation. Endocr Relat Cancer/18 347–56. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velulescu VE, Zhou S, Diaz LA, Kinzler KW 2013. Cancer genome landscapes. Science/339 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM 1991. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med/325 1688–95. [DOI] [PubMed] [Google Scholar]
- Welander J, Larsson C, Bäckdahl M, Hareni N, Sivlér T, Brauckhoff M, Söderkvist P, Gimm O 2012. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet/21 5406–16. [DOI] [PubMed] [Google Scholar]
- Wells SA Jr, Pacini F, Robinson BG, Santoro M 2013. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab/98 3149–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettschureck N, Lee E, Libutti SK, Offermanns S, Robey PG, Spiegel AM 2007. Parathyroid-specific double knockout of Gq and G11 alpha-subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+ -sensing receptor. Mol Endocrinol/21 274–280. [DOI] [PubMed] [Google Scholar]
- Wickman KD, Iñiguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE 1994. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature/368(6468) 255–7. [DOI] [PubMed] [Google Scholar]
- Yabuta T, Miyauchi A, Onoue H, Yoshida h, Hirokawa M, Amino N 2009. a patient with primary hyperparathyroidism associated with familial hypocalciuric hypercalcemia induced by a novel germlineCASR gene mutation. Asian J Surg/32 118–122. [DOI] [PubMed] [Google Scholar]
- Yamauchi M, Sugimoto T, Yamaguchi T, Yano S, Wang J, Bai M, Brown EM, Chihara K 2002. Familial hypocalciuric hypercalcemia caused by an R648stop mutation in the calcium-sensing receptor gene. J Bone Min Res/17 2174–2182112.5. [DOI] [PubMed] [Google Scholar]
- Zenaty D, Aigrain Y, Peuchmaur M, Philippe-Chomette P, Baumann C, Cornelis F, Hugot JP, Chevenne D, Barbu V, Guillausseau PJ, Schlumberger M, Carel JC, Travagli JP, Léger J 2009. Medullary thyroid carcinoma identified within the first year of life in children with hereditary multiple endocrine neoplasia type 2A (codon 634) and 2B. Eur J Endocrinol/160 807–13. [DOI] [PubMed] [Google Scholar]
- Zhou H, Chisari M, Raehal KM 2012. GIRK channel modulation by assembly with allosterically regulated RGS proteins. Proc Nat Acad Sci (USA) / 109 19977–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.