

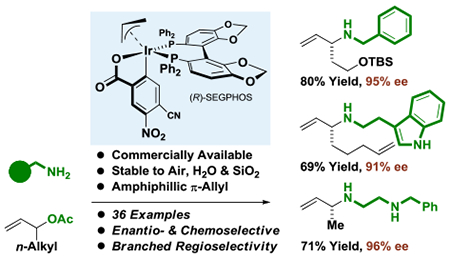
Abstract
The first examples of amphiphilic reactivity in the context of enantioselective catalysis are described. Commercially available π-allyliridium C,O-benzoates, which are stable to air, water and SiO2 chromatographically, and are well known to catalyze allyl acetate-mediated carbonyl allylation, are now shown to catalyze highly chemo-, regio- and enantioselective substitutions of branched allylic acetates bearing linear alkyl groups with primary amines.
Graphical Abstract
Since the discovery of the Tsuji-Trost reaction,1 diverse catalytic systems for metal catalyzed allylic substitution have emerged.2 Among the many useful transformations based on this pattern of reactivity, enantioselective iridium catalyzed aminations figure prominently.3–11 Largely due to the pioneering work of Takeuchi,3,4 Helmchen,5,6 Hartwig,7,8 Carreira9 and You,10,11 a broad range of amine nucleophiles can now be accommodated in highly regio- and enantioselective reactions of linear or branched allylic acetates (and in certain cases allylic alcohols).3–11 Two classes of iridium catalysts may be distinguished on the basis of their ability to promote regio- and enantioselective reactions of either linear or branched allylic acetates (Figure 1). For type I catalysts, linear allyl proelectrophiles are used under basic conditions. For type II catalysts, branched allyl proelectrophiles are used under acidic conditions. While type I catalysts are modified by one phosphoramidite and one external diene ligand, type II catalysts incorporate an internal mono-olefin ligand and two phosphoramidites, which may account for their exceptional electrophilicity. The internal olefin of type II catalysts may enhance enantioselectivity in reactions of branched allyl proelectrophiles by displacing the π-bond of (σ+π)-allyl (enyl) iridium intermediates,12 thus enabling rapid π-facial interconversion in otherwise stereospecific substitutions.13
Figure 1.
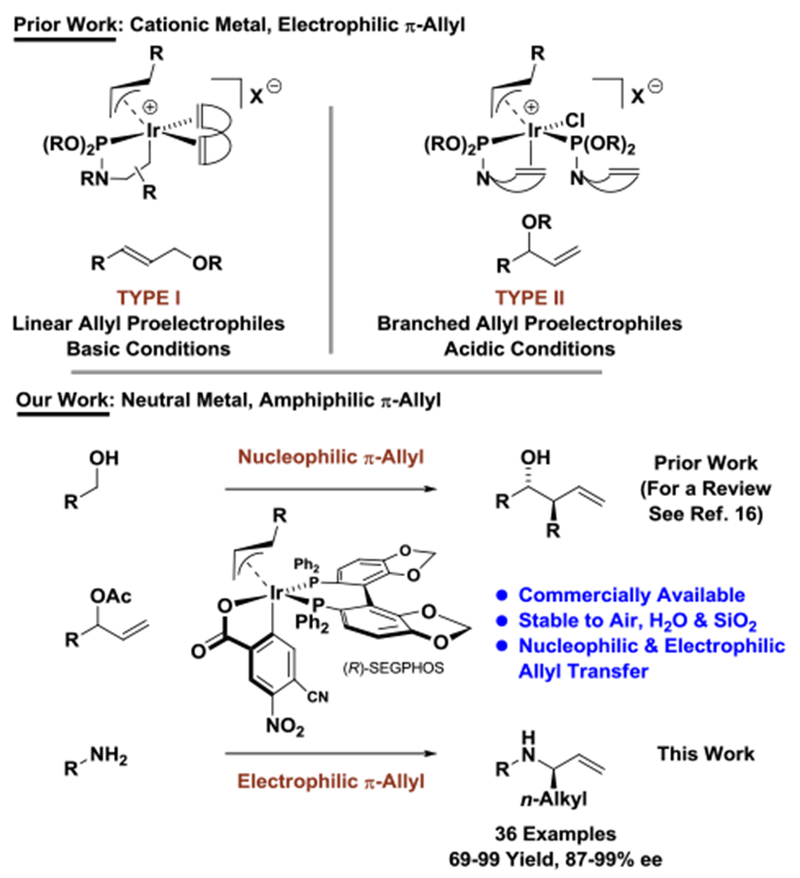
Phosphoramidite modified iridium complexes and amphiphilic π-allyliridium C,O-benzoates for regio- and enantioselective allylic amination.
Both type I and II catalysts bear π-acidic ligands and a formal positive charge at the metal center. These features enforce electrophilic properties of the allyliridium intermediate. In contrast, we have developed π-allyliridium C,O-benzoates, which are neutral and incorporate relatively strong σ-donor ligands (Figure 1).14,15 As demonstrated by their ability to promote diverse allylative carbonyl additions, the supporting ligands of this catalyst confer nucleophilic character onto the allyl moiety.16 However, as observed in our initial studies,14b carbonyl allylation is often accompanied by small quantities of O-allylation, suggesting amphiphilic character of these π-allyl complexes. Pursuant to an accumulation of similar observations by coauthors at Genentech, a systematic attempt to evoke electrophilic behavior was undertaken in the context of allylic amination. Here, we show that the commercially available π-allyliridium C,O-benzoate modified by SEGPHOS catalyzes asymmetric allylic amination. This catalyst overcomes a significant limitation in scope across all known catalytic systems – the ability to engage branched allylic acetates bearing linear alkyl groups with uniformly high levels of regio- and enantioselectivity.3–11,17,18
In an initial set of experiments, α-methyl allyl acetate 1a (100 mol%) was exposed to benzyl amine 2a (200 mol%) in the presence of the SEGPHOS modified π-allyliridium C,O-benzoate (5 mol%) in THF (1 M) at 40 °C (Table 1). In the absence of base, products of amination were not observed. However, after screening various heterogeneous bases, it was found that reactions conducted in the presence of CS2CO3 (120 mol%) provided the desired product of allylic amination 3a exclusively as the branched regioisomer with high levels of enantiomeric enrichment. After further optimization, the allylic amine 3a could be obtained in 90% isolated yield as a single regioisomer in 90% enantiomeric excess. Wet THF solvent is required, perhaps to partially solubilize CS2CO3. Using distilled THF under otherwise optimal conditions, 3a was obtained in only 61% yield. Optimal yields were restored using distilled THF in combination with water (250 mol%). The absolute stereochemistry of 3a was determined by comparison of its optical rotation to that of an authentic sample reported in the literature (and x-ray analysis of 3j′).19
Table 1.
Influence of base in the amination of α-methyl allyl acetate 1a to form allylic amine 3a.a
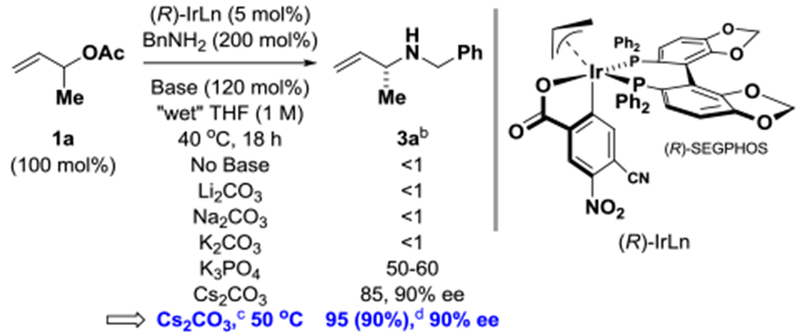 |
All reactions were performed on a 0.44 mmol scale. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Conversion was determined by 1H NMR.
Cs2CO3 (200 mol%).
Yield of material isolated by silica gel chromatography.
Under these optimized conditions, primary benzylic, allylic and aliphatic amines serve as nucleophilic partners in aminations of diverse branched allylic acetates (Table 2). Complete branched-regioselectivity was accompanied by high levels of enantioselectively (1H NMR & HPLC). Additionally, the levels of chemoselectivity in favor of primary amine allylation were sufficiently high that N-benzyl ethylene diamine and tryptamine could be converted to the respective amination products 3l and 3a′ without competing allylation of the secondary amine. As demonstrated by the formation of 3z, 3g′, 3h′, 3i′, primary amines that are branched at the α-position are effective nucleophilic partners. Further, as illustrated by the formation of 3b vs 3c, 3e vs 3f and 3h vs 3i, excellent levels of catalyst-directed stereoinduction are observed. Perhaps the notable feature of this catalytic system, however, resides in the ability to engage branched allylic acetates bearing linear alkyl groups in highly regio- and enantioselective aminations – a capability that complements the scope of previously reported iridium catalysts for allylic amination.3–11,17,18
Table 2.
Regio- and enantioselective iridium-catalyzed amination of branched allylic carboxylates.a
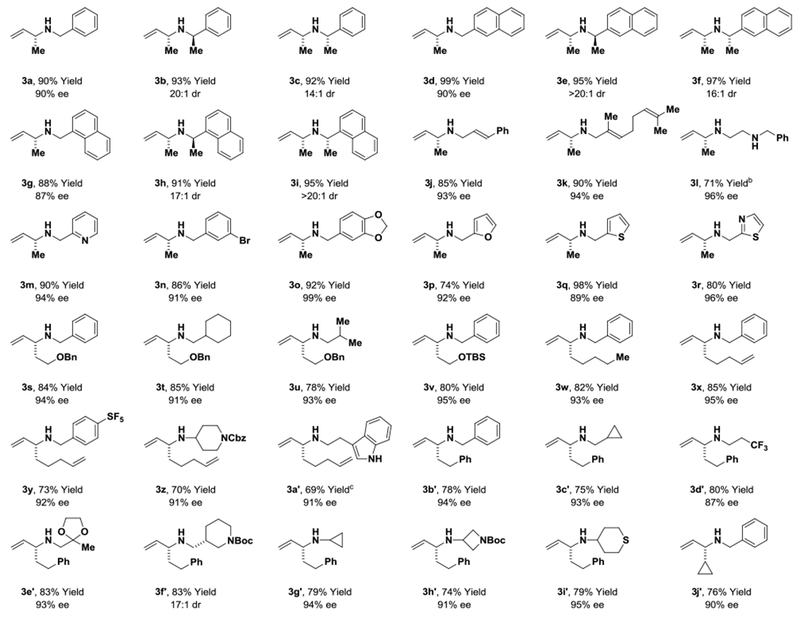 |
Yields of material isolated by silica gel chromatography. Standard conditions: (R)-IrLn (5 mol%), amine (200 mol%), Cs2CO3 (200 mol%), “wet” THF (1 M), 50 °C. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
H2NCH2CH2NHBn (400 mol%), ee% determined at the stage of 4l (eq. 1).
The cyclometallated iridium catalyst modified by the Roche ligand was used.18
To illustrate the utility of the reaction products a series of transformations were performed. The N-benzyl ethylene diamine adduct 3l was subjected to reductive amination with glyoxal to form the piperazine 4l (eq. 1).21 Adducts 3x and 3j′ were converted to 1-(N-benzyl-amino)-2-cyclohexene 4x (eq. 2)22 and the cyclopropyl substituted lactam 4j′ through ring-closing metathesis (eq. 3).5g
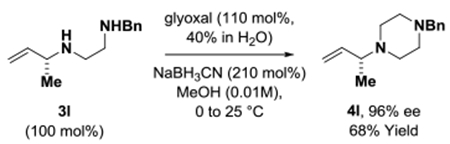 |
(eq. 1) |
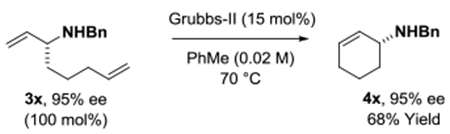 |
(eq. 2) |
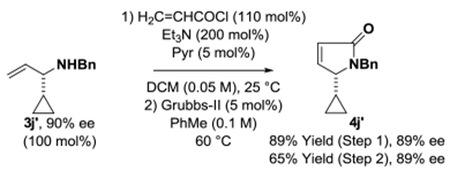 |
(eq. 3) |
While numerous reports of “umpoled allylations” exist,23 the amphiphilic properties displayed by π-allyliridium C,O-benzoates appear quite unique.24 The same catalyst, (R)-IrLn (5 mol%), will promote both nucleophilic and electrophilic allylation under very similar conditions. For example, under standard amination conditions, 4-aminomethyl-benzyl alcohol and α-methyl allyl acetate are directly converted to compound 7 in 36% yield. Alternatively, compound 5 can be converted to compound 7 in a step-wise manner with higher levels of stereoselectivity using the same iridium catalyst (Scheme 1).
Scheme 1.
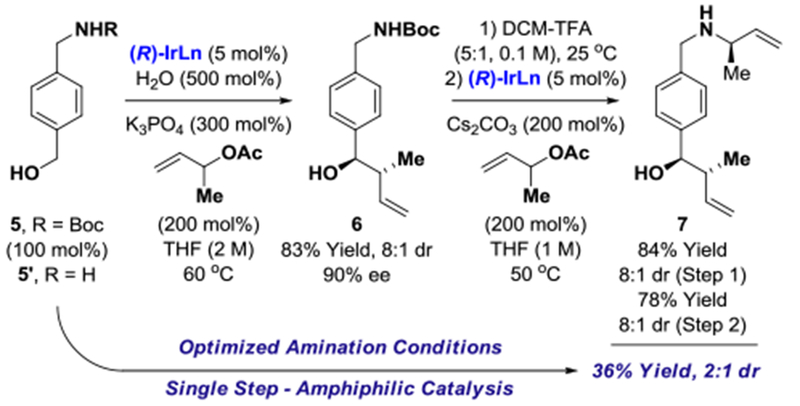
Identical π-allyliridium C,O-benzoate complexes promote both nucleophilic and electrophilic allylation.a
aSee Supporting Information for further experimental details.
The nature of the C-N bond forming event merits discussion. For related enantioselective iridium catalyzed allylic aminations, an outer-sphere mechanism is postulated.3–11 While it is likely such pathways are also operative in aminations catalyzed by π-allyliridium C,O-benzoates, an inner-sphere mechanism cannot be excluded. The more stringent steric constraints of an inner-sphere mechanism may account for the fact that primary amines engage in allylation while secondary amines do not. Furthermore, enantiofacial selectivity for amination through an inner sphere mechanism matches that observed in nucleophilic allylations of α-substituted allylic acetates (Figure 2). Computational studies are underway to evaluate outer vs inner sphere pathways.
Figure 2.
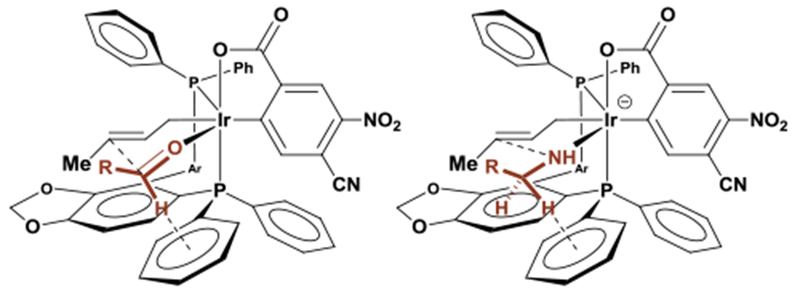
Stereochemical models accounting for equivalent π-facial selectivity in crotylation and amination.
In summary, highly tractable and commercially available π-allyliridium C,O-benzoates, which are well known to catalyze nucleophilic carbonyl allylation, are now shown to catalyze chemo-, regio- and enantioselective electrophilic aminations of branched allylic acetates bearing linear alkyl groups. These processes broaden access to chiral N-containing building blocks25 and establish unique amphiphilic properties of π-allyliridium C,O-benzoates, which should inform the design of new catalytic process. More broadly, this work demonstrates how academic-industrial collaboration accelerates the discovery of robust, innovative methods for chemical synthesis.
Supplementary Material
Acknowledgments.
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445) The Alexander von Humboldt Foundation Feodor-Lynen postdoctoral fellowship program (T. W.) are acknowledged for partial support of this research. Steven T. Staben is thanked for helpful discussions and Baiwei Lin, Kewei Xu and Yanzhou Liu for analytical data.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. HPLC traces of racemic and enantiomerically enriched products. Crystallographic data for 3j′. This material is available free of charge via the internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Tsuji J; Takahashi H; Morikawa M Tetrahedron Lett 1965, 6, 4387. [Google Scholar]; (b) Trost BM; Fullerton TJ J. Am. Chem. Soc 1973, 95, 292. [Google Scholar]
- (2).For selected reviews on metal catalyzed allylic substitution, see: Trost BM; Van Vranken DL Chem. Rev 1996, 96, 395. Trost BM; Crawley ML Chem. Rev 2003, 103, 2921. Graening T; Schmalz H-G Angew. Chem., Int. Ed 2003, 42, 2580. Trost BM J. Org. Chem 2004, 69, 5813. Lu Z; Ma S Angew. Chem., Int. Ed 2008, 47, 258. Trost BM; Zhang T; Sieber JD Chem. Sci 2010, 1, 427. Tosatti P; Nelson A; Marsden SP Org. Biomol. Chem 2012, 10, 3147. Moberg C Top. Organomet. Chem 2011, 38, 209. Sundararaju B; Achard M; Bruneau C Chem. Soc. Rev 2012, 41, 4467. Oliver S; Evans PA Synthesis 2013, 45, 3179. Butt NA; Zhang W Chem. Soc. Rev 2015, 44, 7929. Trost BM Tetrahedron 2015, 71, 5708.
- (3).(a) Takeuchi R; Ue N; Tanabe K; Yamashita K; Shiga N J. Am. Chem. Soc 2001, 123, 9525. [DOI] [PubMed] [Google Scholar]; (b) Onodera G; Watabe K; Matsubara M; Oda K; Kezuka S; Takeuchi R Adv. Synth. Catal 2008, 2725. [Google Scholar]
- (4).Reviews: Takeuchi R Synlett 2002, 1954. Takeuchi R; Kezuka S Synthesis 2006, 3349.
- (5).(a) Bartels B; García-Yebra C; Rominger F; Helmchen G Eur. J. Inorg. Chem 2002, 2569. [Google Scholar]; (b) Lipowsky G; Helmchen G Chem. Commun 2004, 116. [DOI] [PubMed] [Google Scholar]; (c) Welter C; Koch O; Lipowsky G; Helm-chen G Chem. Commun 2004, 896. [DOI] [PubMed] [Google Scholar]; (d) Welter C; Dahnz A; Brunner B; Streiff S; Dübon P; Helmchen G Org. Lett 2005, 7, 1239. [DOI] [PubMed] [Google Scholar]; (e) Weihofen R; Dahnz A; Tverskoy O; Helmchen G Chem. Commun 2004, 3541. [DOI] [PubMed] [Google Scholar]; (f) Weihofen R; Tverskoy O; Helmchen G Angew. Chem. Int. Ed 2006, 45, 5546. [DOI] [PubMed] [Google Scholar]; (g) Speiss S; Berthold C; Weihofen R, Helmchen G Org. Biomol. Chem 2007, 5, 2357. [DOI] [PubMed] [Google Scholar]; (h) Speiss S; Raskatov JA; Gnamm C; Brödner K; Helmchen G Chem. Eur. J 2009, 15, 11087. [DOI] [PubMed] [Google Scholar]; (i) Gärtner M; Jäkel M; Achatz M; SonnenSchein C; Tverskoy O; Helmchen G Org. Lett 2011, 13, 2810. [DOI] [PubMed] [Google Scholar]
- (6).Reviews: Helmchen G; Dahnz A; Dübon P; Schelwies M; Weihofen R Chem. Commun 2007, 675. Helmchen G In Iridium Complexes in Organic Synthesis; Oro LA, Claver C, Eds.; Wiley-VCH: Weinheim, 2009; pp 211–250. Qu J; Helmchen G Acc. Chem. Res 2017, 50, 2539.
- (7).(a) Ohmura T; Hartwig JF J. Am. Chem. Soc 2002, 124, 15164. [DOI] [PubMed] [Google Scholar]; (b) Kiener CA; Shu C; Incarvito C; Hartwig JF J. Am. Chem. Soc 2003, 125, 14273. [DOI] [PubMed] [Google Scholar]; (c) Leitner A; Shu C; Hartwig JF Proc. Nat. Acad. Sci 2004, 101, 5830. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shu C; Leitner A; Hartwig JF Angew. Chem. Int. Ed 2004, 43, 4797. [DOI] [PubMed] [Google Scholar]; (e) Leitner A; Shekhar S; Pouy MJ; Hartwig JF J. Am. Chem. Soc 2005, 127, 15506. [DOI] [PubMed] [Google Scholar]; (f) Leitner A; Shu C; Hartwig JF Org. Lett 2005, 7, 1093. [DOI] [PubMed] [Google Scholar]; (g) Shekhar S; Trantow B; Leitner A; Hartwig JF J. Am. Chem. Soc 2006, 128, 11770. [DOI] [PubMed] [Google Scholar]; (h) Yamashita Y; Gopalarathnam A; Hartwig JF J. Am. Chem. Soc 2007, 129, 7508. [DOI] [PubMed] [Google Scholar]; (i) Markovic D; Hartwig JF J. Am. Chem. Soc 2007, 129, 11680. [DOI] [PubMed] [Google Scholar]; (j) Pouy MJ; Leitner A; Weix DJ; Ueno S; Hartwig JF Org. Lett 2007, 9, 3949. [DOI] [PubMed] [Google Scholar]
- (8).Reviews: Hartwig JF; Stanley LM Acc. Chem. Res 2010, 43, 1461. Hartwig JF; Pouy MJ Top. Organomet. Chem 2011, 38, 169.
- (9).(a) Defieber C; Ariger MA; Moriel P; Carreira EM Angew. Chem. Int. Ed 2007, 46, 3139. [DOI] [PubMed] [Google Scholar]; (b) Roggen M; Carreira EM J. Am. Chem. Soc 2010, 132, 11917. [DOI] [PubMed] [Google Scholar]; (c) Lafrance M; Roggen M; Carreira EM Angew. Chem. Int. Ed 2012, 51, 3470. [DOI] [PubMed] [Google Scholar]; (d) Rossler SL; Krautwald S; Carreira EM J. Am. Chem. Soc 2017, 139, 3603. [DOI] [PubMed] [Google Scholar]
- (10).(a) Ye K-Y; Dai L-X; You S-L Org. Biomol. Chem 2012, 10, 5932. [DOI] [PubMed] [Google Scholar]; (b) Liu W-B; Zhang X; Dai L-X; You S-L Angew. Chem. Int. Ed 2012, 51, 5183. [DOI] [PubMed] [Google Scholar]; (c) Ye K-Y; Zhao Z-A; Lai Z-W; Dai L-X; You S-L Synthesis 2013, 2109. [Google Scholar]; (d) Ye K-Y; Dai L-X; You S-L Chem. Eur. J 2014, 20, 3040. [DOI] [PubMed] [Google Scholar]; (e) Yang Z-P; Wu Q-F; You S-L Angew. Chem. Int. Ed 2014, 53, 6986. [DOI] [PubMed] [Google Scholar]; (f) Zhang X; Yang Z-P; Huang L; You S-L Angew. Chem. Int. Ed 2015, 54, 1873. [DOI] [PubMed] [Google Scholar]; (g) Yang Z-P; Wu Q-F; Shao W; You S-L J. Am. Chem. Soc 2015, 137, 15899. [DOI] [PubMed] [Google Scholar]; (f) Ye K-Y; Cheng Q; Zhuo C-X; Dai L-X; You S-L Angew. Chem. Int. Ed 2016, 55, 8113. [DOI] [PubMed] [Google Scholar]; (g) Zhang X; Liu W-B; Cheng Q; You S-L Organometallics 2016, 35, 2467. [Google Scholar]; (h) Yang Z-P; Zheng C; Huang L; Qian C; You S-L Angew. Chem. Int. Ed 2017, 56, 1530. [DOI] [PubMed] [Google Scholar]
- (11).(a) Liu W-B; Xia J-B; You S-L Top. Organomet. Chem 2011, 38, 155. [Google Scholar]; (b) Zhou C-X; Zhang W; You S-L Angew. Chem. Int. Ed 2012, 51, 12662. [DOI] [PubMed] [Google Scholar]; (c) Wu W-T; Zhang L; You S-L Chem. Soc. Rev 2016, 45, 1570. [DOI] [PubMed] [Google Scholar]
- (12).For spectroscopic and crystallographic evidence of a stable rhodium enyl complex and its role in enabling regio- and stereospecific rhodium catalyzed allylic substitution, respectively, see: Lawson DN; Osborn JA; Wilkinson G J. Chem. Soc. A 1966, 1733. Tanaka I; Jin-no N; Kushida T; Tsutsui N; Ashida T; Suzuki H; Sakurai H; Moro-oka Y; Ikawa T Bull. Chem. Soc. Jpn 1984, 56, 657. Evans PA; Nelson JD J. Am. Chem. Soc 1998, 120, 5581.
- (13).Beyond the rather complex effects of Lewis basic additives (refs. 5a, 6a vs 6b), highly π-acidic ligands are required to preserve stereo-specificity in iridium catalyzed reactions of branched allyl proelectrophiles: Bartels B; Helmchen G Chem. Commun 1999, 741.
- (14).For initial reports, see: Kim IS; Ngai M-Y; Krische MJ J. Am. Chem. Soc 2008, 130, 6340. Kim IS; Han SB; Krische MJ J. Am. Chem. Soc 2009, 131, 2514.
- (15).For preparation and chromatographic purification of the iridium catalyst in 85% yield, see: Gao X; Townsend IA; Krische MJ J. Org. Chem 2011, 76, 2350.
- (16).For a recent overview of enantioselective allylations and propargylations catalyzed by π-allyliridium C,O-benzoates, see: Kim SW; Zhang W; Krische MJ Acc. Chem. Res 2017, 50, 2371.
- (17).For other enantioselective metal catalyzed allylic aminations employing diverse allyl proelectrophiles, see: Cooke ML; Xu K; Breit B Angew. Chem., Int. Ed 2012, 51, 10876. Arnold JS; Nguyen HM J. Am. Chem. Soc 2012, 134, 8380. Chen Q-A; Chen Z; Dong VM J. Am. Chem. Soc 2015, 137, 8392. Xu K; Wang Y-H; Khakyzadeh V; Breit B Chem. Sci 2016, 7, 3313. Mwenda ET; Nguyen HM Org. Lett 2017, 19, 4814. Guo W; Cai A; Xie J; Kleij AW Angew. Chem. Int. Ed 2017, 56, 11797.
- (18).For selected reviews on the catalytic enantioselective synthesis of allylic amines, see: Cannon JS; Overman LE Acc. Chem. Res 2016, 49, 2220. Grange RL; Clizbe EA; Evans PA Synthesis 2016, 2911. Fernandes Rodney A.; Kattanguru Pullaiah; Gholap Sachin P.; Chaudhari Dipali A. Org. Biom. Chem 2017, 15, 2672.
- (19).An enantiomeric enrichment of 82% ee was observed using the SEGPHOS-modified catalyst. For the Roche ligand, see: Schmid R; Cereghetti M; Heiser B; Schönholzer P; Hansen H-J Helv. Chim. Acta 1988, 71, 897. Schmid R; Foricher J; Cereghetti M; Schönholzer P Helv. Chim. Acta 1991, 74, 370.
- (20).(a) Vrienze DC; Hohe GS; Hoerter PZ; van Haistma JT; Samas BM Org. Lett 2009, 11, 3140. [DOI] [PubMed] [Google Scholar]; (b) Höcker J; Rudolf G; Bächlein F; Fleischer S; Lindner BD; Helmchen G Eur. J. Org. Chem 2013, 5149. [Google Scholar]
- (21).Lee BK; Kim MS; Nahm HS; Kim DS; Lee WK; Ha H-J Tetrahedron 2006, 62, 8393. [Google Scholar]
- (22).For a related RCM, see: Edwards AS; Wybrow RAJ; Johnstone C; Adams H; Harrity JPA Chem. Commun, 2002, 1542.
- (23).For selected reviews on carbonyl allylation via umpolung of π-allyls, see: Masuyama Y In Advances in Metal-Organic Chemistry; Liebeskind LS, Ed.; JAI Press: Greenwich, 1994; Vol 3, p. 255. Tamaru Y In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi E.-i., de Meijere A, Eds.; Wiley: New York, 2002; Vol. 2, pp. 1917. Tamaru Y In Perspectives in Organopalladium Chemistry for the XXI Century; Tsuji J Ed.; Elsevier: Amsterdam, 1999, pp. 215. Kondo T; Mitsudo T-A Curr. Org. Chem 2002, 6, 1163. Tamaru Y Eur. J. Org. Chem 2005, 13, 2647. Zanoni G; Pontiroli A; Marchetti A; Vidari G Eur. J. Org. Chem 2007, 22, 3599.
- (24).Amphiphilic properties were postulated in palladium catalyzed conjugate allylation-allylic alkylations of alkylidene malononitriles and related compounds. The requirement of both allyltributylstannane and allylic chloride suggests these reactions do not involve amphiphilic π-allylpalladium intermediates: Nakamura H; Shim J-G; Yamamoto Y J. Am. Chem. Soc 1997, 119, 8113. For related processes, see: Nakamura H; Aoyagi K; Shim J-G; Yamamoto Y J. Am. Chem. Soc 2001, 123, 372. Nakamura H; Shimizu K Tetrahedron Lett 2011, 52, 426. Genady AR; Nakamura H Org. Biomol. Chem 2011, 9, 7180. Masakazu N; Atsushi W; Itami K Chem. Sci 2012, 3, 3474.
- (25).For statistical analysis of the appearance of N-heterocycles in FDA approved drugs, see: Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.