

Abstract
The development of molecularly targeted probes that exhibit high biostability, biocompatibility, and efficient clearance profiles is key to optimizing biodistribution and transport across biological barriers. Further, coupling probes designed to meet these criteria with high-sensitivity, quantitative imaging strategies is mandatory for ensuring early in vivo tumor detection and timely treatment response. These challenges have often only been examined individually, impeding the clinical translation of fluorescent probes. By simultaneously optimizing these design criteria, we created a new generation of near-infrared fluorescent core–shell silica-based nanoparticles (C dots) tuned to hydrodynamic diameters of 3.3 and 6.0 nm with improved photophysical characteristics over the parent dye. A neutral organic coating prevented adsorption of serum proteins and facilitated efficient urinary excretion. Detailed particle biodistribution studies were performed using more quantitative ex vivo fluorescence detection protocols and combined optical-PET imaging. The results suggest that this new generation of C dots constitutes a promising clinically translatable materials platform which may be adapted for tumor targeting and treatment.
Graphical Abstract
Advances in the fundamental science and engineering underlying medical practice are poised to change the patient experience toward a more personalized, predictive, and preventive medicine.1 Explosive growth in the fields of molecular biology, genomics, proteomics, and informatics, in parallel with advances in nanotechnology and imaging, is transforming our understanding of disease at the molecular level2–4 and beginning to create novel solutions to diagnostic and therapeutic challenges.5–7 Nanotechnology platforms have enabled advances in molecular diagnostics and proteomic technologies and have increased our understanding of the molecular mechanisms of diseases, such as cancer. Such platforms promise to improve response assessments and address limitations in the sensitivity and specificity of lesion detection using currently available probes and imaging methodologies.
Optical fluorescence imaging comprises a suite of technologies that are being used in a rapidly growing array of biological and medical applications and offer clear potential for clinical translation,2–4,8 particularly for endoscopic and open-surgical procedures. The combination of high sensitivity and imaging speed, advantageous for high-throughput screening studies, along with low complexity and cost, makes fluorescence imaging very appealing for surface-based diagnostic detection, drug discovery, molecular pathway analysis, intraoperative mapping of tumor margins, and nodal disease spread. Limitations of commonly used planar fluorescence imaging methods include limited depth resolution (<1 cm) due to intrinsic tissue signal attenuation and autofluorescence arising from organic components and absorbed dietary constituents, which reduce signal-to-noise ratios. Moreover, only relative quantification of imaging data sets can be achieved, given the dependence of the measured signal on depth and sample composition, although the data can be validated by rigorous ex vivo analytic procedures.
Although the literature frequently acknowledges a critical need to advance clinically translatable, optically driven platforms which are biostable, demonstrate enhanced photophysical features, and offer improved size- and charge-dependent clearance characteristics, a detailed understanding of how to best achieve these properties in a single particle platform is lacking. Systematic and rigorous particle studies, which are ultimately required for efficiently translating promising particle platforms to the clinic are, at best, sporadic in the literature. Although one or two studies have, in fact, evaluated particular particle parameters of interest (i.e., size), no studies to date have concomitantly investigated and optimized all key platform characteristics for the sole purpose of improving in vivo pharmacokinetic behavior and tumor targeting.
Current state-of-the-art fluorescence imaging probes can be broadly classified as conventional fluorophores (organic dyes,3 fluorescent proteins9), quantum dots (QDs), and hybrid architectures incorporating one or more of these emitters in an inert matrix.10–15 To realize the broad potential of fluorescence imaging, while addressing the above limitations, a new generation of probes, which emit photons in the near-infrared region (NIR, 650–900 nm), have been developed. Compared to visible emitters, NIR-emitting probes exhibit decreased tissue attenuation and autofluorescence from nontarget tissue, thereby maximizing contrast per dose. Moreover, NIR nanoparticle-based probes can be designed to enhance clearance, minimize toxicity, and limit probe interference with other imaging or therapeutic techniques. Probes must be made from inert and biocompatible materials designed to efficiently exit the body, either by biodegradation or intact excretion. To date, these criteria have not been collectively satisfied in a single probe.
Research on probes for optical and other imaging modalities has shown renal clearance to be the most effective route of excretion, as opposed to liver sequestration, subsequent reticuloendothelial macrophage uptake, and eventual hepatobiliary excretion into the intestinal tract.16 By regulating the fluid and chemical homeostatic balance in blood, the kidneys serve as an ideal conduit for the filtration of contrast agents from the bloodstream. Further, this body of work has defined the criteria that determine the glomerular filtration rate of a material, specifically particle size and surface charge.
Combining the dendrimer-based work of Kobayashi and Brechbiel6 with the recently presented quantum dot work of the Bawendi and Frangioni groups,7 the consensus is that the optimal size to balance reasonable circulation lifetime with efficient clearance is between 3 and 7 nm in hydrodynamic diameter. Probes below 3 nm (including molecular dyes) are prone to extravasation and nonspecific tissue dispersal,5,6 which increases background fluorescence and potentially prolongs retention. Larger probes (e.g., most QDs and hybrid architectures) accumulate in the liver, followed by eventual hepatobiliary excretion.6,7,11 While ultimately effective, this mode of clearance prolongs exposure, increasing the potential for toxicity and interference with other imaging assessments.
Materials with a net surface charge tend to become opsonized16 by serum proteins, effectively increasing probe size and preventing renal excretion. Both neutral and zwitterionic materials have been shown to clear effectively via the kidneys,6,7 although chemically neutral surfaces such as poly- or oligo(ethylene glycol) chains are generally more bioinert than zwitterionic surfaces containing reactive amine and/or carboxylate groups.17
Despite the excellent brightness and photostability of QDs for in vivo imaging applications, the risk of systemic toxicity remains high, given their incorporation of heavy metals (e.g., Pb, Cd), precluding their widespread use and ultimate clinical translation. Further, development of QDs of suitable stabilized size in the far-red/near-infrared spectral region has proven difficult given their size-dependent emission wavelengths. The well-known limitations of QDs in their present form have accelerated the development of alternative particle platforms that are high contrast, nontoxic, efficiently excreted, and, most importantly, poised for clinical translation.
We have sought to simultaneously achieve all of these criteria in a single particle platform by synthesizing a new generation fluorescent core–shell silica nanoparticles (C dots, Figure 1a). The hydrodynamic radius of this probe is tunable down to ~3 nm diameter, a size previously thought to be unattainable. These nanoparticles covalently incorporate organic dyes into an amorphous silica matrix to prevent dye leaching (schematically depicted in Figure 1a).10,13,18 Larger C dots (15–40 nm diameter) exhibit significantly enhanced dye quantum yields due to increases in radiative and decreases in nonradiative decay rates resulting from interactions with the silica matrix, as well as enhanced photostability compared to the parent fluorophore.10–14 Prior work has demonstrated ~30 nm diameter particles with bare silica surfaces as imaging agents for in vitro (e.g., antibody-mediated labeling13) and in vivo (e.g., sentinel lymph node mapping11) applications. A particular advantage of this architecture is an ability to control particle size without changing emission wavelength, which led us to develop long-wavelength probes tuned for renal clearance and in vivo imaging.
Figure 1.
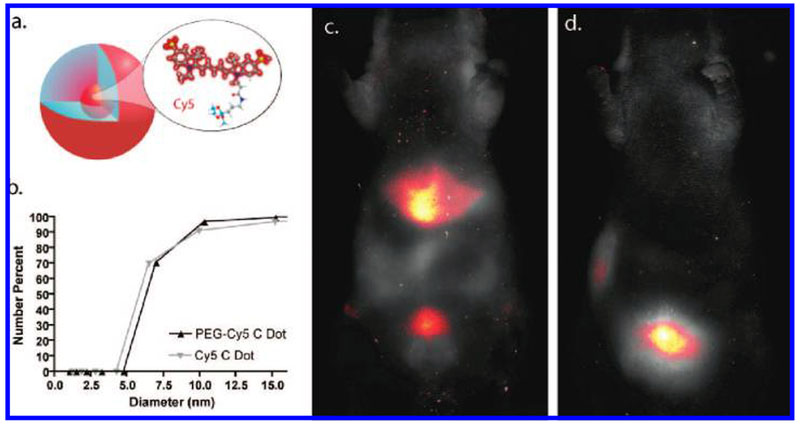
C dot schematic and in vivo imaging (a) A schematic representation of Cy5 reactive dye incorporated into the core of an amorphous silica nanoparticle. (b) DLS (number average) plot of particle size for bare silica (gray) and PEG-coated (black) Cy5-containing silica nanoparticles (C dots). (c) In vivo imaging of spectrally demixed Cy5 particle fluorescence (pseudocolor) overlaid on visible light imaging of nude mice 45 min postinjection with bare silica C dots, showing particle accumulation in the liver and bladder. (d) In vivo imaging of spectrally demixed Cy5 particle fluorescence (pseudocolor) overlaid on visible light imaging of nude mice 45 min postinjection with PEG-ylated Cy5 C dots showing particle accumulation in the bladder.
Initial syntheses derived from methods published elsewhere,12,19,20 using reactive derivatives of the organic dye Cy5 that exhibit emission maxima above 650 nm (Figure 2b), yielded nanoparticles in the 6–10 nm diameter range, as shown by dynamic light scattering (DLS) results in Figure 1b. In vivo whole-body near-infrared fluorescence (NIRF) imaging of these bare silica nanoparticles in nude mice showed considerable renal clearance 45 min postinjection with a significant accumulation remaining in the liver (Figure 1c). Eventual excretion into the enterohepatic circulation occurred during the ensuing 24 h (data not shown). On the basis of these promising results, the particles were covalently coated with methoxy-terminated poly(ethylene glycol) chains (PEG, ~0.5 kDa) to prevent opsonization and further enhance particle clearance while maintaining small hydrodynamic size. This treatment noticeably decreased liver retention and resulted in increased renal filtration into the bladder at 45 min postinjection, as shown by in vivo fluorescence imaging (Figure 1d), with bladder fluorescence visible out to 24 h. These probes were well tolerated, with no adverse effects or animal deaths observed over the course of the study.
Figure 2.
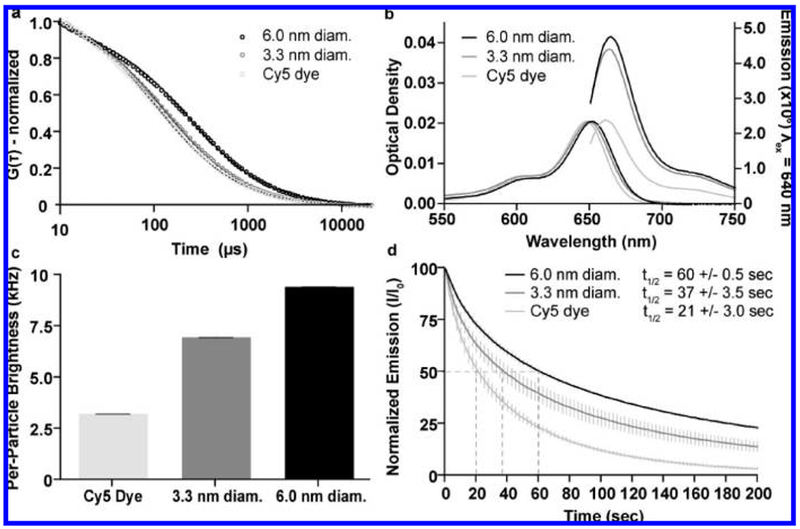
Photophysical characterization. (a) FCS data and single exponential fits for Cy5 dye (light gray), 3.3 ± 0.06 nm diameter (dark gray, mean ± standard deviation n = 9) and 6.0 ± 0.1 nm diameter (black, mean ± standard deviation, n = 6) Cy5-containing PEG-coated C dots showing the differences in diffusion time resulting from the different hydrodynamic sizes of the different species. Particle sizes are significantly larger than dye for both 3.3 and 6.0 nm diameter (one-tail Mann–Whitney U test, p = 6.1 × 10−7). (b) Absorption and emission spectra of Cy5 dye (light gray), 3.3 nm diameter (dark gray) and 6.0 nm diameter (black) PEG-coated C dots, comparing the peak photoemission of the three species at equal peak absorption optical density to demonstrate the enhanced performance of the dye following the encapsulation reaction. (c) Relative brightness comparison of free dye (light gray) with 3.3 nm (dark gray) and 6.0 nm diameter (black) C dots, measured as count rate per molecule/particle as determined from the FCS curves, demonstrating the enhancement effects over free dye that increase with particle size (mean ± standard deviation, n = 15) Both 3.3 and 6.0 nm diameter particles exhibit significant increases in count rate/particle compared to free dye (one-tail Mann–Whitney U test, p = 1.53 × 10−6, 1.53 × 10−6). (d) Photobleaching data for Cy5 dye (light gray), 3.3 nm diameter (dark gray), and 6.0 nm diameter (black) PEG-coated C dots under ~3.5 mW laser excitation, demonstrating the enhanced photostability of the dye following silica encapsulation (mean ± standard deviation, n = 5). The time at which 50% of the photoemission is reduced (t1/2) is indicated for each of the different species. Both 3.3 and 6.0 nm diameter particles exhibit significant increases in photobleaching half-life with respect to free dye (one tail Mann–Whitney U test, p = 0.0104, 0.0104, respectively).
On the basis of these initial in vivo data, a more detailed biodistribution and clearance study of coated core–shell silica nanoparticles was undertaken on two sets of PEG-ylated Cy5-containing core-shell silica particles to assess the effects of probe size on biodistribution. In collaboration with Hybrid Silica Technologies (HST, Ithaca, NY), the particle synthesis was further optimized and new generation particles with hydrodynamic diameters of 3.3 ± 0.06 and 6.0 ± 0.1 nm, as measured by fluorescence correlation spectroscopy (FCS),21 were generated (Figure 2a). Prior to in vivo studies, particle photophysical properties were thoroughly investigated to establish their performance levels versus free dye.
Despite the extremely small size of the particles, the silica-encapsulated dye molecules exhibited several notable photophysical enhancements over free dye.12,13,22 By absorption and emission spectroscopy (ensemble averages, Figure 2b), as well as FCS (single molecule/particle method, Figure 2c), significant enhancements in brightness were observed. Furthermore, compared to the free dye, the 3.3 and 6.0 nm diameter dots exhibited 2- and 3-fold increases in photobleaching half-life, respectively, when irradiated with a high-power 635 nm laser (Figure 2d).23 In all cases, photophysical enhancements scaled with increasing particle size.23 Thus, these new generation C dot probes are both brighter and more photostable than their free dye counterparts, creating an excellent platform for in vivo imaging studies.
Although semiquantitative estimates of in vivo nanoparticle behavior can be generated from whole-body imaging,24 postmortem analysis of tissue homogenates and fluids via fluorescence plate reader enhances sensitivity and allows calibrated quantitation of the variations observed in NIRF imaging. These samples were grouped as “retained” (liver, kidney, lung, spleen homogenates, and blood) and “excreted” (urine) sources of particle fluorescence. All values were background-corrected and converted to percent of the initial dose (% ID) per animal based on calibration curves (Supplementary Figure 1). Tissue analysis showed minimal particle retention in the major organs, with most of the fluorescence attributed to circulating blood (Figure 3, panels a and b) To minimize the difference between NIRF imaging and homogenate analysis, organs were not perfused prior to homogenization. The net particle retention was calculated as the sum of the “retained” components, and was fit with an exponential decay curve to determine the kinetics of excretion (Figure 3c). The larger particles exhibited a longer tissue half-life (t1/2(3.3 nm) = 190 min, t1/2(6.0 nm) = 350 min) and greater initial organ retention, as expected. After 48 h (Figure 3, panels c and d), both particle sizes exhibited minimal retention in the body (Rtotal(3.3 nm) = 1.0 ± 0.1% ID, Rtotal(6.0 nm) = 2.4 ± 0.6% ID).
Figure 3.
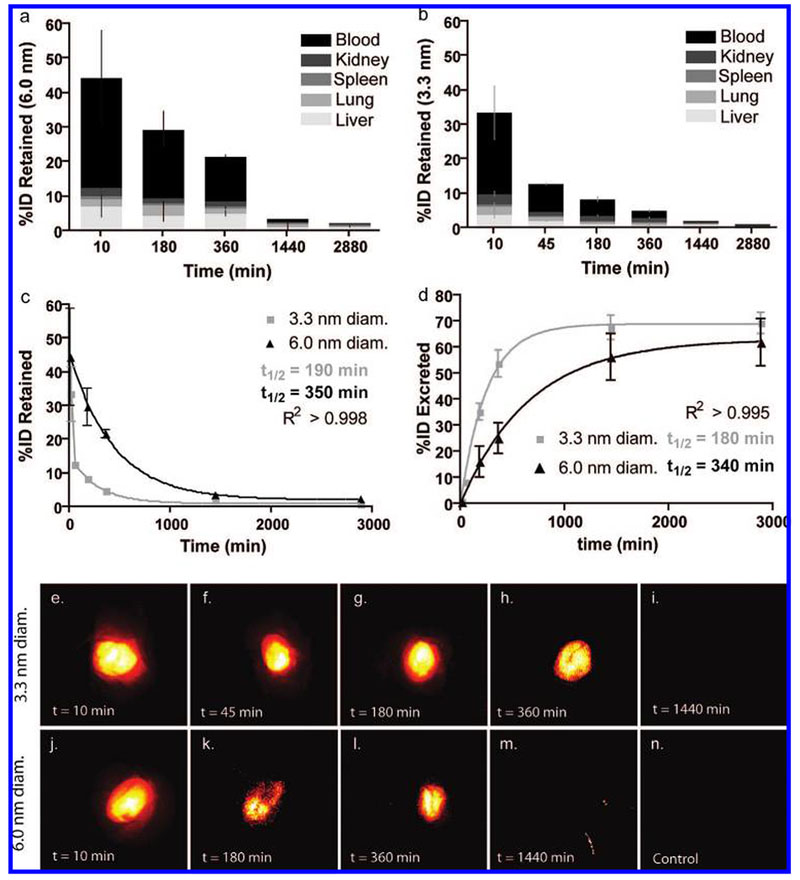
Particle retention and excretion and near-infrared fluorescence imaging. (a, b) Percent of initial particle dose (%ID) retained by blood (black) and tissues: liver (light gray), lung (mid-low gray), spleen (midgray), and kidney (mid-high gray) for 6.0 nm (a) and 3.3 nm (b) diameter C dots at various time points from 10 min to 48 h postinjection (n = 3 mice, mean ± standard deviation). (c) Plot of retained particle concentration for 3.3 nm (light gray) and 6.0 nm (black) diameter C dots and the associated logarithmic decay fits and half-lives. (d) Plot of estimated particle excretion for 3.3 nm (light gray) and 6.0 nm (black) diameter dots and the associated logarithmic fits and half-lives (mean ± standard deviation, n = 9 (three mice, three technical replicates per time point)). (e–i) Pseudocolor images of Cy5 fluorescence (demixed) in intact mouse bladders showing the accumulation of 3.3 nm diameter dots over the course of the first 6 h postinjection (e–h), followed by the negligible particle fluorescence seen at 24 h postinjection (i).(j–m) Pseudocolor images of Cy5 fluorescence (demixed) in intact mouse bladders showing the accumulation of 6.0 nm diameter dots in the first 6 h postinjection (j–l) and at the 24 h end point (m). (n) Pseudocolor image of a control mouse bladder. Further fluorescence and anatomical brightfield images can be found in the Supporting Information, Figures 2 and 3.
The major route of excretion for this new generation of particles is expected to be via renal filtration, given their small size and neutral surface. Thus, urine samples collected at the time of sacrifice were analyzed to determine the particle concentration using a similar serial dilution calibration scheme as that for tissue homogenates (Supplementary Figure 1). This information, in turn, was used to estimate the total renal clearance based on a conservative estimate of the average urine volume excreted per unit time25 (Supplementary Table 1). By this method, the %ID excreted over time was estimated for both particle sizes (Figure 3d). Exponential fits of the resulting curves corroborated the kinetics seen in the tissue analysis, namely, that the smaller (3.3 nm) particles were more quickly and completely excreted. The urine half-lives of the two particle types were quite consistent between the tissue and urine results (t1/2(3.3 nm) = 180 min, t1/2(6.0 nm) = 360 min). Further, the estimated total excreted fraction (Etotal) after 48 h was higher for the smaller particles (Etotal(3.3 nm); 73%ID versus Etotal(6.0 nm); 64%ID), which is consistent with filtration rates expected for similar-sized probes.2,6 To account for variations in urine volume and concentration due to circadian rhythm, animals in each cohort (time-point and treatment) were sacrificed at different times throughout the day.
These results are supported by the whole-body NIRF imaging data collected postmortem for each mouse (representative bladder images, Figure 3, panels e–n, and Supplementary Figures 2 and 3), in which particle fluorescence is visible in animals up to 6 h postinjection (Figure 3, panels e–h, and Figure 3, panels j—l, for 3.3 and 6.0 nm diameter dots, respectively). The images were spectrally demixed based on reference samples to remove background fluorescence (e.g., food fluorescence), as evinced by low background counts from control animals (Figure 3n). Serial positron emission tomography imaging over a 96 h period following intravenous injection of 124I labeled coated C dots corroborated the optical imaging findings (Supplementary Figure 4).
The results of this study suggest that this new generation of C dots may serve as a first-of-its-kind materials platform for translation to the clinic. Thorough particle photophysical characterization (brightness and photobleaching) was performed and more quantitative protocols were established for in vivo evaluation of NIR-emitting nanoparticle probes. This platform has been designed to simultaneously yield high contrast, photostability biocompatibility, and efficient renal excretion. Despite the small particle sizes, dye encapsulation in the silica matrix leads to enhancements in photophysical properties, while concomitantly achieving efficient renal clearance due to the PEG surface coating, particularly relevant for the larger particle size used here.
Optimization of the photophysical and biological characteristics of the nonfunctionalized C dot, as well as the attachment of a suitable radiolabel, serves as an initial step toward the development and deployment of multimodal, targeted probes as early detection and/or therapeutic vehicles for a broad range of biomedical applications. However, even the nontargeted C dot preparations may confer benefits in a clinical setting, particularly for sentinel lymph node mapping, in which the range of nodal sizes interrogated may be expanded to include the subcentimeter regime (i.e., <5 mm), for assessing ureteral pathologies (<10 nm particles) and for improving realtime visualization of microvascular beds and neural structures intraoperatively. The silica surface provides a versatile template for the attachment of small peptides and other targeting moieties that do not substantially increase particle size, promote opsonization or fouling, or substantially alter renal clearance, while allowing for incorporation of metal ions for use in combined imaging experiments. Key considerations during this development process include target selection and specificity, optimization of tumor-to-background ratios, and the size, shape, and surface charge of the functionalized C dot. Such a multimodal platform may provide complementary information at both the tissue and cellular level, offering distinct advantages for translation to the clinic compared with unbound fluorescent dyes, simple radiolabeled constructs, and QDs. The findings of this study pave the way for this and other nanosized probe technologies to address important biological issues.
Methods.
Synthesis of New Generation Core–Shell Silica Nanoparticles (C Dots).
Particles were synthesized by a modified Stöber-type silica condensation as described in detail elsewhere.12,19,20 Bare particles were dialyzed to deionized water, and ethylene glycol-coated particles were dialyzed to physiological saline (0.15 M NaCl), through 3500 MWCO Snakeskin Dialysis Membranes (Pierce) and sterile filtered through 0.2 μm syringe filters. All samples were optical density-matched at their peak absorption wavelength (640 nm) prior to injection.
Hydrodynamic Size Measurements by Dynamic Light Scattering (DLS).
Particles dialyzed to water were measured on a Brookhaven Instruments Company BIC 200SM static/dynamic light scattering system using a HeNe laser (λ = 632.8 nm). Due to overlap of the dye absorption with the excitation source, 15 min integration times were used to achieve acceptable signal-to-noise ratios.
Hydrodynamic Size Measurements by Fluorescence Correlation Spectroscopy (FCS).
Particles dialyzed to water were diluted into physiological saline (0.15 M NaCl in H2O) and measured on a Zeiss LSM 510 Confocor 2 FCS using HeNe 633 nm excitation. The instrument was calibrated for size prior to all measurements.
Spectrophotometry and Spectrofluorometry.
Absorption-matched samples were prepared by appropriate dilution of water-dialyzed particles or water-diluted dye and measured in quartz cuvettes on a Varian Cary 5000 spectrophotometer (Varian, Palo Alto, CA). Fluorescence measurements were performed on a Photon Technologies International Quantamaster spectrofluorometer (PTI, Birmingham, NJ).
Photobleaching.
A custom photobleaching apparatus was constructed in our laboratory, which consisted of an 8 mW (3.5 mW at the sample) 635 nm diode laser coupled to a 3.5× (0.10 NA) objective. The excitation beam was focused on a 0.4 mm internal diameter glass capillary tube aligned parallel to the excitation beam in a custom three-axis, tilt mount. Two microliters of appropriately diluted particles or dye (0.01 peak OD) was loaded into the capillary tube and irradiated for each trial, with all data shown recorded in triplicate. Emitted light was collected by a Perkin-Elmer single photon counting module interfaced with a Correlator.com autocorrelator card. Twenty-minute time series were recorded at 2.5 s intervals, normalized to the peak intensity, and analyzed by Prism Version 4.0c to determine photobleaching kinetics.
Animal Models.
Male Taconic NCRNU-M-M Homozygous (nu/nu) nude mice were purchased from Taconic Laboratories and studied at approximately 10 weeks of age. Mice were provided with water and fed Harlan Teklad Global Diet 2016, ad libitum, a 16% protein rodent diet without soy, alfalfa, fish, or animal protein to decrease background fluorescence in the spectral window of interest. Standard high protein diets containing alfalfa and soy have been shown to contribute to autofluorescence from the gastrointestinal tract and skin of the animal at 674 nm, which has been linked to the products of chlorophyll metabolism.26
Biodistribution and Clearance Measurements.
Mice were individually injected intravenously with 200 μL of fluorescent silica nanoparticles in physiological saline (0.15 M NaCl) at a peak absorption of 0.30 OD (640 nm), to ensure delivery of equal amounts of contrast agent across the test groups. Mice were sacrificed at various time points postinjection (10, 45, 180, 360, 1440, and 2880 min, n = 3 per time point) by CO2 asphyxiation and imaged postmortem in a Cambridge Research Instruments Maestro imager. Given the expected slower kinetics of excretion of the larger 6.0 nm diameter particles, a longer 48 h time point was used in lieu of a 45 min time point. Images for spectral deconvolution were collected at 2 nm intervals from 630 to 850 nm using a 575–605 bandpass excitation filter and a 645 nm long-pass emission filter exposed at f/11 for 278 ms. Spectral deconvolution algorithms were applied to the images collected to separate particle fluorescence from food-based and other intrinsic (background) emissions using Maestro Imaging Software v. 2.4.
Tissue and fluid samples (liver, kidney, spleen, lung, blood, bile, and urine) were collected and frozen immediately on dry ice for later examination. Bile volume varied greatly across the experimental population, so a complete data set could not be generated, though testing of the samples collected showed minimal particle fluorescence above background. Tissue samples were thawed and homogenized by ultrasonication following dilution of 3 μL of H2O/mg of tissue with a Sonics Vibracell CV188 probe sonicator to facilitate pipetting. Fluid samples were thawed and used as collected. All samples were pipetted into black polystyrene 384-well plates (Thermo Scientific Microtiter Cliniplate 384) in triplicate (as sample volume allowed) and measured on a Tecan Safire plate reader (λex = 650 nm, λem = 680 nm, 12 nm bandwidth). Fluids and tissue homogenates from control animals were analyzed to determine background levels. Excess fluid and homogenates were used to generate calibration curves by mixing samples with known volumes of particles, followed by serial dilutions with control homogenate or fluid to ensure that any background fluorescence or absorption/scattering would be taken into account. All plate reader data were analyzed and plotted with GraphPad Prism 4.0c. Following background subtraction, calibration curves were generated for each organ or fluid to determine particle concentration in each well. These calibration curves (Supplementary Figure 1) were applied to the background-corrected sample data to generate the particle per tissue/fluid data shown in Figure 3.
In order to estimate the fraction of particles excreted over time, the urine samples were first analyzed to determine the particle concentration at each time point. With an estimate of average daily mouse urine volume, a graph was generated plotting the urine concentration at each time point to the urine excreted over that time. By calculating the area under the curve generated by these data points, a measure of the %ID of particles removed from the system was determined (concentration/volume × volume = concentration). The circadian variation in urine volume was averaged out by collection of samples from mice at various time points throughout the day.
Particle Radiolabeling.
Radiolabeling of the PEG-ylated C dots was achieved by conjugation of tyrosine residues to the PEG chains using protocols described in the literature.27 An aliquot of the coated silica nanoparticles bearing these tyrosine residues was radiolabeled using the IODOGEN method (Pierce, Rockford, IL).28 Briefly, 25 μL of the coated nanoparticle in deionized water (0.039 mM) was incubated for 10 min in a 0.5 mCi 124I in an Iodogen-coated tube. Final purification was performed using size exclusion chromatography (PD-10 column; GE Healthcare) with phosphate-buffered saline (pH 7.4) as the mobile phase. Nontumor bearing mice (n = 3) were intravenously injected with 8 pmol of particles (200 μL), and serial microPET images were acquired using a dedicated small-animal PET scanner (Focus 120 microPET; Siemens Medical Solutions USA, Inc.). Mice were maintained under 2% isoflurane anesthesia in oxygen at 2 L/min during the entire scanning period. Static acquisitions of 15 min to 1 h were initiated at the time of injection of 7.4–11.1 MBq (200–300 mCi). An energy window of 410–590 keV and a coincidence timing window of 6 ns were used. Images were iteratively reconstructed using the ordered-subset expectation maximization (OSEM) technique. The image data were corrected for nonuniformity of the scanner response, dead time count losses, and physical decay to the time of injection. No correction was applied for attenuation, scatter, or partial-volume averaging. A computed tomography (microCT) scan (microCAT II, Imtek, Inc.) was additionally obtained 24 h after injection using routine image acquisition parameters (70 kVp, 90 mA s with 2 mm aluminum filtration). The microCT study was acquired immediately following the microPET study for anatomic coregistration purposes using in-house coregistration software and a custom-built imaging bed for immobilizing and fixing a small animal.29
Supplementary Material
Acknowledgment.
We thank Dr. Bradley Beattie for technical support. A.B. would like to thank Erin Hill for helpful discussions. Technical services were additionally provided by the MSKCC Small Animal Imaging Core Facility, supported in part by NIH Small-Animal Imaging Research Program (SAIRP). M.B. would like to gratefully acknowledge support by Grant UL1RR024996 of the Clinical and Translation Science Center at Weill Cornell Medical College. A. B. would like to thank the Cornell Nanobiotechnology Center (NBTC) and STC Program of the NSF under Agreement No. ECS-9876771 for funding and infrastructure. E. H. acknowledges funding by NEI Corporation under a contract with the U.S. Army Research Office.
Footnotes
Supporting Information Available: Calibration curves, in vivo whole body imaging of mice injected with dots, and urine concentration and excretion data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Hood L; Heath J; Phelps M; Lin B Science 2004, 306, 640–643. [DOI] [PubMed] [Google Scholar]
- (2).Frangioni JV Curr. Opin. Chem. Biol 2003, 7 (5), 626–634. [DOI] [PubMed] [Google Scholar]
- (3).Sevick-Muraca E; Houston J; Gurfinkel M Curr. Opin. Chem. Biol 2002, 6, 642–650. [DOI] [PubMed] [Google Scholar]
- (4).Wagner V; Dullaart A; Bock A-K; Zweck A Nat. Biotechnol 2006, 24 (10), 1211–1217. [DOI] [PubMed] [Google Scholar]
- (5).Ohnishi S; Lomnes S; Laurence R; Gogbashian A; Mariani G; Frangioni J Mol. Imaging 2005, 4 (3), 172–181. [DOI] [PubMed] [Google Scholar]
- (6).Kobyashi H; Brechbiel M AdV. Drug DeliVery ReV 2005, 57, 2271–2286. [DOI] [PubMed] [Google Scholar]
- (7).Choi HS; Liu W; Misra P; Tanaka E; Zimmer JP; Ipe BI; Bawendi MG; Frangioni JV Nat. Biotechnol 2007, 25 (10), 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bridot J-L; Faure A-C; Laurent S; Riviere C; Billotey C; Hiba B; Janier M; Josserand V; Coll J-L; Elst LV; Muller R; Roux S; Perriat P; Tillement OJ Am. Chem. Soc 2007, 129, 5076–5084. [DOI] [PubMed] [Google Scholar]
- (9).Sherbo D; Merzlyak E; Chepurnykh T; Fradkov A; Ermakova G; Sollovieva E; Lukyanov K; Bogdanova E; Zaraisky A; Lukyanov S; Chudakov D Nat. Methods 2007, 4 (9), 741–746. [DOI] [PubMed] [Google Scholar]
- (10).Burns A; Ow H; Wiesner U Chem. Soc. ReV 2006, 35 (11), 1028–1042. [DOI] [PubMed] [Google Scholar]
- (11).Choi J; Burns A; Williams R; Zhou Z; Flesken-Nikitin A; Zipfel W; Wiesner U; Nikitin AJ Biomed. Opt 2007, 12 (6), 064007(1–13). [DOI] [PubMed] [Google Scholar]
- (12).Larson D; Ow H; Vishwasrao H; Heikal A; Wiesner U; Webb W Chem. Mater. 2008, 20 (8), 2677–2684. [Google Scholar]
- (13).Ow H; Larson DR; Srivastava M; Baird BA; Webb W; Wiesner U Nano Lett 2005, 5 (1), 113–117. [DOI] [PubMed] [Google Scholar]
- (14).Zhao X; Hilliard L; Mechery S; Wang Y; Bagwe R; Jin S; Tan W Proc. Natl. Acad. Sci. U.S.A 2004, 101 (42), 15027–15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Piao Y; Burns A; Kim J; Wiesner U; Hyeon T AdV. Funct. Mater 2008, 18, 3745–3758. [Google Scholar]
- (16).Moghimi SM; Hunter AC; Murray JC Pharmacol. ReV 2001, 53 (2), 283–318. [PubMed] [Google Scholar]
- (17).Nel A; Xia T; Madler L; Li N Science 2006, 311, 622–627. [DOI] [PubMed] [Google Scholar]
- (18).Blaaderen AV; Vrij A J. Colloid Interface Sci 1993, 156, 1–18. [Google Scholar]
- (19).Bogush GH; Tracy MA; Zukoski ICF J. Non-Cryst. Solids 1988, 104 (1), 95–106. [Google Scholar]
- (20).Sadasivan S; Dubey A; Li Y; Rasmussen DJ Sol-Gel Sci. Technol 1998, 12, 5–14. [Google Scholar]
- (21).Hess ST; Huang S; Heikal AA; Webb WW Biochemistry 2002, 41 (3), 697–705. [DOI] [PubMed] [Google Scholar]
- (22).Herz E; Burns A; Lee S; Sengupta P; Bonner D; Ow H; Liddell C; Baird B; Wiesner U Proc. of SPIE, 2006, 6096, 609605. [Google Scholar]
- (23).Song L; Hennink EJ; Young IT; Tanke HJ Biophys. J 1995, 68, 2588–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cai W; Chen K; Li Z-B; Gambhir S; Chen XJ Nucl. Med 2007, 48 (11), 1862–1870. [DOI] [PubMed] [Google Scholar]
- (25).Drickamer LC J. Chem. Ecol 1995, 21 (10), 1481–1493. [DOI] [PubMed] [Google Scholar]
- (26).Weagle G; Paterson PE; Kennedy J; Pottier RJ Photochem. Photobiol., B 1988, 2, 313–320. [DOI] [PubMed] [Google Scholar]
- (27).Hermanson G Bioconjugate Techniques, 2nd ed.; Academic Press: New York, 2008; p 1323. [Google Scholar]
- (28).Piatyszek M; Jarmolowski A; Augustyniak J Anal. Biochem 1988, 172, 356–359. [DOI] [PubMed] [Google Scholar]
- (29).Beattie B; Forster G; Govantes R; Le C; Longo V; Zanzonico P; Bidaut L; Blasburg R; Koutcher J Mol. Imaging 2007, 6, 108–120. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.