

Abstract
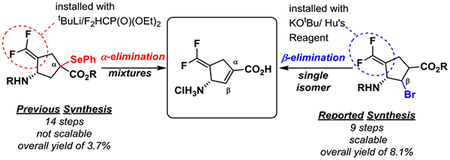
(S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (OV329, 1) is being developed for the treatment of addiction and epilepsy. The previous 14-step synthesis of OV329 was low yielding, involved an unselective α-elimination to form the cyclopentene, required the use of tert-butyllithium, and produced toxic selenium by-products in the penultimate step. A new synthesis, which avoids the aforementioned issues, was carried out on large scale, reducing the step count from 14 to 9 steps and increasing the overall yield from 3.7% to 8.1%.
Graphical Abstract
γ-Aminobutyric acid (GABA) is a major inhibitory neurotransmitter in the central nervous system.1 When GABA concentrations in the brain diminish below a threshold level, convulsions can occur; increasing GABA levels has been shown to stop convulsions and has been clinically indicated for the treatment of epilepsy.1–3 Additionally, increased concentrations of GABA antagonize the release of dopamine from the nucleus accumbens, a region of the hypothalamus associated with reward and motivation, and has been suggested as a possible treatment of addiction.4 Unfortunately, systemic administration of GABA is not viable as GABA does not cross the blood brain barrier under ordinary conditions. However, GABA concentrations in the brain can be increased by inhibiting GABA aminotransferase (GABA-AT), a pyridoxal-5’-phosphate (PLP)-dependent enzyme that degrades GABA to succinic semialdehyde. 4-Aminohex-5-enoic acid, also known as vigabatrin (marketed as Sabril), currently is the only FDA-approved inhibitor of GABA-AT for the treatment of infantile spasms and has been shown to be a possible treatment for addiction.4–6 However, when vigabatrin is taken in large doses (1–3 g/day), it inhibits multiple GABA receptors/enzymes and, with prolonged use, causes retinal damage in 25–40% of patients.7
Because of the drawbacks of vigabatrin, we recently reported the design, synthesis, and evaluation of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (OV329, 1), a potent inactivator of GABA-AT (Figure 1).8 OV329 inhibits GABA-AT via enzyme-catalyzed hydrolysis of the difluoromethenyl group, resulting in dicarboxylate metabolite 2, which binds tightly to the enzyme via electrostatic interactions. In vivo studies in rats indicated that OV329 was vastly superior to previous inhibitors of GABA-AT at suppressing the release of dopamine in the corpus striatum after exposure to cocaine or nicotine. Additionally, OV329 does not inhibit off-target aminotransferase enzymes such as alanine aminotransferase and aspartate aminotransferase. It additionally does not inhibit the hERG potassium ion channel or various microsomal cytochrome P450 enzymes. Furthermore, on the basis of the Irwin test, the no observed adverse effect level in mice was 6 mg/kg by oral administration. Given the potency (effective in vivo at 0.01–0.1 mg/kg), lack of off-target activity, low plasma protein binding, high microsome stability, lack of activity against the Cerep SpectrumScreen panel of 176 pharmacological targets, negative Ames test, and promising in vivo pharmacokinetics and toxicology of 1, we look to further study the possibility of OV329 as a treatment for addiction and epilepsy.
Figure 1.

OV329 (1) inhibits GABA-AT through hydrolysis of the 1,1’-difluoromethylene unit, resulting in metabolite 2 and an increase in the concentration of GABA, which is beneficial in the treatment of epilepsy and addiction. PLP: pyridoxal-5’-phosphate; PMP: pyridoxamine-5’-phosphate; GABA: γ-aminobutyric acid.
The major hindrance in moving forward with advanced preclinical studies is straightforward synthetic access to OV329. Currently, OV329 has been synthesized in six steps from CPP-115 (3), an inhibitor of GABA-AT that we previously designed, currently in Phase 1 clinical trials for the treatment of epilepsy (Figure 2a).5,9 Given that CPP-115 requires an 8-step synthesis,10 the total synthetic step count from commercial material to OV329 is 14 with an overall yield of 3.7%. The synthesis of CPP-115, itself, involves the use of pyrophoric tert-butyllithium (on gram scale) to install the 1,1’-difluoroolefin, which limits the scale at which the reaction can be run. Furthermore, the current synthesis relies of the introduction of the cyclopentene through selenoxide elimination. Protected CPP-115 (not shown) is selenated in 70% yield, although yields can vary depending on scale (Figure 2a). Elimination of the oxidized selenide in 4 yields a mixture of chromatographically inseparable isomers, 1 and 5, in a 5:3 ratio, favoring 1. Isomer 5 is then selectively degraded using thiosalicylic acid to produce solely 1 in an overall yield of 36% from 4. Currently, only small amounts (<20 mg) of OV329 can be obtained. Additionally, given that selenium is regulated by the FDA to levels below 80–150 μg/day, the production of selenol in the penultimate step complicates the synthesis and future FDA consideration of OV329.11
Figure 2.

(a) The previous synthesis of OV329 (1) resulted in a mixture of isomers (1 and 5) as the result of an unselective α-elimination of a phenyl selenyl ether. (b) This nonselectively and use of toxic selenium was a major drawback of the current synthesis. A new synthesis, through a β-elimination, whose retrosynthesis is shown, addresses these drawbacks.
We therefore developed a second-generation synthesis that: (a) is scalable and high yielding, (b) avoids the use of selenium and tert-butyllithium, and (c) does not form multiple isomers from an α-elimination (Figure 2b). We propose that elimination of a leaving group from the β-position would preclude the possibilty of a mixture of isomers and streamline the synthesis. Thus, an intermediate such as 7 would be desirable. Intermediate 7 would be derived from the hydrolysis of bicyclic compound 8, which could come from known intermediate 9.
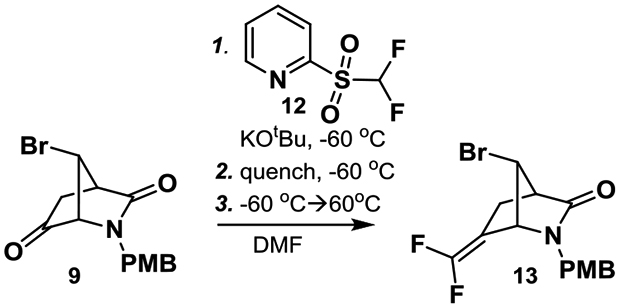
Starting from the chiral Vince lactam (10) and following a slight modification of the literature steps,12 11 was obtained in good yield on multi-gram scale (Scheme 1). Methanolysis of the acetate and oxidation yielded ketone 9. Slight modifications of these steps, such as the use of PMBOH/HCl, allowed us to run them on multi-gram scale. The first key step involved the difluoro-Horner-Wadsworth-Emmons olefination of ketone 9. The standard conditions, which were identical to those used in the synthesis of CPP-115, of tert-BuLi and F2CHP(O)(OEt)2, did not yield the product.10 Alteration of temperature, base, and addition method did not produce 13. A more traditional Wittig reaction with Ph3PCHF2Br13,14 also failed to provide 13. When Hu’s reagent, 12,15,16 was employed with KOtBu as base, following Hu’s reported conditions, 13 was obtained in small amounts (<10% yield). Given the complex procedure, a more in-depth optimization was carried out. On the basis of Hu’s mechanism of this reaction, multiple intermediates form during the course of the reaction.15,16 Intermediate 17, which forms first, rearranges via cyclic intermediate 18 to form sulfonate 19, which is then protonated, triggering elimination and formation of the olefin (Scheme 2). If the reaction was quenched at −60 °C with 6 M HCl five minutes after the addition of KOtBu to a mixture of 9 and 12, then only 17 was observed by LC/MS (entry 1, Table 1). 17 did not rearrange to 13 or 19, even upon heating to 100 °C. Further heating caused hydrolysis of the lactam. Addition of KOtBu followed by a 6 M HCl quench at −60 °C (entry 2), and subsequent heating at 60 °C for 1 h provided 13 in 9% yield along with starting material and intermediate 17. Quenching after 1 hour with a saturated NH4Cl solution, followed by 6 M HCl, slightly improved the yield (entry 3). We thought that a slow infusion of base would limit degradation of 12, considering its reported instability even at low temperatures.17 Slow infusion of base with a syringe pump over 30 min with an NH4Cl/6 M HCl quench dramatically increased the yield to 45%. Prolonging infusion of base to one hour increased the yield to 58%. The reaction did not seem greatly affected by scale as an outside company was able to scale the reaction to over 100 g without a decrease in yield.
Scheme 1: Improved synthesis of OV329.

Reagents and conditions: (a) PMBOH (1.5 equiv.), HCl, then 10, NaH (1.1 equiv.) 0 °C, THF/DMF (1:1), 6 h, 73%; (b) DBDMH (0.6 equiv), AcOH, 23 °C, 6 h, 90%; (c) K2CO3 (3 equiv), MeOH 1 h; (d) (COCl)2 (1.4 equiv), DMSO (2.3 equiv), Et3N (7 equiv), −78 °C, DCM, 60% over two steps; (e) see Table 1, 12 (1.2 equiv), KOtBu (1.5 equiv), DMF, −60 °C, 30 min, then NH4Cl, then 6 M HCl, then 23 °C, then 60 °C, 1 h, 58%; (f) CAN (3 equiv), MeCN, H2O, 0 °C, 1 h, 80%; (g) Boc2O (1.2 equiv), DMAP (0.1 equiv), Et3N (1.5 equiv), CH2Cl2, 1 h; (h) K2CO3 (3 equiv), MeOH, 6 h, 52% over two steps; (i) HCl (6 M), dioxane, 80 °C, 2 h, 97%. Abbreviations: PMBOH: 4-methoxybenzyl alcohol; DMF: N-N-dimethylformamide; DBDMH: 1,3-dibromo-5,5-dimethylhydantoin; CAN: ceric ammonium nitrite; DMAP: N,N-dimethylaminopyridine
Scheme 2.

Proposed Mechanism of Fluorination
Table 1.
Optimization of Fluorination a
| entry | Scale (g) | base addition method | quench (time)b | yieldc |
|---|---|---|---|---|
| 1 | 0.05 | dropwise over 5 min | 6 M HCl (5 min) | 0 |
| 2 | 0.05 | dropwise over 5 min | 6 M HCl (1 h) | 9% |
| 3 | 0.05 | dropwise over 5 min | a. sat. NH4Cl (1 h) b. 6 M HCl (1 min) |
15% |
| 4 | 0.13 | infusion over 30 min | a. sat. NH4Cl (1 h) b. 6 M HCl (2 min) |
45% |
| 5 | 1 | infusion over 60 min | a. sat. NH4Cl (1 h) b. 6 M HCl (2 min) |
58% |
| 6 | 3.5 | infusion over 90 min | a. sat. NH4Cl (1 h) b. 6 M HCl (2 min) |
50% |
| 7 | 120 | dropwise over 400 min | a. sat. NH4Cl (0.5 h) b. 6 M HCl (10 min) |
58% |
Conditions: 9 (1 equiv), 12 (1.2 equiv), DMF (0.3 M) −60 °C, then KOtBu (1.5 equiv) in DMF (0.5 M), then quench at −60 °C, then 23 °C, then 60 °C for 1 h;
time before quenching solution was added;
isolated yield after chromatography.
With 13 in hand, the next step was the methanolysis of the lactam and elimination. Deprotection of 13 proceeded smoothly to yield 14 in 80% yield. A small amount of 4-methoxybenzoyl-protected lactam also was isolated. Boc protection of 14 activated the lactam for methanolysis with K2CO3 and methanol, leading to subsequent elimination of the bromide. This reaction proceeded smoothly in a 53% yield over two steps with no observable isomerization of the olefin. Final deprotection at 80 °C in 6 M HCl yielded 1 in 97% yield with no observable isomerization or degradation. Overall, the yield from Vince lactam (10) to OV329 was 8.1%. The reaction scheme sequence was repeated with little to no modification by an outside company on kilogram scale resulting in over 40g of OV329 with a total yield of 3.7%.
In conclusion, we have developed a new method for the synthesis of OV329 (1), a potent inactivator of GABA-AT for the potential treatment of epilepsy and addiction. This method reduces the number of synthetic steps from 14 to 9, while increasing the overall yield for the synthesis from 3.7% to 8.1%. Furthermore, the synthesis does not involve the use of toxic selenium in the penultimate step or the use of tert-butyllithium. The elimination to form the cyclopentene is selective resulting in a single isomer, 1, and the entire synthesis can be run on kilogram scale. The key step involves the use of Hu’s reagent (12) to furnish a 1,1’-difluoroalkene followed by methanolysis and subsequent elimination. With an increased amount of OV329 in hand, we can now move into advanced preclinical studies for the treatment of epilepsy and addiction.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (Grant R01 DA030604 to R.B.S.) for financial support. The authors would like to acknowledge Dr. Wei Zhu and Dr. Sida Shen for helpful discussions. We would also like to acknowledge Wuxi Apptec for providing the large scale data. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource(NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN).
Footnotes
Supporting Information
Supporting Information is available free of charge on the ACS publications website.
Procedures, characterization, spectra (PDF)
REFERENCES
- (1).Karlsson A; Fonnum F; Malthe-Sorenssen D; Storm-Mathisen J Biochem. Pharmacol 1974, 23, 3053–3061. [DOI] [PubMed] [Google Scholar]
- (2).Yogeeswari P; Sriram D; Vaigundaragavendran J Curr. Drug Metab 2005, 6, 127–139. [DOI] [PubMed] [Google Scholar]
- (3).Doumlele K; Conway E; Hedlund J; Tolete P; Devinsky O Epilepsy Behav. Case Rep 2016, 6, 67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dewey SL; Morgan AE; Ashby CR; Horan B; Kushner SA; Logan J; Volkow ND; Fowler JS; Gardner EL; Brodie JD Synapse 1998, 30, 119–129. [DOI] [PubMed] [Google Scholar]
- (5).Pan Y; Gerasimov MR; Kvist T; Wellendorph P; Madsen KK; Pera E; Lee H; Schousboe A; Chebib M; Brauner-Osborne H; Craft CM; Brodie JD; Schiffer WK; Dewey SL; Miller SR; Silverman RB J. Med. Chem 2012, 55, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kushner SA; Dewey SL; Kornetsky CJ Pharmacol. Exp. Ther 1999, 290, 797–802. [PubMed] [Google Scholar]
- (7).Wild JM; Chiron C; Ahn H; Baulac M; Bursztyn J; Gandolfo E; Goldberg I; Goñi FJ; Mercier F; Nordmann J-P; Safran AB; Schiefer U; Perucca E CNS Drugs 2009, 23, 965–982. [DOI] [PubMed] [Google Scholar]
- (8).Juncosa JI; Takaya K; Le HV; Moschitto MJ; Weerawarna PM; Mascarenhas R; Liu D; Dewey SL; Silverman RB J. Am. Chem. Soc 2018, 140, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Silverman RB J. Med. Chem 2012, 55, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pan Y; Qiu J; Silverman RB J. Med. Chem 2003, 46, 5292–5293. [DOI] [PubMed] [Google Scholar]
- (11).“Q3D Elemental Impurities, Guidance for Industry”, US Food and Drug Administration, 2015, Accessed on: 3/13/2018, https://www.fda.gov/downloads/drugs/guidances/ucm371025.pdf.
- (12).Qiu J; Silverman RB J. Med. Chem 2000, 43, 706–720. [DOI] [PubMed] [Google Scholar]
- (13).Deng Z; Lin JH; Cai J; Xiao JC Org. Lett 2016, 18, 3206–3209. [DOI] [PubMed] [Google Scholar]
- (14).Li Q; Lin J-H; Deng Z-Y; Zheng J; Cai J; Xiao J-CJ Fluor. Chem 2014, 163, 38–41. [Google Scholar]
- (15).Zhao Y; Huang W; Zhu L; Hu J Org. Lett 2010, 12, 1444–1447. [DOI] [PubMed] [Google Scholar]
- (16).Gao B; Zhao Y; Hu J; Hu J Org. Chem. Front 2015, 2, 163. [Google Scholar]
- (17).Gao B; Zhao Y; Hu M; Ni C; Hu J Eur. J. Chem 2014, 20, 7803–7810 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.