

Abstract
T98G cells have been shown to support long-term human cytomegalovirus (HCMV) genome maintenance without infectious virus release. However, it remains unclear whether these viral genomes could be reactivated. To address this question, a recombinant HCMV (rHCMV) containing a GFP gene was used to infect T98G cells, and the infected cells absent of infectious virus production were designated T98G-LrV. Upon dibutyryl cAMP plus IBMX (cAMP/IBMX) treatment, a serial of phenomena were observed, including GFP signal increase, viral genome replication, lytic genes expression and infectious viruses release, indicating the reactivation of HCMV in T98G-LrV cells from a latent status. Mechanistically, HCMV reactivation in the T98G-LrV cells induced by cAMP/IBMX was associated with the PKA-CREB signaling pathway. These results demonstrate that HCMV was latent in T98G-LrV cells and could be reactivated. The T98G-LrV cells represent an effective model for investigating the mechanisms of HCMV reactivation from latency in the context of neural cells.
Keywords: Human cytomegalovirus, Reactivation, Latent infection, T98G cells, Latent cell model of brain origin
1. Introduction
Human cytomegalovirus (HCMV) is a member of the beta-herpesvirinae. It is a ubiquitous pathogen with the serum positive rate as high as 95% in China. In immunocompetent individuals, primary HCMV infection is typically asymptomatic, but establishes a lifelong persistent/latent infection in its host. However, in immunocompromised individuals, such as AIDS patients and transplant recipients, primary infection or reactivation/recurrent infection of HCMV results in serious diseases, including pneumonia, gastrointestinal disease, retinitis and nephritis (Ljungman et al., 2002). In addition, congenital HCMV infection caused by a maternal primary-infection or reactivation causes 5–10% of infected neonates to suffer microcephaly or periventricular calcification at birth. 10–15% of the subclinically infected infants subsequently develop late-onset sequelae including sensorineural hearing loss, mental retardation and learning disabilities within 3 years (Leung et al., 2003). HCMV reactivation has largely focused on hematopoietic stem cells (Goodrum et al., 2002; Hahn et al., 1998a; Khaiboullina et al., 2004; Kondo et al., 1994; Mendelson et al., 1996; Soderberg-Naucler et al., 2001). Certainly late-onset neurodevelopmental disorders could be caused by virus reactivation from the myeloid reservoirs, persistent infection, or a new lytic infection. However, were they to exist in vivo, reactivation from latently infected neural cells could also contribute. In order to study the potential mechanism(s) of late-onset neurodevelopmental disorders caused by congenital HCMV infection, an effective latent-reactivation HCMV infection model in the context of neural cell type is crucial.
HCMV infection is characterized as lytic or persistent/latent infection. During lytic infection in permissive cells (such as fibroblasts, endothelial cells, epithelial cells and macrophages), viral genes are expressed in an temporal cascade (Wathen et al., 1981; Wathen and Stinski, 1982). The major immediate early (IE) genes are the first viral genes to be transcribed, resulting in abundant proteins (such as IE1 and IE2). These IE proteins activate the expression of early genes (such as UL44 and UL54), which are required for viral DNA replication and eventually lead to the expression of late genes (such as UL94 and UL99). Finally, the progeny viruses are assembled and released.
After primary infection, HCMV establishes a latent infection in specific sites within the hosts. Latent infection is defined as a type of persistent infection and characterized as maintenance of the viral genome without shedding infectious virus except for intermittent episodes of reactivation (Knipe et al., 2013). At present, latent HCMV is commonly accepted to reside within hematopoietic stem cells in vivo, particularly in undifferentiated myeloid lineage and monocytes, such as CD34+ progenitor cells (Hahn et al., 1998b), granulocyte-macrophage progenitors (GM-Ps) (Kondo et al., 1994), and CD14+ monocytes (Bolovan-Fritts et al., 1999; Taylor-Wiedeman et al., 1991). In vitro HCMV latent cell models, including embryonic stem cell lines (Penkert and Kalejta, 2013), myeloid progenitor cell line Kasumi-3 (O’Connor and Murphy, 2012), monocytic THP-1 cells (Bego et al., 2005; Weinshenker et al., 1988), and human teratocarcinoma Nera-2 (NT2) cells (Gonczol et al., 1984), have been well established. These models are useful for exploring HCMV pathogenesis in immunocompromised individuals, but not suitable for investigating the mechanism of HCMV reactivation in the context of neural cells.
Our previous studies have demonstrated that T98G glioblastoma cells are semi-permissive for HCMV infection as viral protein expression is delayed. Moreover, HCMV-infected T98G cells harbor viral genomes but without detectable infectious virus following passaging (Duan et al., 2014; Luo and Fortunato, 2007). This suggested the T98G cells might serve as an HCMV latent-infection cell model. However, the latency status of HCMV in T98G cells can only be confirmed upon successful reactivation, evidenced by viral replication and release of infectious viruses, which remains unclear so far.
The stimuli and the corresponding mechanisms involved in HCMV reactivation are not fully understood. Previous reports demonstrate that HCMV lytic infection is dependent on the status of cellular differentiation. Treatments with cellular differentiation associated agents, such as phorbol ester (TPA), retinoic acid, cyclic AMP (cAMP) or cAMP plus 3-isobutyl-1-methylxanthine (IBMX), resulted in differentiating cells into HCMV-permissive cells, inducing the viral lytic genes transcription and/or promoting infectious virions release (Matsukage et al., 2006; Meier, 2001; Poland et al., 1994; Stamminger et al., 1990; Weinshenker et al., 1988). Moreover, it is becoming increasingly evident that chromatin remodeling (as histone modifications present on the MIEP, resulting in chromatin structure loosening) affects the transcriptional activity in HCMV reactivation (Ioudinkova et al., 2006; Reeves, 2011; Sinclair, 2010). A series of inhibitors specific for epigenetic regulation, including histone deacetylase (HDACs) inhibitors trichostatin A and sodium butyrate, and DNA methylation inhibitor 5′-aza-2′- deoxycytidine, have been proved to be capable of activating the CMV promoter (Choi et al., 2005; Grassi et al., 2003; Murphy et al., 2002).
Here we used a Towne strain derived recombinant HCMV (rHCMV) containing an SV40 promoter driven GFP gene in the viral genome. rHCMV can be visualized by fluorescence microscopy and analyzed by flow cytometry, allowing determination of rHCMV infection status in the T98G cells. The infected cells absent of infectious virus production were designated as T98G-LrV. We further observed that dibutyryl cAMP plus IBMX (cAMP/IBMX) treatment effectively reactivated the latent rHCMV in T98G-LrV potentially via the PKA-CERB signaling pathway. This cell model of brain origin should offer a significant improvement to understand the pathogenesis of HCMV-caused late-onset neurodevelopmental disorders.
2. Materials and methods
2.1. Cells culture and virus infection
Human T98G glioblastoma cell line (ATCC CRL-1690) and human foreskin fibroblasts (HFFs) were cultured in growth medium of minimal essential medium (MEM) (Gibco BRL) supplemented with 10% fetal bovine serum (FBS) (Gibco BRL), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco BRL).
Recombinant HCMV virus (rHCMV) was derived from Towne strain by inserting a GFP gene driven by an SV40 promoter into the viral genome (Marchini et al., 2001). Virus was propagated in HFFs and virus titers were determined by plaque forming assay as described previously (Compton et al., 1992).
T98G cells were infected with rHCMV as previously reported (Duan et al., 2014; Luo and Fortunato, 2007). Briefly, serum starved T98G cells were plated in 100 mm cell culture dishes (5 × 105 cells/dish) and infected with rHCMV at an multiplicity of infection (MOI) of 10. At 16 h post infection, the inoculum was removed, and the cells were refed with growth medium after washing with phosphate-buffered saline (PBS, pH 7.4). Then the infected T98G cells were reseeded to new 100 mm dishes at a density of 5 × 105 cells/dish, cultured for 7 days and passaged. Notably, the interval of passaging was modified to 7 days instead of 3 days as previously reported, which is to ensure the virus completing a full replication cycle. The original rHCMV-infected T98G cells were designated as passage 0 (P0), the following subculture cells were designated as P1, P2,….. Cells at P5 which maintained viral genome with undetectable infectious virus were designated as T98G-LrV cells.
2.2. Treatment with chemical reagents
T98G-LrV cells were plated in T25 flasks (5 × 105 cells/flask) or 6-well plates (1.5 × 106 cells/plate). To reactivate the latent virus in T98G-LrV cells, the growth medium was replaced with maintenance medium of MEM supplemented with 2% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), 100 μM dibutyryl cAMP, and 100 μM IBMX. The cells were cultured in the presence of the treatment reagents with refreshing the medium every 4 days, and harvested until the indicated times. Where indicated, T98G-LrV cells were pretreated with phosphonoacetic acid (PAA, 50 μg/ ml) and H89 (5 μM or 10 μM). All chemical reagents used in the study were purchased from Sigma (Sigma-Aldrich, USA).
2.3. Determination of infectious virus
Supernatant samples or cells were harvested. The cell pellets were resuspended in the growth medium. Aliquots of the cell suspension were repeatedly freeze-thawed to prepare cell lysate samples. The supernatants and cell lysates were inoculated on monolayers of HFFs to evaluate the infectious virus which was judged by GFP signals. At 14 days post incubation, the cells were observed with a Nikon Eclipse Ti-S fluorescence microscope (Nikon, Tokyo, Japan).
After reactivation treatment, infectious cell-associated viruses were determined by coculture HFFs with treated cells directly or after sonication. DMSO or cAMP/IBMX treated T98G-LrV cells were harvested, and resuspended to 1 × 105 cells/ml. A portion of the cell suspension was sonicated to prepare the sonicated cell samples. Treated T98G-LrV cells (1 × 105) or the sonicated cells (1 × 105) were inoculated onto monolayers of HFFs (5 × 105). Three hours later, soft agar (0.4% W/V) was added to characterize the ability of cells to form GFP plaques. After 10 days post coculture, GFP plaques on HFFs were observed with a Nikon Eclipse Ti-S fluorescence microscope (Nikon, Tokyo, Japan).
2.4. Flow cytometry (FACS)
Aliquots of cells were harvested after trypsinization and resuspended in PBS. GFP-positive cells were determined on a FACScan flow cytometer using the CellQuest software package (Becton Dickinson, Franklin Lakes, N.J.). Twenty thousand live cell events were acquired for each analysis, and the GFP gate was set according to the mock-infected T98G cells.
2.5. PCR and qPCR
Cellular DNA was extracted using a Tissue/Cells/Blood Extraction Kit (TianGen, China). Viral genomic DNA was detected by nested PCR for IE1 gene. Viral genome copy numbers were determined by absolute quantitative PCR (qPCR). The plasmids pcDNA3.0-UL83 and pcDNA3.0-GAPDH were constructed and used to generate the standard curve for quantification as previously described (Fu et al., 2015). Viral genome copy number was calculated and presented as “copies/ cell” according to normalization with GAPDH.
Total RNA was extracted from the cells using RNAiso reagent (Invitrogen). After treatment with DNase I, equal amounts of RNA were subjected to reverse transcription PCR (RT-PCR) using the RT Primer Mix (TAKARA) according to the manufacturer’s instruction. The resulted cDNA from passages of infected T98G cells was subjected to PCR to detect the transcription of the indicated viral genes, including viral lytic gene UL44 and viral latency associated genes (latency unique nuclear antigen (LUNA), US28, vIL10) (Duan et al., 2014).
cDNA from drug treated cells were subjected to qPCR to analyze the relative transcription level of the representative viral lytic genes on a Bio-Rad CFX Manager (Bio-Rad, Hercules, USA). The examined viral lytic genes include IE genes (IE1, IE2), E genes (UL44 and UL54), and L genes (UL94 and UL99). Cycle conditions were as follows: 3 min at 95 °C, 40 cycles of 95 °C for 30 s, 63 °C for 10 s. The results were presented as the fold change compared to the calibrators (the DMSO treated T98G-LrV cells) following normalization to the reference gene GAPDH. The specific gene primers were shown in Table 1.
Table 1.
Nucleotide sequences of primers used in the study.
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| aUL83a | GCGAGACCGTGGAACTGC | TTGCCCTGGATGCGATACTG |
| aGAPDHa | GACCCCTTCATTGACCTCAACTA | TCCTGGAAGATGGTGATGGG |
| IE1 outerb | CTGTATGTGACCCATGTGCTT | GGGGCAACTTCCTCTATCTCA |
| IE1 innerb | TCAGCCTTCCCTAAGACCAC | ACATGGTCATCATACAAGCG |
| RNA-LUNAb | CGCGGTTATTATCAACGTCT | GCTCGTCATCCCCTCGTG |
| RNA-US28b | ACTGCCTGTTTCTACGTGGCTA | ATAAAGACAAGCACGACCGCTA |
| RNA-vIL10b | ATTGCAAGATCTCCGCGTCACC | CCGCAACCTAACAGAGGGCATT |
| RNA-UL44 | CGGCGGTGGCGGCAAGAAG | AGAATCCTCGCTGTCGCTCTCCTC |
| qIE1 | GCCTTCCCTAAGACCACCAAT | ATTTTCTGGGCATAAGCCATAATC |
| qIE2 | ATGGTTTTGCAGGCTTTGATG | ACCTGCCCTTCACGATTCC |
| qUL44 | GTCGGGAACAGCGGCAAGTC | CGCCACCTCCTCCCAGACC |
| qUL54 | CCGTGTCACCTAACGCCGCTATC | CGCCGACGCTGCTACTACTGTTAC |
| qUL94 | TGACGCAGTCGCTCATCCACAAC | CCAGGTCCCACACATCGGTCTTG |
| qUL99 | GGGTCGCCAGGTGTCTCTACG | GCTGCCGCTACTATTGTCGTTTCC |
| GAPDHc | TCTCTGCTCCTCCTGTTCGAC | GACAAGCTTCCCGTTCTCAG |
The primers are used for determination of viral genome copies and are the same as that used in our previous report (Fu et al., 2015).
The primers are the same as that used in our previous report (Duan et al., 2014).
The primers are used in the assay of RT-PCR and qPCR.
2.6. Immunofluorescent assay (IFA)
Cells were plated on coverslips in 100 mm cell culture dishes (5 × 105 cells/dish) or 12-well plates (1.5 × 106 cells/plate). At indicated time point, coverslips were harvested and subjected to IFA as described previously (Duan et al., 2012). Briefly, cells were collected, fixed by 4% formaldehyde, and permeabilized with 0.1% Triton X-100. After blocking with 30% FBS, samples were incubated with mouse monoclonal antibody against UL44 or pp28 respectively, and subsequently reacted with a combination of specific anti-mouse secondary antibody and Hoechst. Coverslips were then mounted onto slides. Images were acquired using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan) equipped with a NIS-elements viewer (version 4.20). For each experiment, minimum three coverslips were used, and at least five random fields were observed on each coverslip.
2.7. High content analysis (HCA)
DMSO or cAMP/IBMX treated T98G-LrV cells were trypsinized, plated into black 96-well plates (2 × 104 cells/well), and cultured in growth medium containing DMSO or cAMP/IBMX for two days. Cells were subjected to high content imaging and high content analysis for determination of the GFP positive cells using a Operetta high content screening system (PerkinElmer, Waltham, MA) (Buchser et al., 2014).
2.8. Western blotting
Cells were lysed with RIPA buffer (Beyotime, Shanghai, China) following the manufacturer’s instructions. The protein concentration was measured using BCA (Beyotime, Shanghai, China). Equal amount of protein samples were separated by SDS-PAGE electrophoresis and then transferred to PVDF membranes (Millipore). After blocking with 5% (w/v) milk-TBST, the blots were incubated with the primary antibodies against the indicated targets, followed by the corresponding horseradish peroxidase-conjugated secondary antibody. The primary antibodies include mouse monoclonal antibodies against IE1/IE2, UL44 (Virusys), actin (Abclonal), and rabbit monoclonal antibodies against CREB and phospho-CREB (Cell signaling technology). The protein bands were detected using Enhanced Chemiluminescence (Thermo) according to the manufacturer’s instructions.
2.9. Statistical analysis
All data shown were either the representative results or the average from at least 3 independent experiments. The average results were presented as the mean ± standard deviation (SD). Statistical analysis was performed with student’s t-test using the SPSS Statistics software package (Version 17.0). P values were calculated and statistical significance was reported as significant using *p < 0.05, ** p < 0.01, or *** p < 0.001.
3. Results
3.1. Latent infection T98G cell model is established with rHCMV
HCMV-infected T98G cells have been previously reported to harbor viral genome and express a subset of viral genes at limited levels, but lack detectable levels of infectious virus (Duan et al., 2014; Luo and Fortunato, 2007). To establish a latent HCMV infection cell model with a visible reporter for conveniently monitoring virus reactivation, a Towne strain derived recombinant HCMV (rHCMV) containing a SV40 promoter driven GFP gene in the viral genome (Marchini et al., 2001) was used to infect T98G cells.
To verify whether the GFP was a reliable reporter for monitoring virus infection, rHCMV-infected T98G cells were analyzed by IFA to determine the expression of GFP and representative viral protein pp28. As shown in Fig. 1, the presence of pp28 protein was always associated with GFP expression, and vice versa. These data demonstrated that GFP was an accurate marker for virus infection in the T98G cells.
Fig. 1. GFP is a reliable marker for monitoring rHCMV infection in T98G cells.
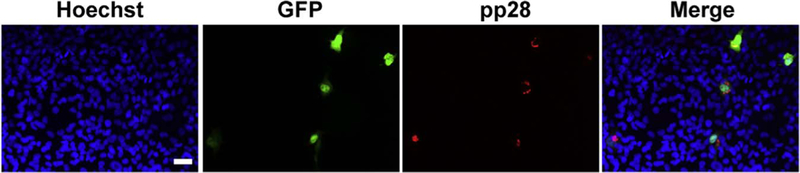
T98G cells cultured on coverslips were infected with rHCMV at an MOI of 10. At 7 days post infection (dpi), cells were analyzed for expression of GFP and late protein pp28 by IFA. Representative images from three independent experiments are shown. Scale bar: 50 µm.
rHCMV infection in the T98G cells was further characterized during passages. Because of the delayed viral proteins expression in T98G cells, rHCMV-infected T98G cells were passaged every 7 days in this study, instead of every 3 days as described previously (Luo and Fortunato, 2007), to allow the virus to complete its entire replication cycle. Cell-free and cell-associated virus were examined by inoculating the supernatants or freeze-thaw cells onto HFFs. HFFs inoculated with samples from the first 4 passages were infected by rHCMV, as evidenced by the obvious GFP signals, indicating the existence of infectious virions both in the supernatant and in the cells. However, no GFP signal was observed in HFF inoculated with samples after passage 5 (P5), which suggested the absence of infectious virus since P5 (Fig. 2A). The infectious virus in the supernatant was further titrated by plaque forming assay. Virus titers showed that cell-free virus became barely detectable after P3, and were completely undetectable from P5 (Fig. 2B).
Fig. 2. Establishment of latently infected T98G cells with rHCMV (T98G-LrV).
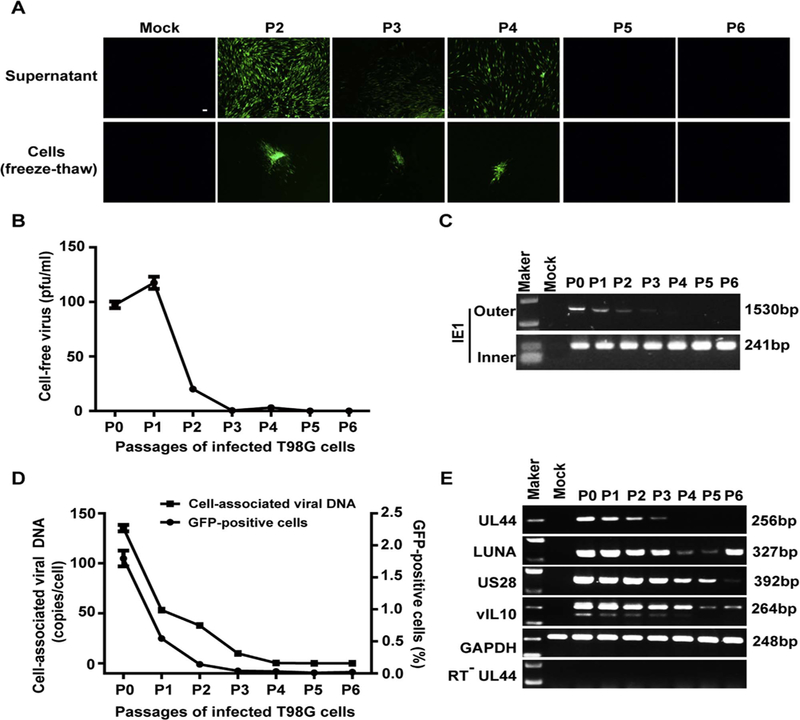
T98G cells were infected with rHCMV at an MOI of 10 and passaged every 7 days. Supernatants and cells of each passage were harvested before subculture. (A) Infectious virus. HFFs monolayers were inoculated with the supernatants (upper panel) or the infected cells (frozen-thawed cells, lower panel). GFP-positive foci were observed 14 days post inoculation. Scale bar: 100 µm. (B) Cell-free virus titer. Virus titers in the supernatants were determined by plaque forming assay. (C) Viral DNA. Cellular DNA was extracted from the infected cells of each passage, and viral genomic DNA was examined by nested PCR based on IE1 gene. Shown are the representative results of the first (Outer) and nested (Inner) amplification of PCR, respectively. (D) Cell-associated viral DNA and GFP-positive cells. Viral genome copy numbers in the infected cells were determined by qPCR. Serial dilution of T98G cells was isolated for DNA and subjected to qPCR for determination the GAPDH copy number of each cell. Viral genome copies were calculated as “copies/cell” with adjusting to GAPDH. GFP-positive cells at each passage was analyzed by FACS. (E) Transcription of representative viral genes. Total RNA was extracted from each set of the passaged cells, reversely transcribed to cDNA, and assayed for the indicated genes by PCR. The mRNA without reverse transcription (RT−) were assayed for UL44 as control to exclude viral DNA contamination.
Viral genome replication levels in the passaged cells were determined by PCR and qPCR. The results from nested PCR showed that the viral genomes persistently existed in the infected T98G cells (Fig. 2C). Moreover, qPCR results also indicated that viral genomes were persistently maintained during passaging (Fig. 2D). These results are consistent with our previous reports (Duan et al., 2014; Luo and Fortunato, 2007). To further determine whether GFP expression was associated with virus replication, GFP-positive cells were evaluated by FACS. The results showed that GFP-positive cells gradually decreased from 1.795% at P0 to 0.019% at P6, which well mirrored the change patterns of cell-associated viral DNA (Fig. 2D). Furthermore, RT-PCR confirmed the transcription of three representative latency associated genes, LUNA, US28 and vIL10, were observed throughout the passages. Importantly, the transcription of lytic gene UL44 was gradually reduced in the first 3 passages and no longer evident from P4 (Fig. 2E).
These results indicate that infectious viruses gradually decreased along with passaging and became undetectable, while viral genomes were maintained in rHCMV infected T98G cells, indicating a switch to latency. This infection profile was consistent with our previous reports (Duan et al., 2014; Luo and Fortunato, 2007). The potentially latent rHCMV-infected T98G cells at P5, which maintained viral genomes but absent of infectious virus production, were designated as T98G-LrV cells.
3.2. cAMP/IBMX reactivates latent HCMV
HCMV latency is defined as maintenance of the viral genome but absent of infectious virus, with these genomes capable of being reactivated in the presence of stimuli. Having established that rHCMV genomes were maintained in T98G-LrV cells without detect-able infectious virus, we next considered whether the latent virus was capable of being reactivated upon relevant stimulation. We found that combination of cAMP and IBMX (cAMP/IBMX) potentially reactive the latent virus. Compared to DMSO treated control, the GFP plaques were significantly increased by cAMP/IBMX (Fig. 3A upper panel b, left), representing an increase of 13.5-fold (Fig. 3A upper panel, right). Magnified images display that the GFP plaques were clearly enlarged by cAMP/IBMX treatment (Fig. 3A, a1 versus b1–2). Notably, single GFP-positive cells were also observed in the distant area from the GFP plaques upon cAMP/IBMX treatment (Fig. 3A b3). Whereas, there was no single GFP-positive cell presented in the DMSO treated control (Fig. 3A a2–3). These phenomena might be a consequence of subsequent infection by released progeny virions. To confirm this possibility, cell-free supernatants of the treated T98G-LrV cells were inoculated onto HFFs monolayers. As shown in Fig. 3B, GFP signal were only observed in the HFFs incubated with supernatant from cAMP/IBMX treated cells, but not from DMSO treated cells, indicating that cell-free viruses were recovered in T98G-LrV cells upon cAMP/IBMX treatment. All together, these data indicated that cAMP/IBMX potentially reactivated the latent HCMV in T98G-LrV cells.
Fig. 3. cAMP/IBMX reactivates latent HCMV in T98G-LrV cells.
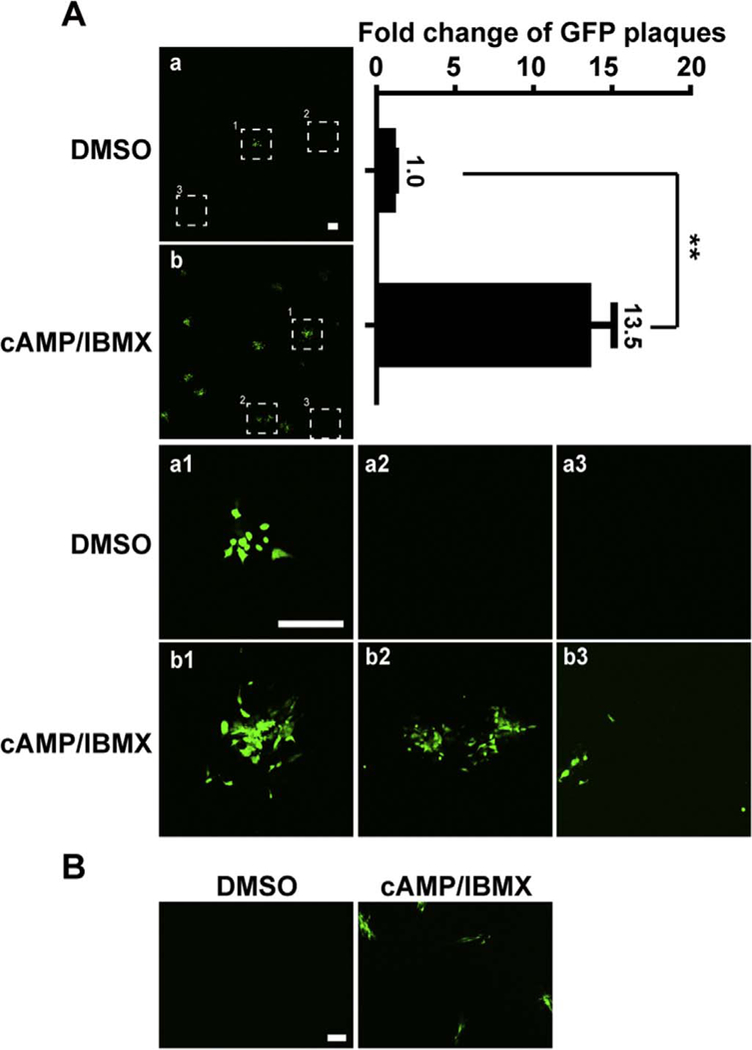
T98G-LrV cells were treated with DMSO or cAMP/IMBX for 30 days. (A) GFP signal in the treated T98G-LrV cells. GFP signal in cells was observed using a Nikon A1 confocal microscope. The numbers of GFP foci were counted and the fold changes compared to the DMSO control were calculated. Representative images (upper panel, left) and the fold change (upper panel, right) from 3 independent experiments are shown. The magnified images of the indicated sites are shown in a1–3 and b1–3, respectively. Scale bar: 500 µm. ** p < 0.01. (B) Infectious virions release. The cell-free supernatants from the treated T98G-LrV cells were harvested, and inoculated onto HFFs monolayers for 14 days to detect infectious virions. Scale bar: 100 µm.
3.3. cAMP /IBMX increases GFP signal in T98G-LrV cells
The reactivation efficacy of cAMP/IMBX was further determined by FACS and HCA. DMSO and cAMP/IBMX treated T98G-LrV cells were subjected to FACS to analyze the GFP positive frequency. Results showed that 0.027% of the DMSO-treated T98G-LrV cells expressed GFP; and the frequency increased to 1.54% after cAMP/IBMX treatment (Fig. 4A). The increase of GFP positive cells upon cAMP/IBMX treatment was further confirmed by HCA (Fig. 4B, upper panel). The quantification analysis of HCA showed an about 15-fold increase of GFP positive cells by cAMP/ IBMX treatment compared to the DMSO control (Fig. 4B, lower panel). Taken together, cAMP/IBMX significantly increased the GFP-positive cells.
Fig. 4. cAMP/IBMX increases GFP signal.
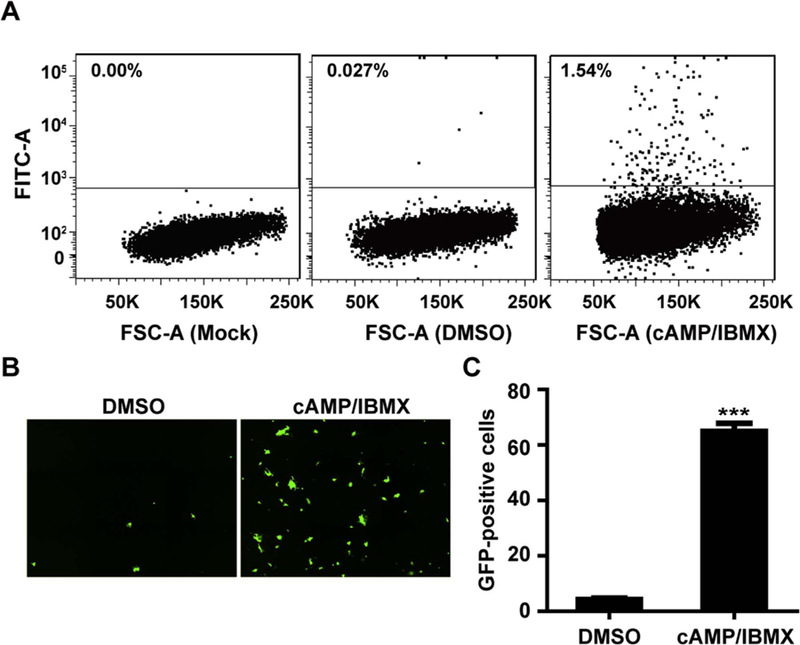
T98G-LrV cells were treated with DMSO or cAMP/IBMX for 30 days to detect the GFP signal. (A) Flow cytometry analysis of GFP-positive cells. Representative results are shown from 3 independent experiments. (B-C) High content analysis of GFP-positive cells. Representative high content images and the average GFP positive cells per well from 10 independent replicates are shown. *** p < 0.001.
3.4. cAMP /IBMX initiates HCMV replication
Although GFP expression was coincident with that of pp28 (Fig. 1), whether the increased GFP signal was associated with viral replication needed to be further investigated. UL44, a CMV DNA polymerase accessory protein, found in the viral replication compartments (Pari et al., 1993; Park et al., 2006; Weiland et al., 1994), was used to elucidate viral replication and GFP expression. cAMP/IBMX treatment increased the GFP-positive cells, which were also UL44 positive. Notably, more advanced single large replication foci were contained in cAMP/IBMX-treated cells than that in the DMSO control (Fig. 5A). These data indicated that the increased GFP signal was coincident with UL44 expression and viral replication center development.
Fig. 5. cAMP/IBMX initiates HCMV replication.
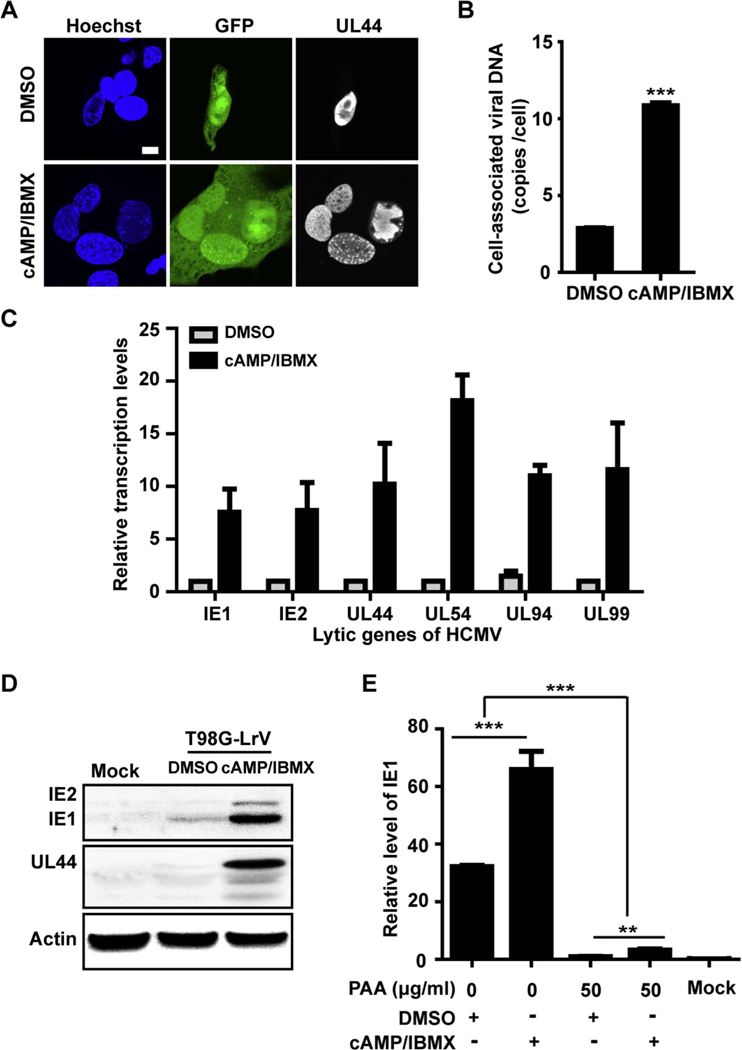
T98G-LrV cells were treated with DMSO or cAMP/IBMX for about 30 days. Cells and supernatants were collected for following assays. (A) Viral replication centers. The cells were plated onto coverslips and examined the expression of GFP and UL44 by IFA. Scale bar: 10 µm. (B) Viral genome copy number. Total DNA was extracted from the collected cells, and viral genome copy number per cell was determined by qPCR. *** p < 0.001. (C) Transcription levels of the representative lytic viral genes. RNAs were extracted from the collected cells followed by RT-PCR, and relative transcription levels of the indicated viral genes were assessed by qPCR with GAPDH as the reference gene. (D) Viral proteins expression. The collected cells were lysed and subjected to western blotting for examination of the indicated viral proteins. Actin serves as the loading control. (E) Effect of PAA on IE1 transcription. T98G-LrV cells were pretreated with PAA for 12 days, then cultured for additional 8 days in the presence of DMSO or cAMP/IBMX. Total RNA was extracted, and IE1 transcription level was quantified by qPCR. The relative level of IE1 was calibrated to that in the DMSO treated cells in the presence of PAA, with GAPDH as the reference gene, and the uninfected T98G cells as the mock.
To further confirm virus replication, T98G-LrV cells treated with DMSO or cAMP/IBMX for 30 days were examined for viral genome copy number, viral genes transcription and expression. After treatment with cAMP/IBMX, cell-associated virus genomes showed a 3.79-fold increase compared to DMSO control (Fig. 5B). Transcription levels of the representative lytic genes, including IE genes (IE1, IE2), early genes (UL44, UL54) and late genes (UL94, UL99), were also significantly increased upon cAMP/IBMX treatment (Fig. 5C). Moreover, despite of the low level expression in the controls, viral proteins of IE1/ IE2 and UL44 were also dramatically induced in cAMP/IBMX treated T98G-LrV cells (Fig. 5D). PAA inhibits DNA polymerase but does not affect IE expression unless virus replication (Huang, 1975). The transcription of IE1 was significantly suppressed upon PAA treatment, further confirming that cAMP/IBMX treatment induced viral genome replication. While the IE1 transcription in the DMSO treated cells was barely detectable after PAA treatment, indicating there was limited virus genome replication, which was coincident with the low level expression of IE1/IE2 and UL44, in the DMSO treated T98G-LrV cells (Fig. 5E).
3.5. cAMP /IBMX induces the production of infectious viruses
Since the characteristic of HCMV reactivation is the recovery of infectious virus, the cell-free and cell-associated viruses were further examined in the treated cells. As Fig. 3B showed cAMP/IBMX but not DMSO treatment recovered the infectious virus. Here, to monitor the dynamics of cell-free infectious virus release, supernatants of the treated cells were collected during treatment process and subjected to virus titration by plaque forming assay. Recovery infectious viruses were observed in the T98G-LrV cells treated with cAMP/IBMX, but not in the DMSO control (Fig. 6A), which is coincidence with the result of Fig. 3B. Interestingly, virus titers displayed an obvious increase at 12 days post treatment, then decreased and maintained a gradual increase during treatment process, which might be associated with the delayed expression of viral genes and asynchronous reactivation of latent virus (Fig. 6A).
Fig. 6. cAMP/IBMX increases infectious viruses.
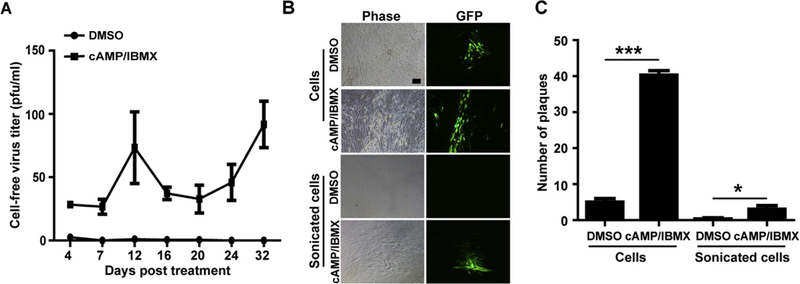
(A) Cell-free virus titer. Supernatant samples were harvested at the indicated times and cell-free virus was titrated by plaque forming assay. The results were presented as mean ± SD from 3 biological replicates. (B-C) Cell-associated virus. T98G-LrV cells were treated with DMSO or cAMP/IBMX for 30 days. Then intact cells or sonicated cells were cocultured with HFFs monolayers in 6-well plates. GFP signals were observed at 10 days post coculture and the numbers of GFP plaque per well were counted. Representative images of GFP plaques and the average results of GFP plaque presented as mean ± SD from 3 biological replicates are shown. Scale bar: 100 µm. * p < 0.05; *** p < 0.001.
Cell-associated virus was examined by directly explanting the treated T98G-LrV cells onto the HFFs monolayer. Although GFP signal presented in coculture with DMSO treated cells, more GFP plaques were observed in coculture with cAMP/IBMX treated cells (Fig. 6B upper panel and 6C). Intact cell explanation resulted it is difficult to differentiate whether the GFP-positive plaques were caused by infectious virions or GFP-positive T98G cells. To further confirm the existence of infectious virus, same amounts of the treated cells were sonicated prior to coculture with HFFs. Although the amount was dramatically decreased, the GFP positive plaques were still observed in the HFFs cocultured with sonicated cAMP/IBMX treated cells, but not with the sonicated DMSO treated cells (Fig. 6B lower panel and 6C). These data confirmed that cAMP/IBMX treatment recovered the infectious virus production in T98G-LrV cells. In addition, despite of the low level viral gene expression and genome replication, the absence of infectious virus indicated that no virus reactivation were induced by DMSO treatment.
Taken together, these results demonstrated that the latent HCMV in T98G-LrV cells was reactivated by cAMP/IBMX treatment.
3.6. HCMV reactivation induced by cAMP/IBMX is associated with the PKA-CREB signaling pathway
It has been reported that cAMP elevation stimulates the protein kinase A (PKA) signaling pathway, which thereby activates IE gene expression and induces HCMV lytic replication from latent infection (Keller et al., 2007; Yuan et al., 2009). The pathway is mediated by phosphorylating the cAMP response element-binding transcription factor (CREB) and activating transcription factor 1 (ATF-1) (Montminy, 1997; Shaywitz and Greenberg, 1999), However, HCMV reactivation via the PKA signaling pathway is cell type dependent (Prösch et al., 2000). To evaluate whether PKA-CREB signaling is involved in HCMV reactivation in the T98G-LrV cells, PKA inhibitor H89 was applied to treat the cells. H89 treatment significantly reduced the recovered viruses (Fig. 7A), and suppressed the transcription level of IE1 (Fig. 7B), which were both induced by cAMP/ IBMX. These inhibitions of H89 were in dose dependent manners. Although the steady-state protein levels of CREB were not changed by either cAMP/IBMX or H89 treatment, the levels of phosphorylated CREB and ATF-1 were clearly elevated by cAMP/IBMX, but inhibited by H89 treatment in a dose dependent manner. These inhibitions were mirrored by similar changes in IE1 protein levels (Fig. 7C). These results indicated that cAMP/IBMX activated the PKA-CREB signaling pathway, which was potentially involved in HCMV reactivation in T98G-LrV cells.
Fig. 7. HCMV reactivation in T98G-LrV cells is associated with the PKA-CREB signaling pathway.
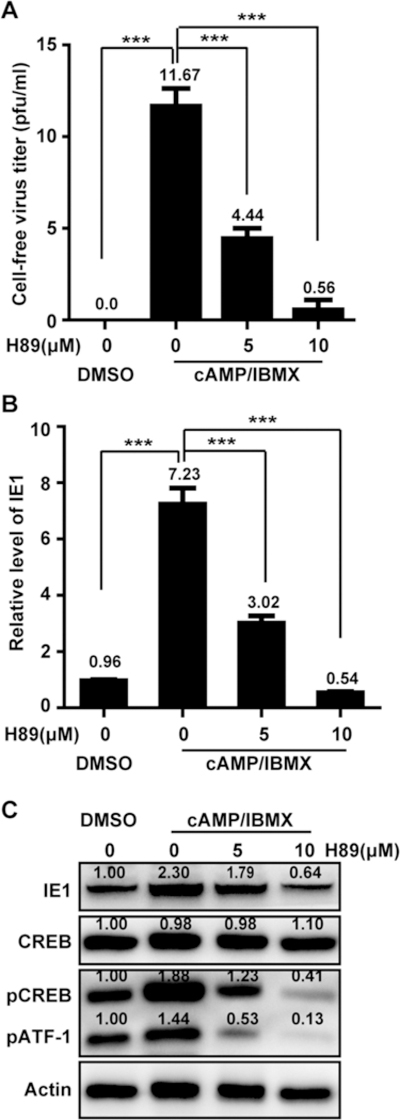
T98G-LrV cells were pretreated with H89 for 1 h, and then treated with cAMP/IBMX in the presence of H89 for 8 days. Medium was changed every 4 days. Supernatants and cells were collected for the subsequent assays. (A) Cell-free virus titer. Supernatants were subjected to the plaque forming assay to determine the cell-free virus. The results were presented as mean ± SD from 3 biological replicates. (B) Relative level of IE1. Total RNA was extracted from the collected cells, and relative transcription level of IE1 was examined by qPCR with GAPDH as the reference gene. *** p < 0.001. (C) Protein levels of IE1 and representative components of PKA-CREB signaling pathway. Cell lysates were prepared and equal amount of proteins were subjected to western blotting. The protein levels were quantitated by densitometry using Image J software (National Institutes of Health) and presented as the relative level to that of DMSO control after normalization to actin.
4. Discussion
It is well known that herpesviruses latently reside in specific sites after primary infection and recurrent after reactivation in accompanying with associated diseases. For example, latent herpes simplex virus 1 (HSV1) establishes latency in neurons of the dorsal root ganglia and trigeminal ganglia, and its reactivation leads to orofacial infections and encephalitis (Whitley et al., 1998; Whitley and Roizman, 2001). Latent Epstein-Barr virus (EBV) has been found primarily in B lymphocytes and leads to lympho-proliferative syndrome after reactivation (Cohen, 2000). Kaposi’s sarcoma -associated herpes virus (KSHV) reactivation within the lymphoid reservoir disseminates to recruit endothelial cell targets and encodes numerous molecules to promote neoangiogenesis, which is associated with carcinoma (Arvanitakis et al., 1997; Bais et al., 1998). Substantial evidence has accumulated that latent HCMV resides in myeloid lineages (Schrier et al., 1985; Sinclair, 2008), and there have been no published reports of latency in cells of neural origin. However, a tractable in vitro model for a latent-like infection of neural cells provides an additional experimental option to explore possible mechanisms that might contribute to late-onset neurodevelopmental disorders caused by congenital HCMV infection (Stagno et al., 1982), and adult neurological damages occur upon suppression of immunity (Egbert et al., 1980; Jacobson et al., 1997).
Due to the intrinsic difficulty of obtaining primary human neural cultures, much work has been done with established neoplastic cells of human brain origin. Those established cells include human astrocytoma/glioblastoma (Poland et al., 1990), neuroblastoma cells (Jault et al., 1994), and oligodendroglioma cell line (Spiller et al., 1997). However, most of those works focused on studying the permissiveness for HCMV infection, but not HCMV latency/reactivation. Our previous studies have also shown that T98G glioblastoma cells are semi-permissive for HCMV infection, and HCMV-infected T98G cells harbor viral genomes, but lack detectable infectious virions through passaging (Duan et al., 2014; Luo and Fortunato, 2007). Here, we further demonstrated that T98G cells are capable of supporting HCMV reactivation from latency upon cAMP/IBMX treatment, as demonstrated by HCMV genome replication, lytic gene transcription and expression, and infectious virions release. The T98G-LrV cells provide a proper cell model of human brain origin for revealing the neuropathogenesis of congenital HCMV infection.
HCMV strains include clinical isolates (also known as low passage strains) and laboratory strains (also known as high passage strains), which exhibit different competencies in tropism and virulence. The clinical isolates are more relevant to be used to study the HCMV pathogenesis, but their propagation in fibroblasts is less efficiently than the laboratory strains (Prichard et al., 2001). Therefore, the high passaged HCMV strains, Towne and AD169, were widely used in laboratory studies. However, the genetic content as well as the biological properties of the laboratory strains have been changed as the extensively passaging (Cha et al., 1996; Prichard et al., 2001). Especially for the HCMV latency study, the clinical isolates commonly entered and exited latency in hematopoietic cells, but AD169 failed to become latent in CD34+ cells (Goodrum et al., 2007) and CD14+ cells (Hargett and Shenk, 2010). Whereas Towne has been shown to successfully establish latency in the NT2 cells (Gonczol et al., 1984; Liu et al., 2010) and GM-Ps (Kondo et al., 1994), as well as in T98G cells as demonstrated in the present study. The different abilities to establish latency possibly contributed by the genetic differences among different strains.
The application of rHCMV, a recombinant HCMV containing a GFP gene in the genome, which visualizes the virus reactivation from latency and offers the opportunity to obtain informative data during the reactivation process. According to the GFP signal, FACS and HCA analysis indicated a 57- and 15-fold increase, respectively, of GFP positive cells upon cAMP/IBMX treatment. Infectious viruses, as demonstrated by GFP plaques formation, were recovered from the reactivated cells by cAMP/IBMX. The GFP-reporter greatly enhances this strain’s utility in determining reactivation efficiencies. And these observations provide strong evidence that the rHCMV infected T98G cell model is well suited for studying the mechanism of HCMV reactivation and associated diseases.
Beta-herpesvirinae have more restricted host ranges and protracted growth cycle in vitro than alpha-herpesvirinae. These limitations are associated with heterogeneity or asynchrony in the timing of events in individual cells of the population. After cAMP/IBMX treatment, latent rHCMV was reactivated and viral replication was initiated, resulting in the infected cell lyse and viruses release, thereby increased the cell-free viruses. As the semi-permissiveness of T98G cells for HCMV infection, entry and replication of the recovered viruses are delayed in the newly infected cells (Luo and Fortunato, 2007; Randall et al., 1985), which further protracted the viral replication process. Coupled with the heterogeneity or asynchrony of reactivated cells, the cell-free viruses were decreased. With induction of cAMP/IBMX, viral replication was enhanced, the release of virus was gradually increased, leading to cell-free viruses increase.
It is well accepted that cellular differentiation and epigenetic regulation play important roles in HCMV reactivation, but the mechanisms underlying HCMV latency and reactivation remain poorly understood. Some cellular signaling pathway have been demonstrated to involve in HCMV latency and reactivation. Epidermal growth factor receptor (EGFR), its downstream phosphatidylinositol-3-kinase (PI(3) K) Kim et al., 2016), and HCMV-encoded miR-UL148D mediated immediate early response gene 5 (IER5)-cell division cycle 25B (CDC25B) (Pan et al., 2016) promote HCMV latency in CD34+ cells. PKA-CREB-TORC2 signaling cascade (Yuan et al., 2009), PKC mediated cellular CREB and NF-κB (Liu et al., 2010), and mitogen and stress activated kinases coupled with CREB (Kew et al., 2014) have been reported to activate IE gene expression from latency. The cellular signaling pathway involved in the HCMV reactivation is dependent on the stimuli and cell type. In the present study, PKA-CREB signaling pathway have been demonstrated to involve in the HCMV reactivation from latency in T98G cells. Others mechanism(s) need to be further revealed.
In summary, HCMV enters latency in infected T98G cells through passaging, which could be reactivated upon cAMP/IBMX treatment via PKA-CREB signaling pathway. The establishment of latency in HCMV- infected T98G cells provides a useful platform for studying the mechanism of HCMV reactivation and understanding the neuropatho-genesis of HCMV-associated neuropathy.
Acknowledgements
We thank Juan Min and Ding Gao for technical support on the flow cytometry and confocal microscopy. We thank John O’Dowd for critical reading of the manuscript. The study was supported by the National Natural Science Foundation of China (81601770), Applied Basic Research Program of Wuhan City (2016060101010050) and Sino-Africa Joint Center (SAJC201605).
References
- Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E, 1997. Human Herpesvirus KSHV Encodes a Constitutively Active G-protein-coupled Receptor Linked to Cell Proliferation [DOI] [PubMed]
- Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gerhengorn MC, Mesri EA, 1998. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391, 86–89. [DOI] [PubMed] [Google Scholar]
- Bego M, Maciejewski J, Khaiboullina S, Pari G, Jeor S, 2005. Characterization of an antisense transcript spanning the UL81–82 locus of human cytomegalovirus. J. Virol 79, 11022–11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolovan-Fritts CA, Mocarski ES, Wiedeman JA, 1999. Peripheral blood CD14(+) cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood 93, 394–398. [PubMed] [Google Scholar]
- Buchser W, Collins M, Garyantes T, Guha R, Haney S, Lemmon V, Li Z, Trask OJ, 2014. Assay Development Guidelines for Image-based High Content Screening, High Content Analysis and High Content Imaging
- Cha T. a., Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR, 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol 70, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Basma H, Singh J, Cheng P-W, 2005. Activation of CMV promoter-controlled glycosyltransferase and β-galactosidase glycogenes by butyrate, tricostatin A, and 5-Aza-2′-deoxycytidine. Glycoconj. J 22, 63–69. [DOI] [PubMed] [Google Scholar]
- Cohen JI, 2000. Epstein–Barr virus infection. N. Engl. J. Med 343, 481–492. [DOI] [PubMed] [Google Scholar]
- Compton T, Nepomuceno RR, Nowlin DM, 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191, 387–395. [DOI] [PubMed] [Google Scholar]
- Duan Y, Miao L, Ye H, Yang C, Fu B, Schwartz PH, Rayner S, Fortunato EA, Luo M-H, 2012. A faster immunofluorescence assay for tracking infection progress of human cytomegalovirus. Acta Biochim. Biophys. Sin 44, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan YL, Ye HQ, Zavala AG, Yang CQ, Miao LF, Fu BS, Seo KS, Davrinche C, Luo MH, Fortunato EA, 2014. Maintenance of large numbers of virus genomes in human cytomegalovirus-infected T98G glioblastoma cells. J. Virol 88, 3861–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egbert PR, Pollard RB, Gallagher JG, Merigan TC, 1980. Cytomegalovirus retinitis in immunosuppressed hosts. II. Ocular manifestations. Ann. Intern. Med 93, 664–670. [DOI] [PubMed] [Google Scholar]
- Fu YR, Liu XJ, Li XJ, Shen ZZ, Yang B, Wu CC, Li JF, Miao LF, Ye HQ, Qiao GH, Rayner S, Chavanas S, Davrinche C, Britt WJ, Tang QY, Mcvoy M, Mocarski E, Luo MH, 2015. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J. Virol 89, 1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczol E, Andrews PW, Plotkin SA, 1984. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 224, 159–161. [DOI] [PubMed] [Google Scholar]
- Goodrum F, Reeves M, Sinclair J, High K, Shenk T, 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum FD, Jordan CT, High K, Shenk T, 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. USA 99, 16255–16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Maccaroni P, Meyer R, Kaiser H, D’ambrosio E, Pascale E, Grassi M, Kuhn A, Di Nardo P, Kandolf R, 2003. Inhibitors of DNA methylation and histone deacetylation activate cytomegalovirus promoter-controlled reporter gene expression in human glioblastoma cell line U87. Carcinogenesis 24, 1625–1635. [DOI] [PubMed] [Google Scholar]
- Hahn G, Jores R, Mocarski ES, 1998a. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 95, 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn G, Jores R, Mocarski ES, 1998b. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 95, 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargett D, Shenk TE, 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 107, 20039–20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, 1975. Human cytomegalovirus. IV. Specific inhibition of virus-induced DNA polymerase activity and viral DNA replication by phosphonoacetic acid. J. Virol 16, 1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C, 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384, 120–128. [DOI] [PubMed] [Google Scholar]
- Jacobson MA, Zegans M, Pavan PR, O’donnell JJ, Sattler F, Rao N, Owens S, Pollard R, 1997. Cytomegalovirus retinitis after initiation of highly active antiretroviral therapy. Lancet 349, 1443–1445. [DOI] [PubMed] [Google Scholar]
- Jault FM, Spector SA, Spector DH, 1994. The effects of cytomegalovirus on human immunodeficiency virus replication in brain-derived cells correlate with permissiveness of the cells for each virus. J. Virol 68, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MJ, Wu AW, Andrews JI, McGonagill PW, Tibesar EE, Meier JL, 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol 81, 6669–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew VG, Yuan J, Meier J, Reeves MB, 2014. Mitogen and stress activated kinases act co-operatively with CREB during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog 10, e1004195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaiboullina SF, Maciejewski JP, Crapnell K, Spallone PA, Dean Stock A, Pari GS, Zanjani ED, Jeor SS, 2004. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br. J. Haematol 126, 410–417. [DOI] [PubMed] [Google Scholar]
- Kim JH, Collins-McMillen D, Buehler JC, Goodrum FD, Yurochko AD, 2016. HCMV requires EGFR signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J. Virol, 01206–01216. [DOI] [PMC free article] [PubMed]
- Knipe D, Howley P, Cohen J, Griffin D, Lamb R, Martin M, Racaniello V, Roizman B, 2013. Fields Virology Lippincott Williams & Wilkins, Philadelphia. [Google Scholar]
- Kondo K, Kaneshima H, Mocarski ES, 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 91, 11879–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Sauve RS, Davies HD, 2003. Congenital cytomegalovirus infection. J. Natl. Med. Assoc 95, 213. [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yuan J, Wu AW, McGonagill PW, Galle CS, Meier JL, 2010. Phorbol ester-induced human cytomegalovirus major immediate-early (MIE) enhancer activation through PKC-delta, CREB, and NF-κB desilences MIE gene expression in quiescently infected human pluripotent NTera2 cells. J. Virol 84, 8495–8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman P, Griffiths P, Paya C, 2002. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin. Infect. Dis.: Off. Publ. Infect. Dis. Soc. Am 34, 1094–1097. [DOI] [PubMed] [Google Scholar]
- Luo MH, Fortunato EA, 2007. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J. Virol 81, 10424–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini A, Liu H, Zhu H, 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol 75, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukage S, Kosugi I, Kawasaski H, Miura K, Kitani H, Tsutsui Y, 2006. Mouse embryonic stem cells are not susceptible to cytomegalovirus but acquire susceptibility during differentiation. Birth Defects Res. Part A, Clin. Mol. Teratol 76, 115–125. [DOI] [PubMed] [Google Scholar]
- Meier JL, 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol 75, 1581–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson M, Monard S, Sissons P, Sinclair J, 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol 77 (Pt 12), 3099–3102. [DOI] [PubMed] [Google Scholar]
- Montminy M, 1997. Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem 66, 807–822. [DOI] [PubMed] [Google Scholar]
- Murphy JC, Fischle W, Verdin E, Sinclair JH, 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor CM, Murphy EA, 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol 86, 9854–9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C, Zhu D, Wang Y, Li L, Li D, Liu F, Zhang C-Y, Zen K, 2016. Human cytomegalovirus miR-UL148D facilitates latent viral infection by targeting host cell immediate early response gene 5. PLoS Pathog 12, e1006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pari GS, Kacica M, Anders D, 1993. Open reading frames UL44, IRS1/TRS1, and UL36–38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol 67, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M-Y, Kim Y-E, Seo M-R, Lee J-R, Lee CH, Ahn J-H, 2006. Interactions among four proteins encoded by the human cytomegalovirus UL112–113 region regulate their intranuclear targeting and the recruitment of UL44 to prereplication foci. J. Virol 80, 2718–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert RR, Kalejta RF, 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. MBio 4, e00298–00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland SD, Costello P, Dekaban GA, Rice G, 1990. Cytomegalovirus in the brain: in vitro infection of human brain-derived cells. J. Infect. Dis 162, 1252–1262. [DOI] [PubMed] [Google Scholar]
- Poland SD, Bambrick LL, Dekaban GA, Rice GP, 1994. The extent of human cytomegalovirus replication in primary neurons is dependent on host cell differentiation. J. Infect. Dis 170, 1267–1271. [DOI] [PubMed] [Google Scholar]
- Prichard MN, Penfold ME, Duke GM, Spaete RR, Kemble GW, 2001. A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Rev. Med. Virol 11, 191–200. [DOI] [PubMed] [Google Scholar]
- Prösch S, Wendt CE, Reinke P, Priemer C, Oppert M, Krüger DH, Volk H-D, Döcke W-D, 2000. A novel link between stress and human cytomegalovirus (HCMV) infection: sympathetic hyperactivity stimulates HCMV activation. Virology 272, 357–365. [DOI] [PubMed] [Google Scholar]
- Randall R, Newman C, Honess R, 1985. Asynchronous expression of the immediate-early protein of herpesvirus saimiri in populations of productively infected cells. J. Gen. Virol 66, 2199–2213. [DOI] [PubMed] [Google Scholar]
- Reeves MB, 2011. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res 157, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier RD, Nelson JA, Oldstone MB, 1985. Detection of human cytomegalovirus in peripheral blood lymphocytes in a natural infection. Science 230, 1048–1051. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME, 1999. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem 68, 821–861. [DOI] [PubMed] [Google Scholar]
- Sinclair J, 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J. Clin. Virol.: Off. Publ. Pan Am. Soc. Clin. Virol 41, 180–185. [DOI] [PubMed] [Google Scholar]
- Sinclair J, 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech 1799, 286–295. [DOI] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA, 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol 75, 7543–7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller OB, Borysiewicz LK, Morgan BP, 1997. Development of a model for cytomegalovirus infection of oligodendrocytes. J. Gen. Virol 78, 3349–3356. [DOI] [PubMed] [Google Scholar]
- Stagno S, Pass RF, Dworsky ME, Henderson RE, Moore EG, Walton PD, Alford CA, 1982. Congenital cytomegalovirus infection: the relative importance of primary and recurrent maternal infection. N. Engl. J. Med 306, 945–949. [DOI] [PubMed] [Google Scholar]
- Stamminger T, Fickenscher H, Fleckenstein B, 1990. Cell type-specific induction of the major immediate early enhancer of human cytomegalovirus by cyclic AMP. J. Gen. Virol 71, 105–113. [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J, Sissons JGP, Borysiewicz LK, Sinclair JH, 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol 72, 2059–2064. [DOI] [PubMed] [Google Scholar]
- Wathen M, Thomsen D, Stinski M, 1981. Temporal regulation of human cytomegalovirus transcription at immediate early and early times after infection. J. Virol 38, 446–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathen MW, Stinski MF, 1982. Temporal patterns of human cytomegalovirus transcription: mapping the viral RNAs synthesized at immediate early, early, and late times after infection. J. Virol 41, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland KL, Oien NL, Homa F, Wathen MW, 1994. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res 34, 191–206. [DOI] [PubMed] [Google Scholar]
- Weinshenker BG, Wilton S, Rice G, 1988. Phorbol ester-induced differentiation permits productive human cytomegalovirus infection in a monocytic cell line. J. Immunol 140, 1625–1631. [PubMed] [Google Scholar]
- Whitley RJ, Roizman B, 2001. Herpes simplex virus infections. Lancet 357, 1513–1518. [DOI] [PubMed] [Google Scholar]
- Whitley RJ, Kimberlin DW, Roizman B, 1998. Herpes simplex viruses. Clin. Infect. Dis, 541–553. [DOI] [PubMed]
- Yuan J, Liu X, Wu AW, McGonagill PW, Keller MJ, Galle CS, Meier JL, 2009. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol 83, 6391–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]