

Abstract
mGlu5 receptors are involved in mechanisms of activity-dependent synaptic plasticity, and are targeted by drugs developed for the treatment of CNS disorders. We report that mGlu3 receptors, which are traditionally linked to the control of neurotransmitter release, support mGlu5 receptor signaling in neurons and largely contribute to the robust mGlu5 receptor-mediated polyphosphoinositide hydrolysis in the early postnatal life. In cortical pyramidal neurons, mGlu3 receptor activation potentiated mGlu5 receptor-mediated somatic Ca2+ mobilization, and mGlu3 receptor-mediated long-term depression in the prefrontal cortex required the endogenous activation of mGlu5 receptors. The interaction between mGlu3 and mGlu5 receptors was also relevant to mechanisms of neuronal toxicity, with mGlu3 receptors shaping the influence of mGlu5 receptors on excitotoxic neuronal death. These findings shed new light into the complex role played by mGlu receptors in physiology and pathology, and suggest reconsideration of some of the current dogmas in the mGlu receptor field.
Keywords: Metabotropic glutamate receptors, Polyphosphoinositide hydrolysis, Synaptic plasticity, Receptor-receptor cross-talk, Neurodevelopment, Long-term depression, Neuronal death, G-protein βγ subunits
1. Introduction
mGlu5 receptors mediate processes of activity-dependent synaptic plasticity underlying learning and memory, and are candidate drug targets for disorders of the central nervous system (CNS) (Vinson and Conn, 2012; Nicoletti et al., 2015). Abnormalities in the expression or function of mGlu5 receptors are associated with schizophrenia (Ohnuma et al., 1998; Matosin et al., 2016), focal cortical dysplasia (DuBois et al., 2017), absence epilepsy (D’Amore et al., 2015), Down’s syndrome (Iyer et al., 2014), neuropathic pain (Lin et al., 2015), Fragile-X syndrome, and other types of monogenic autism (Huber et al., 2002; Giuffrida et al., 2005; Ronesi et al., 2012; D’Antoni et al., 2014; Pignatelli et al., 2014). Canonical mGlu5 receptor signaling proceeds through the hydrolysis of polyphosphoinositides (PI). PI hydrolysis precipitates the formation of inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol, which stimulate intracellular Ca2+ release and protein kinase C (PKC), respectively (Nicoletti et al., 2011). mGlu5 receptor-mediated PI hydrolysis is dramatically high during early postnatal life and progressively declines across CNS maturation (Nicoletti et al., 1986a; Casabona et al., 1997). Not surprisingly, mGlu5 receptor signaling has been shown to regulate cortical development and has been implicated in neuronal migration, dendritic outgrowth, and synaptic formation and elimination (Reid et al., 1998; Hannan et al., 2001; Wijetunge et al., 2008; Ballester-Rosado et al., 2016). Mice lacking mGlu5 receptors show developmental abnormalities in the prefrontal cortex (PFC) and hippocampus that are typically associated with schizophrenia, such as reduced expression of reelin, glutamate decarboxylase-65 and −67, and parvalbumin mRNA levels (Luoni et al., 2016). Intracellular mechanisms underpinning mGlu5 receptor signaling have been extensively characterized. Scaffolding proteins, such as Homer and Shank, link mGlu5 receptors to a variety of effector proteins (Brakeman et al., 1997; Xiao et al., 1998; Tu et al., 1999; Ango et al., 2000; Rong et al., 2003; Kammermeier and Worley, 2007), and disruptions in mGlu5 receptor-Homer interactions underlie abnormalities associated with several disease states (Giuffrida et al., 2005; Ronesi et al., 2012; D’Antoni et al., 2014; Guo et al., 2016). In contrast, less is known about how other membrane receptors regulate mGlu5 receptor signaling. Activation of NMDA receptors has been shown to potentiate mGlu5 receptor function by restraining mGlu5 receptor desensitization via activation of protein phosphatase 2B/calcineurin (Alagarsamy et al., 1999, 2002; 2005). In addition, a reciprocal interaction between A2A adenosine receptors and mGlu5 receptors has been consistently observed in striatal projection neurons (Díaz-Cabiale et al., 2002; Ferré et al., 2002; Nishi et al., 2003; Coccurello et al., 2004; Domenici et al., 2004). Despite these reports, very little is known about how mGlu5 receptor function is regulated by other subtypes of mGlu receptors.
We now report that activation of mGlu3 receptors amplifies mGlu5 receptor signaling and that the functional partnership between mGlu3 and mGlu5 receptors mediates the robust stimulation of PI hydrolysis induced by excitatory amino acids in the developing brain. In addition, we demonstrate that mGlu3 activation potentiates mGlu5 receptor-mediated somatic Ca2+ mobilization in cortical pyramidal neurons and mGlu5 receptor activity is required for mGlu3 receptor-dependent long-term depression (LTD) in the PFC. Finally, we demonstrate that neuronal mGlu3 receptors shape mGlu5 receptor function in mechanisms of neurodegeneration, with mGlu5 receptors switching from neurotoxic to neuroprotective when mGlu3 receptors are selectively inhibited. Together, these data provide compelling evidence for a novel functional interaction between mGlu3 and mGlu5 receptors. This functional interaction underlies the dramatic developmental changes in mGlu receptor signaling as well as neuronal mGlu receptor function and synaptic plasticity in the adult PFC.
2. Materials and methods
(RS)-3,5-dihydroxyphenylglycine (DHPG), (−)-2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268) and 6-cyano-7-nitroquinoxaline-2,3-dione, 3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl-(cis-4-methoxycyclohexyl)-methanone (JNJ16259685), 2-methyl-6-(phenylethynyl) pyridine (MPEP) and N-methyl-D-aspartate (NMDA) were purchased from Tocris Bioscience (Bristol, UK). 3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP), VU0469650, VU0650786 and VU6001966 were synthesized in house. Tetrodotoxin was purchased from Sigma. Myo-[3H]inositol (18 Ci/mmol) was purchased from Perki-nElmer (Milan, Italy). JNJ16259685, VU0650786, VU0469650 and VU6001966 were dissolved in dimethyl sulfoxide (DMSO) at the initial concentration of 10 mM. The final concentration of DMSO applied to the cultures was 0.1%. NMDA, MPEP and MTEP were dissolved in distilled H2O at the initial concentration of 10 mM. DHPG was dissolved in Krebs buffer at the initial concentration of 10 mM. LY379268 was dissolved in distilled H2O + 1.1 mEq NaOH at the initial concentration of 10 mM and diluted in Krebs at the final concentration.
2.1. HEK 293 cells for assessment of mGlu receptor-stimulated PI hydrolysis
Human embryonic kidney (HEK) 293 cells (ATCC, Cat# CRL-1573) were cultured in DMEM containing 10% fetal calf serum, and transfected as described previously (Iacovelli et al., 2014). The plasmid encoding for the C-terminal domain of GRK2 (Gly495-Leu689) (GRK2-Cter) was kindly provided by C. Scorer (GlaxoSmithKline, Uxbridge, Middlesex, UK); mGlu2 receptor cDNA was kindly provided by J. Blahos (Academy of Science, Prague, Czech Republic). mGlu3 receptor cDNA was kindly provided by F. Ferraguti (Innsbruck Medical University, Innsbruck, Austria); human mGlu1 receptor cDNA and mGlu5 receptor cDNA were kindly provided by M. Corsi (Glaxo Wellcome, Verona, Italy).
2.2. Preparation of pure primary neuronal cell cultures
Experiments were performed according to the Institutional Animal Care and Use Committee of the University of Catania. Cultures of pure cortical neurons were prepared from male and female rats at embryonic day 15. Briefly, cortices were dissected in Ca2+/Mg2+-free buffer and mechanically dissociated. Cortical cells were plated at a density of 0.45 × 106/well on 48-well multiplates (Costar, Corning Inc., NY) pre-coated with 0.1 μg/ml poly-D-lysine (Sigma-Aldrich, Milan, Italy) in NeuralQ Basal Medium (GSM-9420, MTI-Global Stem, Gaithersburg, MD), a serum-free basal medium optimized for maximum growth and survival of primary neurons in culture, supplemented with GS21Tm Neural Supplement (GSM-3100, MTI-Global Stem) and 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cytosine-D-arabinofuranoside (10 μM) was added to the cultures 18 h after plating to avoid the proliferation of non-neuronal elements and maintained for 3 days before partial replacement of the medium. This method yielded nearly pure neuronal cultures with <2% of glial cells (Copani et al., 1999).
2.3. Preparation of primary reactive astrocytes
Primary cultures of mouse cortical astrocytes were prepared as described previously (Caraci et al., 2011). Briefly, astrocyte cell cultures were prepared from male and female CD1 mice at 1–3 days after birth. Dissociated cortical cells were grown in 15-mm multiwell vessels (Falcon Primaria, Lincoln Park, NJ) using a plating medium of MEM-Eagle’s salts supplemented with fetal bovine serum (10%), horse serum (10%), glutamine (2 mM), glucose (21 mM) and 100 U/ml penicillin, and 100 mg/ml streptomycin. For the induction of a reactive phenotype, we followed the protocol described in Miller et al. (1995). In brief, cultured astrocytes at 6 DIV were shaken overnight to remove microglia and oligodendrocytes, and then switched into serum-free medium containing growth factors (Caraci et al., 2011).
2.4. Immunoblotting of mGlu receptors in transfected HEK 293 cells
Co-transfected HEK 293 cells were rinsed in ice-cold PBS and solubilized in Triton X-lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM sodium orthovanadate, 50 mM sodium fluoride, and 10 mM β-glycerophosphate). Protein cell lysates (80 μg) were separated by SDS-PAGE electrophoresis, blotted onto nitrocellulose, and probed using a mouse anti-mGlu1 receptor antibody (1:1000; BD Bioscience, Milano, Italy; cat# 556389) or a rabbit anti-mGlu5 receptor antibody (1:1000; Millipore; Billerica, MA; cat# AB5675). The immunoreactive bands were visualized by enhanced chemiluminescence (GE Health care, Buckinghamshire, UK) using horseradish peroxidase-conjugated secondary antibodies.
2.5. Assessment of mGlu receptor-stimulated PI hydrolysis in HEK 293 cells
Forty-eight hr after transfection, HEK 293 cells were seeded in 12-well plates and exposed to 2 μCi/ml of myo-[3H]inositol for 24 h. Afterwards, cells were washed in Krebs-Hensleit buffer equilibrated with 95% O2, 5% CO2 to pH 7.4, containing 10 mM LiCl, and DHPG (300 μM) and/or LY379268 (5 μM) were added and maintained in the incubation buffer for additional 30 min. The reaction was stopped with ice-cold methanol. After scraping, the cells were collected and methanol, water, and chloroform were added (1:1:1, final). The [3H]inositolmonophosphate (InsP) present in the aqueous phase was separated by anion exchange chromatography in 10-ml columns containing 1.5 ml of Dowex 1-X-8 resin (formate form, 100e200 mesh, Bio-Rad, Milan, Italy). Columns were washed twice with water, once with a solution of 5 mM sodium tetraborate and 40 mM sodium formate, and the [3H]InsP was eluted with 6.5 ml of 0.2 M ammonium formate and 0.1 M of formic acid.
2.6. Assessment of NMDA toxicity in pure cultures of rat cortical neurons
Mature cell cultures (13 DIV) were exposed for 20 min to low (75 μM) or high (300 μM) concentrations of NMDA and/or DHPG (100 μM) and VU0650786 (10 μM) in a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered salt solution containing 0.8 mM Mg2+, 10 μM L-glycine, and JNJ16259685 (10 μM) to prevent the activation of mGlu1 receptors. After the 20-min pulse, cell cultures were maintained for 22–24 h in NeuralQ Basal Medium and 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, without GS21Tm. Neuronal injury was measured by either trypan blue staining (0.4% for 10 min) or the methyltetrazolium test (MTT) assay (Caraci et al., 2016).
2.7. RNA isolation, reverse transcription and quantitative real-time PCR
Total RNA was extracted from primary reactive astrocytes with Trizol reagent according to manufacturer’s protocol. The RNA was further treated with DNase (Qiagen, Hilden, Germany) and single strand cDNA was synthesized from 2 μg of total RNA using superscript III (Invitrogen, Carlsbad, CA) and random hexamers. Real-time PCR was performed on 20 ng of cDNA by using specific primers and Ssoadvanced Universal SYBR Green on an Applied Biosystems Step-One instrument. Thermal cycler conditions were as follows: 10 min at 95 °C, 40 cycles of denaturation (15 s at 95 °C), and combined annealing/extension (1 min at 60 °C). Primers used were as follows: Grm1 Forw AAATCTACAGCAATGCTGGCGA and Rev CTTCGATGACTTCATCTCTGTC; Grm2 Forw ACCAGAAGGGTGGCCC AGCA and Rev GCACCCCAGGCAGCAGATGG; Grm3 Forw CAGCAAGCTCCCTCTTTTGT and Rev GCTAAAAGAGCCCGTCACTG; Grm5 Forw ACGAAGACCAACCGTATTGC and Rev AGACTTCTCGGATGCTT GGA; and GAPDH Forw CGTCCCGTAGACAAAATGGT and Rev TCAA TGAAGGGGTCGTTGAT.
mRNA copy number of each gene analyzed was calculated from serially diluted standard curves simultaneously amplified with the samples and normalized against GAPDH copy number.
2.8. Assessment of PI hydrolysis in primary reactive astrocytes
Reactive astrocyte cultures seeded in 24-multiwells were labeled with 1 μCi/well of myo-[3H]inositol overnight, and then challenged with DHPG (20 or 100 μM) and/or LY379268 (1 μM) and VU0650786 (10 μM) in the presence of 10 mM LiCl for 1 h. PI hydrolysis was measured as described above.
2.9. Animals used for preparation of brain slices for PI hydrolysis measurements
Studies were performed in accordance with the national and international guidelines and regulations on animal care and use and were approved by the Neuromed Institutional Animal Care and Use Committee. All efforts were made to minimize animal suffering and to reduce the number of animals used. Animals were housed under controlled conditions (temperature, 22 °C; humidity, 40%) with a 12 h light/dark cycle and food and water ad libitum. CD1 male and female mice at different ages (from postnatal day (PND)8/9 to PND13/14) and CD1 male mice at 30 and 60 PND (Charles River Laboratories, Calco, Italy). mGlu5 receptor knockout (mGlu5−/−) male mice and their male wild-type counterparts at PND15. mGlu5 heterozygous mice were originally purchased from the Jackson Laboratories (Bar Harbor, ME, B6.129-Grm5tm1Rod/J, stock # 003558). Mice heterozygous for the targeted mutation were back-crossed to homozygosity and all mice used were generated by heterozygous breeding. Crv4 male mice lacking mGlu1 receptors at PND 15 and their male wild-type counterparts. Crv4 mice are mice of a BALB/c/Pas inbred strain carrying a spontaneous recessive mutation that consists of a LTR insertion that disrupts the splicing of the Gm1 gene encoding for mGlu1 receptors (Conti et al., 2006). These mice were kindly provided by A. Puliti (University of Genoa, Italy). mGlu2 receptor knockout (mGlu2−/−) and mGlu3 receptor knockout (mGlu2−/−) male and female mice at PND9/10 and PND13/14. mGlu2−/− mice, mGlu3−/− mice, and their CD1 wild-type counterparts were kindly provided by Eli Lilly & Company (Indianapolis, IN) (Linden et al., 2005).
2.10. Animals used for preparation of brain slices for Ca2+ imaging and LTD experiments
Adult (8–12 week) male C57Bl6/J mice were used. Mice were housed in groups (2–5 per cage) on a standard 12-hr light cycle (lights on at 6:00 a.m.) with food and water ad libitum. All experimental protocols were approved by the Vanderbilt Institutional Animal Care and Use Committee.
2.11. Measurement of PI hydrolysis in brain slices
Measurements of PI hydrolysis in cortical slices were performed as described in detail (Nicoletti et al., 1986b). Cortical slices were prepared from CD1 male and female mice at different ages (from PND8/9 to PND13/14); from CD1 male mice at PND30 and PND60; from mGlu5−/− male mice and their male wild-type counterparts at PND15; from crv4 male mice at PND 15 and their male wild-type counterparts; and from mGlu2−/− and mGlu3−/− male and female mice at PND9/10 and PND13/14. Animals were killed by decapitation, and brain regions were dissected out and transferred in icecold Krebs-Henseleit buffer (NaCl 118 mM, KCl 4.7 mM, MgSO4 1.18 mM, KH2PO4 1.18 mM, NaHCO3 24.8 mM, CaCl2 1.2 mM, D-glucose 10 mM) that had been pre-gassed with 95% O2 and 5% CO2 to pH 7.4. Slices (350 × 350 μm) were prepared by a Mc Ilwain tissue chopper. Forty ml of gravity packed slices were then incubated for 60 min in 350 μl buffer containing 1 μCi of myo-[3H] inositol. Slices were incubated with LiCl (10 mM, for 10 min) followed by mGlu receptor ligands. One hr later, the incubation was stopped by the addition of 900 μl methanol:chloroform (2:1). After further addition of 300 μl chloroform and 600 μl water, samples were centrifuged at low speed to facilitate phase separation and the [3H]InsP present in the supernatant was separated by anion exchange chromatography as detailed above. For protein measurements samples were dryed after removal of the water phase and incubated with 0.5 N NaOH at 50 °C for 2 h. Proteins were measured as described by Lowry et al. (1951).
2.12. Ca2+ imaging and LTD in the mouse PFC
For Ca2+ imaging and LTD, all drugs were prepared at 1000X, aliquoted, frozen, and thawed on the day of use. LY379268, DHPG, and MTEP were prepared in purified water. VU0650786 and VU0469650 were prepared in DMSO. LY279268, DHPG, and tetrodotoxin were purchased from Abcam. MTEP, VU0650786, and VU0469650 were synthesized in-house.
Mice were deeply anesthetized with isoflurane and decapitated for acute brain slice preparation. The brain was quickly removed and placed in ice-cold cutting/recovery solution (in mM): 93 N-methyl-D-glucamine, 20 HEPES, 2.5 KCl, 0.5 CaCl2, 10 MgCl2, 1.2 NaH2PO4, 25 glucose, 5 Na-ascorbate, and 3 Na-pyruvate. Coronal slices (300 μM) containing the PFC were prepared using a Leica VT1000S vibratome and immediately transferred to a heated (34 ± 1 °C) bath containing the recovery solution for 10 min. Slices were then transferred to a room-temperature (23 ± 1 C) holding chamber filled with artificial cerebrospinal fluid (aCSF) (in mM): NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 11 glucose, and 26 NaHCO3. After at least 60 min, slices were placed in the recording chamber and perfused with fresh, warm (30 ± 1 °C) aCSF at 2 ml/min. All solutions were continuously oxygenated (95% O2/5% CO2).
Pyramidal cells in layer 5 of the PFC were visually identified by differential interference contrast on an upright microscopy (Olympus, BX51WI). Borosilicate glass electrodes (2–4 MΩ) were pulled using a Flaming/Brown micropipette puller (Sutter, P-1000) and filled with a potassium-based internal solution (in mM): 125 K-gluconate, 4 NaCl, 10 HEPES, 4 MgATP, 0.3 NaGTP, 10 Trisphosphocreatine. Before all experiments, cells were verified as regular-spiking by their characteristic spike-firing adaptation in response to increasing current injections. Fast-spiking neurons (putative interneurons) were discarded. Local glutamate release was elicited at 0.1 Hz by 0.1–0.15 ms electrical stimulation from a concentric bipolar electrode (CBARC75, FHC Inc) placed slightly medial to the recording electrode in layer 5. To preclude recording inhibitory post-synaptic currents, all long-term recordings were performed in voltage-clamp configuration at –70 mV. Excitatory post-synaptic currents (EPSCs) were completely blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (data not shown). After acquiring a stable baseline recording, 100 nM LY379268 and/or 100 μM DHPG was applied to the slice for 3 or 10 min to elicit LTD. NAM application occurred for at least 5 min prior to and co-terminated with LY379268/DHPG. Recordings were acquired with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz and digitized at 10 kHz. Data acquisition and analysis were performed using pClamp 10.4 software (Axon Instruments, Union City, CA).
Ca2+ imaging experiments were performed as described (Foster et al., 2014). The membrane-impermeable fluorescent Ca2+ indicator Fluo-4 (0.1 mM pentapotassium salt) was added to the internal solution and cells were dialyzed for at least 15 min prior to imaging. The relative difference in fluorescence between the soma and background was quantified and monitored at 1 Hz with 5–50 ms light stimulation from a mercury light source (Olympus, U-ULH) and shutter (Sutter, Lambda 10–2) operated with Metamorph 10.4 software. LY379268 and/or DHPG were applied for 2 min and the maximum increase in fluorescence was reported. All cells included in the analysis exhibited a depolarization-induced increase in fluorescence at the end of each experiment to ensure proper whole-cell access and potential for somatic Ca2+ mobilization. All Ca2+ imaging experiments were performed at –70 mV in the presence of tetrodotoxin (0.5–1 μM) and the mGlu1 receptor NAM, VU0469650 (10 μM), to isolate neuron-autonomous, mGlu5 receptor-mediated responses.
2.13. Quantification and statistical analysis
Significance was assessed using the parametric Student’s t-test, for single comparisons, or ANOVA and post hoc Fisher’s LSD test or Bonferroni post-test, for multiple comparisons. In the text, values are reported as means ± S.E.M.; in the Figure legends, values are reported as means ± S.E.M., significance is reported either at the p < 0.05 or p < 0.01 level and n values, t values and F values are also reported.
3. Results
3.1. Functional interaction between group-I and group-II mGlu receptors in heterologous expression systems
HEK 293 cells were co-transfected with either mGlu1α or mGlu5 receptor cDNA and/or mGlu2 or mGlu3 receptor cDNA. Co-transfection did not change expression levels of mGlu1α or mGlu5 receptor protein, as detected by immunoblotting (Fig. 1A). In cells expressing mGlu1α or mGlu5 receptors alone, the mGlu1/5 agonist, (RS)-3,5-dihydroxyphenylglycine (DHPG, 300 μM), stimulated PI hydrolysis by about 3–5 fold. This stimulation was unaffected by co-expression of either mGlu2 or mGlu3 receptors. The mGlu2/3 receptor agonist, LY379268 (5 μM), which was inactive on its own, enhanced DHPG-stimulated PI hydrolysis when mGlu2 or mGlu3 receptors were co-expressed with either mGlu1α or mGlu5 receptors. The extent of potentiation by LY379268 was similar regardless of receptor combinations (Fig. 1B). To examine whether the facilitation of group-I mGlu receptor signaling by group-II mGlu receptors was mediated by the βγ subunits of Gi/o proteins we used HEK 293 cells co-expressing mGlu1 or mGlu5 receptors, mGlu3 receptors, and the C-terminal domain of type-2 G-protein coupled receptor kinase (GRK2-Cter), which buffers the βγ subunit (Premont et al., 1995). In these cells, LY379268 failed to amplify DHPG-stimulated PI hydrolysis (Fig. 1C).
Fig. 1. Functional cross-talk between group-I and group-II mGlu receptors in transfected HEK 293 cells.
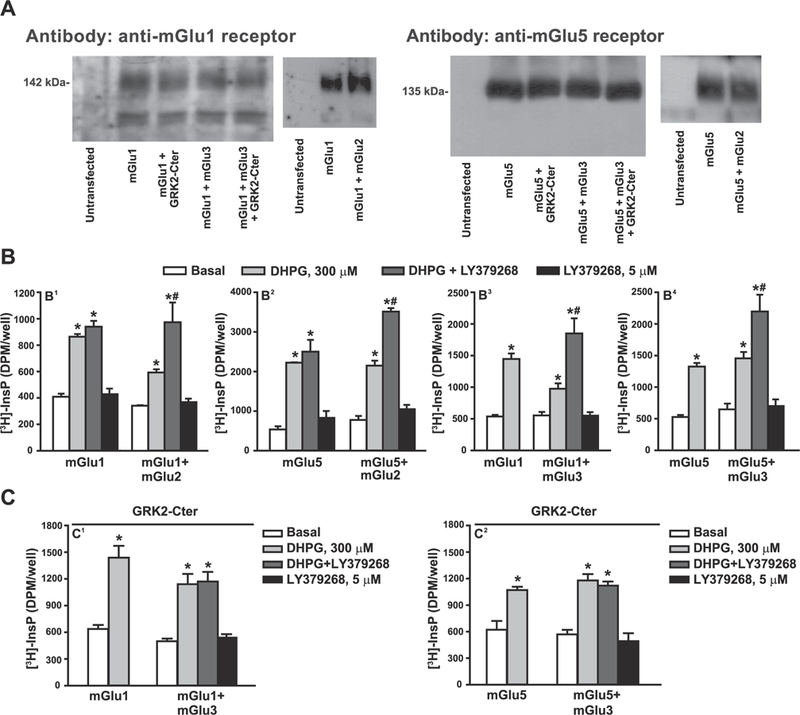
Expression of mGlu1 and mGlu5 receptors in cells transfected with mGlu1 or mGlu5 cDNA in the absence or presence of mGlu2, mGlu3 receptors or GRK2-C-ter cDNA is shown in (A). Stimulation of PI hydrolysis by DHPG and/or LY379268 in cells expressing mGlu1 or mGlu5 receptors with or without mGlu2 or mGlu3 receptors is shown in (B). The effect of GRK2-C-ter on the synergism between mGlu3 and mGlu1 or mGlu5 receptors is shown in (C). In (B) and (C), values are mean ± S.E.M. of 3–9 determinations. p < 0.05 (Two-way ANOVA + Bonferroni test) * vs. the respective basal and # vs. the respective DHPG alone (B 1: F(1,16) = 4.69 for transfection factor and F(3,16) = 43.93 for treatment factor; B2: F(1,16) = 11.39 for transfection factor and F(3,16) = 112.73 for treatment factor; B3: F(1,25) = 2.17 for transfection factor and F(3,25) = 27.00 for treatment factor; B4: F(1,47) = 1.04 for transfection factor and F(3,47) = 41.17 for treatment factor; C1: F(1,25) = 4.89 for transfection factor and F(3,25) = 28.97 for treatment factor; C2: F(3,29) = 30.78 for treatment factor).
3.2. Functional interaction between native group-I and group-II receptors is restricted to mGlu3 and mGlu5 receptors
Pharmacological activation of group-II mGlu receptors with non-subtype selective agonists potentiates group-I mGlu receptor-mediated PI hydrolysis in rat hippocampal slices (Nicoletti et al., 1993; Genazzani et al., 1994; Schoepp et al., 1996). We reproduced these findings in mouse cortical slices, where LY379268 amplified DHPG-stimulated PI hydrolysis in a concentration-dependent fashion (Fig. 2A). Knowing that mGlu receptor-mediated PI hydrolysis is developmentally regulated (Nicoletti et al., 1986a), we examined the interaction between group-I and group-II mGlu receptors at different postnatal ages. At PND9/10, 100 μM DHPG stimulated PI hydrolysis by as much as 14-fold. This response was saturating, and, therefore, could not be amplified by LY379268 (Fig. 2B). Potentiation by LY379268 at this age could be demonstrated by using lower concentrations of DHPG (10 μM), which stimulated PI hydrolysis by about 4–5 fold (see Fig. 3E). At PND11/12, PND13/14, PND30 and PND60, LY379268 significantly enhanced the stimulation of PI hydrolysis induced by 100 μM DHPG (Fig. 2B). Unexpectedly, LY379268 alone could stimulate PI hydrolysis by less than two-fold at PND9/10 and PND11/12, but not at later developmental ages (Fig. 2B, reproduced on a different scale in Fig. 2B’).
Fig. 2. Functional partnership between group-II and group-I mGlu receptors in mouse cortical slices at different developmental ages.
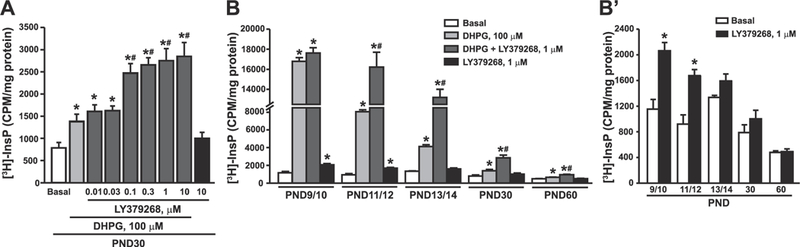
Concentration-dependent amplification of DHPG-stimulated PI hydrolysis by LY379268 in cortical slices of mice at PND30 is shown in (A). The age-dependent profile of DHPG-stimulated PI hydrolysis in the absence or presence of LY379268 is shown in (B). Data with LY379268 alone are highlighted in (B′). Values are mean ± S.E.M. of 3–4 determinations. p < 0.05 (One-way ANOVA + Fisher’s LSD in A and B and Student’s t-test in B′) vs. the respective basal (*), DHPG (#). (A): F(8,27) = 22.49; (B): F(3,11) = 840.6 (PND9/10); F(3, 8) = 132.27 (PND11/12); F(3,8) = 263.84 (PND13/14); F(3,12) = 29.46 (PND30); F(3,12) = 45.25 (PND60); (B′): t(5) = −4.84 (PND9/10); t(4) = −5.32 (PND11/12).
Fig. 3. Functional partnership between native group-I and group-II mGlu receptors is mediated by mGlu3 and mGlu5.

Stimulation of PI hydrolysis by DHPG and/or LY379268 in cortical slices prepared from wild-type, mGlu2−/− and mGlu3−/− mice are shown in (A–C), where wild-type data at PND9/10 and 60 are the same data as in Fig. 2B for comparative purposes. Data with LY379268 at PND9/10 are highlighted in (A′). The effect of the mGlu3 receptor NAM, VU0650786, on DHPG/LY379268-stimulated PI hydrolysis is shown in (D–F). The effect of the mGlu2 receptor NAM, VU6001966, on DHPG/LY379268-stimulated PI hydrolysis is shown in (G). The effect of the mGlu1 receptor NAM, JNJ16259685, and the mGlu5 receptor NAM, MPEP, on DHPG/LY379268-stimulated PI hydrolysis is shown in (H), where control data are the same data as in Fig. 2B at PND13/14 for comparative purposes. Stimulation of PI hydrolysis by DHPG/LY379268 in cortical slices from mGlu5−/− or crv4 mice and their wild-type counterparts is shown in (I) and (J), respectively. Values are mean ± S.E.M. of 3–5 determinations. p < 0.05 (Two-way ANOVA + Bonferroni test) * vs. the respective basal values and # vs. the DHPG alone or DHPG + LY379268 values obtained in wild-type and mGlu2−/− mice (A: F(2,33) = 110.73 for genotype factor and F(3,33) = 693.82 for treatment factor; A’: F(2,15) = 36.15 for genotype factor and F(3,15) = 77.46 for treatment factor; B: F(2,30) = 36.33 for genotype factor and F(3,30) = 235.35 for treatment factor; C: F(2,35) = 24.79 for genotype factor and F(2,35) = 40.05 for treatment factor). p < 0.05 (Two-way ANOVA + Bonferroni test) * vs. the respective basal values and # vs. the DHPG values (D: F(1,26) = 913.61 for Basal/DHPG factor and F(4,26) = 13.95 for VU0650786 concentrations factor; E: F(1,14) = 70.67 for Basal/DHPG factor and F(3,14) = 57.33 for drugs factor; F: F(3,31) = 29.60 for drugs factor and F(2,31) = 8.56 for VU0650786 concentrations factor; G: F(3,28) = 234.24 for drugs factor and F(1,28) = 9.89 for VU6001966 concentrations factor; H: F(2,24) = 93.54 for drugs - JNJ16259685 and MPEP - factor and F(3,24) = 284.48 for treatment factor. p < 0.05 (Two-way ANOVA + Bonferroni test) * vs. the respective basal values and # vs. the respective DHPG alone values (I: F(1,32) = 2826.74 for genotype factor and F(3,32) = 1763.58 for treatment factor; J: F(1,22) = 160.79 for genotype factor and F(3,22) = 236.83 for treatment factor).
To unravel the identity of the group-II mGlu receptor subtype that functionally interacts with group-I receptors, we used mice with genetic deletion of either mGlu2 or mGlu3 receptors. At PND13/14 and PND60, potentiation of DHPG-stimulated PI hydrolysis by LY379268 was maintained in cortical slices prepared from mGlu2−/− mice, but was lost in slices from mGlu3−/− mice (Fig. 3B and C). At PND9/10, stimulation of PI hydrolysis by saturating concentrations of DHPG (100 μM) was similar in wild-type and mGlu2−/−mice, but was halved in mGlu3−/− mice (Fig. 3A). In mGlu3−/− mice at PND9/10, LY379268 did not potentiate DHPG-stimulated PI hydrolysis, although the PI response was submaximal (Fig. 3A). Stimulation of PI hydrolysis by LY379268 alone in PND9/10 cortical slices was maintained in mGlu2−/− mice, but not in mGlu3−/− mice (Fig. 3A shown in a different scale in Fig. 3A’). In different sets of experiments, we treated cortical slices from wild-type mice with the selective mGlu3 receptor negative allosteric modulator (NAM), VU0650786 (Engers et al., 2015) or with the selective mGlu2 receptor NAM, VU6001966 (Bollinger et al., 2017). In slices from PND8/9 mice, stimulation of PI hydrolysis by saturating concentrations of DHPG (100 μM) was reduced by VU0650786 in a concentration-dependent fashion (Fig. 3D). Maximally effective concentrations of VU0650786 halved the PI response to DHPG similarly to what found in slices prepared from mGlu3−/− mice (compare Fig. 3D and A). VU0650786 inhibited the enhancing effect of LY379268 on PI hydrolysis induced by sub-saturating concentrations of DHPG (10 μM) at PND8/9 (Fig. 3E) or by saturating concentrations of DHPG (100 μM) at PND13/14 (Fig. 3F). We also examined whether stimulation of PI hydrolysis by DHPG alone or combined with LY379268 was affected by co-incubation with the mGlu2 NAM VU6001966. VU6001966 was applied to cortical slices from PND13/14 mice at concentration of 10 μM, which is more than 10-fold greater that the IC50 for mGlu2 receptors, and does inhibit mGlu3 receptors ((Bollinger et al., 2017). In contrast to the mGlu3 receptor NAM, VU0650786, addition of VU6001966 did not attenuate the amplification of DHPG-stimulated PI hydrolysis by LY379268 (Fig. 3G), and unexpectedly induced a modest enhancement.
We next examined which of the two group-I subtypes (i.e., mGlu1 or mGlu5) were positively modulated by mGlu3 receptors using either subtype-selective drugs or mice lacking mGlu5 or mGlu1 receptors. The ability of LY379268 to potentiate DHPG-stimulated PI hydrolysis was preserved in slices treated with the mGlu1 receptor NAM, JNJ16259685 (10 μM) (2.5 fold potentiation in both control and JNJ16259685-treated slices), whereas it was lost in slices treated with MPEP (Fig. 3H).
Stimulation of PI hydrolysis by 100 μM DHPG was halved in cortical slices from mGlu5−/− mice at PND15, as compared to their wild-type counterparts. LY379268 amplified the PI response to DHPG in slices from wild-type mice but not in slices from mGlu5−/−mice (Fig. 3I). In contrast, the lack of mGlu1 receptors in crv4 mice did not affect the ability of LY379268 to enhance DHPG-stimulated PI hydrolysis (Fig. 3J).
3.3. mGlu3 receptor activation potentiates mGlu5 receptor-mediated calcium mobilization in cortical pyramidal cells
To examine whether the mGlu3 and mGlu5 receptors interact in neurons we performed Ca2+ imaging experiments in acute slices prepared from the mouse PFC. Individual, regular-spiking pyramidal cells were identified and loaded with Fluo-4. To isolate neuron-autonomous, mGlu5 receptor-specific, Ca2+ responses, all experiments were performed in the presence of tetrodotoxin and the selective mGlu1 receptor NAM, VU0469650 (Lovell et al., 2013). Strong activation of the pyramidal cells with DHPG (100 μM) generated immediate, sharp (4–5 s) peaks in Fluo-4 fluorescence that often recurred with a 30–40-sec interval (Fig. 4A). On average, neither a threshold concentration of DHPG (30 μM) nor a high concentration of LY379268 (100 nM) was sufficient to generate large spikes in somatic Ca2+ mobilization on its own. However, the combination of threshold DHPG and LY379268 induced a robust Ca2+ response following co-application. The synergy between LY379268 and DHPG was abrogated when experiments were performed in the presence of the mGlu3 receptor NAM, VU0650786 (10 μM) (Fig. 4B), confirming that mGlu3 receptor activation potentiates postsynaptic neuronal signaling mediated by mGlu5 receptors.
Fig. 4. mGlu 3 receptor activation enhances mGlu5 receptor-mediated somatic Ca2+ mobilization in pyramidal cells of the mouse PFC.
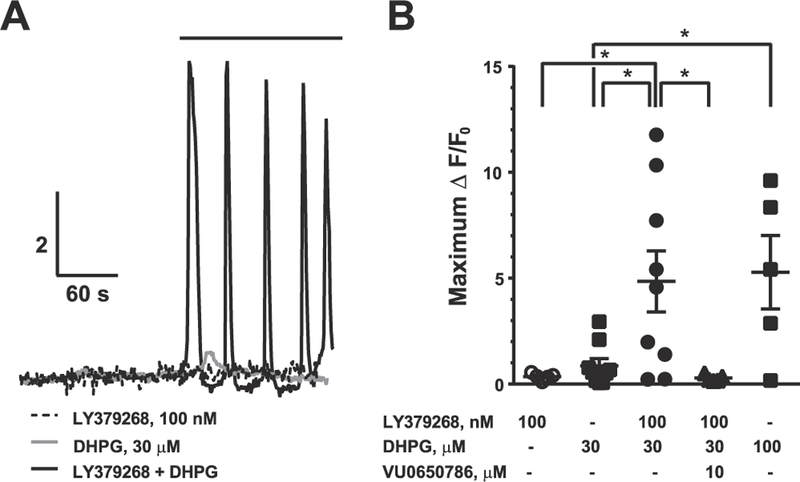
(A) Representative calcium imaging fluorescence experiments. To isolate neuron-autonomous, mGlu5 receptor-specific calcium responses, all experiments were performed in the presence of tetrodotoxin and the selective mGlu1 receptor NAM, VU0490650 (10 μM). Pyramidal cells were loaded with the cell-impermeable fluorescent calcium indicator Fluo-4. (B) Summary of calcium imaging experiments. Values are mean ± S.E.M. *p < 0.05, **p < 0.01, Bonferroni post-tests.
3.4. Pharmacological activation of mGlu3 receptors slightly enhanced DHPG-stimulated PI hydrolysis in astrocytes
We also assessed PI hydrolysis in primary cultures of astrocytes prepared from the mouse cerebral cortex. In confluent astrocytes grown in the presence of serum, DHPG failed to stimulate PI hydrolysis when applied either alone or in combination with LY379268. When astrocytes were switched from a serum-containing medium to a serum-free medium supplemented with a cocktail of trophic factors, cells adopted a reactive-like phenotype characterized by increased expression of glial fibrillary acid protein. Reactive astrocytes expressed the transcript of mGlu3 and mGlu5 receptors, but not the transcript of mGlu1 and mGlu2 receptors, as assessed by real-time PCR (not shown). In these cells DHPG stimulated PI hydrolysis to an extent that depended on the percentage of astrocytes bearing a reactive phenotype. Treatment with LY379268 (1 μM) did not affect stimulation of PI hydrolysis by saturating concentrations of DHPG (100 μM) (Supplementary Fig. 1A). Using lower concentrations of DHPG (20 μM), we could observe a slightly potentiation by LY379268, which was abrogated by the mGlu3 NAM, VU0650786 (10 μM) (Supplementary Fig. 1B). DHPG did not stimulate PI hydrolysis in resting astrocytes when applied alone or in combination with LY379268 (Supplementary Fig. 1A).
3.5. mGlu3 and mGlu5 receptors synergize to induce LTD in the PFC
To study the functional relevance of interactions between mGlu3 and mGlu5 receptors in neurons, we measured drug-induced changes in excitatory synaptic transmission in the mouse PFC. Pharmacological activation of mGlu3 receptors has previously been shown to induce LTD of excitatory transmission in the PFC (Walker et al., 2015). We used whole-cell voltage-clamp recordings from PFC layer V pyramidal cells to determine whether activation of mGlu5 receptors can enhance induction of mGlu3 receptor-mediated LTD in this region. To evaluate the effect of mGlu5 receptor activation on threshold LTD, a low concentration of LY379268 (100 nM) was applied to PFC slices for 3 min. This relatively brief application of LY379268 was not sufficient to induce LTD (Fig. 5A, grey bar, 102 ± 14% baseline, n = 4), and application of DHPG (100 μM) induced a transient depression of synaptic transmission, but did not induce LTD in the PFC (Fig. 5B, 108 ± 15% baseline, n = 4). We then co-applied both agonists for the same 3-min period and observed a persistent LTD at least 30 min following drug wash-out (Fig. 5C,D, 65 ± 4% baseline, n = 5). To confirm that mGlu3 and mGlu5 receptors mediate this LTD, we used the selective mGlu3 and mGlu5 receptor NAMs, VU0650786 and MTEP, respectively. Application of either VU0650786 (Fig. 5E, 94 ± 11% baseline, n = 4) or MTEP (Fig. 5F, 100 ± 12% baseline, n = 4) blocked LTD, suggesting that mGlu3 and mGlu5 receptors act synergistically in native tissue to generate long-lasting changes in excitatory synaptic transmission.
Fig. 5. mGlu3 and mGlu5 receptors synergize to induce LTD in the mouse PFC.
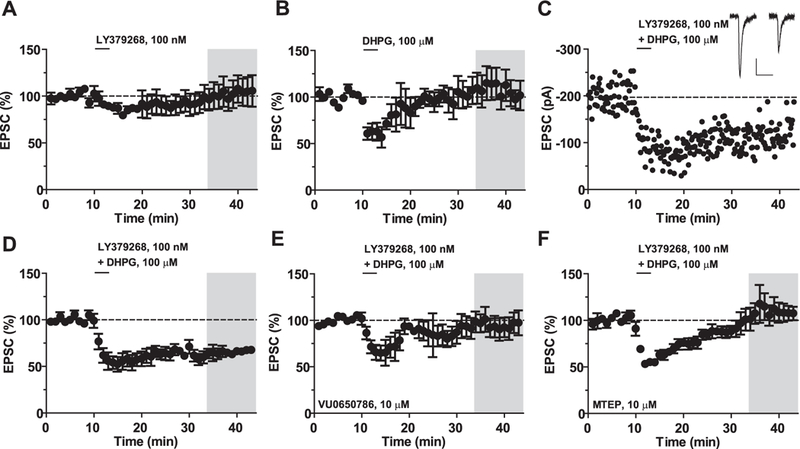
(A) Application of the mGlu2/3 receptor agonist LY379268 (100 nM) for 3 min does not generate a long-term change in the amplitude of the excitatory postsynaptic current (EPSC) (102 ± 14% baseline, n = 4). (B) Application of the mGlu1/5 receptor agonist DHPG (100 μM) for 3 min does not alter long-term excitatory transmission (108 ± 15% baseline, n = 4). (C) Representative experiment showing LTD induced by the combined bath application of LY379268 and DHPG. Inset, representative EPSC traces before and after LTD. Scale bars denote 50 ms and 50 pA. (D) Summary time course of LTD experiments (65 ± 4% baseline, n = 5). (E) VU0650786 blocked the induction of LTD (94 ± 11% baseline, n = 4). (F) MTEP blocked the induction of LTD (100 ± 12% baseline, n = 4). Values are mean ± S.E.M. in A-F.
As we and others have shown (Walker et al., 2015; Huang and Hsu, 2008; Kasanetz et al., 2013), a more prolonged 10-min bath application of LY379268 induces saturating LTD (Fig. 6A,B, 54 ± 7% baseline, n = 5). Through selective pharmacology and genetic studies, we previously demonstrated that this LTD is mediated by activation of mGlu3, and not mGlu2 receptors (Walker et al., 2015). Moreover, others have implicated several molecules related to Gq-signaling in this mGlu3 receptor-dependent LTD, including PKC and IP3 receptors (Otani et al., 2002; Huang et al., 2007). These previous studies were somewhat perplexing in light of the fact that mGlu3 receptor was not thought to activate Gq-signaling. However, based on the present findings, we postulated that co-activation of mGlu3 and mGlu5 receptors may be required for induction of mGlu3 receptor-LTD in the PFC. Consistent with this hypothesis, the mGlu5 receptor NAM MTEP blocked the induction of mGlu3 receptor-LTD (Fig. 6C,D, 89 ± 10% baseline, n = 5), suggesting that constitutive mGlu5 receptor activity, or activation of mGlu5 receptors by endogenous glutamate, is necessary for mGlu3 receptor-mediated synaptic plasticity. A synopsis of all LTD data is shown in Supplementary Fig. 2.
Fig. 6. Saturating mGlu3 receptor-LTD requires the activation of mGlu5 receptors.
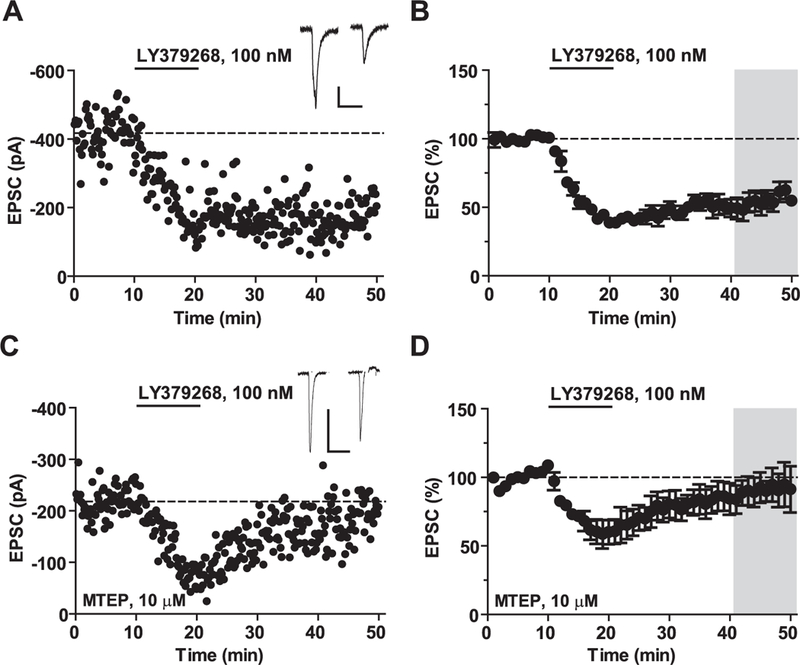
mGlu3 receptor-dependent-LTD requires activation of mGlu5 receptors. (A) Representative experiment displaying LTD of excitatory transmission following bath application of the mGlu2/3 receptor agonist LY379268 (100 nM) for 10 min. Inset, representative excitatory postsynaptic current (EPSC) before and after LTD. All scale bars denote 100 ms and 100 pA. (B) Summary of LTD experiments. LY379268 induces LTD (54 ± 7% baseline, n = 5). (C) Representative experiment following application of the selective mGlu5 receptor NAM, MTEP (10 μM). (D) MTEP blocked the induction of LTD (89 ± 10% baseline, n = 5). Values are mean ± S.E.M. in A-D.
3.6. mGlu5 receptor-mediated enhancement of excitotoxic neuronal death requires the activation of mGlu3 receptors
To study the relevance of the functional partnership between mGlu3 and mGlu5 receptors in mechanisms of neurodegeneration, we used pure cultures of rat cortical neurons challenged with toxic concentrations of NMDA. Neurons were exposed for 20 min to NMDA, and neuronal death was assessed after 22–24 h by either trypan blue staining or the MTT assay. DHPG (100 μM) and the mGlu3 receptor NAM, VU0650786 (10 μM), were added to the medium during the NMDA pulse. All experiments were carried out in the presence of the mGlu1 receptor NAM, JNJ16259685 (10 μM). DHPG had a small effect on neuronal viability on its own, but amplified excitotoxic neuronal death when combined with either 75 or 300 μM NMDA. DHPG no longer amplified NMDA toxicity but became neuroprotective when endogenous activation of mGlu3 receptors was prevented by co-treatment with VU0650786 (Fig. 7). Of note, VU0650786 had no effect per se on NMDA toxicity (Fig. 7), suggesting that neuronal mGlu3 receptors influence neuro-degeneration by shaping the activity of mGlu5 receptors.
Fig. 7. Amplification of NMDA toxicity by DHPG in rat cortical neurons requires the endogenous activation of mGlu3 receptors.
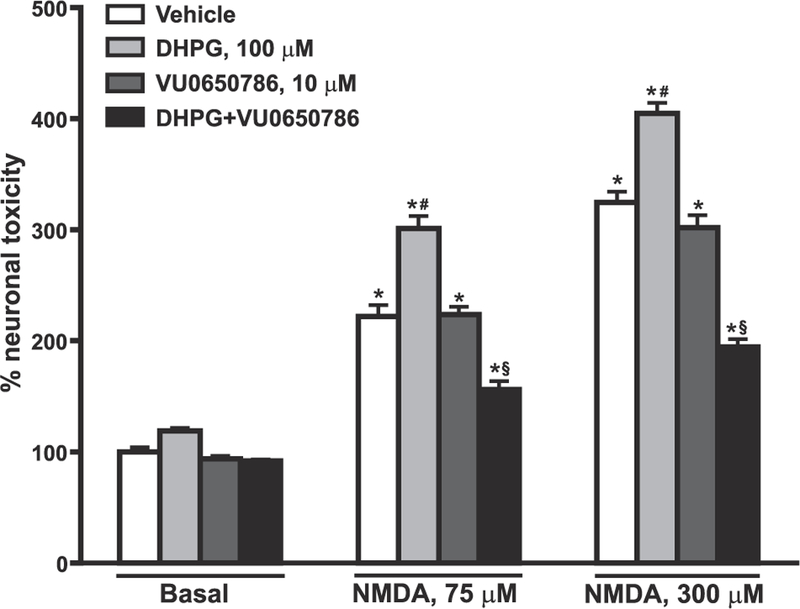
Pure cultures of rat cortical neurons were challenged with either 875 or 300 μM NMDA for 20 min in the absence or presence of DHPG and/or the mGlu3 receptor NAM, VU0650786. The experiment was carried out in the presence of the mGlu1 receptor NAM, JNJ16259685 (10 μM), to avoid the stimulation of mGlu1 receptors by DHPG. Neuronal toxicity was assessed by trypan blue staining. Values are means ± S.E.M. of 4 determinations. p < 0.05 (One-way ANOVA + Fisher’s LSD) vs. the respective basal values (*), the vehicle values of the same group (NMDA alone) (#), or vs. the respective DHPG values of the same group (§). F(11,36) = 177.91. This experiment was repeated twice and data were confirmed by using the MTT assay.
4. Discussion
We have shown for the first time that mGlu3 and mGlu5 receptors specifically interact in the CNS and that endogenous activation of mGlu3 receptors is required for maximal mGlu5 receptor signaling. In addition, endogenous activation of mGlu5 receptors is required for the induction of mGlu3 receptor-dependent LTD in the PFC. The demonstration of a functional partnership between mGlu3 and mGlu5 receptors sheds new light into the complex role played by mGlu receptors in mechanisms of synaptic plasticity during development and in adulthood, and provides compelling rationale for evaluating mGlu3 receptor modulators as potential treatments in CNS disorders associated with altered mGlu5 receptor function.
There are multiple examples in which mGlu receptor subtypes coupled to Gi have been shown to interact with other receptors coupled to Gq and vice versa (Aghajanian and Marek, 2000; Marek et al., 2000; Tabata et al., 2004; González-Maeso et al., 2008; Rives et al., 2009; Iacovelli et al., 2017). However, this is the first time that a partnership between two mGlu receptor subtypes coupled to different G proteins is shown in neurons.
mGlu receptor subtypes form homo- and heterodimers (Romano et al., 2001; Goudet et al., 2005; El Moustaine et al., 2012; Kammermeier, 2012; Yin and Niswender, 2014), but mGlu5 receptors are not thought to form inter-group heterodimers with either mGlu2 or mGlu3 receptors (Doumazane et al., 2011; Sevastyanova and Kammermeier, 2014). Thus, the interactions between mGlu3 and mGlu5 receptors shown here are likely mediated by cross-talk of signaling pathways and not by heterodimer interactions. Consistent with this, in HEK 293 cells functional interaction between mGlu3 and mGlu5 receptors was abrogated by expression of the C-terminal domain of GRK-2, which buffers the βγ subunits of G proteins (Premont et al., 1995). This suggests that mGlu3 receptors boost mGlu5 receptor signaling via the βγ subunits released from Gi proteins, which act synergistically with Gq in activating phospholipase-Cβ (Wang et al., 1999).
In heterologous expression systems, activation of both mGlu2 and mGlu3 receptors could amplify PI hydrolysis stimulated by either mGlu1 or mGlu5 receptors, suggesting that any group-II mGlu receptor (coupled to Gi) can functionally interact to any group-I mGlu receptor (coupled to Gq) when receptors are artificially co-expressed in same cells. This implies that under native conditions the interaction between mGlu3 and mGlu5 receptors is specific because the two receptors are co-localized in subcellular compartments, or that cell type-specific scaffolding proteins tightly regulate mGlu receptor interactions. This contrasts with the general belief that neuronal mGlu3 receptors are predominantly (albeit not exclusively) localized in presynaptic terminals, whereas mGlu5 receptors are found in postsynaptic densities (Nicoletti et al., 2011). We could demonstrate the synergism between mGlu3 and mGlu5 receptors in layer V pyramidal neurons of the PFC under conditions in which transynaptic action potential-driven events were blocked by tetrodotoxin. These findings suggest that postsynaptic mGlu3 receptors amplify mGlu5 receptor signaling in neurons, consistent with recent electron microscopy data showing that mGlu3 receptors are concentrated in postsynaptic locations in the PFC (Jin et al., 2017). Activation of postsynaptic mGlu3 receptors induces LTD in the medial PFC (Walker et al., 2015), a particular form of activity-dependent synaptic plasticity that plays a key role in medial PFC-dependent cognitive functions (Walker et al., 2015; Kasanetz et al., 2013; Otani et al., 1999). Accordingly, selective blockade of mGlu3 receptors disrupts learning in a medial PFC-fear extinction task (Walker et al., 2015), and polymorphic variants of the GRM3 (the gene encoding mGlu3 receptors) are associated with poor performance in PFC-dependent cognitive tasks (Egan et al., 2004; Harrison et al., 2008). We demonstrated here that induction of mGlu3 receptor-dependent LTD requires the endogenous activity of mGlu5 receptors. This finding is consistent with previous reports demonstrating that PFC mGlu3-LTD is blocked by inhibitors of proteins involved in canonical Gq signaling, such as PLC and PKC (Otani et al., 2002; Huang et al., 2007). On the other hand, Otani et al. (2002) concluded that group-I mGlu receptors are not involved in PFC mGlu3-LTD. Several mechanistic and technical differences may account for the discrepancy between that interpretation and ours. For one, Otani et al. (2002) performed recordings in slices prepared from the juvenile rat PFC. In light of the pronounced developmental changes observed in the present study, it would not be surprising if mechanistic differences in mGlu receptor synaptic plasticity exist at these different stages. Consistent with that possibility, Otani et al. (2002) implicated NMDA receptor activation in mGlu3-LTD, whereas recent studies from our lab and others show that NMDA receptor activity is not required (Walker et al., 2015; Huang and Hsu, 2008). Furthermore, Otani et al. (2002) used the mGlu1-preferring antagonist AIDA, which may not have efficiently blocked mGlu5 receptor activation. Alternatively, we utilized highly selective NAMs to inhibit mGlu5 function. In addition to selectivity, the contrasting modes of inhibition (i.e. orthosteric antagonist vs. allosteric inverse agonist) could factor into the difference in findings. While further investigation into the mechanism is warranted, the finding that the mGlu5 receptor is required for mGlu3-mediated synaptic plasticity is of great relevance in physiology and pathology. Cognitive dysfunction caused by abnormalities in synaptic plasticity in the PFC lies at the core of schizophrenia and other psychiatric disorders (Carpenter and Buchanan, 1994; Tamminga and Holcomb, 2005; Falkai et al., 2015). Our findings suggest that the functional partnership between mGlu5 and mGlu3 receptors cannot be ignored in the design of new nootropic drugs that target mGlu5 receptors. Moreover, mGlu3 receptors might modulate synaptic plasticity in brain regions that express prominent mGlu5-mediated plasticity. With that in mind, modulators of mGlu3 receptor signaling may hold therapeutic potential in a host of disease states that have yet to be considered.
One of the most striking features of mGlu5 receptors is that agonist-stimulated PI hydrolysis is extraordinarily large during the first 10 days of postnatal life, and progressively declines afterwards (Nicoletti et al., 1986a; Casabona et al., 1997). mGlu5 receptors are involved in mechanisms of developmental plasticity, and drive key events in the formation of the cortical somatosensory map (Hannan et al., 2001; Wijetunge et al., 2008; Loerwald et al., 2015; Ballester-Rosado et al., 2016). mGlu5 receptors are necessary for the development of fast-spiking GABAergic interneurons, which regulate the synchronous firing of pyramidal neurons and are critical for the generation of network oscillations in the PFC (Barnes et al., 2015; Luoni et al., 2016). We were surprised to find that endogenous activation of mGlu3 receptors was required for the full efficiency of mGlu5 receptor-mediated PI hydrolysis in the first 10 days of postnatal life. In addition, pharmacological activation of mGlu3 receptors with LY379268 was sufficient to stimulate PI hydrolysis in the first 12 days of postnatal development. This unexpected effect might be explained by amplification of greater constitutive activity of mGlu5 receptors during this early developmental stage. The finding that LY379268 alone did not stimulate PI hydrolysis at later stages might reflect the developmental decline of mGlu5 receptors (Casabona et al., 1997; Catania et al., 1994). We suggest that the functional partnership between mGlu3 and mGlu5 receptors is instrumental for the large stimulation of PI hydrolysis in the early phases of postnatal development, when high levels of intracellular Ca2+ are required for mechanisms of developmental plasticity. Of note, expression of both mGlu3 and mGlu5 receptors is high in the early postnatal brain, and progressively declines across postnatal development (Catania et al., 1994; Lopez-Bendito et al., 2002). Genetic variants of GRM3 are associated with schizophrenia (Fujii et al., 2003; Egan et al., 2004; Chen et al., 2005; Norton et al., 2005; Bishop et al., 2007; Schwab et al., 2008), and dimerization of mGlu3 receptors is altered in the PFC of patients affected by schizophrenia (Corti et al., 2007). Abnormalities in mGlu3 receptor expression or function may disrupt the functional partnership between mGlu3 and mGlu5 receptors, with detrimental consequences during critical time windows of postnatal development. Preclinical studies indicate that mGlu5 receptor positive allosteric modulators (PAM) are putative candidate drugs in the treatment of schizophrenia (Kinney et al., 2005; Rodriguez et al., 2010; Vinson and Conn, 2012). Group-II mGlu receptor agonists have also shown antipsychotic-like effects in preclinical models, but selective mGlu2 PAMs have failed to reach the clinic. Together, these data suggest that selective PAMs of mGlu3 receptors may hold promise in treating schizophrenia and confer antipsychotic or nootropic benefits by potentiating mGlu5 receptor signaling. Moreover, our data suggest that the efficacy of mGlu5 receptor PAMs may be suboptimal in schizophrenic patients with a defective expression or function of mGlu3 receptors, and perhaps a multimodal drug amplifying both mGlu3 and mGlu5 receptors would be highly valuable in the treatment of cognitive dysfunction associated with schizophrenia.
Our data may also have a strong impact on the study of mGlu receptors in mechanisms of neurodegeneration/neuroprotection (Bruno et al., 2017). Activation of mGlu5 receptors may either support or dampen neuronal viability depending on the experimental paradigm of neurotoxicity (Nicoletti et al., 1999; Bruno et al., 2001, 2017), whereas pharmacological blockade of mGlu5 receptors is consistently neuroprotective in a variety of models of neurodegenerative disorders (Battaglia et al., 2002, 2004; Masilamoni et al., 2011; Hamilton et al., 2014). Glial mGlu3 receptors are neuroprotective via a paracrine mechanism mediated by the production of neurotrophic factors, (Bruno et al., 1998; Battaglia et al., 2009, 2015; Caraci et al., 2011), whereas the role of neuronal mGlu3 receptors in neurodegeneration/neuroprotection is unknown. Using pure cultures of cortical neurons, we have shown that pharmacological activation of mGlu5 receptors amplified excitotoxic neuronal death, and this effect was abrogated by the mGlu3 receptor NAM, VU0650786. Interestingly, activation of mGlu5 receptors became neuroprotective in the presence of VU0650786, which had no effect on NMDA toxicity on its own. This suggests that mGlu5 receptors may either amplify or restrain neuronal toxicity and the balance between these two functions is critically regulated by mGlu3 receptors. Thus, the expression and function of mGlu3 receptors may shape the role of mGlu5 receptors in mechanisms of neurodegeneration/neuroprotection. This novel mechanism raises concern about the use of mGlu3 receptor agonists or PAMs as potential neuroprotective agents in the treatment of Parkinson’s disease and other chronic neurodegenerative disorders (Bruno et al., 2017). Perhaps the combination of mGlu3 PAMs and mGlu5 NAMs might represent an optimal therapeutic option to slow the progression of chronic neurodegenerative disorders.
In conclusion, the demonstration of the functional partnership between mGlu3 and mGlu5 receptors may have important implications in physiology and pathology and stimulates reconsideration of the existing literature on mGlu3 and mGlu5 receptors from a different angle. The high expression of mGlu3 and mGlu5 receptors during early postnatal development may be an absolute requirement to ensure an efficient stimulation of PI hydrolysis that is essential for mechanisms of developmental plasticity, and CNS disorders which are related to a defective activation of mGlu5 receptors, such as schizophrenia, might be caused by a disrupted functional interaction between the two receptors. On the other hand, mGlu3 receptors might have a role in disorders characterized by an over-activity of mGlu5 receptors, such as Fragile X and other types of monogenic autism. It remains to be determined whether the pharmacodynamic boosting mediated by mGlu3 receptors is restricted to mGlu5 receptor-stimulated PI hydrolysis or is extended to other intracellular transduction mechanisms.
Supplementary Material
Acknowledgments
This work was supported by the 5‰ grant.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.neuropharm.2017.10.026.
References
- Aghajanian GK, Marek GJ, 2000. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain Res. Brain Res. Rev 31, 302–312. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S, Marino MJ, Rouse ST, Gereau RW 4th, Heinemann SF, Conn PJ, 1999. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat. Neurosci 2, 234–240. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S, Rouse ST, Junge C, Hubert GW, Gutman D, Smith Y, Conn PJ, 2002. NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol. Biochem. Behav 73, 299–306. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S, Saugstad J, Warren L, Mansuy IM, Gereau RW 4th, Conn PJ, 2005. NMDA-induced potentiation of mGluR5 is mediated by activation of protein phosphatase 2B/calcineurin. Neuropharmacology 49 (Suppl. 1), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, Pin JP, Tu JC, Xiao B, Worley PF, Bockaert J, Fagni L, 2000. Dendritic and axonal targeting of type 5 metabotropic glutamate receptor is regulated by homer1 proteins and neuronal excitation. J. Neurosci 20, 8710–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester-Rosado CJ, Sun H, Huang JY, Lu HC, 2016. mGluR5 exerts cell-autonomous influences on the functional and anatomical development of layer IV cortical neurons in the mouse primary somatosensory cortex. J. Neurosci 36, 8802–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SA, Pinto-Duarte A, Kappe A, Zembrzycki A, Metzler A, Mukamel EA, Lucero J, Wang X, Sejnowski TJ, Markou A, Behrens MM, 2015. Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neurodevelopmental disorders. Mol. Psychiatry 20, 1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A, Melchiorri D, Nicoletti F, 2002. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J. Neurosci 22, 2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Busceti CL, Molinaro G, Biagioni F, Storto M, Fornai F, Nicoletti F, Bruno V, 2004. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J. Neurosci 24, 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Molinaro G, Riozzi B, Storto M, Busceti CL, Spinsanti P, Bucci D, Di Liberto V, Mudo G, Corti C, Corsi M, Nicoletti F, Belluardo N, Bruno V, 2009. Activation of mGlu3 receptors stimulates the production of GDNF in striatal neurons. PLoS One 4, e6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Riozzi B, Bucci D, Di Menna L, Molinaro G, Pallottino S, Nicoletti F, Bruno V, 2015. Activation of mGlu3 metabotropic glutamate receptors enhances GDNF and GLT-1 formation in the spinal cord and rescues motor neurons in the SOD-1 mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis 74, 126–136. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Wang K, Moline J, Ellingrod VL, 2007. Association analysis of the metabotropic glutamate receptor type 3 gene (GRM3) with schizophrenia. Psychiatr. Genet 17, 358. [DOI] [PubMed] [Google Scholar]
- Bollinger AK, Felts SA, Brassard JC, Engers LJ, Rodriguez LA, Weiner LR, Cho PH, Chang S, Bubser M, Jones KC, Blobaum LA, Niswender MC, Conn PJ, Emmitte AK, Lindsley WC, 2017. Design and Synthesis of mGlu2 NAMs with Improved Potency and CNS Penetration Based on a Truncated Picolinamide Core https://doi.org/10.1021/acsmedchemlett.7b00279. Publication Date (Web): August 3, 2017. [DOI] [PMC free article] [PubMed]
- Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, Worley PF, 1997. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F, 1998. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J. Neurosci 18, 9594–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Copani A, D’Onofrio M, Di Iorio P, De Blasi A, Melchiorri D, Flor PJ, Nicoletti F, 2001. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow. Metab 21, 1013–1033. [DOI] [PubMed] [Google Scholar]
- Bruno V, Caraci F, Copani A, Matrisciano F, Nicoletti F, Battaglia G, 2017. The impact of metabotropic glutamate receptors into active neurodegenerative processes: a “dark side” in the development of new symptomatic treatments for neurologic and psychiatric disorders. Neuropharmacology 115, 180–192. [DOI] [PubMed] [Google Scholar]
- Caraci F, Molinaro G, Battaglia G, Giuffrida ML, Riozzi B, Traficante A, Bruno V, Cannella M, Merlo S, Wang X, Heinz BA, Nisenbaum ES, Britton TC, Drago F, Sortino MA, Copani A, Nicoletti F, 2011. Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer’s disease: selective activation of mGlu2 receptors amplifies beta-amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol. Pharmacol 79, 618–626. [DOI] [PubMed] [Google Scholar]
- Caraci F, Tascedda F, Merlo S, Benatti C, Spampinato SF, Munafo A, Leggio GM, Nicoletti F, Brunello N, Drago F, Sortino MA, Copani A, 2016. Fluoxetine prevents Ab(1–42)-induced toxicity via a paracrine signaling mediated by transforming-growth-factor-b1. Front. Pharmacol 7, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter WT Jr., Buchanan RW, 1994. Schizophrenia. N. Engl. J. Med 330, 681–690. [DOI] [PubMed] [Google Scholar]
- Casabona G, Knopfel T, Kuhn R, Gasparini F, Baumann P, Sortino MA, Copani A, Nicoletti F, 1997. Expression and coupling to polyphosphoinositide hydrolysis of group I metabotropic glutamate receptors in early postnatal and adult rat brain. Eur. J. Neurosci 9, 12–17. [DOI] [PubMed] [Google Scholar]
- Catania MV, Landwehrmeyer GB, Testa CM, Standaert DG, Penney JB Jr., Young AB, 1994. Metabotropic glutamate receptors are differentially regulated during development. Neuroscience 61, 481–495. [DOI] [PubMed] [Google Scholar]
- Chen Q, He G, Chen Q, Wu S, Xu Y, Feng G, Li Y, Wang L, He L, 2005. A case-control study of the relationship between the metabotropic glutamate receptor 3 gene and schizophrenia in the Chinese population. Schizophr. Res 73, 21–26. [DOI] [PubMed] [Google Scholar]
- Coccurello R, Breysse N, Amalric M, 2004. Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology 29, 1451–1461. [DOI] [PubMed] [Google Scholar]
- Conti V, Aghaie A, Cilli M, Martin N, Caridi G, Musante L, Candiano G, Castagna M, Fairen A, Ravazzolo R, Guenet JL, Puliti A, 2006. Crv4, a mouse model for human ataxia associated with kyphoscoliosis caused by an mRNA splicing mutation of the metabotropic glutamate receptor 1 (Grm1). Int. J. Mol. Med 18, 593–600. [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella A.M., Canonico PL, Nicoletti F, Sortino MA, 1999. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J 13, 2225–2234. [PubMed] [Google Scholar]
- Corti C, Crepaldi L, Mion S, Roth AL, Xuereb JH, Ferraguti F, 2007. Altered dimerization of metabotropic glutamate receptor 3 in schizophrenia. Biol. Psychiatry 62, 747–755. [DOI] [PubMed] [Google Scholar]
- D’Amore V, von Randow C, Nicoletti F, Ngomba RT, van Luijtelaar G, 2015. Anti-absence activity of mGlu1 and mGlu5 receptor enhancers and their interaction with a GABA reuptake inhibitor: effect of local infusions in the somatosensory cortex and thalamus. Epilepsia 56, 1141–1151. [DOI] [PubMed] [Google Scholar]
- D’Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, Nicoletti F, Huber KM, Catania MV, 2014. Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with intellectual disability and autism. Neurosci. Biobehav. Rev 46, 228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Cabiale Z, Vivo M, Del Arco A, O’Connor WT, Harte MK, Müller CE, Martínez E, Popoli P, Fuxe K, Ferre S, 2002. Metabotropic glutamate mGlu5 receptor-mediated modulation of the ventral striopallidal GABA pathway in rats. Interactions with adenosine A(2A) and dopamine D(2) receptors. Neurosci. Lett 324, 154–158. [DOI] [PubMed] [Google Scholar]
- Domenici MR, Pepponi R, Martire A, Tebano MT, Potenza RL, Popoli P, 2004. Permissive role of adenosine A2A receptors on metabotropic glutamate receptor 5 (mGluR5)-mediated effects in the striatum. J. Neurochem 90, 1276–1289. [DOI] [PubMed] [Google Scholar]
- Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, Pin JP, 2011. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J 25, 66–77. [DOI] [PubMed] [Google Scholar]
- DuBois JM, Rousset OG, Guiot MC, Hall JA, Reader AJ, Soucy JP, Rosa-Neto P, Kobayashi E, 2017. Metabotropic glutamate receptor type 5 (mGluR5) cortical abnormalities in focal cortical dysplasia identified in vivo with [11C] ABP688 positron-emission tomography (PET) imaging. Cereb. Cortex 26, 4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, Mattay VS, Bertolino A, Hyde TM, Shannon-Weickert C, Akil M, Crook J, Vakkalanka RK, Balkissoon R, Gibbs RA, Kleinman JE, Weinberger DR, 2004. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl. Acad. Sci. U. S. A 101, 12604–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Moustaine D, Granier S, Doumazane E, Scholler P, Rahmeh R, Bron P, Mouillac B, Baneres JL, Rondard P, Pin JP, 2012. Distinct roles of metabo-tropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc. Natl. Acad. Sci. U. S. A 109, 16342–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers JL, Rodriguez AL, Konkol LC, Morrison RD, Thompson AD, Byers FW, Blobaum AL, Chang S, Venable DF, Loch MT, Niswender CM, Daniels JS, Jones CK, Conn PJ, Lindsley CW, Emmitte KA, 2015. Discovery of a selective and CNS penetrant negative allosteric modulator of metabotropic glutamate receptor subtype 3 with antidepressant and anxiolytic activity in rodents. J. Med. Chem 58, 7485–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkai P, Rossner MJ, Schulze TG, Hasan A, Brzozka MM, Malchow B, Honer WG, Schmitt A, 2015. Kraepelin revisited: schizophrenia from degeneration to failed regeneration. Mol. Psychiatry 20, 671–676. [DOI] [PubMed] [Google Scholar]
- Ferré S, Karcz-Kubicha M, Hope BT, Popoli P, Burgueño J, Gutierrez MA, Casado V, Fuxe K, Goldberg SR, Lluis C, Franco R, Ciruela F, 2002. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc. Natl. Acad. Sci. U. S. A 99, 11940–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, Gentry PR, Lizardi-Ortiz JE, Bridges TM, Wood MR, Niswender CM, Sulzer D, Lindsley CW, Xiang Z, Conn PJ, 2014. M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor’s location. J. Neurosci 34, 3253–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Shibata H, Kikuta R, Makino C, Tani A, Hirata N, Shibata A, Ninomiya H, Tashiro N, Fukumaki Y, 2003. Positive associations of polymorphisms in the metabotropic glutamate receptor type 3 gene (GRM3) with schizophrenia. Psychiatr. Genet 13, 71–76. [DOI] [PubMed] [Google Scholar]
- Genazzani AA, L’Episcopo MR, Casabona G, Shinozaki H, Nicoletti F, 1994. (2S,1’R,2’R,3’R)-2-(2,3-dicarboxycyclopropyl) glycine positively modulates metabotropic glutamate receptors coupled to polyphosphoinositide hydrolysis in rat hippocampal slices. Brain Res 659, 10–16. [DOI] [PubMed] [Google Scholar]
- Giuffrida R, Musumeci S, D’Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV, 2005. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci 25, 8908–8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC, 2008. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, Blahos J, Prezeau L, Pin JP, 2005. Asymmetric functioning of dimeric metabotropic glutamate receptors disclosed by positive allosteric modulators. J. Biol. Chem 280, 24380–24385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Molinaro G, Collins KA, Hays SA, Paylor R, Worley PF, Szumlinski KK, Huber KM, 2016. Selective disruption of metabotropic glutamate receptor 5-Homer interactions mimics phenotypes of Fragile X syndrome in mice. J. Neurosci 36, 2131–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Esseltine JL, DeVries RA, Cregan SP, Ferguson SS, 2014. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC, 2001. PLC-beta1, activated via mGluRs, mediatesactivity-dependent differentiation in cerebral cortex. Nat. Neurosci 4, 282–288. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Lyon L, Sartorius LJ, Burnet PW, Lane TA, 2008. The group II metabotropic glutamate receptor 3 (mGluR3, mGlu3, GRM3): expression, function and involvement in schizophrenia. J. Psychopharmacol 22, 308–322. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS, 2008. The role of NMDA receptors in regulating group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. Neuropharmacology 54, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yang PC, Lin HJ, Hsu KS, 2007. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J. Neurosci 27, 2958–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF, 2002. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U. S. A 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli L, Felicioni M, Nistico R, Nicoletti F, De Blasi A, 2014. Selective regulation of recombinantly expressed mGlu7 metabotropic glutamate receptors by G protein-coupled receptor kinases and arrestins. Neuropharmacology 77, 303–312. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Di Menna L, Peterlik D, Stangl C, Orlando R, Molinaro G, DeBlasi A, Bruno V, Battaglia G, Flor PJ, Uschold-Schmidt N, Nicoletti F, 2017. Type-7 metabotropic glutamate receptors negatively regulate α(1)-adrenergic receptor signalling. Neuropharmacology 113, 343–353. [DOI] [PubMed] [Google Scholar]
- Iyer AM, van Scheppingen J, Milenkovic I, Anink JJ, Lim D, Genazzani AA, Adle-Biassette H, Kovacs GG, Aronica E, 2014. Metabotropic glutamate receptor 5 in Down’s syndrome hippocampus during development: increased expression in astrocytes. Curr. Alzheimer Res 11, 694–705. [DOI] [PubMed] [Google Scholar]
- Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AF, Paspalas CD, 2017. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: postsynaptic mGluR3 strengthen working memory networks. Cereb. Cortex https://doi.org/10.1093/cercor/bhx005, 2017 Jan 19. [DOI] [PMC free article] [PubMed]
- Kammermeier PJ, 2012. Functional and pharmacological characteristics of metabotropic glutamate receptors 2/4 heterodimers. Mol. Pharmacol 82, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ, Worley PF, 2007. Homer 1a uncouples metabotropic glutamate receptor 5 from postsynaptic effectors. Proc. Natl. Acad. Sci. U. S. A 104, 6055–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, Berson N, Balado E, Fiancette JF, Renault P, Piazza PV, Manzoni OJ, 2013. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol. Psychiatry 18, 729–737. [DOI] [PubMed] [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL Jr., 2005. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J. Pharmacol. Exp. Ther 313, 199–206. [DOI] [PubMed] [Google Scholar]
- Lin TB, Lai CY, Hsieh MC, Wang HH, Cheng JK, Chau YP, Chen GD, Peng HY, 2015. VPS26A-SNX27 interaction-dependent mGluR5 recycling in dorsal horn neurons mediates neuropathic pain in rats. J. Neurosci 35, 14943–14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden AM, Shannon H, Baez M, Yu JL, Koester A, Schoepp DD, 2005. Anxiolytic-like activity of the mGLU2/3 receptor agonist LY354740 in the elevated plus maze test is disrupted in metabotropic glutamate receptor 2 and 3 knock-out mice. Psychopharmacology 179, 284–291. [DOI] [PubMed] [Google Scholar]
- Loerwald KW, Patel AB, Huber KM, Gibson JR, 2015. Postsynaptic mGluR5 promotes evoked AMPAR-mediated synaptic transmission onto neocortical layer 2/3 pyramidal neurons during development. J. Neurophysiol 113, 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Fairen A, Lujan R, 2002. Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb. Cortex 12, 625–638. [DOI] [PubMed] [Google Scholar]
- Lovell KM, Felts AS, Rodriguez AL, Venable DF, Cho HP, Morrison RD, Byers FW, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Emmitte KA, 2013. N-Acyl-N’-arylpiperazines as negative allosteric modulators of mGlu1: identification of VU0469650, a potent and selective tool compound with CNS exposure in rats. Bioorg. Med. Chem. Lett 23, 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ, 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem 193, 265–275. [PubMed] [Google Scholar]
- Luoni A, Gass P, Brambilla P, Ruggeri M, Riva MA, Inta D, 2016. Altered expression of schizophrenia-related genes in mice lacking mGlu5 receptors. Eur. Arch. Psychiatry Clin. Neurosci. 2016 Aug 31, [Epub ahead of print]. [DOI] [PubMed]
- Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK, 2000. Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J. Pharmacol. Exp. Ther 292, 76–87. [PubMed] [Google Scholar]
- Masilamoni GJ, Bogenpohl JW, Alagille D, Delevich K, Tamagnan G, Votaw JR, Wichmann T, Smith Y, 2011. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134, 2057–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matosin N, Fernandez-Enright F, Lum JS, Engel M, Andrews JL, Gassen NC, Wagner KV, Schmidt MV, Newell KA, 2016. Molecular evidence of synaptic pathology in the CA1region in schizophrenia. NPJ Schizophr 2, 16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Romano C, Cotman CW, 1995. Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. J. Neurosci 15, 6103–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Iadarola MJ, Wroblewski JT, Costa E, 1986a. Excitatory amino acid recognition sites coupled with inositol phospholipid metabolism: developmental changes and interaction with alpha 1-adrenoceptors. Proc. Natl. Acad. Sci. U. S. A 83, 1931–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Meek JL, Iadarola MJ, Chuang DM, Roth BL, Costa E, 1986b. Coupling of inositol phospholipid metabolism with excitatory amino acid recognition sites in rat hippocampus. J. Neurochem 46, 40–46. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Casabona G, Genazzani AA, L’Episcopo MR, Shinozaki H, 1993. (2s,1′R,2′R,3′R)-2-(2,3-Dicarboxycyclopropyl) glycine enhances quisqualate-stimulated inositol phospholipid hydrolysis in hippocampal slices. Eur. J. Pharmacol 245, 297–308. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Catania MV, Battaglia G, Copani A, Barbagallo G, Ceña V, Sanchez-Prieto J, Spano PF, Pizzi M, 1999. Group-I metabotropic glutamate receptors: hypotheses to explain their dual role in neurotoxicity and neuro-protection. Neuropharmacology 38, 1477–1484. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP, 2011. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60, 1017–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Ngomba RT, Gradini R, Battaglia G, 2015. Metabotropic glutamate receptors as drug targets: what’s new? Curr. Opin. Pharmacol 20, 89–94. [DOI] [PubMed] [Google Scholar]
- Nishi A, Liu F, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P, 2003. Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc. Natl. Acad. Sci. U. S. A 100, 1322–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton N, Williams HJ, Dwyer S, Ivanov D, Preece AC, Gerrish A, Williams NM, Yerassimou P, Zammit S, O’Donovan MC, Owen MG, 2005. No evidence for association between polymorphisms in GRM3 and schizophrenia. BMC Psychiatry 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC, 1998. Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the PFC from normal individuals and patients with schizophrenia. Brain Res. Mol. Brain Res 56, 207–217. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin MP, Crepel F, 1999. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J. Neurosci 19, 9788–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F, 2002. Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J. Neurosci 22, 3434–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli M, Piccinin S, Molinaro G, Di Menna L, Riozzi B, Cannella M, Motolese M, Vetere G, Catania MV, Battaglia G, Nicoletti F, Nistico R, Bruno V, 2014. Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling Angelman syndrome. J. Neurosci 34, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Inglese J, Lefkowitz RJ, 1995. Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J 9, 175–182. [DOI] [PubMed] [Google Scholar]
- Reid SN, Romano C, Hughes T, Daw NW, 1998. Developmental and sensory-dependent changes of phosphoinositide-linked metabotropic glutamate receptors. J. Comp. Neurol 389, 577–583. [DOI] [PubMed] [Google Scholar]
- Rives ML, Vol C, Fukazawa Y, Tinel N, Trinquet E, Ayoub MA, Shigemoto R, Pin JP, Prezeau L, 2009. Crosstalk between GABAB and mGlu1a receptors reveals new insight into GPCR signal integration. EMBO J 28, 2195–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ, 2010. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and anti-psychotic activity. Mol. Pharmacol 78, 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano C, Miller JK, Hyrc K, Dikranian S, Mennerick S, Takeuchi Y, Goldberg MP, O’Malley KL, 2001. Covalent and noncovalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol. Pharmacol 59, 46–53. [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM, 2012. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci 15, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K, 2003. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat. Neurosci 6, 1153–1161. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Salhoff CR, Wright RA, Johnson BG, Burnett JP, Mayne NG, Belagaje R, Wu S, Monn JA, 1996. The novel metabotropic glutamate receptor agonist 2R,4R-APDC potentiates stimulation of phosphoinositide hydrolysis in the rat hippocampus by3,5-dihydroxyphenylglycine: evidence for a synergistic interaction between group 1 and group 2 receptors. Neuropharmacology 35, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Schwab SG, Plummer C, Albus M, Borrmann-Hassenbach M, Lerer B, Trixler M, Maier W, Wildenauer DB, 2008. DNA sequence variants in the metabotropic glutamate receptor 3 and risk to schizophrenia: an association study. Psychiatr. Genet 18, 25–30. [DOI] [PubMed] [Google Scholar]
- Sevastyanova TN, Kammermeier PJ, 2014. Cooperative signaling between homodimers of metabotropic glutamate receptors 1 and 5. Mol. Pharmacol 86, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Araishi K, Hashimoto K, Hashimotodani Y, van der Putten H, Bettler B, Kano M, 2004. Ca2+ activity at GABAB receptors constitutively promotes metabotropic glutamate signaling in the absence of GABA. Proc. Natl. Acad. Sci. U. S. A 101, 16952–16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminga CA, Holcomb HH, 2005. Phenotype of schizophrenia: a review and formulation. Mol. Psychiatry 10, 27–39. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF, 1999. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. [DOI] [PubMed] [Google Scholar]
- Vinson PN, Conn PJ, 2012. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology 62, 1461–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, Lindsley CW, Conn PJ, 2015. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial PFC and fear extinction. Proc. Natl. Acad. Sci. U. S. A 112, 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Pentyala S, Rebecchi MJ, Scarlata S, 1999. Differential association of the pleckstrin homology domains of phospholipases C-beta 1, C-beta 2, and C-delta 1 with lipid bilayers and the beta gamma subunits of heterotrimeric G proteins. Biochemistry 38, 1517–1524. [DOI] [PubMed] [Google Scholar]
- Wijetunge LS, Till SM, Gillingwater TH, Ingham CA, Kind PC, 2008. mGluR5 regulates glutamate-dependent development of the mouse somatosensory cortex. J. Neurosci 28, 13028–13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD, Ruggiero A, Lanahan AA, Wenthold RJ, Worley PF, 1998. Homer regulates the association of group 1metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 21, 707–716. [DOI] [PubMed] [Google Scholar]
- Yin S, Niswender CM, 2014. Progress toward advanced understanding of metabotropic glutamate receptors: structure, signaling and therapeutic indications. Cell. Signal 10, 2284–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.