

Abstract
Brain endocannabinoid system is proposed to play a role in the pathogenesis of affective disorders. In the present study, we analyzed the functionality of the cannabinoid receptor type 1 (CBa receptor) at different transduction levels in prefrontal cortex (PFC) of depressed suicide victims. We examined stimulation of [35S]GTPγS binding, activation of Gα protein subunits and inhibition of adenylyl cyclase by the cannabinoid agonist WIN55,212-2, as well as [3H]CP55,940 binding, in PFC homogenates from suicide victims with major depression (MD) and matched control subjects. CB1 receptor-stimulated [35S]GTPγS binding was significantly greater in the PFC of MD compared with matched controls (23%, p < 0.05). This increase was most evident in the PFC from MD subgroup with negative blood test for antidepressants (AD) at the time of death (AD-free) (38%, p < 0.05), being absent when comparing the AD-treated MD cases with their controls. The density of CB1 receptors and their coupling to adenylyl cyclase were similar between MD and control cases, regardless of the existence of AD intake. Analysis of [35S]GTPγS-labelled Gα subunits allowed for the detection of upregulated CB1 receptor coupling to Gαo, but not to Gαi1, Gαi2, Gαi3, Gαz subunits, in the PFC from AD-free MD suicides. These results suggest that increased CB1 receptor functionality at the Gαi/o protein level in the PFC of MD subjects is due to enhanced coupling to Gαo proteins and might be modulated by AD intake. These data provide new insights into the role of endocannabinoid neurotransmission in the pathobiology of MD and suggest its regulation by ADs.
Keywords: CB1 receptors, Gα subunits, Adenylyl cyclase, Major depression, Prefrontal cortex
1. Introduction
Major depression (MD) is a chronic, recurrent and prevalent disorder with major impacts in quality of life and cost of health care. Despite decades of intense research, there is a relative lack of knowledge regarding the aetiology of depression. In this sense, the majority of research in animal models and postmortem human brain suggests that alterations in the number and/or functionality of 5-hydroxytryptamine (5-HT) and norepinephrine (NE) receptors, reflecting a deficit in these neurotransmission systems, are involved in the pathogenesis of MD [1–3]. Indeed, depression is commonly treated with antidepressant compounds (ADs) that increase the synaptic content of 5-HT and/or NE [4]. Nevertheless, the clinical improvement associated to ADs intake results evident only after several weeks of treatment, suggesting that the acute enhancement of monoaminergic neurotransmission is not responsible for the therapeutic efficacy of these drugs [5]. More importantly, less than 50% of the patients with depression experience complete remission following antidepressant treatment [4]. Current research towards the development of more effective ADs is aimed at improving our understanding of the mechanisms that contribute to the pathophysiology of depression, and to the efficacy of nowadays available antidepressant medications.
Converging evidence suggest that targeting brain endocannabinoid system may be a useful strategy for the development of new antidepressant medications [4,6]. Cannabinoid CB1 receptor is the main subtype in the central nervous system, and its transduction mechanisms include the inhibition of adenylyl cyclase (AC) as well as the modulation of ionic currents via Gi/o proteins [7], and the activation of the mitogen-activated protein kinase (MAPK) pathway [8]. CB1 receptors are expressed in high levels in brain areas relevant to depression, such us the prefrontal cortex (PFC) or the hippocampus [9]. Thus, the pharmacological or genetic blockade of CB1 cannabinoid receptors in animal studies result in behavioral responses that resemble human depressive symptoms [10,11]. On contrast, other studies indicate that CB1 receptor mRNA, protein expression and/or signaling in the PFC are upregulated in animal models of the disease [12–15]. Similar discrepancies have been described in association to antidepressant treatment. Thus, the acute CB1 receptor activation produces antidepressant responses and enhances the effects of ADs in rodents [12,16], although antidepressant effects resulting from CB1 receptor blockade have also been reported [17]. Finally, a number of studies have shown that chronic administration of ADs modulates the activity of brain endocannabinoid system [13,14,18,19]. All these data support a role of the endocannabinoid system in MD.
Despite these accumulating data from research using animal models, limited information that supports a role for CB1 receptors in depression is nowadays available from human studies [20]. In this sense, variation of the gene coding for the CB1 receptor has been shown to influence both the susceptibility to MD and the response to AD treatment [20–24]. With regard to postmortem studies, we [25] and others [26,27] have demonstrated increased CB1 receptor mediated activation of Gi/o proteins in the PFC of depressed suicide victims. However, the mechanisms underlying this observed upregulation of CB1 receptor signaling in MD subjects, its possible modulation by ADs, and the resulting consequences downstream Gi/o proteins, have not been investigated in detail. The aim of this study was to examine specific molecular mechanisms linked to the activation of CB1 receptors in MD. Specifically, we addressed CB1 receptor-mediated activation of different Gα protein subunits and regulation of adenylyl cyclase in the postmortem prefrontal cortex of subjects with major depressive disorder. The study was designed to assess the possible influence of antidepressant treatment on these parameters. These data would provide new insights to the participation of CB1 receptors in the pathophysiology of depression, and strengthen the idea that targeting brain endocannabinoid system may be a useful strategy for the treatment of this psychiatric illness.
2. Materials and methods
2.1. Subject selection and toxicological screening
Human brain PFC samples (Brodmann’s area 9) were obtained at autopsies performed in the Basque Institute of Legal Medicine, Bilbao, and in the Service of Pathologic Anatomy of the “Marqués de Valdecilla” Universitary Hospital, Santander, Spain. Brain tissue collection was performed in accordance with the approved protocols of the Basque Institute of Legal Medicine and the “Marqués de Valdecilla” University Hospital for postmortem human studies. All the deaths were subjected to retrospective careful search for previous medical diagnosis and treatment using examiner’s information and records of hospitals and mental health centers. This searching was blind to the biochemical findings. After searching for antemortem information, the brains from suicide victims who fulfilled the DSM-IV criteria of the American Psychiatric Association [28] for MD were selected. Serum samples from all the MD and control subjects included in the study were assayed for the presence of antidepressant, antipsychotic and anxiolytic drugs, as well as for psychotropic drugs (including delta-9-tetrahidrocannabinol, Δ9-THC) and alcohol. The laboratory analyses were performed at the National Institute of Toxicology, Madrid, Spain. Control subjects with a positive toxicological test for psychotropic drugs were excluded from the study. Toxicological screening was positive for antidepressants in six suicide victims (cases 2, 4, 7, 8, 9 and 10 from Table 1). These cases will be referred as “antidepressant-treated subjects” (AD-treated). The demographic characteristics, prescribed treatments and laboratory screening results of the MD and control subjects included in the study are detailed in Table 1. Due to tissue availability limitations, all experimental procedures could not be carried out in the same cohorts of MD and control cases. Nevertheless, the MD PFC samples included in each experimental technique were always carefully matched in terms of gender, age at death and postmortem delay (PM) with control samples obtained from subjects without evidence of neurological or psychiatric disorder and who died by a non-suicide mechanism. All the assays were carried out in a parallel design, so that in a given experiment, PFC samples from a MD brain were processed and incubated in parallel with the ones obtained from a matched control.
Table 1.
Demographic characteristics, diagnoses, prescribed treatment and toxicological analysis of individual cases of suicide victims with depressive disorders and their respective control subjects.
| Case | Gender1 | Age2 | PM3 | Cause of death | Treatment4 | Drug blood levels5 | Assay6 |
|---|---|---|---|---|---|---|---|
| Case 1 | F | 35 | 39 | Caustic intoxication | TMT, SMZ | negative | FB, B, AC, IP |
| Control 1 | F | 33 | 43 | Vehicle accident | negative | FB, B, AC, IP | |
| Case 2 | M | 73 | 60 | Gunshot wound | ATD, BZD | CIT | FB, B, AC |
| Control 2 | M | 79 | 66 | Vehicle accident | negative | FB, AC | |
| Control 2b | M | 67 | 61 | Cardiac arrest | not performed | B | |
| Case 3 | F | 72 | 49 | Jumping from height | Untreated | negative | FB, B, AC, IP |
| Control 3 | F | 79 | 39 | Vehicle accident | negative | FB, B, AC | |
| Control 3b | F | 68 | 30 | Breast Cancer | Negative | IP | |
| Case 4 | M | 65 | 30 | Drug intoxication | ATD, BZD | IMI, TIA, BZD, SUL, EtOH | FB, B, AC, IP |
| Control 4 | M | 65 | 50 | Vehicle accident | negative | B, AC, IP | |
| Control 4b | M | 68 | 36 | Breast Cancer | negative | FB | |
| Case 5 | F | 58 | 27 | Hanging | ATD, BZD | negative | FB, B, AC, IP |
| Control 5 | F | 58 | 37 | Vehicle accident | BZD, PC | negative | FB, B, AC, IP |
| Case 6 | M | 42 | 19 | Jumping from height | ATD, APS | CLO; BDZ, MET | FB, B, AC, IP |
| Control 6 | M | 41 | 19 | Laboral accident | negative | FB, B, AC, IP | |
| Case 7 | F | 88 | 9 | Jumping from height | ATD, BZD | positive: SER | FB, B, AC, IP |
| Control 7 | F | 81 | 19 | Cardiac arrest | negative | FB, B, AC, IP | |
| Case 8 | F | 68 | 25 | Jumping from height | ATD | SER | FB, B, AC, IP |
| Control 8 | F | 68 | 38 | Vehicle accident | ASA | negative | FB, AC, IP |
| Control 8b | F | 73 | 8 | Gastric cancer | ASA | not performed | B |
| Case 9 | F | 64 | 27 | Jumping from height | ATD, BZD | MIA; CIT | IP |
| Control 9 | F | 66 | 16 | Vehicle accident | negative | ||
| Case 10 | F | 64 | 25 | Jumping from height | ATD, BZD | CIT | IP |
| Control 10 | F | 67 | 35 | Cardiac arrest | negative | ||
| Case 11 | F | 71 | 19 | Jumping from height | TEO, ASA | negative | IP |
| Control 11 | F | 70 | 18 | Vehicle accident | negative | ||
| Case 12 | F | 53 | 17 | Jumping from height | BZD | BZD | FB, B |
| Control 12 | F | 54 | 18 | Lung cancer | Negative | B | |
| Control 12b | F | 63 | 20 | Cardiac arrest | Negative | FB |
Gender, F: female; M: male.
Age at death (years).
PM: Postmortem delay (hours).
Treatment prescription coded at the time of death: antidepressants (ATD), benzodiazepines (BDZ), antipsychotics (APS), trimethoprim (TMT), sulfamethoxazole (SMZ), theophylline (TEO), acetylsalicylic acid (ASA) and piroxicam (PC).
Drugs, blood levels detected coded as: citalopram (CIT), imipramine (IMI), tiapride (TIA), benzodiazepines and metabolites (BDZ), sulpiride (SUL), ethanol (EtOH), clotiapine (CLO), metamizole (MET), sertraline (SER), mianserine (MIA).
Assay carried out in each case and matching control(s): FB: Agonist-stimulation of [35S]GTPγS binding; B: [3H]CP55,940 saturation binding assay; AC: agonist-inhibition of AC activity; IP: immunoprecipitation of [35S]GTPγS labeled Gα subunits.
2.2. Agonist-stimulation of [35S]GTPγS binding
PFC samples were homogenized (1:20 w/v) in ice-cold buffer (50 mM Tris-HCl, 3 mM MgCl2, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM dithiothreitol (DTT) and 250 mM sucrose; pH, 7.4) using a Teflon tissue grinder (10 s, 800 rpm). The homogenates were centrifuged (1000g, 15 min, 4 °C) and the resulting supernatants were then centrifuged at 40,000g for 15 min (4 °C). The obtained pellets were resuspended in the same buffer and centrifuged again (50,000g, 15 min, 4 °C). Pellets were again resuspended and membrane aliquots (50 μg protein/ml in the assay) were incubated for 120 min at 30 °C in assay buffer (50 mM Tris-HCl, 3mM MgCl2, 1 mM EGTA, 100 mM NaCl, 100 μM guanosine diphosphate (GDP), 0.2 mM DTT, 0.5% bovine serum albumin (BSA); pH, 7.4) containing 0.05 nM [35S]GTPγS (specific activity 1250 Ci/mmol, New England Nuclear/Dupont, Boston, MA, USA). CB1 receptor mediated-stimulation of [35S]GTPγS binding was measured using the cannabinoid agonist WIN55,212-2 (1 nM-100 μM) (Tocris Cookson, Bristol, UK). The specificity of the cannabinoid agonist was verified by incubation of 10 μM WIN55,212-2 with 1 μM of the CB1 receptor selective antagonist SR141716A (kindly supplied by Sanofi Reserche, Montpellier, France). Basal binding was determined in the absence of agonist and non-specific binding was measured by coincubation with 10 μM GTPγS. The experiments were terminated by rapid filtration under vacuum (Cell Harvester M-12R, Brandel, Gaithersburg, MD, USA) through GF/C glass fiber filters, followed by three washes in ice-cold buffer (50 mM Tris-HCl, 1 mg/ml BSA; pH, 7.4). Bound radioactivity was determined using a Beckman LS6000 liquid scintillation counter (Beckman Instruments Inc., Fullerton, CA, USA), after overnight extraction in 5 ml Ecolite scintillation fluid (MP Biomedicals, LLC, Solon, OH, USA). All assays were performed in triplicate Sigmacote (Sigma-Aldrich Quimica SL, Madrid, Spain)-treated borosilicate tubes, and the results were confirmed in two independent experiments.
2.3. [3H]CP55,940 saturation binding assay
Frozen PFC samples were homogenized (1:20 w/v) in ice-cold buffer (50 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and 250 mM sucrose; pH, 7.4) using a motor driven Teflon and glass tissue grinder (10 s, 800 rpm). Homogenates were first centrifuged (1000g, 10 min, 4 °C) and the supernatants were centrifuged (50000g, 15 min, 4 °C). The obtained pellets were resuspended in assay buffer (50 mM Tris-HCl, 1 mM EDTA, 3mM MgCl2; pH, 7.4), incubated for 15 min at 37 °C and centrifuged again (50000g, 15 min, 4 °C). The pellets were resuspended (50 μg protein/ml in the assay) and incubated for 60 min at 37 °C in assay buffer with the cannabinoid agonist [3H]CP55,940 (0.0125-3.2 nM) (New England Nuclear/Dupont, Boston, MA, USA). Non-specific binding was measured in the presence of WIN55,212-2 (1 μM). The experiments were terminated as detailed for [35S]GTPγS binding experiments. All assays were performed in duplicate Sigmacote-treated borosilicate tubes, and the results were confirmed in three independent experiments.
2.4. Agonist-inhibition of adenylyl cyclase activity
PFC samples were homogenized (1:70 w/v) in ice-cold buffer (20 mM Tris-HCl, 1 mM EGTA, 5 mM EDTA, 1 mM DTT, 25 μM leupeptine and 300 mM sucrose; pH, 7.4) using a Teflon tissue grinder (10 s, 800 rpm) and centrifuged (1500g, 5 min, 4 °C), with the resulting supernatants centrifuged again (13,000g, 15 min, 4 °C). The obtained pellets were resuspended (150 μg protein/ml) in ice-cold assay buffer (80 mM Tris-HCl, 0.2 mM EGTA, 1 mM EDTA, 2 mM MgCl2, 100 mM NaCl, 60 mM sucrose, 1 mM DTT, 10 μM guanosine triphosphate (GTP), 0.5 mg/ml BSA, 0.5 mM 3-isobutyl-l-methylxanthine (IBMX), 5mM phosphocreatine, 50U/ml creatine phosphokinase, 5U/ml myokinase and 5 μM forskolin; pH, 7.4), and incubated for 5 min at 37 °C in the presence of WIN55,212-2 (10 nM-100 μM). The specificity of the cannabinoid agonist was verified by incubation of WIN55,212-2 (10 μM) with the selective CB1 receptor antagonist SR141716A (10 μM). The enzymatic reaction was started by addition of ATP to a final concentration of 200 μM. The mixture was then incubated for 10 min at 37 °C, and the reaction was rapidly terminated by 5 min incubation at 100 °C. The samples were centrifuged (5 min, 13,000g) and cyclic AMP (cAMP) concentration was determined in the supernatants using the [3H]cAMP assay kit (TRK 432) from GE Amersham International PLC (Amersham, Buckinghamshire, UK). The assays were performed in triplicate Sigmacote-treated borosilicate tubes, and the results were confirmed in two independent experiments.
2.5. Immunoprecipitation of [35S]GTPγS labeled Gα subunits
Membrane homogenates were obtained as reported for agonist-stimulated [35S]GTPγS binding assays. Resuspended pellets (500 μg protein/ml in the assay) were incubated with 2 nM [35S]GTPγS and 10 μM WIN55,212-2 in a final 100 μl assay volume for 30 min at 30 °C. Non-specific binding was determined in the presence of 10 μM of GTPγS. Membrane suspensions were then solubilized on ice with 1% Igepal® (Sigma-Aldrich Quimica SL, Madrid, Spain), 0.5% sodium deoxycholate, 0.1% SDS, 2.5 mM CHAPS, 0.1 mM phenylmethylsulfonyl-fluoride (PMSF), 0.01 M aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μl/ml antipain, 10 μg/ml chymostatin (Sigma-Aldrich Quimica SL, Madrid, Spain) for 30 min. Solubilized membranes were incubated for 3 h at room temperature with 15 μl of specific rabbit anti-Gαi2, anti-Gαi3, anti-Gαz, anti-Gαo antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA) and immobilized to superparamagnetic Dynabeads® Protein A (Thermo Fisher Scientific Baltics, UAB, Vilnius, Lithuania) overnight at 4 °C. After three washes with 1 ml de phosphate buffered saline (PBS) the beads were pelleted and the entrapped radioactivity was counted in 4 ml of Ecolite scintillation cocktail. Antibody specificity was confirmed in our experimental conditions by Western blot as reported previously [29].
2.6. Protein content determination
Membrane protein content was determined with the Bio-Rad Protein Assay Kit (Bio-Rad, Munich, Germany), using γ-globulin as standard.
2.7. Data analysis
The effect of each concentration of cannabinoid agonist was expressed as percentage of stimulation (% = (agonist effect × 100)/(basal activity-100)) in [35S]GTPγS assays and percentage of inhibition in cAMP assays (% = (agonist effect × 100)/(forskolin effect-100)). Analysis of [3H]CP55,940 saturation binding and WIN55,212-2 concentration-effect curves was conducted by nonlinear regression using GraphPad Prism Software (GraphPad, La Jolla, CA, USA) in order to estimate the theoretical maximal binding sites (Bmax), dissociation constants (Kd), agonist maximal effect (Emax, Imax) and potency (EC50, IC50) values in [35S]GTPγS binding and AC assays. Kd, EC50 and IC50 values were normalized as their -log values (pKd, pEC50 and pIC50) for comparison. Levels of coupling of CB1 receptors by WIN55,212-2 to the diverse Gα protein subunits were calculated as the percentage over the value in the absence of agonist.
The statistical analysis was performed in two phases. Initially, and in order to confirm that the MD and control PFC samples included in each experimental procedure were properly matched in terms of age and PM, these demographic parameters were compared between both groups of cases by means of unpaired Student’s t-tests [30,31] (Table 2). In addition, MD and control groups assayed in the four different techniques were comparable in terms of age and PM, as evidenced by one-way ANOVA analysis performed for each variable (age or PM) in both MD and control groups.
Table 2.
Comparison of the demographic characteristics between the major depression suicide victims and control cohorts included in the different experimental procedures. MD, major depression; AC, adenylyl cyclase; F, female; M, male.
| MD | Control | p | |
|---|---|---|---|
| Stimulation of [35SJGTPγS binding (n = 9, 6F/3M) | |||
| Age1 | 61.6 ± 5.5 | 63.3 ± 5.6 | 0.82 |
| Postmortem delay2 | 30.6 ± 5.4 | 34.1 ± 5.1 | 0.64 |
| Inhibition of AC activity (n = 8, 5F/3M) | |||
| Age1 | 62.6 ± 6.1 | 63.0 ± 6.4 | 0.97 |
| Postmortem delay2 | 32.3 ± 5.8 | 38.8 ± 5.4 | 0.43 |
| [3H]CP55,940 binding (n = 9, 6F/3M) | |||
| Age1 | 61.6 ± 5.5 | 60.0 ± 5.1 | 0.84 |
| Postmortem delay2 | 30.6 ± 5.4 | 31.6 ± 5.8 | 0.90 |
| Immunoprecipitation of Gα subunits (n = 10, 7F/3M) | |||
| Age1 | 62.7 ± 4.8 | 62.8 ± 4.8 | 0.99 |
| Postmortem delay2 | 24.9 ± 4.0 | 31.9 ± 3.9 | 0.25 |
Group values are mean ± SEM and p values correspond to non-paired Student’s t-tests.
Age at death (years).
Postmortem delay (hours).
Secondly, data obtained in each experimental technique for MD and matched control groups were compared using two-sided paired Student’s t-tests [31–33]. Additionally, results from MD subjects with negative or positive toxicology for ADs at the time of death (AD-free and AD-treated, respectively) and their respective control subgroups were analyzed using two-way ANOVA (disease and treatment) followed by Bonferroni post-hoc test. The statistical analysis test used for each experimental set are indicated in the results section and figure and table legends. Differences were taken as statistically significant when p < 0.05. Data are presented as mean ± SEM.
3. Results
3.1. Demographic characteristics of the samples
Because the MD and control subjects were matched in terms of gender, age at death and PM (Table 1), we detected no differences when comparing these parameters between the cases included in each experimental technique (Table 2). Between 8 and 10 MD subjects out of a total number of twelve, and their matched controls, were included in each of the biochemical assays performed (receptor binding, [35S]GTPγS binding, immunoprecipitation of [35S]GTPγS labeled Gα subunits and adenylyl cyclase activity assays). Consistently, one-way ANOVA indicated that the MD cohorts tested in the four biochemical assays were comparable in terms of age (F = 0.01; p = 0.99) and PM (F = 0.41; p = 0.75), and so were their respective control groups (F = 0.08; p = 0.97 for age; and F = 0.45; p = 0.72 for PM).
3.2. CB1 receptor-mediated stimulation of [35S]GTPγS binding
The cannabinoid agonist WIN55,212-2 stimulated the binding of [35S]GTPγS in PFC homogenates from MD and control subjects in a concentration-dependent manner (Fig. 1A). The maximal ability of WIN55,212-2 to stimulate [35S]GTPγS binding (%Emax) was increased by 23% in MD PFC (282.4 ± 18.3% MD vs 230.1 ± 18.5% control; p = 0.016, paired Student’s t-test) (Fig. 1A and B). On the contrary, basal [35S]GTPγS binding (in fmol/mg protein: 18.7 ± 2.1 MD vs 18.8 ± 2.2 control; p = 0.97, paired Student’s t-test) and the potency of the cannabinoid agonist in these assays (normalized pEC50 values were 5.75 ± 0.05 MD vs 5.80 ± 0.04 control; p = 0.57, paired Student’s t-test) were similar between MD and control cases. We next evaluated possible differences in the efficacy of the cannabinoid agonist to stimulate [35S]GTPγS binding between MD subjects with negative and positive antidepressant toxicology at the time of death (AD-free and AD-treated, respectively) and their control cases. A two-way ANOVA analysis of the %Emax values showed a significant effect of disease [F(1,14) = 6.529, p < 0.05] and treatment [F(1,14) = 13.30, p < 0.01]. AD-free depressed suicide victims presented a 38% increase in the %Emax of WIN55,212-2 as compared to their matched controls (260.7 ± 13.2 AD-free MD vs 188.4 ± 14.5% control; p < 0.05) (Fig. 1B). By contrast, we did not detect differences in the %Emax of the cannabinoid agonist when comparing AD-treated MD subjects to their matched controls (309.7 ± 33.5 AD-treated-MD vs 282 ± 8.7% control) (Fig. 1B). Similarly, two-way ANOVA detected no differences between experimental groups concerning basal [35S]GTPγS binding or the potency of WIN55,212-2 binding in these assays.
Fig. 1.
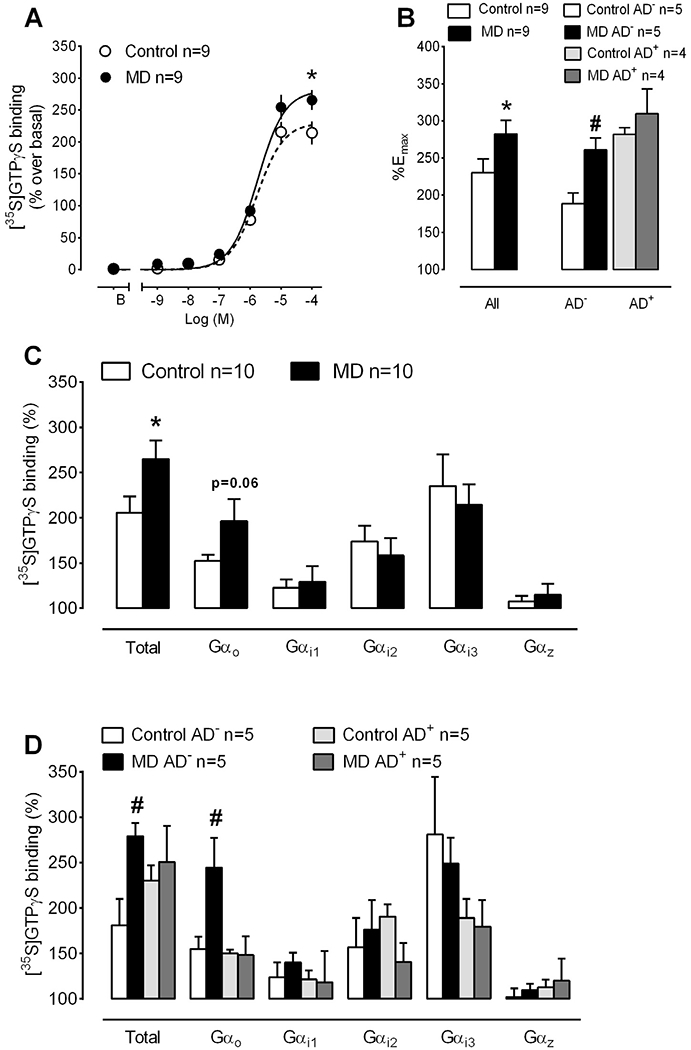
CB1 receptor-mediated activation of Gi/o proteins in PFC homogenates from MD subjects and matched controls. (A) Average concentration-response curves for the stimulation of [35S]GTPγS binding by the cannabinoid agonist WIN55,212-2 in MD and control cases. The maximal ability of WIN-55,212-2 to stimulate [35S]GTPγS binding was significantly enhanced in the MD group (n = 9 MD and matched control cases; *p < 0.05 vs control group, paired Student’s t-test). (B) Comparison of the maximal effect of WIN55,212-2 (%Emax) in [35S]GTPγS binding assays in MD cases with relation to AD intake. The increased efficacy of the cannabinoid agonist in MD subjects is restricted to the AD-free depressed suicide victims (n = 5 AD-free + 4 AD-treated MD and control cases; #p < 0.05, two-way ANOVA followed by Bonferroni post-hoc test vs control cases).(C)Stimulation of Gαo, Gαi1, Gαi2, Gαi3 and Gαz protein subunits by the cannabinoid agonist WIN55,212-2 (10 μM). The activation of total Gα proteins by the cannabinoid agonist was increased in MD (n = 10 MD and matched control cases; *p < 0.05 vs control group, paired Student’s t-test). (D)Comparison of the effect of WIN55,212-2 to activate Gα subunits in MD cases with relation to AD intake. The increased activation of Gα proteins by WIN55,212-2 in depressed subjects is due to a selective enhancement in the ability of the cannabinoid agonist to stimulate Gαo subunits in AD-free cases (n = 5 AD-free + 5 AD-treated MD and control cases; #p < 0.05, two-way ANOVA followed by Bonferroni post-hoc test vs control cases). Control AD−: matching control of MD AD-free subjects; control AD+ : matching control of MD AD-treated subjects.
3.3. CB1 receptor expression and agonist inhibition of adenylyl cyclase activity
Previous data have suggested that the upregulation of CB1 receptor-stimulated [35S]GTPγS binding in the PFC of depressed suicide victims reflects an increased number of CB1 proteins [26]. This possibility was assessed in our Spanish cohort of MD suicide cases by means of [3H]CP55,940 binding assays. The Bmax of [3H]CP55,940 binding to PFC homogenates was similar between MD subjects and their matched controls (in fmol/mg wet tissue: 42.6 ± 4.8 MD vs 41.9 ± 4.3 control; p = 0.82, paired Student’s t-test), and so were pKd values (9.77 ± 0.09 MD vs 9.79 ± 0.09 control; p = 0.66, paired Student’s t-test). Consistently, a two-way ANOVA analysis revealed no differences between AD-treated and AD-free depressed subgroups and their respective control cohorts neither in the Bmax, nor in the pKd values of [3H]CP55,940 binding.
We next tested whether the observed upregulation of CB1 coupling to Gαi/o in the PFC of MD suicide victims could be detected at the adenylyl cyclase signal transduction level. Similar basal adenylyl cyclase activity and forskolin effect values were measured in PFC from the MD and control brains (Table 3). Comparison of the maximal inhibitory effect (%Imax) and the potency (pEC50) of the cannabinoid agonist WIN55,212-2 in cAMP assays using paired Student’s t-test yielded no difference between depressed suicide victims and their matched control cases (Table 3). Similarly, a two-way ANOVA analysis revealed no differences between AD-free and AD-treated MD cases and their respective control subgroups regarding basal and forskolin-stimulated adenylyl cyclase activity, %Imax and pEC50 of WIN55,212-2 values in cAMP experiments
Table 3.
CB1receptor mediated inhibition of adenylyl cyclase activity in major depression suicide victims (MD) and matched control subjects.
| MD | Control | p | |
|---|---|---|---|
| Basal (pmol cAMP/min/mg protein) | 11.1 ± 5.5 | 12.1 ± 2.2 | 0.66 |
| Forskolin (pmol cAMP/min/mg protein) | 26.9 ± 2.1 | 26.7 ± 3.4 | 0.95 |
| %Imax | 67.6 ± 3.4 | 67.8 ± 4.2 | 0.97 |
| pEC50 | 6.44 ± 0.1 | 6.48 ± 0.1 | 0.79 |
Imax and pEC50 values correspond to the estimated maximal inhibitory effect (%) and potency of the cannabinoid agonist WIN55,212-2 in adenylyl cyclase assays. Group values are mean ± SEM of two independent experiments performed in triplicate including brain samples from 8 MD subjects and 8 matched controls, p values correspond to paired Student’s t-tests.
3.4. CB1 receptor-mediated immunoprecipitation of [35S]GTPγS labeled Gα subunits
In the absence of changes in CB1 receptor density, the lack of parallelism between the enhanced activation of Gαi/o proteins and the unaltered inhibition of AC activity by WIN55,212-2 may suggest altered receptor coupling ability to specific Gα subunits in the PFC of depressed suicide victims. In order to test this hypothesis, we performed immunoprecipitation of WIN55,212-2-stimulated [35S]GTPγS labelled Gα proteins. Noteworthy, similar to the above reported increased Emax of WIN55,212-2 in [35S]GTPγS binding assays, total G protein activation in response to a single concentration of the cannabinoid agonist was significantly higher in the MD cases included in the immunoprecipitation assays when compared to their matched controls (% stimulation over basal activity: 264.9 ± 20.5 MD vs 205.7 ± 17.8 control, p = 0.03; paired Student’s t-test) (Fig. 1C). The coupling efficiency of CB1 receptors at Gα proteins, as determined by the ability of the cannabinoid agonist WIN55,212-2 to induce the activation of specific protein subunits, in the PFC of control subjects was: Gαi3 (235% ± 35%) > Gαi2 (174% ± 17%) > Gαo (153% ± 7%) > Gαi1 (123% ± 9%) > Gαz (108% ± 6%) (Fig. 1C). We detected a non-significant increase in the activation of Gαo subunits by WIN55,212-2 in the MD group (197 ± 24% MD vs 153 ± 7% control, p = 0.06). However, the coupling to Gαi1 (129 ± 17% MD vs 123 ± 9% control, p = 0.38), Gαi2 (158 ± 19% MD vs 174 ± 17% control, p = 0.33), Gαi3 (235 ± 35% MD vs 214 ± 22 control%, p = 0.29) or Gαz (115 ± 12% MD vs 108 ± 6% control, p = 0.31) was similar between MD and control subjects (paired Student’s t-tests) (Fig. 1C). A two-way ANOVA analysis of the total G proteins activated by WIN55,212-2 in AD-free and AD-treated subgroups showed a significant effect of the disease [F(1,16) = 4.807, p < 0.05] and no significant changes for treatment and interaction (Fig. 1D). AD-free depressed suicide subjects showed a higher total Gα protein binding compared to their matched controls (279 ± 15% AD-free MD vs 181 ± 29% control; p < 0.05, Bonferroni post-hoc test) (Fig. 1D). Moreover, a two-way ANOVA analysis of the Gαo protein subunits showed a significant effect of the disease [F(1,16) = 4.549, p < 0.05], the treatment [F(1,16) = 6.042, p < 0.05], and the interaction disease × treatment [F(1,16) = 4.957, p < 0.05]. AD-free depressed suicide victims presented higher Gαo protein binding when compared to their matched controls (245 ± 33% AD-free MD vs 155 ± 14% control; p < 0.05, Bonferroni post-hoc test) (Fig. 1D). This increase was not observed when comparing the MD AD-treated group with their respective control cases (Fig. 1D). By contrast, two-way ANOVA revealed no differences in the efficacy of WIN55,212-2 to activate Gαi1, Gαi2, Gαi3 and Gαz between AD-free or AD-treated depressed subjects and their respective control cases (Fig. 1D).
4. Discussion
Increasing our current knowledge on the neurobiological basis of MD is a necessary step towards the development of more efficacious antidepressant medications. During the last decade, evidence has accumulated from studies in animal models that consistently suggest the participation of the endocannabinoid system in the pathophysiology of depression and in the long-term effects of ADs [6]. In this regard, the biological relevance of CB1 receptors in human depression is highlighted by reports of psychiatric adverse reactions, including depression and suicidal behaviour, associated to the use of the CB1 receptor antagonist rimonabant in clinical trials for the prevention of cardiovascular risk [34,35].
The results of the present study show increased CB1 receptor-mediated stimulation of [35S]GTPγS binding in the PFC of depressed suicides compared to matched controls, in agreement with previous reports [26,27]. Nevertheless, our data also suggest that this augmented activation of Gi/o proteins by the cannabinoid agonist WIN55,212-2 does not result from increased 0Β3 receptor density in the PFC of MD cases. Consistently, enhanced CB3 receptor-mediated inhibition of AC, which is likely to be expected from an elevated density of the receptor protein, was not detected in the PFC of the MD subjects included in the present study. Together with the data of Hungund et al. [26] and Choi et al. [27], our data strengthen the idea that enhanced CB1 receptor signaling at the Gi/o protein level in the PFC is a consistent finding in MD, although the underlying mechanisms may differ, probably due to the heterogeneity of this disease. Further evidence of the complex role of endocannabinoid signalling in MD comes from results showing association of lower CB1 receptor density in cannabis abusers [36] to both higher incidence [37] and lower risk [38] of suffering MD.
Stimulation of CB1 receptors by WIN55,212-2 resulted in the activation of at least five different Gαi/o protein subunits in human PFC, in agreement with previous studies [39]. These results differ from own observations in rodent PFC, where we detected no significant coupling to Gαi1 using the same anti-Gα subunit antibodies [19] while Gαz proteins are more significantly activated [40]. Regarding MD cases, agonist-stimulated immunoprecipitation assays revealed augmented CB1 receptor activation of Gαo protein subunits in the PFC of depressed suicide victims with negative blood test for ADs at the time of death (AD-free), as underlying mechanism for the observed increase in WIN55,212-2 stimulation of [35S]GTPγS binding. This result would suggest that, as previously reported in the mouse brain [41], [35S]GTPγS binding assays in human PFC mainly detect the activation of Gαo subunits, likely due to a significant excess in Gαo density over Gαi [42,43]. It is also noteworthy that, in contrast to the observed upregulation of CB1 receptor coupling to Gαo proteins, similar stimulation of Gαi1, Gαi2, Gαi3 and Gαz proteins was detected when comparing our cohort of MD subjects versus matched controls. Both CB1 receptor coupling to the inhibition of AC and to the regulation of different K+ and Ca2+ conductances in neural tissue have been shown to occur through pertussis toxin-sensitive Gαi/o proteins [7], although the specific Gα subunits involved in each response have not been characterized in detail. Proteins of the Gαi and Gαz subfamilies are responsible for the inhibitory regulation of adenylyl cyclase activity by G protein-coupled receptors, whereas the Gαo subtypes are more consistently involved in the modulation of ion channel function [44–46]. Moreover, Gαo protein subtype has no inhibitory effect on adenylyl cyclase activity [44,47]. In this scenario, the observed profile of CB1 receptor coupling to Gα subunits in the PFC of depressed suicides is consistent with the reported absence of changes at the AC transduction level, and may point to an altered regulation of ion channel function by neural CB1 receptors in MD.
A possible explanation for the increased activation of Gαo subunits by CB1 receptors reported here could be the existence of an enhanced expression of these G proteins in the brain of depressed patients [2]. Although the limited availability of tissue did not allow us to perform immunoblotting assays for these proteins in our samples, the majority of postmortem studies indicate unchanged levels of Gαi1/2 and Gαo proteins in the brain of suicide victims [3,48–50]. Consistently, previous observations suggest normal levels of Gi/o protein activation by other neurotransmitter receptors in the PFC of suicide victims with mood disorders [3]. These evidences suggest that the functional upregulation of CB1 receptors reported here is neither a common feature for other receptor systems coupled to Gαo proteins, nor related to enhanced expression of Gαo subunits in the PFC of subjects with MD.
An interesting finding of the present study is that both the augmented stimulation of [35S]GTPγS binding and the increased activation of Gαo subunits by CB1 receptors were selectively found in the PFC of AD-free depressed suicides, being absent when comparing AD-treated depressed suicides with their matched controls. This fact suggest that the enhanced signaling of CB1 receptors at the Gαo protein level in the brain of MD subjects is related to the pathobiology of depression, and not to the intake of antidepressant medication, as previously suggested in the study by Hungund et al. [26]. In second place, these results support the idea that antidepressant medications may modulate endocannabinoid neurotransmission in the human brain, as suggested from animal studies [13,14]. In this regard, repeated administration of ADs to naïve rodents has been shown to upregulate CB1 receptor expression and/or functionality in different brain areas [18,19,51]. By contrast, the increased CB1 receptor expression and coupling to Gαi/o proteins observed in the PFC of rats exposed to depression models is normalized by chronic ADs [13,14]. Collectively, these results indicate that the consequences of antidepressant medications at the CB1 receptor signaling level in the rodent brain depend on the pre-existence of a “depressive-like” state. In this study, the absence of modifications regarding CB1 receptor coupling to Gαo proteins in AD-treated MD cases is in marked contrast with our findings in AD-free suicides (present report and Hungund et al. [26]) and resembles the normalization of CB1 receptor function in rodent models of depression following chronic AD treatment [13,14]. Although caution is needed when evaluating these results, as only acute AD intake can be asserted from the positive blood testing at the time of death, our findings support the hypothesis that antidepressant medications modulate the activity of endocannabinoid system in the human brain.
Previous work has suggested that different biological and experimental parameters may modulate the expression and functionality of CB1 receptors and other neurotransmitter receptor proteins, in postmortem human brain [52,53]. The possible confounding effect of these variables in our results was avoided by matching the brain samples included in each experimental procedure in terms of age, sex and PM. A second limitation of the present study might be the fact that different cohorts of MD and matched controls were included in the four different assays carried out in order to address CB1 receptor expression and functionality. Nevertheless, the possibility that this fact is behaving as a confounding factor seems unlikely for several reasons. First, seven out of a total number of twelve MD cases were common between all the experimental procedures, and always compared to matched controls. Consistently, the different MD and cohorts were similar in terms of age and PM. Finally, the observed increase of CB1 receptor coupling to Gαi/o protein in the AD-free depressed suicides included in [35S]GTPγS binding experiments is consistent with the enhanced coupling to Gαo subunits detected in immunoprecipitation assays, and so are the lack of modifications in CB1 receptor expression and agonist-inhibition of adenylyl cyclase activity.
In summary, the present data suggest that enhanced CB1 receptor signaling in the brain of depressed suicides involves augmented coupling to Gαo, and not to Gαi/z protein subunits, providing additional insights into the participation of dysregulated endocannabinoid signaling in the pathophysiology of MD. In addition, this study provides the first evidence that antidepressant medications may modulate endocannabinoid neurotransmission in the brain of depressed subjects. Although additional research is necessary in order to unveil the biological significance of upregulated CB1 receptor function in MD, the present findings strengthen the idea that targeting brain endocannabinoid system could be a useful strategy for the clinical management of this devastating disorder.
Acknowledgements
This study was partially supported by the Spanish Ministry of Economy and Competitiveness, Spain (SAF2007/61862, SAF2011-25020 and SAF2015-67457-R, MINECO/FEDER) to AP, SAF 2009-08460 to JJM, and the Basque Government (IT-616-13 to JJM). The technical assistance of the staff from the Basque Institute of Legal Medicine, as well as of Lourdes Lanza, Josefa Castillo and Beatriz Romero, is kindly acknowledged. The authors would like to thank Professor Elsa Valdizán (deceased on 18th March 2016) for her contribution and support.
Abbreviations:
- MD
major depression
- 5-HT
5-hydroxytryptamine
- NE
norepinephrine
- AD
antidepressant
- CB1
cannabinoid receptor 1
- AC
adenylyl cyclase
- PFC
prefrontal cortex
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- DTT
dithiothreitol
- BSA
bovine serum albumin
- EDTA
ethylenediaminetetraacetic acid
Footnotes
Financial disclosures
Prof. Crespo-Facorro has received unrestricted research funding from AstraZeneca, Pfizer, Bristol-Myers Squibb, and Johnson & Johnson that was deposited into research accounts at the University of Cantabria.
Prof. Crespo-Facorro has received honoraria for his participation as a speaker at educational events from Pfizer, Bristol-Myers Squibb, and Johnson & Johnson and consultant fees from Pfizer.
References
- [1].Arango V, Underwood MD, Gubbi AV, Mann JJ, Localized alterations in pre- and postsynaptic serotonin binding sites in the ventrolateral prefrontal cortex of suicide victims, Brain Res 688 (1–2) (1995) 121–133, https://doi.org/10.1016/0006-8993(95)00523-S. [DOI] [PubMed] [Google Scholar]
- [2].Garcia-Sevilla JA, Escriba PV, Ozaita A, La Harpe R, Walzer C, Eytan A, et al. , Up-regulation of immunolabeled alpha2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides, J. Neurochem 72 (1) (1999) 282–291, https://doi.org/10.1046/j.1471-4159.1999.0720282.x. [DOI] [PubMed] [Google Scholar]
- [3].Gonzalez-Maeso J, Rodriguez-Puertas R, Meana JJ, Garcia-Sevilla JA, Guimon J, Neurotransmitter receptor-mediated activation of G-proteins in brains of suicide victims with mood disorders: selective supersensitivity of alpha(2A)-adrenoceptors, Mol. Psychiatry 7 (7) (2002) 755–767, https://doi.org/10.1038/sj.mp.4001067. [DOI] [PubMed] [Google Scholar]
- [4].Berton O, Nestler EJ, New approaches to antidepressant drug discovery: beyond monoamines, Nat. Rev. Neurosci 7 (2) (2006) 137–151, https://doi.org/10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- [5].Wong ML, Licinio J, Research and treatment approaches to depression, Nat. Rev. Neurosci 2 (5) (2001) 343–351, https://doi.org/10.1038/35072566. [DOI] [PubMed] [Google Scholar]
- [6].Micale V, Di Marzo V, Sulcova A, Wotjak CT, Drago F, Endocannabinoid system and mood disorders: priming a target for new therapies, Pharmacol. Ther 138 (1) (2013) 18–37, https://doi.org/10.1016/j.pharmthera.2012.1012.1002. [DOI] [PubMed] [Google Scholar]
- [7].Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. , International Union of Pharmacology. XXVII. Classification of cannabinoid receptors, Pharmacol. Rev 54 (2) (2002) 161–202. [DOI] [PubMed] [Google Scholar]
- [8].Childers SR, Pacheco MA, Bennett BA, Edwards TA, Hampson RE, Mu J, et al. , Cannabinoid receptors: G-protein-mediated signal transduction mechanisms, Biochem. Soc. Symp 59 (1993) 27–50. [PubMed] [Google Scholar]
- [9].Mechoulam R, Parker LA, The endocannabinoid system and the brain, Annu. Rev. Psychol 64 (2013) 21–47, https://doi.org/10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- [10].Hill MN, Gorzalka BB, Is there a role for the endocannabinoid system in the etiology and treatment of melancholic depression? Behav. Pharmacol 16 (5–6) (2005) 333–352. [DOI] [PubMed] [Google Scholar]
- [11].Valverde O, Torrens M, CB1 receptor-deficient mice as a model for depression, Neuroscience 204 (2012) 193–206, https://doi.org/10.1016/j.neuroscience.2011.09.031. [DOI] [PubMed] [Google Scholar]
- [12].Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, et al. , Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress, Biol. Psychiatry 62 (10) (2007) 1103–1110, https://doi.org/10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- [13].Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, et al. , Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment, J. Neurochem 106 (6) (2008) 2322–2336, https://doi.org/10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rodriguez-Gaztelumendi A, Rojo ML, Pazos A, Diaz A, Altered CB receptor-signaling in prefrontal cortex from an animal model of depression is reversed by chronic fluoxetine, J. Neurochem 108 (6) (2009) 1423–1433, https://doi.org/10.1111/j.1471-4159.2009.05898.x. [DOI] [PubMed] [Google Scholar]
- [15].Smaga I, Jastrzębska J, Zaniewska M, Bystrowska B, Gawliński D, Faron-Górecka A, et al. , Changes in the brain endocannabinoid system in rat models of depression, Neurotox. Res 31 (3) (2017) 421–435, https://doi.org/10.1007/s12640-017-9708-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bambico FR, Katz N, Debonnel G, Gobbi G, Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex, J. Neurosci 27 (43) (2007) 11700–11711, https://doi.org/10.1523/JNEUROSCI.1636-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Witkin JM, Tzavara ET, Davis RJ, Li X, Nomikos GG, A therapeutic role for cannabinoid CB1 receptor antagonists in major depressive disorders, Trends Pharmacol. Sci 26 (12) (2005) 609–617, https://doi.org/10.1016/j.tips.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [18].Hill MN, Ho WS, Hillard CJ, Gorzalka BB, Differential effects of the antidepressants tranylcypromine and fluoxetine on limbic cannabinoid receptor binding and endocannabinoid contents, J. Neural Transm 115 (12) (2008) 1673–1679, https://doi.org/10.1007/s00702-008-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mato S, Vidal R, Castro E, Diaz A, Pazos A, Valdizan EM, Long-term fluoxetine treatment modulates cannabinoid type 1 receptor-mediated inhibition of adenylyl cyclase in the rat prefrontal cortex through 5-hydroxytryptamine 1A receptor-dependent mechanisms, Mol. Pharmacol 77 (3) (2010) 424–434, https://doi.org/10.1124/mol.1109.060079. [DOI] [PubMed] [Google Scholar]
- [20].Ibarra-Lecue I, Pilar-Cuéllar F, Muguruza C, Florensa-Zanuy E, Díaz Á, Urigüen L, et al. , The endocannabinoid system in mental disorders: evidence from human brain studies, Biochem. Pharmacol (2018), https://doi.org/10.1016/j.bcp.2018.07.009. [DOI] [PubMed] [Google Scholar]
- [21].Domschke K, Dannlowski U, Ohrmann P, Lawford B, Bauer J, Kugel H, et al. , Cannabinoid receptor 1 (CNR1) gene: impact on antidepressant treatment response and emotion processing in major depression, Eur. Neuropsychopharmacol 18 (10) (2008) 751–759, https://doi.org/10.1016/j.euroneuro.2008.05.003. [DOI] [PubMed] [Google Scholar]
- [22].Monteleone P, Bifulco M, Maina G, Tortorella A, Gazzerro P, Proto MC, et al. , Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression, Pharmacol. Res 61 (5) (2010) 400–404, https://doi.org/10.1016/j.phrs.2010.1001.1002. [DOI] [PubMed] [Google Scholar]
- [23].Mitjans M, Gasto C, Catalan R, Fananas L, Arias B, Genetic variability in the endocannabinoid system and 12-week clinical response to citalopram treatment: the role of the CNR1, CNR2 and FAAH genes, J. Psychopharmacol 26 (10) (2012) 1391–1398, https://doi.org/10.1177/0269881112454229. [DOI] [PubMed] [Google Scholar]
- [24].Mitjans M, Serretti A, Fabbri C, Gastó C, Catalán R, Fañanás L, et al. , Screening genetic variability at the CNR1 gene in both major depression etiology and clinical response to citalopram treatment, Psychopharmacology 227 (3) (2013) 509–519, https://doi.org/10.1007/s00213-013-2995-y. [DOI] [PubMed] [Google Scholar]
- [25].Mato S, Rodríguez-Puertas R, González-Maeso J, Meana J, Salles J, Pazos A, Cannabinoid receptors in postmortem human brain: a radiometric and transductional study in major depression, Br. J. Pharmacol 134 (Suppl 1) (2001) 161 abstract.11522608 [Google Scholar]
- [26].Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, et al. , Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims, Mol. Psychiatry 9 (2) (2004) 184–190, https://doi.org/10.1038/sj.mp.4001376. [DOI] [PubMed] [Google Scholar]
- [27].Choi K, Le T, McGuire J, Xing G, Zhang L, Li H, et al. , Expression pattern of the cannabinoid receptor genes in the frontal cortex of mood disorder patients and mice selectively bred for high and low fear, J. Psychiatr. Res 46 (7) (2012) 882–889, https://doi.org/10.1016/j.jpsychires.2012.1003.1021. [DOI] [PubMed] [Google Scholar]
- [28].American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders (DSM III-R), 3th ed,, American Psychiatric Press, Washington, DC, 1987. [Google Scholar]
- [29].Valdizán EM, Castro E, Pazos A, Agonist-dependent modulation of G-protein coupling and transduction of 5-HT1A receptors in rat dorsal raphe nucleus, Int. J. Neuropsychopharmacol 13 (7) (2010) 835–843, https://doi.org/10.1017/S1461145709990940. [DOI] [PubMed] [Google Scholar]
- [30].Yatham LN, Liddle PF, Shiah IS, Scarrow G, Lam RW, Adam MJ, et al. , Brain serotonin2 receptors in major depression: a positron emission tomography study, Arch. Gen. Psychiatry 57 (9) (2000) 850–858. [DOI] [PubMed] [Google Scholar]
- [31].Shelton RC, Hal Manier D, Lewis DA, Protein kinases A and C in post-mortem prefrontal cortex from persons with major depression and normal controls, Int. J. Neuropsychopharmacol 12 (9) (2009) 1223–1232, https://doi.org/10.1017/S1461145709000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Arango V, Underwood MD, Boldrini M, Tamir H, Kassir SA, Hsiung S, et al. , Serotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims, Neuropsychopharmacology 25 (6) (2001) 892–903, https://doi.org/10.1016/S0893-133X(01)00310-4. [DOI] [PubMed] [Google Scholar]
- [33].Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G, Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression, Neuropsychopharmacology 29 (11) (2004) 2088–2096, https://doi.org/10.1038/sj.npp.1300525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A, Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials, Lancet 370 (9600) (2007) 1706–1713, https://doi.org/10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- [35].Topol EJ, Bousser MG, Fox KA, Creager MA, Despres JP, Easton JD, et al. , Rimonabant for prevention of cardiovascular events (CRESCENDO): a randomised, multicentre, placebo-controlled trial, Lancet 376 (9740) (2010) 517–523, https://doi.org/10.1016/S0140-6736(1010)60935-X. [DOI] [PubMed] [Google Scholar]
- [36].Hirvonen J, Goodwin RS, Li CT, Terry GE, Zoghbi SS, Morse C, et al. , Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers, Mol. Psychiatry 17 (6) (2012) 642–649, https://doi.org/10.1038/mp.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wright NE, Scerpella D, Lisdahl KM, Marijuana use is associated with behavioral approach and depressive symptoms in adolescents and emerging adults, PLoS One 11 (11) (2016) e0166005, , https://doi.org/10.1371/journal.pone.0166005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Denson TF, Earleywine M, Decreased depression in marijuana users, Addict. Behav 31 (4) (2006) 738–742, https://doi.org/10.1016/j.addbeh.2005.05.052. [DOI] [PubMed] [Google Scholar]
- [39].Erdozain AM, Diez-Alarcia R, Meana JJ, Callado LF, The inverse agonist effect of rimonabant on G protein activation is not mediated by the cannabinoid CB1 receptor: evidence from postmortem human brain, Biochem. Pharmacol 83 (2) (2012) 260–268, https://doi.org/10.1016/j.bcp.2011.1010.1018. [DOI] [PubMed] [Google Scholar]
- [40].Diez-Alarcia R, Ibarra-Lecue I, Lopez-Cardona AP, Meana J, Gutierrez-Adán A, Callado LF, et al. , Biased agonism of three different cannabinoid receptor agonists in mouse brain cortex, Front. Pharmacol 7 (2016) 415, https://doi.org/10.3389/fphar.2016.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L, Most central nervous system D2 dopamine receptors are coupled to their effectors by Go, Proc. Natl. Acad. Sci. U.S.A 98 (6) (2001) 3577–3582, https://doi.org/10.1073/pnas.051632598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sternweis PC, Robishaw JD, Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain, J. Biol. Chem 259 (22) (1984) 13806–13813. [PubMed] [Google Scholar]
- [43].Spicher K, Nuernberg B, Jager B, Rosenthal W, Schultz G, Heterogeneity of three electrophoretically distinct Go alpha-subunits in mammalian brain, FEBS Lett 307 (2) (1992) 215–218, https://doi.org/10.1016/0014-5793(92)80770-H. [DOI] [PubMed] [Google Scholar]
- [44].Wong YH, Conklin BR, Bourne HR, Gz-mediated hormonal inhibition of cyclic AMP accumulation, Science 255 (5042) (1992) 339–342, https://doi.org/10.1126/science.1347957. [DOI] [PubMed] [Google Scholar]
- [45].Hille B, Modulation of ion-channel function by G-protein-coupled receptors, Trends Neurosci 17 (12) (1994) 531–536, https://doi.org/10.1016/0166-2236(94),90157-0. [DOI] [PubMed] [Google Scholar]
- [46].Taussig R, Gilman AG, Mammalian membrane-bound adenylyl cyclases, J. Biol. Chem 270 (1) (1993) 1–4, https://doi.org/10.1074/jbc.270.1.1. [DOI] [PubMed] [Google Scholar]
- [47].Katada T, Oinuma M, Two guanine nucleotide-binding proteins in rat brain serving as the specific substrate of islet-activating protein, pertussis toxin. Interaction of the alpha-subunits with beta gamma-subunits in development of their biological activities, J. Biol. Chem 261 (18) (1986) 8182–8191. [PubMed] [Google Scholar]
- [48].Cowburn RF, Marcusson JO, Eriksson A, Wiehager B, O’Neil C, Adenylyl cyclase activity and G-protein subunit levels in postmortem frontal cortex of suicide victims, Brain Res 633 (1–2) (1994) 297–304, https://doi.org/10.1016/0006-8993(94),91552-0. [DOI] [PubMed] [Google Scholar]
- [49].Dowlatshahi D, MacQueen GM, Wang JF, Reiach JS, Young LT, G Protein-coupled cyclic AMP signaling in postmortem brain of subjects with mood disorders: effects of diagnosis, suicide, and treatment at the time of death, J. Neurochem 73 (3) (1999) 1121–1126, https://doi.org/10.1046/j.1471-4159.1999.0731121.x. [DOI] [PubMed] [Google Scholar]
- [50].Gonzalez-Maeso J, Meana JJ, Heterotrimeric g proteins: insights into the neurobiology of mood disorders, Curr. Neuropharmacol 4 (2) (2006) 127–138, https://doi.org/10.2174/157015906776359586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB, Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic-pituitary-adrenal axis, Neuropsychopharmacology 31 (12) (2006) 2591–2599, https://doi.org/10.1038/sj.npp.1301092. [DOI] [PubMed] [Google Scholar]
- [52].Mato S, Pazos A, Influence of age, postmortem delay and freezing storage period on cannabinoid receptor density and functionality in human brain, Neuropharmacology 46 (5) (2004) 716–726, https://doi.org/10.1016/j.neuropharm.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [53].González-Maeso J, Torre I, Rodríguez-Puertas R, García-Sevilla JA, Guimón J, Meana JJ, Effects of age, postmortem delay and storage time on receptor-mediated activation of G-proteins in human brain, Neuropsychopharmacology 26 (4) (2002) 468–478, https://doi.org/10.1016/S0893-133X(01)00342-6. [DOI] [PubMed] [Google Scholar]