

Abstract
Inhibition of cytochrome P-450 1A2 (CYP1A2)-mediated activation of procarcinogens may be an important chemopreventive mechanism. Consumption of apiaceous vegetables (rich in furanocoumarins) inhibits CYP1A2 in humans. Because many furanocoumarins are potent inhibitors of several CYP, we characterized the effects of three furanocoumarins from apiaceous vegetables on human CYP1A2 (hCYP1A2). We assessed hCYP1A2 methoxyresorufin O-demethylase (MROD) activity using microsomes from Saccharomyces cerevisiae expressing hCYP1A2. Isopimpinellin exhibited mechanism-based inactivation (MBI) of hCYP1A2 (Ki = 1.2 μM, kinact = 0.34 min−1, and partition ratio = 8). Imperatorin and trioxsalen were characterized as mixed inhibitors with Ki values of 0.007 and 0.10 μM, respectively. These results indicate that even if present at low levels in apiaceous vegetables, imperatorin, trioxsalen and isopimpinellin may contribute significantly to CYP1A2 inhibition and potentially decreased procarcinogen activation. Moreover, the in vivo effect of isopimpinellin on CYP1A2 may be longer lasting compared to reversible inhibitors.
Keywords: CYP1A2, inhibition, isopimpinellin, imperatorin, trioxsalen
1. Introduction
Cytochrome P-450 1A2 (CYP1A2) is one of the major phase I enzymes and accounts for approximately 13% of the total cytochrome P-450 (CYP) content in the human liver [3]. CYP1A2 plays an important role in carcinogenesis, where it mediates the formation of highly reactive carcinogenic intermediates from a variety of procarcinogenic parent compounds such as aflatoxins, heterocyclic aromatic amines, nitrosamines, and polycyclic aromatic hydrocarbons [16, 35, 48]. Metabolic activation of procarcinogens by CYP1A2 may ultimately result in the formation of DNA adducts when the reactive metabolites are not appropriately detoxified by phase II enzymes such as glutathione S-transferases and UDP-glucuronosyltransferase [39, 48]. Even modest inhibition of CYP1A2-mediated activation of procarcinogens to prevent the rate of activation from exceeding detoxification may be a potential chemopreventive approach. Various dietary factors modulate CYP1A2 activity which may consequently affect carcinogenesis.
Dietary intake of apiaceous vegetables, which includes carrot, celery, dill, cilantro, parsnip and parsley, inhibits CYP1A2 activity in humans [27] perhaps due to the furanocoumarin content of the vegetables [36]. Of the phytochemicals in apiaceous vegetables, the furanocoumarins 5-methoxypsoralen (5-MOP), 8-methoxypsoralen (8-MOP) and psoralen are potent inhibitors of human CYP1A2 (hCYP1A2) and hCYP1A2-mediated mutagenicity of aflatoxin in yeast expressing hCYP1A2 [10, 28, 34, 37]. Further, intake of 5-MOP and 8-MOP in drug forms decreases CYP1A2-mediated caffeine metabolism in human subjects [2, 30]. Other furanocoumarins in apiaceous vegetables, such as imperatorin and isopimpinellin, have not been studied in as much detail as psoralen, 5-MOP, and 8-MOP. However, imperatorin and isopimpinellin exhibit inhibitory effects on CYP1A2 in human cDNA-expressed P450 microsomal systems [21].
A number of furanocoumarins in apiaceous vegetables have been identified as mechanism-based inactivators (MBIs) of various CYPs. 8-MOP is an MBI of human CYP2A6, human CYP2A13, mouse CYP1A1 and rat CYP2B1 [6, 11, 24, 25, 50]. 5-MOP and psoralen are MBIs of both human CYP2A6 and rat CYP2B1 [11, 24, 25]. A key feature of MBI is that the inactivation involves the formation of a covalent adduct within the active site, which occurs without the release of the reactive intermediate from the active site and results in irreversible loss of activity [20]. Hence, it is plausible that despite a short-term exposure to a furanocoumarin food source, there could be a prolonged reduction in activity of the aforementioned CYPs because the decreased activity could only be regained by synthesis of new enzyme. Given the role of CYP1A2 in the activation of several procarcinogens and the number of CYPs irreversibly inactivated by furanocoumarins, characterizing the interaction of furanocoumarins with CYP1A2 is warranted. Furanocoumarins that exhibit reversible inhibition may also have important, albeit more transient, effects regarding decreased activation of procarcinogenic species and thus may also affect the carcinogenesis process.
Our objective here was to characterize the effects of the furanocoumarins imperatorin, isopimpinellin, angelicin, and trioxsalen (Fig. 1) on hCYP1A2 activity in vitro. We hypothesized that the four compounds are MBIs of hCYP1A2. We chose to pursue these studies using a yeast strain expressing recombinant hCYP1A2 in order to isolate and more clearly identify the effects of these furanocoumarins specifically on hCYP1A2 without interference from other enzyme systems [13].
Fig. 1.
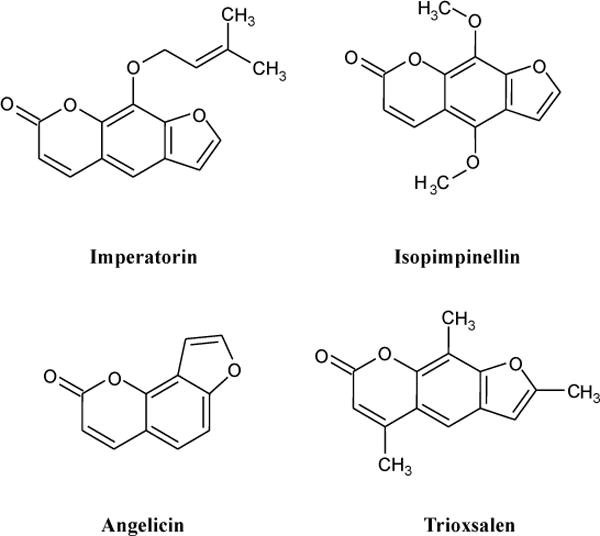
Chemical structures of furanocoumarins in apiaceous vegetables tested in this study.
2. Materials and methods
2.1. Chemicals
Imperatorin, isopimpinellin, and trioxsalen were purchased from Indofine Chemical Company (Hillsborough, NJ, USA). Angelicin, dicumarol, dimethyl sulfoxide (DMSO), glutathione (GSH), glycerol, and β-nicotinamideadenine dinucleotidephosphate (NADPH) and resorufin were purchased from Sigma (St. Louis, MO, USA). Methoxyresorufin was obtained from AnaSpec (Fremont, CA). All phytochemicals were of ≥ 98% purity (per Indofine Chemical Company and Sigma product specifications). HEPES and Hank’s Balanced Salt Solution (HBSS) were purchased from Invitrogen Corporation (Carlsbad, CA, USA). Superoxide dismutase (SOD) was purchased from MP Biomedicals (Solon, OH, USA).
2.2. Yeast microsomes
A transformed yeast strain expressing recombinant hCYP1A2 (pHE36) was used for our assays, and a wild-type yeast strain (pDP34) was also prepared to subtract the background activities in this yeast strain. The plasmid construct for these strains has been described previously [13]. In brief, yeast were cultured overnight at 30 °C in minimal synthetic media that lacked uracil, and grown to an optical density of ~0.5 (~8 × 107 cells/ml) at 600 nm, and the microsomes were prepared using a previously described method [14, 43]. The protein concentration of yeast microsomes was determined according to the method proposed by Bradford using bovine serum albumin as a standard [4]. Microsomal CYP content was determined spectrophotometrically [33].
2.3. Methoxyresorufin O-demethylase (MROD) assay
In order to screen for potent inhibitors of hCYP1A2, the activities of microsomal hCYP1A2 were determined spectrofluorimetrically by monitoring the formation of resorufin from methoxyresorufin [23]. Substrate concentration and incubation times were determined previously [37]. The experiments were conducted at 37 °C in 96-well plates in a Biotek Synergy HT microplate reader (excitation: 540 nm; emission: 580 nm; extinction coefficient: 73,000 mM−1 cm−1 at 572 nm) [23, 37]. For all samples (including controls), the HBSS reaction mixture (pH 7.05) consisted of 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES, 5 μM methoxyresorufin, and 250 μM NADPH. Dicumarol was added to inhibit the potential effect of quinine oxidoreductase which may cause the reduction of resorufin to a nonfluorescent product [17]. We confirmed that the concentration of dicumarol in this study had no significant effect on MROD activity (data not shown). Furanocoumarins were dissolved in DMSO and were added to supplemented HBSS at the following concentrations: 0.005, 0.015, 0.05, 0.15, 0.5, and 1.5 μM for imperatorin and isopimpinellin, and 0.5, 1.5, 5.0, 15, 50, and 150 μM for angelicin and trioxsalen. The final DMSO concentration was 0.5%, at which no substantial inhibitory effects have been observed; and existing literature indicates no significant inhibition up to 3% DMSO [5, 12]. Yeast microsomal hCYP1A2 was added to the HBSS reaction mixture to achieve 2 pmol CYP/ml. After prewarming for 5 min at 37 °C, the plates were read eight times over a 10 min interval. Standard curves for resorufin were used to determine activity. Each assay included the following two controls: (1) hCYP1A2-negative (wild type yeast), NADPH-positive, 0.5% DMSO, and (2) hCYP1A2-positive, NADPH-positive, 0.5% DMSO. Approximate IC50 values were determined from duplicate independent assays with six replicates per assay of each of the six concentrations of the furanocoumarins. IC50 values were used to evaluate which compounds to further characterize as inhibitors of hCYP1A2. An IC50 ≤ 10 μM for a given compound was considered physiologically relevant and warranting further investigation.
2.4. Mechanism-based inactivation of hCYP1A2
To determine whether a compound was an MBI of hCYP1A2, the following experiments were conducted: time- and concentration-dependent inactivation, NADPH-dependent and GSH- and SOD-independent inactivation, irreversibility of inactivation, determination of partition ratio, and evaluation of substrate protection. Each experiment included three independent assays with more than three replicates per assay.
2.4.1. Time- and concentration-dependent inactivation
IC50 values were used to guide concentration ranges for each compound. Isopimpinellin at six different concentrations (0.25, 0.5, 1.0, 2.0, 4.0, and 8.0 μM) or DMSO (vehicle control, 1% v/v final concentration) was preincubated with microsomes (120 pmol CYP/ml) at 37 °C for various lengths of time (0, 1, 2, and 3 min) in the supplemented HBSS reaction mixture containing 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES and 250 μM NADPH. Subsequently, an aliquot (25 μl) of the preincubation mixture was diluted 120-fold into an enzyme activity assay mixture prewarmed to 37 °C containing 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES, 250 μM NADPH, and 5 μM methoxyresorufin. After incubation at 37 °C for 10 min, the reaction was quenched by 50% ethanol and the activity of hCYP1A2 was determined spectrofluorimetrically as described above. Imperatorin (0.02 – 0.09 μM) and trioxsalen (0.1 – 15 μM) did not result in both time- and concentration-dependent inhibition (data not shown), and were further characterized using an inhibition matrix method (see Methods section 2.5).
The observed inhibition rate constants, (kobs), were calculated from the slopes of each line representing the natural logarithm of the percent remaining enzyme activity vs. time. The rate constant for maximal inactivation (kinact), and the concentration of isopimpinellin needed to achieve ½ maximal rate of hCYP1A2 inactivation (KI) were derived from the following equation [45]:
The time required for half the number of hCYP1A2 molecules to be inactivated was calculated from the following equation [7]:
2.4.2. NADPH-dependent and GSH- and SOD-independent inactivation
The NADPH-dependence of isopimpinellin-mediated inactivation was determined at 8 μM isopimpinellin using the same assay described in the abovementioned time- and concentration-dependence tests, albeit with slight modifications. The preincubation mixture contained 8 μM isopimpinellin, 20 μM dicumarol, 5 mM MgCl2 and 15 mM HEPES with and without 250 μM NADPH. At 0, 1, 2 and 3 min, aliquots (25 μl) of the preincubation mixture were added to the prewarmed (37 °C) secondary reaction mixture containing 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES, 250 μM NADPH and 5 μM methoxyresorufin. The remaining hCYP1A2 activity was determined as described above. Similarly, the GSH- and SOD-independence was determined in the presence or absence of GSH (250 μM) or SOD (1000 units) with 8 μM isopimpinellin.
2.4.3. Irreversibility
Using 8 μM isopimpinellin, microsomes (120 pmol CYP/ml) were pre-incubated in supplemented HBSS as described above at 37 °C for 8 min. The mixtures were then dialyzed in 50 mM potassium phosphate buffer (pH 7.4) with 20% glycerol and 100 μM EDTA overnight for 20 h at 4 °C using a 0.5–3 ml Slide-A-Lyzer cassette with a molecular mass cutoff of 10 kDa (Pierce, Rockford. IL, USA). Before and after the overnight dialysis, the hCYP1A2 activity was determined as described above. The percent remaining hCYP1A2 activity was calculated from comparison to uninhibited control that was also dialyzed. The activity of the post-dialysis uninhibited control was 94 ± 3% of the pre-dialysis uninhibited control.
2.4.4. Partition ratio
The partition ratio for hCYP1A2 inactivation by isopimpinellin was determined by titration of the enzyme with the inactivator [46]. Isopimpinellin at nine concentrations (0.25, 0.5, 1.0, 2.0, 4.0, 8.0, 16, 32 and 64 μM) or DMSO (vehicle control, 1% v/v final concentration) was preincubated with microsomes (120 pmol CYP/ml) at 37 °C until the maximum inactivation was achieved (30 min) in the HBSS mixture described earlier. The remaining hCYP1A2 activity was monitored using the above MROD assay.
2.4.5. Substrate protection
In order to determine if a CYP1A2 substrate provided protection against inactivation, methoxyresorufin at three different concentrations (0.4, 2.0 and 10 μM) was preincubated with microsomes (120 pmol CYP/ml) at 37 °C for 8 min in the HBSS reaction mixture supplemented with 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES and 250 μM NADPH. A 25 μl aliquot of the preincubation mixture was transferred to a 3 ml enzyme activity assay mixture (prewarmed to 37 °C) containing HBSS, 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES, 5 μM methoxyresorufin, and 250 μM NADPH to achieve 1 pmol CYP/ml, final concentration. After incubation at 37 °C for 10 min, the reaction was terminated by the addition of 50% ethanol and the activity of hCYP1A2 was determined spectrofluorimetrically as described above. The following 5 conditions were included: (1) hCYP1A2-negative (wild-type yeast mircosomes), NADPH-positive, 1% DMSO, (2) hCYP1A2-positive, NADPH-positive, 1% DMSO, (3) hCYP1A2-positive, NADPH-positive, 1.17 μM isopimpinellin, (4) hCYP1A2-positive, NADPH-positive, 1.17 μM isopimpinellin, 0.4 μM methoxyresorufin, (5) hCYP1A2-positive, NADPH-positive, 1.17 μM isopimpinellin, 2.0 μM methoxyresorufin, and (6) hCYP1A2-positive, NADPH-positive, 1.17 μM isopimpinellin, 10 μM methoxyresorufin. The remaining activity of hCYP1A2 was detected by MROD activity as described above.
2.5 Inhibition Matrix Assay
For kinetics assessment of imperatorin and trioxsalen, we estimated the Km of hCYP1A2-mediated methoxyresorufin demethylase activity to be 0.35 ± 0.02 μM. In order to assess the modes of inhibition, six substrate concentrations (0.14, 0.21, 0.42, 0.63, 1.05, and 2.52 μM) were matched with five concentrations of imperatorin (0, 0.00125, 0.0025, 0.005, and 0.0075 μM) and trioxsalen (0, 0.05, 0.1, 0.2, and 0.3 μM) in 96-well plates to create 5 × 6 matrices. HBSS was supplemented with 20 μM dicumarol, 5 mM MgCl2, 15 mM HEPES, and 250 μM NADPH. Microsomes from hCYP1A2-expressing yeast were added to the supplemented HBSS to achieve 2 pmol P-450/ml. After warming at 37 °C for 5 min, plates were read six times over a 10 min interval. Data represent three independent assays with triplicate replicates of each condition within each assay. Dixon plots of the kinetics data were utilized to determine the mechanisms of inhibition. Inhibition models of kinetics data for different types of enzyme inhibition were also compared using Graph Pad Prism 5.0 (San Diego, CA, USA).
2.6 Statistical analysis
All data are presented as mean ± SD (standard deviation). For comparison of concentration curves from the MROD assay to the uninhibited positive control we used linear regression; for concentration comparisons within each compound we used one-way ANOVA with Duncan’s test. IC50 values, Ki values and comparisons of best fit of enzyme inhibition model type were calculated using GraphPad Prism 5.0 (San Diego, CA, USA). For the substrate protection assay, one-way ANOVA with Duncan’s test was used to detect significant difference from control (0 μM methoxyresorufin). All statistical analyses were performed using SAS Version 9.1 (SAS Institute Inc., Cary, NC, USA). In all analyses, p <0.05 was considered significant.
3. Results
3.1. IC50 assessment
While developing the assay, we confirmed that hCYP1A2 did not interact with the furanocoumarins to produce fluorescent products, and that the furanocoumarins did not interact with resorufin, which is the product of the reaction between hCYP1A2 and methoxyresorufin. The concentrations tested for imperatorin and isopimpinellin were lower (0.005, 0.015, 0.05, 0.15, 0.5, and 1.5 μM) than those of angelicin and trioxsalen (0.5, 1.5, 5.0, 15, 50, and 150 μM) because at concentrations greater than 5 μM the remaining activity (%) of hCYP1A2 had already plateaued. Compared to DMSO control, imperatorin, isopimpinellin and trioxsalen were potent hCYP1A2 inhibitors. Imperatorin and isopimpinellin significantly inhibited hCYP1A2 at concentrations ≥ 0.005 μM (p < 0.05) and ≥ 0.05 μM (p < 0.05), respectively (Fig. 2A). Trioxsalen and angelicin exhibited significant hCYP1A2 inhibition at all tested concentrations (p < 0.05) (Fig. 2B). The IC50 values of imperatorin, isopimpinellin, trioxsalen and angelicin were 0.02, 0.46, 0.79, and 9.6 μM respectively. Angelicin did not exhibit a physiologically relevant IC50 (≤ 10μM) and was not considered for further analysis. IC50 values for imperatorin and trioxsalen were used to determine the concentrations used in the inhibition matrix assay.
Fig. 2.
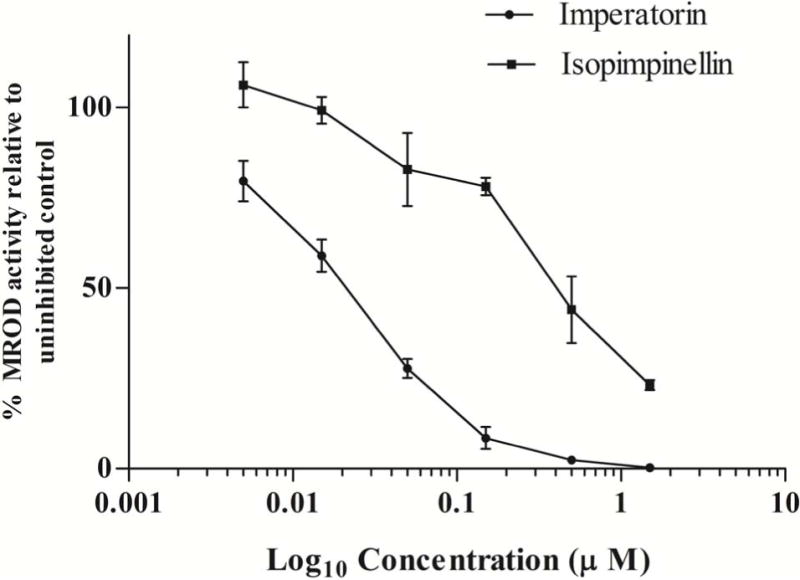
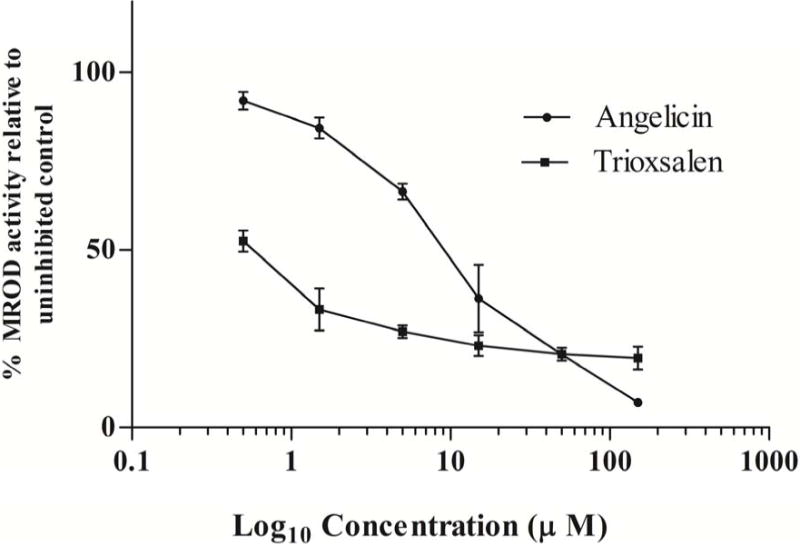
Effects of imperatorin and isopimpinellin (A) and trioxsalen (B) on MROD activity in microsomes from yeast expressing hCYP1A2. Values are means ± SD. The average MROD activity in the uninhibited controls was 0.87 pmol resorufin/min/pmol P450 (imperatorin), 0.87 pmol resorufin/min/pmol P450 (isopimpinellin), 1.07pmol resorufin/min/pmol P450 (trioxsalen), and 0.99 pmol resorufin/min/pmol P450 (angelicin).
3.2. Time- and concentration-dependent inhibition of hCYP1A2
Isopimpinellin inhibited hCYP1A2 in a time- and concentration-dependent manner, and the plots of the natural logarithm of the enzyme activity versus time were highly linear (Fig. 3A). The kobs values were derived from the slopes of each line. We calculated the rate constant for maximal inactivation (kinact) and the concentration of isopimpinellin required to achieve one-half of the maximal rate of CYP1A2 inactivation (KI). A non-linear fit of kobs vs. isopimpinellin concentration (Fig. 3B) was used to estimate the parameters. The kinact value of CYP1A2 inactivation by isopimpinellin was 0.34 ± 0.04 min−1, and KI was 1.2 ± 0.27 μM. The ratio of kinact to KI, which is used to assess the efficiency of enzyme inactivation, was 0.30 ± 0.04 min−1 μM−1. The time required for half the number of enzyme molecules to be inactivated (t1/2) was 2.05 ± 0.22 min. Imperatorin and trioxsalen did not inhibit hCYP1A2 in a time-dependent manner (data not shown).
Fig. 3.
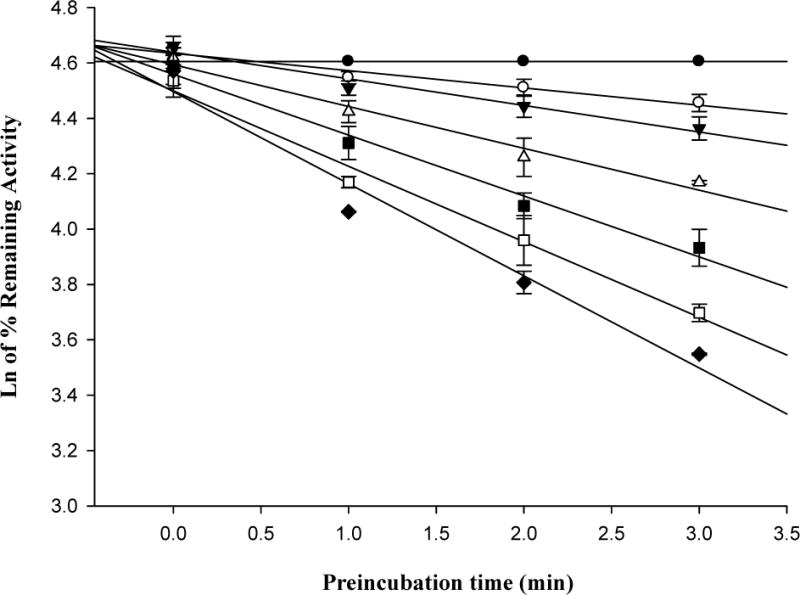
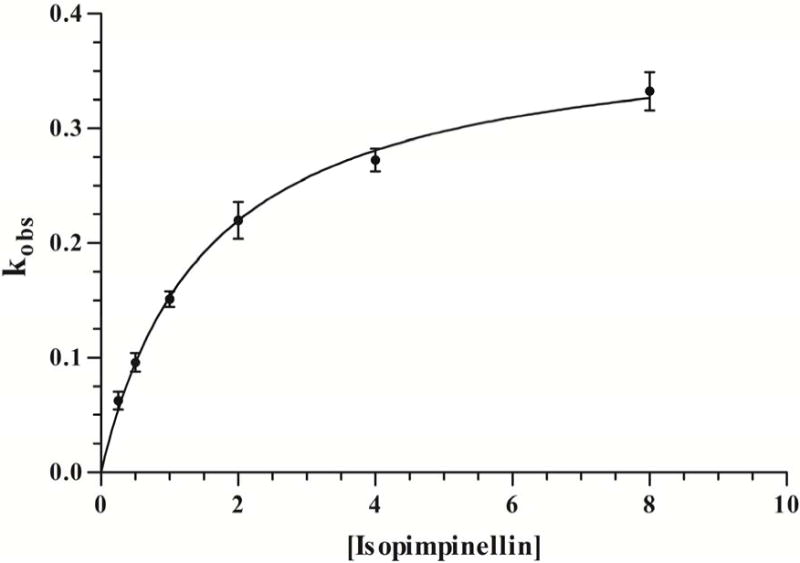
(A) Time- and concentration-dependent loss of microsomal hCYP1A2 activity mediated by 0 μM (●), 0.25 μM (○), 0.5 μM (▼), 1.0 μM (▽), 2.0 μM (■), 4.0 μM (□) and 8.0 μM (◆) of isopimpinellin. (B) Non-linear fit of the relationship between kobs and isopimpinellin concentration (μM). kobs is calculated from the slope of each line in (A). Values are means ± SD. The average activity in the uninhibited control was 3.61 ± 0.16 pmol resorufin/min/pmol P450 at 0 min.
3.3. NADPH-dependent and GSH- and SOD-independent inactivation of hCYP1A2 by isopimpinellin
To confirm the observed inactivation of hCYP1A2 by isopimpinellin required NADPH, the preincubation step was conducted in the presence and absence of NADPH. As demonstrated in Fig. 4A, the lack of NADPH results in the loss of the inhibitory effect, whereas the presence of NADPH with isopimpinellin led to a reduction in enzyme activity.
Fig. 4.
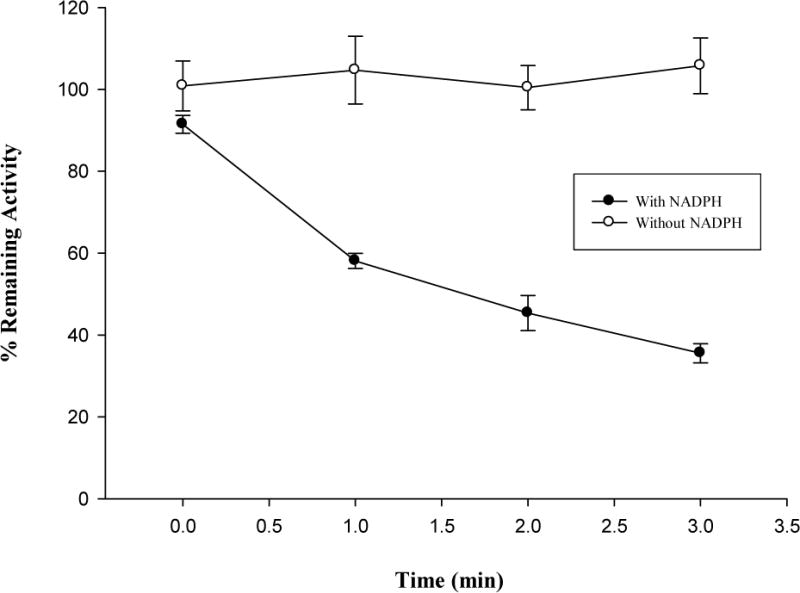
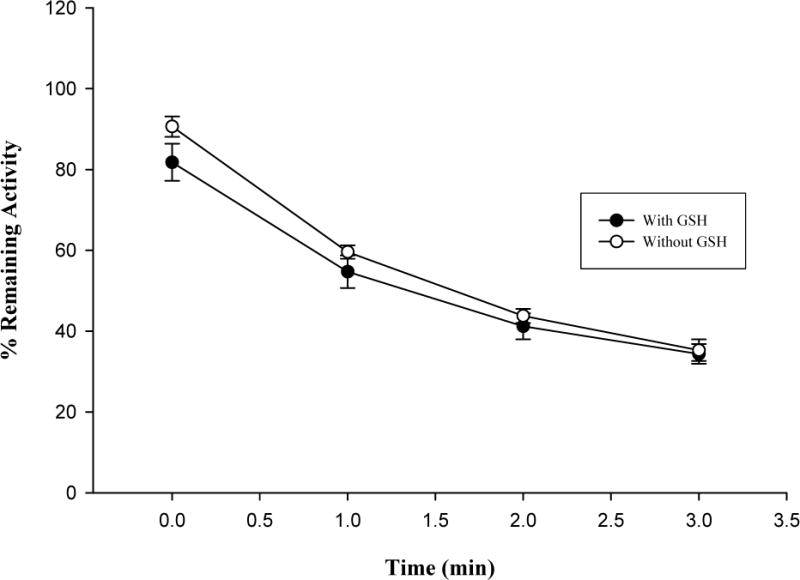
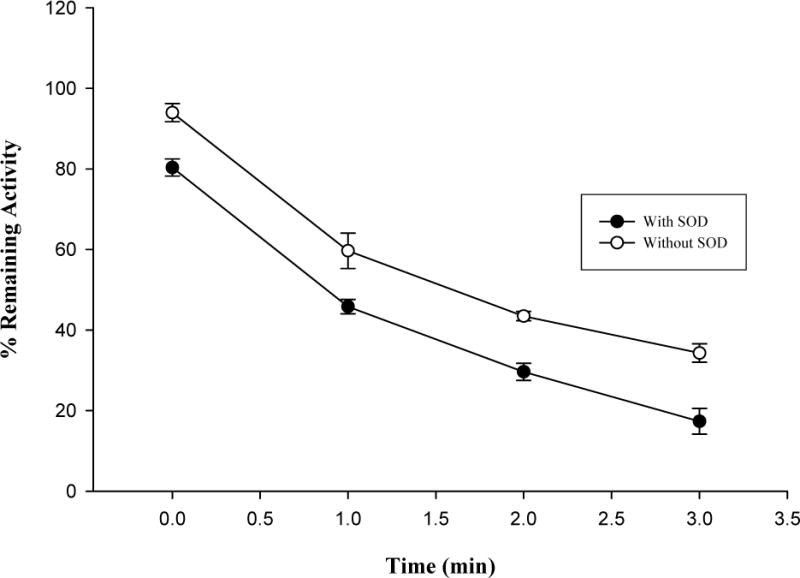
Effects of NADPH (A), GSH (B) and SOD (C) on hCYP1A2 inactivation by isopimpinellin. Values are means ± SD. The average activity of uninhibited controls with NADPH but without GSH or SOD was 3.2 ± 0.11 pmol resorufin/min/pmol P450 at 0 min.
GSH was added to assess the effect of a nucleophilic-trapping agent on the inactivation of the enzyme. As shown in Fig. 4B, GSH failed to protect the hCYP1A2 inactivation mediated by isopimpinellin. Similarly, SOD was added to determine the contribution of reactive oxygen species to the inactivation of the enzyme. As shown in Fig. 4C, SOD did not prevent hCYP1A2 from inactivation, but rather enhanced the inactivation. During the assay, it was found that SOD independently inhibited hCYP1A2 without isopimpinellin (data not shown).
3.4. Irreversibility of inactivation of hCYP1A2 by isopimpinellin
To examine whether the inactivation of hCYP1A2 by isopimpinellin was irreversible, inhibited and uninhibited hCYP1A2 were dialyzed for 20 h. Compared to uninhibited hCYP1A2, the percent remaining activities of hCYP1A2 before and after dialysis were 18 ± 3% and 39 ± 6%, respectively. Percent remaining activity of CYP1A2 was significantly different before dialysis compared to after dialysis (p < 0.001).
3.5. Substrate protection from isopimpinellin
To establish that inactivation by isopimpinellin requires access to the active site of the enzyme, the ability of substrate (methoxyresorufin) to protect against inactivation was evaluated. There was a significant (p < 0.05) reduction in the inhibition of activity of hCYP1A2 at 10 μM methoxyresorufin. The reduction in activity of hCYP1A2 at 10 μM methoxyresorufin was not likely due to excess presence of substrate in the activity assay due to the 1:120 dilution factor. Therefore, the protection of enzyme activity was more likely due to the substrate occupying the enzyme active site. The activities of hCYP1A2 at 0, 0.4, 2.0 and 10 μM of methoxyresorufin with 1.2 μM (KI) of isopimpinellin were 31.8 %, 33.9 %, 36.9 % and 65.4 %, respectively. The average activity of the uninhibited control was 2.34 pmol resorufin/min/pmol CYP1A2.
3.6. Partition ratio of isopimpinellin
The partition ratio is a measure of efficiency for inactivation, and is defined as the number of molecules of inactivator metabolized per molecule of enzyme inactivated. To determine the partition ratio for hCYP1A2 inactivation by isopimpinellin, a plot of the percentage remaining activity of hCYP1A2 versus the ratio of isopimpinellin concentration to enzyme concentration was constructed (Fig. 5). The lower ratio data points were used to extrapolate a straight line, with a resulting partition ratio of 8. However, given that our measured enzyme concentration reflects total CYP content in microsomes, which includes native CYP, the isopimpinellin to hCYP1A2 ratio is likely lower than we anticipated and the partition ratio might be greater than 8. We observed no remaining CYP1A2 activity at the five highest concentration ratios of isopimpinellin to hCYP1A2. Hence, for clearer visual representation, the three highest ratios are not shown in Fig. 5.
Fig. 5.
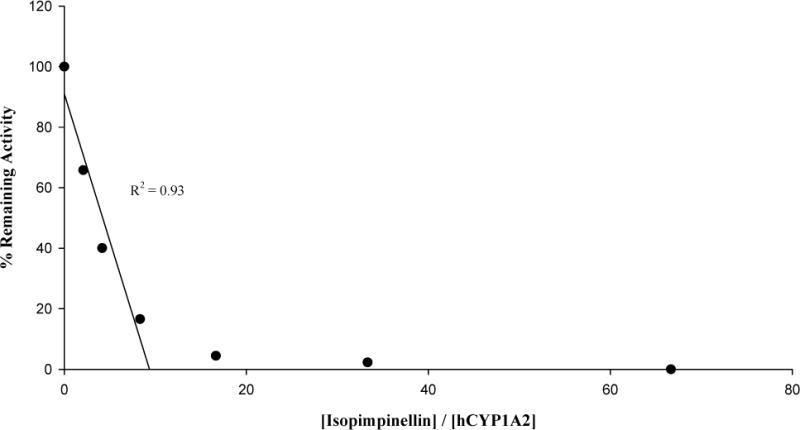
Loss of hCYP1A2 activity as a function of the ratio of hCYP1A2 to isopimpinellin. The average activity in the uninhibited control was 0.90 pmol resorufin/min/pmol P450.
3.7 Inhibition matrix assay
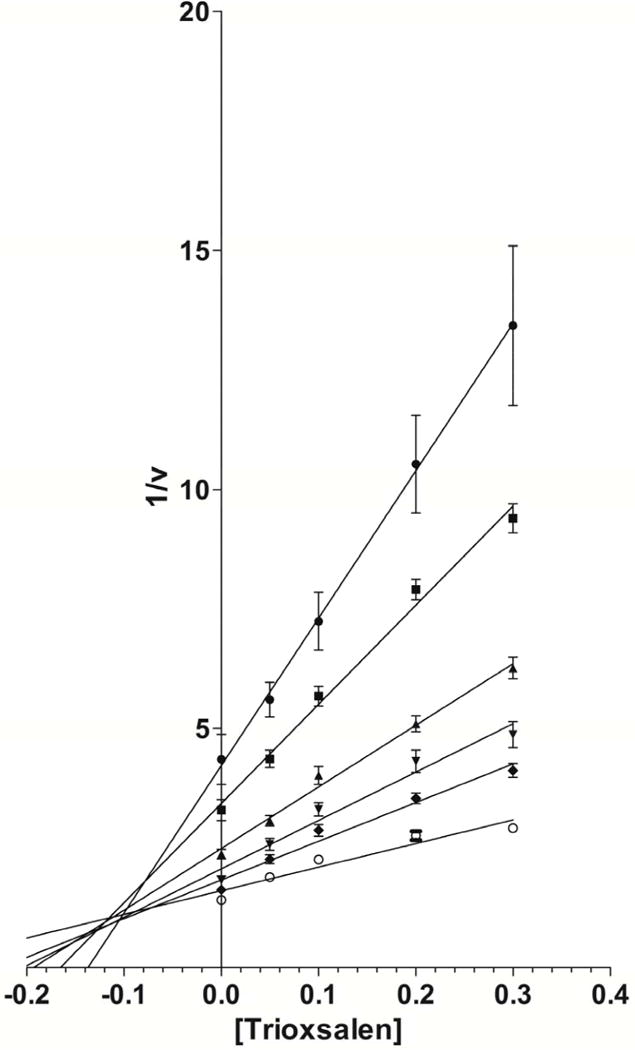
Modes of hCYP1A2 inhibition by imperatorin and trioxsalen were characterized using the inhibition matrix assay. Imperatorin (Fig 6A) and trioxsalen (Fig 6B) exhibited mixed inhibition according to Dixon plots of the kinetics data. The Ki values were 0.007 and 0.10 μM for imperatorin and trioxsalen, respectively.
Fig. 6.
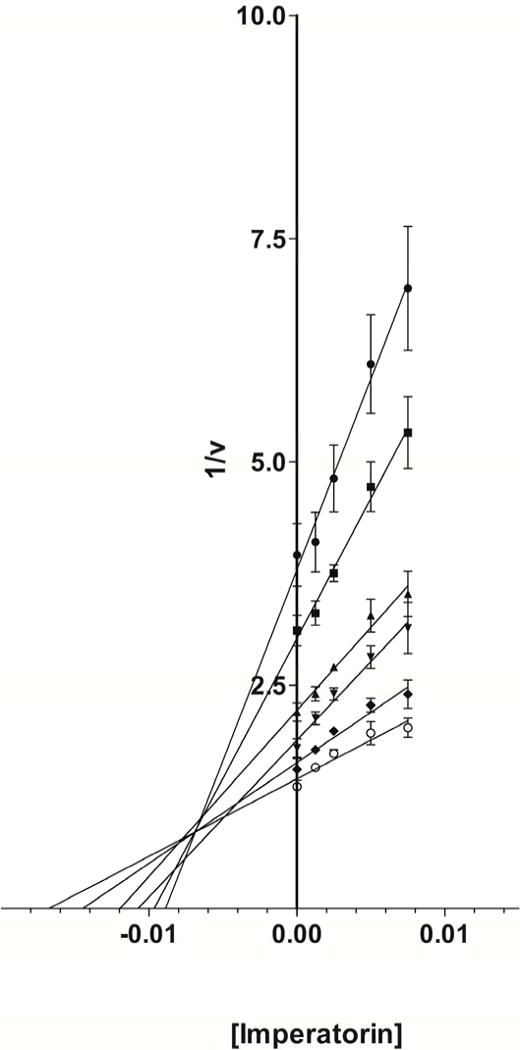
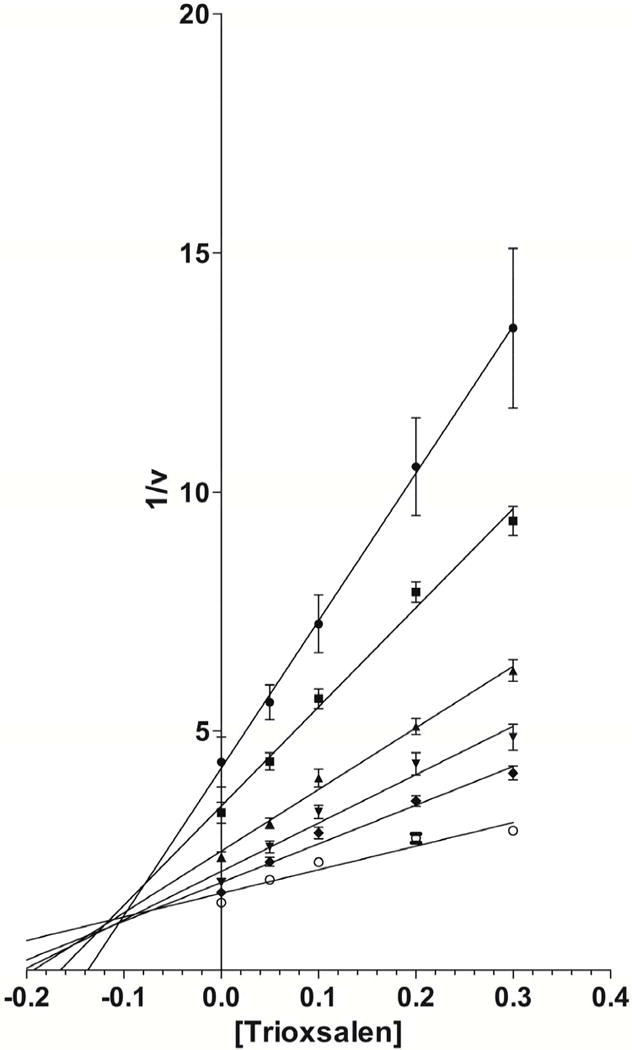
Dixon plots for imperatorin (Fig. 6A) and trioxsalen (Fig. 6B) at substrate concentrations of 0.14 μM (●), 0.21 μM (■), 0.42 μM (▲), 0.63 μM (▼), 1.05 μM (◆), and 2.52 μM (○).
4. Discussion
The major goals of our study were to screen for potent inhibitors of hCYP1A2 in apiaceous vegetables and to characterize the interaction of these compounds with hCYP1A2. Using microsomes from yeast expressing hCYP1A2, we found that the furanocoumarins imperatorin, isopimpinellin and trioxsalen are potent hCYP1A2 inhibitors. Further, the present work also indicated that isopimpinellin is an efficient MBI of hCYP1A2. Imperatorin and trioxalen were characterized as mixed inhibitors of hCYP1A2. To our knowledge, this is the first study that has determined the effects of angelicin and trioxsalen on hCYP1A2. Our results with isopimpinellin and imperatorin are consistent with those of a previous study which reported potent inhibition of CYP1A2 in human liver microsomes by imperatorin and isopimpinellin with IC50 values of 0.38 ± 0.12 and 0.86 ± 0.18 μM, respectively, using ethoxyresorufin as substrate [21]. Additionally, our findings are consistent with the generality that linear furanocoumarins are more potent inhibitors of CYPs than angular furanocoumarins [6]. A number of structural characteristics may predispose an inhibitor to behave as an MBI; however the one commonality amongst them is the capability of forming a reactive intermediate which either alkylates the heme component of the enzyme or forms an adduct with an amino acid residue situated near the active site of the enzyme [18]. A number of molecules categorized as acetylenes, arylamines, quinones, furanocoumarins and others may be MBIs [18]; however as we have demonstrated here, it is likely that not all members of a class are MBIs.
It has been suggested that the inhibition by MBIs lasts longer than the inhibitory effect of reversible inhibitors in vivo [29]. When enzyme activity is lost due to the MBI, the only means by which the activity can be restored is by de novo biosynthesis of the enzyme. In the present study, isopimpinellin met all of the criteria [20] for an MBI. The inhibition of hCYP1A2 by isopimpinellin was concentration-, time-, and NADPH-dependent, and was not prevented by the addition of GSH or SOD. Although the addition of SOD enhanced the inhibition by isopimpinellin, this observation does not interfere with our conclusion since we found that SOD independently inhibited hCYP1A2 in the absence of isopimpinellin. In addition, enzyme inactivation by an MBI should be active-site directed. Not only should the MBI be processed at the catalytic site of the enzyme, but the resulting reactive moiety should also react at the active site [9]. Protection by substrate is evidence for active-site directedness, and the addition of methoxyresorufin decreased the extent of enzyme inactivation by isopimpinellin, indicating that hCYP1A2 inactivation by isopimpinellin is active-site directed.
In our study of isopimpinellin, although approximately 60% inhibition was still observed after 20 h of dialysis which indicates irreversibility, approximately 21% of the activity was recovered after dialysis. Therefore partial reversibility is likely. For dialysis, we tested the highest concentration of isopimpinellin (8 μM) used in the concentration- and time-dependent tests; thus, there is a possibility that the amount of isopimpinellin relative to the amount of enzyme (120 pmol/ml) was excessive. In such a case, it is possible that at higher concentrations of the inactivator, the enzyme active site is saturated and the conversion to reactive intermediate slows down. Indeed, during assay development, there was no recovery of hCYP1A2 activity after dialysis using the lower concentration of 6.4 μM (24 ± 2% activity remaining pre-and post-dialysis in one assay with six replicates at this concentration). Secondly, there is a possibility that isopimpinellin might bind to more than one residue in the enzyme active site and that binding to one residue may be less stable than binding to the other, particularly under the extreme conditions of dialysis. Of the three modes by which MBIs can inactivate CYPs: (1) formation of reactive intermediates coordinating with the heme prosthetic group that subsequently produces a catalytically inactive metabolite-inhibitor complex; (2) alkylation or arylation of the heme group resulting in heme destruction; and (3) binding covalently to the apoprotein [19], the first mode is called quasi-irreversible inactivation since it is reversible after dialysis. Hence, it is also possible that isopimpinellin might use more than one of the three different MBI binding modes including the quasi-irreversible binding. With regard to in vivo applications, conditions are likely to be less severe as compared to those in dialysis conditions, thus quasi-irreversible binding may be more stable in vivo than in dialysis conditions. Importantly, after 20 h of dialysis, we still observed approximately 60% inhibition, indicating that a considerable amount of irreversible inactivation by isopimpinellin occurred.
The inhibitory activity of isopimpinellin is comparable to that of furafylline, a well-known potent mechanism-based CYP1A2 inactivator [26, 44]. By performing experiments on human liver microsomes with various substrates, furafylline has been found to be an efficient MBI of CYP1A2 (KI: 0.8–23 μM; kinact: 0.16–0.87 min−1) [8, 15, 26, 32]. In our study, isopimpinellin was comparable: KI = 1.2 μM and kinact = 0.34 min−1. The partition ratio of CYP1A2 inactivation by isopimpinellin is also comparable to that of furafylline. The partition ratio of furafylline-mediated CYP1A2 inactivation in previous studies was 3–6 [26], and in the present study, the partition ratio of isopimpinellin was 8, indicating that isopimpinellin is an efficient MBI. Similarly, relatively low amounts of imperatorin and trioxsalen are needed to inhibit hCYP1A2 evidenced by their Ki values of 0.007 and 0.10 μM, respectively.
The concentrations of furanocoumarins required here for significant inhibition of hCYP1A2 may be physiologically relevant and achievable from dietary consumption of apiaceous vegetables; however little data exists on the levels of furanocoumarins in vivo following vegetable consumption. Two studies reported undetectable levels of furanocoumarins following furanocoumarin-containing vegetable intake in humans [1, 40], although the furanocoumarins 5-MOP and 8-MOP (1.2 and 0.6 mg/kg, respectively) administered in purified form as a treatment for certain skin disorders such as psoriasis resulted in detectable levels of the compounds in blood samples [51, 52]. In comparison to the pharmacological dose of furanocoumarins above, the consumption of a single serving of 100 g of parsnips could deliver 0.4–14.5 mg of total furanocoumarins Our observations here suggest that even if present at low levels in apiaceous vegetables, imperatorin, isopimpinellin, and trioxsalen may contribute significantly to CYP1A2 inhibition in vivo, although further investigation in humans is warranted.
Utilization of microsomes from the yeast expression system allowed the initial investigation of hCYP1A2 activity more specifically without the complexity of other enzymes competing for the same substrates. We confirmed that the MROD activity was due to hCYP1A2 in the strain.
While we propose that inhibition of CYP1A2 may impart chemopreventive effects, CYP enzymes are also responsible for the metabolism of a wide range of endogenous and exogenous substrates prior to conjugation and excretion. A previous study in humans indicates that the change in activity of CYP enzymes achievable with dietary intervention is in the range of 14 – 24% [38]. Consumption of furanocoumarin containing foods and concomitant decrease in CYP1A2 activity may lead to a shift in the balance between activation and conjugation that might favor more complete conjugation and excretion of potential carcinogens.
Consumption of furanocoumarin-containing vegetables is not without risk since furanocoumarins can cause photosensitization, particularly in individuals who handle fresh produce such as celery [41]. Individuals treated for psoriasis and other skin conditions with pharmacological doses of furanocoumarins (specifically 8-methoxypsoralen) combined with ultraviolet A light (PUVA therapy) may be at increased risk of skin cancers due to the ability of furanocoumarin molecules to form complexes with DNA upon irradiation with ultraviolet light [49]. As with many bioactive molecules, toxicity is many times a matter of dose. Furanocoumarins encountered in the diet are far less concentrated than those used in PUVA therapy. Moreover, a substantial literature investigating the chemopreventive potential of furanocoumarins is accumulating [22, 31, 42, 47].
Because we have demonstrated inhibition of hCYP1A2 in yeast microsomes by furanocoumarins, subsequent studies in human liver microsomes and cell culture are warranted. Identification and characterization of covalent adducts formed by isopimpinellin will help to elucidate specifically how isopimpinellin modifies and inhibits CYP1A2. Additional animal and human studies could further elucidate any differences in effect from individual purified furanocomarins, combinations of the purified compounds, and their whole food sources. Because CYP1A2 acts on a broad range of endogenous and exogenous substrates, the comprehensive effects of diet-induced inhibition of CYP1A2 need to be elucidated.
In summary, we completed an initial characterization of the effects of four furanocoumarins on the inhibition of hCYP1A2. We found isopimpinellin, imperatorin and trioxsalen to be potent inhibitors at low concentrations. This implies that even low exposure to furanocoumarins, such as the levels present in apiaceous vegetables, may confer biologically relevant inhibition of CYP1A2. In particular, the effect of isopimpinellin as an MBI of hCYP1A2 may last longer than the effects of other furanocoumarins despite rapid clearance. Hence it is plausible that more than episodic exposure to certain furanocoumarins in a habitual diet is not necessary to have a meaningful impact on CYP1A2.
Acknowledgments
The authors gratefully acknowledge and thank Dr. Patrick Hanna (Department of Medicinal Chemistry, University of Minnesota) and Dr. Linda von Weymarn (Masonic Cancer Center, University of Minnesota) for their expert guidance. The authors also thank Li Liu for assistance with development of assays. This work has been supported in part by the Minnesota Agricultural Experiment Station and by Award Number T32CA132670 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations
- 5-MOP
5-methoxypsoralen
- 8-MOP
8-methoxypsoralen
- CYP
Cytochrome P-450
- CYP1A2
Cytochrome P-450 1A2
- DMSO
Dimethyl sulfoxide
- HBSS
Hank’s balanced salt solution
- hCYP1A2
Human cytochrome P-450 1A2
- MBI
Mechanism-based inactivator
- MROD
Methoxyresorufin O-demethylase
- NADPH
β-nicotinamideadenine dinucleotidephosphate
References
- 1.Beattie PE, Wilkie MJ, Smith G, Ferguson J, Ibbotson SH. J Am Acad Dermatol. 2007;56:84–87. doi: 10.1016/j.jaad.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Bendriss EK, Bechtel Y, Bendriss A, Humbert PH, Paintaud G, Magnette J, Agache P, Bechtel PR. Br J Clin Pharmacol. 1996;41:421–424. doi: 10.1046/j.1365-2125.1996.33311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berthou F, Flinois JP, Ratanasavanh D, Beaune P, Riche C, Guillouzo A. Drug Metab Dispos. 1991;19:561–567. [PubMed] [Google Scholar]
- 4.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Busby WF, Jr, Ackermann JM, Crespi CL. Drug Metab Dispos. 1999;27:246–249. [PubMed] [Google Scholar]
- 6.Cai Y, Bennett D, Nair RV, Ceska O, Ashwood-Smith MJ, DiGiovanni J. Chem Res Toxicol. 1993;6:872–879. doi: 10.1021/tx00036a018. [DOI] [PubMed] [Google Scholar]
- 7.Chang TK, Chen J, Lee WB. J Pharmacol Exp Ther. 2001;299:874–882. [PubMed] [Google Scholar]
- 8.Clarke SE, Ayrton AD, Chenery RJ. Xenobiotica. 1994;24:517–526. doi: 10.3109/00498259409043254. [DOI] [PubMed] [Google Scholar]
- 9.Copp LJ. In: Enzyme kinetics: A modern approach. Marangoni AG, editor. John Wiley & Son, Inc.; New York: 2003. pp. 158-158–173. [Google Scholar]
- 10.Dierks EA, Stams KR, Lim HK, Cornelius G, Zhang H, Ball SE. Drug Metab Dispos. 2001;29:23–29. [PubMed] [Google Scholar]
- 11.Draper AJ, Madan A, Parkinson A. Arch Biochem Biophys. 1997;341:47–61. doi: 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- 12.Easterbrook J, Lu C, Sakai Y, Li AP. Drug Metab Dispos. 2001;29:141–144. [PubMed] [Google Scholar]
- 13.Eugster HP, Sengstag C. Toxicology. 1993;82:61–73. doi: 10.1016/0300-483x(93)90060-6. [DOI] [PubMed] [Google Scholar]
- 14.Eugster HP, Sengstag C, Meyer UA, Hinnen A, Wurgler FE. Biochem Biophys Res Commun. 1990;172:737–744. doi: 10.1016/0006-291x(90)90736-7. [DOI] [PubMed] [Google Scholar]
- 15.Fairman DA, Collins C, Chapple S. Drug Metab Dispos. 2007;35:2159–2165. doi: 10.1124/dmd.107.017236. [DOI] [PubMed] [Google Scholar]
- 16.Gallagher EP, Kunze KL, Stapleton PL, Eaton DL. Toxicol Appl Pharmacol. 1996;141:595–606. doi: 10.1006/taap.1996.0326. [DOI] [PubMed] [Google Scholar]
- 17.Heinonen JT, Sidhu JS, Reilly MT, Farin FM, Omiecinski CJ, Eaton DL, Kavanagh TJ. Environ Health Perspect. 1996;104:536–543. doi: 10.1289/ehp.96104536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollenberg PF, Kent UM, Bumpus NN. Chem Res Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- 19.Kalgutkar AS, Obach RS, Maurer TS. Curr Drug Metab. 2007;8:407–447. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]
- 20.Kent UM, Juschyshyn MI, Hollenberg PF. Curr Drug Metab. 2001;2:215–243. doi: 10.2174/1389200013338478. [DOI] [PubMed] [Google Scholar]
- 21.Kleiner HE, Reed MJ, DiGiovanni J. Chem Res Toxicol. 2003;16:415–422. doi: 10.1021/tx025636d. [DOI] [PubMed] [Google Scholar]
- 22.Kleiner HE, Vulimiri SV, Miller L, Johnson WH, Jr, Whitman CP, DiGiovanni J. Carcinogenesis. 2001;22:73–82. doi: 10.1093/carcin/22.1.73. [DOI] [PubMed] [Google Scholar]
- 23.Klotz AV, Stegeman JJ, Walsh C. Anal Biochem. 1984;140:138–145. doi: 10.1016/0003-2697(84)90144-1. [DOI] [PubMed] [Google Scholar]
- 24.Koenigs LL, Trager WF. Biochemistry. 1998;37:13184–13193. doi: 10.1021/bi981198r. [DOI] [PubMed] [Google Scholar]
- 25.Koenigs LL, Trager WF. Biochemistry. 1998;37:10047–10061. doi: 10.1021/bi980003c. [DOI] [PubMed] [Google Scholar]
- 26.Kunze KL, Trager WF. Chem Res Toxicol. 1993;6:649–656. doi: 10.1021/tx00035a009. [DOI] [PubMed] [Google Scholar]
- 27.Lampe JW, King IB, Li S, Grate MT, Barale KV, Chen C, Feng Z, Potter JD. Carcinogenesis. 2000;21:1157–1162. [PubMed] [Google Scholar]
- 28.Lee H, Yeom H, Kim YG, Yoon CN, Jin C, Choi JS, Kim BR, Kim DH. Biochem Pharmacol. 1998;55:1369–1375. doi: 10.1016/s0006-2952(97)00644-8. [DOI] [PubMed] [Google Scholar]
- 29.Lin JH, Lu AY. Clin Pharmacokinet. 1998;35:361–390. doi: 10.2165/00003088-199835050-00003. [DOI] [PubMed] [Google Scholar]
- 30.Mays DC, Camisa C, Cheney P, Pacula CM, Nawoot S, Gerber N. Clin Pharmacol Ther. 1987;42:621–626. doi: 10.1038/clpt.1987.209. [DOI] [PubMed] [Google Scholar]
- 31.Miyazaki M, Yamazaki H, Takeuchi H, Saoo K, Yokohira M, Masumura K, Nohmi T, Funae Y, Imaida K, Kamataki T. Carcinogenesis. 2005;26:1947–1955. doi: 10.1093/carcin/bgi156. [DOI] [PubMed] [Google Scholar]
- 32.Obach RS, Walsky RL, Venkatakrishnan K. Drug Metab Dispos. 2007;35:246–255. doi: 10.1124/dmd.106.012633. [DOI] [PubMed] [Google Scholar]
- 33.OMURA T, SATO R. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 34.Ono S, Hatanaka T, Hotta H, Satoh T, Gonzalez FJ, Tsutsui M. Xenobiotica. 1996;26:681–693. doi: 10.3109/00498259609046742. [DOI] [PubMed] [Google Scholar]
- 35.Patten CJ, Smith TJ, Murphy SE, Wang MH, Lee J, Tynes RE, Koch P, Yang CS. Arch Biochem Biophys. 1996;333:127–138. doi: 10.1006/abbi.1996.0373. [DOI] [PubMed] [Google Scholar]
- 36.Peroutka R, Schulzova V, Botek P, Hajslova J. J Sci Food Agri. 2007;87:2152-2152–2163. [Google Scholar]
- 37.Peterson S, Lampe JW, Bammler TK, Gross-Steinmeyer K, Eaton DL. Food Chem Toxicol. 2006;44:1474–1484. doi: 10.1016/j.fct.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Peterson S, Schwarz Y, Li SS, Li L, King IB, Chen C, Eaton DL, Potter JD, Lampe JW. Cancer Epidemiol Biomarkers Prev. 2009;18:3118–3125. doi: 10.1158/1055-9965.EPI-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polychronaki N, Wild CP, Mykkanen H, Amra H, Abdel-Wahhab M, Sylla A, Diallo M, El-Nezami H, Turner PC. Food Chem Toxicol. 2008;46:519–526. doi: 10.1016/j.fct.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 40.Schlatter J, Zimmerli B, Dick R, Panizzon R, Schlatter C. Food Chem Toxicol. 1991;29:523–530. doi: 10.1016/0278-6915(91)90044-8. [DOI] [PubMed] [Google Scholar]
- 41.Seligman PJ, Mathias CG, O’Malley MA, Beier RC, Fehrs LJ, Serrill WS, Halperin WE. Arch Dermatol. 1987;123:1478–1482. [PubMed] [Google Scholar]
- 42.Sellers EM, Ramamoorthy Y, Zeman MV, Djordjevic MV, Tyndale RF. Nicotine Tob Res. 2003;5:891–899. doi: 10.1080/14622200310001615231. [DOI] [PubMed] [Google Scholar]
- 43.Sengstag C, Wurgler FE. Mol Carcinog. 1994;11:227–235. doi: 10.1002/mc.2940110408. [DOI] [PubMed] [Google Scholar]
- 44.Sesardic D, Boobis AR, Murray BP, Murray S, Segura J, de la Torre R, Davies DS. Br J Clin Pharmacol. 1990;29:651–663. doi: 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. Boca Raton: CRC Press; 1988. [Google Scholar]
- 46.Silverman RB. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi H, Saoo K, Matsuda Y, Yokohira M, Yamakawa K, Zeng Y, Miyazaki M, Fujieda M, Kamataki T, Imaida K. Cancer Lett. 2006;234:232–238. doi: 10.1016/j.canlet.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 48.Turesky RJ, Guengerich FP, Guillouzo A, Langouet S. Mutat Res. 2002;506–507:187–195. doi: 10.1016/s0027-5107(02)00165-3. [DOI] [PubMed] [Google Scholar]
- 49.Viola G, Facciolo L, Vedaldi D, Disaro S, Basso G, Dall’Acqua F. Photochem Photobiol Sci. 2004;3:237–239. doi: 10.1039/b313729e. [DOI] [PubMed] [Google Scholar]
- 50.von Weymarn LB, Zhang QY, Ding X, Hollenberg PF. Carcinogenesis. 2005;26:621–629. doi: 10.1093/carcin/bgh348. [DOI] [PubMed] [Google Scholar]
- 51.Yeo UC, Shin JH, Yang JM, Park KB, Kim MM, Bok HS, Lee ES. Br J Dermatol. 2000;142:733–739. doi: 10.1046/j.1365-2133.2000.03419.x. [DOI] [PubMed] [Google Scholar]
- 52.Zucchi A, Raho E, Marconi B, Nicoli S, Santini M, Allegra F, Colombo P, Bettini R, Santi P. J Invest Dermatol. 2001;117:379–382. doi: 10.1046/j.0022-202x.2001.01419.x. [DOI] [PubMed] [Google Scholar]