

Abstract
Alzheimer disease (AD) is a devastating neurodegenerative disorder with high unmet medical need. Drug development is hampered by limited understanding of the disease and its driving factors. Quantitative Systems Pharmacology (QSP) modeling provides a comprehensive quantitative framework to evaluate the relevance of biological mechanisms in the context of disease and to predict the efficacy of novel treatments. Here, we report a QSP model for AD with a particular focus on investigating the relevance of dysregulation of cholesterol and sphingolipids. We show that our model captures the modulation of several biomarkers in subjects with AD, as well as the response to pharmacological interventions. We evaluate the impact of targeting the sphingosine‐1‐phosphate 5 receptor (S1PR5) as a potential novel treatment option for AD, and model predictions increase our confidence in this novel disease pathway. Future applications for the QSP model are in validation of further targets and identification of potential treatment response biomarkers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The relevance of specific molecular changes and their interplay is AD during disease is currently not well understood. Specific mechanisms have been studied by several published mathematical models.
WHAT QUESTION DID THIS STUDY ADDRESS?
We developed a QSP model for AD as a comprehensive quantitative framework with a particular focus on the dysregulation of cholesterol and sphingolipids, and their relationship to Aβ aggregation in the brain.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Our model captures the modulation of several biomarkers in patients with AD and pharmacological responses published in the literature. Using the model, we predict a relevant treatment response in patients with AD by targeting the S1PR5 receptor.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
We anticipate that the model can be used in early discovery for target validation and selection, as well as late discovery and development to guide translational and clinical decisions.
Despite the considerable clinical efforts in the search for treatments for Alzheimer disease (AD), no new medicines that prevent disease development have been discovered and a number of negative results have been reported recently.1 These disappointing findings called into question many of the apparently intuitive therapeutic hypotheses pursued to date. Nevertheless, the pressing unmet medical need remains for the millions affected by dementia. Currently, it is estimated that worldwide there are 44 million sufferers and this is projected to rise to 130 million in 2050.2
In order to more effectively find treatments for AD, there is a need to improve the understanding of the underlying biology and pathology of this complex disease. Given the multifaceted nature of AD, it is likely that selecting the best targets will be nontrivial, combinations of drugs will be required, and the timing of therapy initiation vs. disease progression will be critical to success. Quantitative Systems Pharmacology (QSP) models are one tool to tackle this seemingly intractable complex problem.3, 4, 5 AD is good substrate for a QSP approach as useful mechanistic models,6, 7, 8, 9, 10 as well as statistical and pharmacokinetic/pharmacodynamic PK/PD models,11, 12 of some aspects of AD exist. There is a substantial database of clinical data observations, including pharmacology and biomarker data, that can be used to calibrate and validate QSP models.
Cholesterol and sphingolipids are essential lipids responsible for cell functioning, cell survival, and cell‐cell interactions. Within several neurodegenerative disorders, a dysregulation in these lipids is observed in the central nervous system (CNS) and cerebrospinal fluid (CSF).13, 14 Altered lipid metabolism is also associated with several key genetic risk factors for AD, such as APOE, ABCA1, and ABCA7.15, 16 Several attempts have been made at targeting lipid metabolism and/or synthesis as treatments for AD, but most have focused on cholesterol as the entry point and have proven unsuccessful either due to severe side effects hindering further development (i.e., liver X receptor agonists17) or due to lack of sufficient brain penetration and/or achieved effect size on cholesterol lowering (i.e., simvastatin).18, 19
A potential alternative targeting mechanism is through sphingolipid modulation. At AbbVie, we developed several compounds, including A‐971432,20 which specifically activate sphingosine‐1‐phosphate 5 receptor (S1PR5). The S1PR5 is almost exclusively expressed in the CNS. The hypothesis is that through modulation of S1PR5 activation in the brain, lipid dysregulation in AD could be reversed to healthy levels, providing a potential novel treatment option for AD. The aim of the current work is to use a QSP model to explore (i) how different lipid pathways are linked to amyloid‐beta (Aβ) as a relevant AD disease marker, (ii) to describe quantitatively changes in pathway dynamics to capture the AD disease state compared to the healthy state, and (iii) to explore whether pharmacologically targeting sphingolipid metabolism could significantly modulate the AD disease state. We generate additional in vivo evidence for a treatment effect on lipid modulation and Aβ in an animal model for AD, which is are line with our in silico predictions for patients with AD.
Methods
Quantification of tissue proteins
Human brain samples were obtained via the Neurobiobank Munchen (n = 12 control subjects and n – 12 subjects with AD – Braak stage V/VI, 1:1 gender division, and 74–78 years of age). The samples, originating from the superior and medial frontal gyrus of the brain, were analyzed for oxysterol and sterol content as described in the Proceedings of the National Academy of Sciences.21 In short, samples were prepared and measured for lipid content by a gas‐liquid‐chromatography tandem mass spectrometry (LC‐MS/MS) method. Human CSF and plasma samples were purchased at Precision Med and included n = 20 young adult subjects, n = 25 age‐matched (to AD) controls, and n = 25 subjects with AD. Samples were aliquoted and measured for cholesterol and oxysterol content as listed above. Another aliquot was analyzed by an LC‐MS/MS based quantification of C18:0 ceramide (Cer), C24:0 Cer, sphingosine, and sphingosine‐1‐phosphate (S1P). The measurement includes a chloroform‐based extraction of tissue followed by LS‐MS quantification with internal standards for the different measurements. Animal tissue, CSF, and plasma were collected and processed for various experiments. Species and strains included were: Sprague Dawley rats, Wistar rats, and C57Bl6J mice. Plasma and CSF samples were drawn under either short anesthesia by isoflurane or terminally, snap‐frozen in liquid nitrogen, and stored at −80°C until use. CNS samples were collected under terminal anesthesia, snap‐frozen in liquid nitrogen, and stored at −80°C until use. Samples were processed identical to the human samples for the different measurements. Human and rat CSF samples were checked for hemoglobin content before analysis, mouse CSF samples were visually scored for blood contamination.
In vivo study in SAMP8 mice
Ten‐month‐old male SAMP8 mice (an AbbVie sponsored study in collaboration with S. Farr at Saint Louis University, Saint Louis, MO) were administered A‐971432 (dissolved in water containing 0.5% HPMC + 0.1% Tween 80) via daily oral gavage 2 mL/kg at 0, 0.03, 1, or 3 mg/kg for the duration of the study. A young SAMP8 control group received vehicle for 10 weeks via oral gavage at 2 mL/kg. After 10 weeks of treatment, animals were euthanized and brain tissue was collected. Brain samples were further processed for determination of exposure levels as well as Cer C18:0 and S1P levels per the LC‐MS/MS method. Moreover, the same samples were analyzed for soluble and insoluble amyloid beta 1–40 and 1–42 levels. For the latter, samples were processed in three buffers resulting in the TBS/RAB, radioimmunoprecipitation assay, and FA fraction and analyzed by the MSA V‐PLEX Aβ peptide 1 (4G8) panel (catalog no. 15199G‐1). All samples of the radioimmunoprecipitation assay and FA fraction were below the lower limit of detection for Aβ 1–40 and 1–42 (lower limit of detection was 1.81 pg/mL and 0.8 pg/mL for Aβ 1–40 and 1–42, respectively) and, thus, not further analyzed. Results were expressed as means with their SEM. All data were analyzed by one‐way analysis of variance for each group followed by the appropriate post hoc analysis. Data are marked by significance levels: P < 0.05*, P < 0.01**, and P < 0.001*** or by difference between young and aged SAMP8 mice P < 0.05^.
Pharmacokinetics and fraction unbound quantification
Male Sprague‐Dawley rats were given 2 mg/kg of A‐971432 orally (2 mL/kg in ethanol/PEG400/dextrose5% 10:50:40 v/v/v). Animals were euthanized at various timepoints from 30 minutes up to 48 hours postdosing. Brain and plasma samples were collected and stored at −20°C until analysis by LC/MS/MS for A‐971432 total level quantification. The fraction unbound of A‐971432 in rat and human plasma and rat brain homogenate was determined in vitro using the Rapid Equilibration Dialysis method, described elsewhere.22
Results
QSP model framework
Our QSP model for AD is composed of two main components: a model for the physiology, including brain and CSF, and a model for the pharmacological interventions. Figure 1 shows the Matlab/Simbiology (The Mathworks, Natick, MA) implementation of the complete QSP model, and the following subsections describe the details of our modeling approach. Additional information on the model components are given in the Supplementary Information (Tables S1‐S6).
Figure 1.
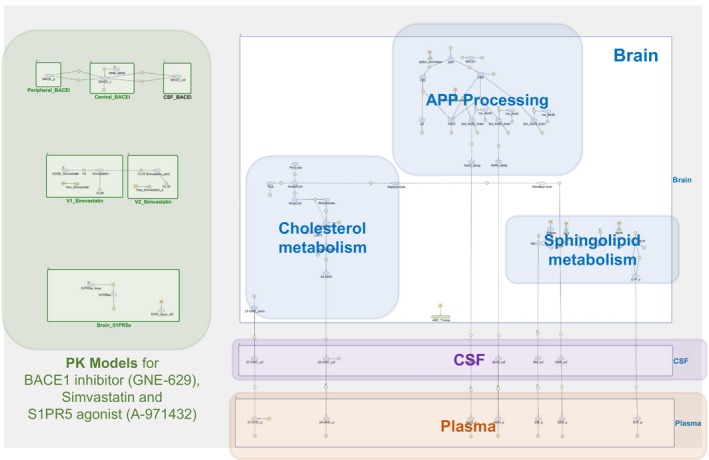
Quantitative systems pharmacology model for lipid dysregulation in Alzheimer disease and pharmacological interventions. Relevant physiology is captured assuming three compartments: plasma, cerebrospinal fluid (CSF), and the brain. Reactants associated with cholesterol and sphingolipid metabolism, amyloid precursor protein (APP) processing/metabolism and sphingosine‐1‐phosphate receptor 5 (S1PR5) binding are described in the brain. They interact and/or distribute between the compartments according to their properties. Pharmacological interventions are implemented using empirical models for pharmacokinetic (PK) and pharmacological effect, as described in the text.
Physiology model
As shown in Figure 1, we considered three physiological compartments in the model, namely the brain, CSF, and plasma, to describe relevant biological mechanisms and transport of different molecular species through different tissues. In the brain compartment, three different submodels were implemented, adapted from published models,7, 8, 9, 10 to describe relevant biological mechanisms: (i) amyloid precursor protein (APP) processing and Aβ aggregation, (ii) cholesterol metabolism, and (iii) sphingolipid metabolism. The integrated QSP model considers that all the mechanisms inside the brain share the same compartmental volume (i.e., brain volume), as adding granularity by including additional brain compartments (e.g., cellular compartment and extracellular brain fluid), produced more complexity in the model without providing better simulations results; the implementation of additional brain subcompartments will be part of future extensions of the model. The details of the submodels are described in Supplementary Information S1, and schematics of the submodels are shown in Figure 2 a–c. We integrated the submodels into our complete model for brain physiology by adding additional molecular interactions described in the literature between molecular species in the submodels (Figure 2 d). Table 1 summarizes these additional interactions with their respective supporting reference, and Table S6 lists the quantitative relationships used in the model. When available, parameter values were taken from the published models (assuming these values do not change when integrating all submodels). However, some parameters needed to be recalibrated and others, where there is no link to another published model, were estimated using the data reported in this work. Tables S4 and S5 show all parameter values used in the model, including the literature reference where applicable.
Figure 2.
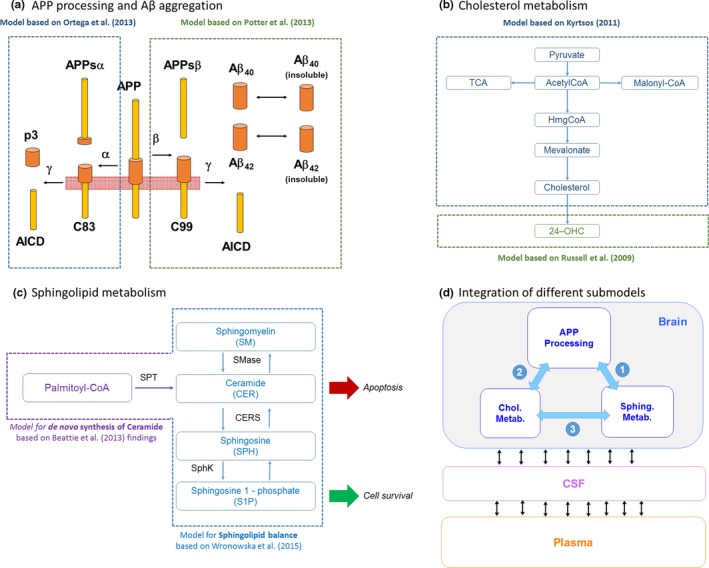
Details of submodels and their integration within quantitative systems pharmacology model. (a) Amyloid precursor protein (APP) processing model based on two published models to describe the production of amyloid beta (Aβ), p3, and AICD. (b) Cholesterol metabolism model diagram. Model mainly based on the production of cholesterol from pyruvate and acetyl‐CoA through the synthesis of HmgCoA and mevalonate as proposed by ref. 7. The 24‐OHC production is modeled using kinetic information reported by ref. 29. (c) Sphingolipid metabolism in the brain. Ceramide is proposed as a key apoptotic stimulator and is derived from palmitoyl‐coA from a reaction catalyzed by serine palmitoyltransferase. Ceramide equilibrates with sphingomyelin and sphingosine, catalyzed by sphingomyelinase and ceramide synthase. In turn, sphingosine can equilibrate with sphingosine‐1‐phosphate (S1P) catalyzed by sphingosine kinase. S1P is proposed to contribute to increased cell survival. (d) High‐level diagram of integration of the different sub‐models within the quantitative systems pharmacology model. CSF, cerebrospinal fluid.
Table 1.
Additional molecular interactions included in the brain physiology model (detailed references are given in Table S6)
| Models being connected | Description of the interaction | References |
|---|---|---|
| 1. Connecting APP processing and sphingolipid metabolism models | Increasing Aβ levels increases SMase activity (increasing ceramide levels) | Tanabe (2013)38, Geekiyanage (2011)39, Grimm (2005)40 |
| Increasing ceramide increases BACE‐1 activity | Puglielli (2003)41 | |
| S1P modulates BACE‐1 | Jesko (2014)42 | |
| S1P inhibits APP secretion | Jesko (2014)42 | |
| Increasing SM decreases α‐secretase activity | Grimm (2005)40 | |
| Increasing AICD downregulates SPT expression | Grimm (2011)43 | |
| Aβ downregulates SphK | Gassowska (2014)44 | |
| S1P blocks SMase activity | Malaplate‐Armand (2006)45 | |
| 2. Connecting APP processing and cholesterol metabolism models | Increasing 27‐OHC levels, increases BACE‐1 activity | Dias (2014)46, Marwarha (2010)47, Marwarha (2013)48, Prasanthi (2009)31, Prasanthi (2015)49, Famer (2007)50 |
| Increasing brain cholesterol increases BACE‐1 activity | Prasanthi (2009)31, Xiong (2008)30 Grimm (2008)51, von Arnim (2008)52, Zhu (2011)53, Malnar (2012)54, Thirumangalakudi (2008)55, Cui (2011)56 | |
| Increasing 24‐OHC levels increases α‐secretase activity | Prasanthi (2009)31, Famer (2007)50 | |
| Increasing brain cholesterol increases γ‐secretase activity | Xiong (2008)30 | |
| Reducing intracellular cholesterol increases APP processing (through α‐secretase) | Cole (2005)57, Fewlass (2004)58 | |
| Increasing 27‐OHC levels decreases LRP1 levels | Marwarha (2010)47, Prasanthi (2009)31 | |
| Increasing Aβ levels decreases de novo synthesis of cholesterol (inhibiting HMGCR activity) | Grimm (2005)40 | |
| Increasing brain 27‐OHC decreases Aβ degradation | Sharman (2013)59 | |
| Increasing AICD inhibits LRP1 | Liu (2007)60 | |
| 3. Connecting sphingolipid metabolism and cholesterol metabolism models | Increasing 24‐OHC levels increases ABC transporter expression | Prasanthi (2009)31 |
| ABC transporter expression increases SM synthesis | Davis (2014)61 | |
| ABC transporter expression increases ceramide production | Davis (2014)61 | |
| Increasing 27‐OHC increases SMase activity | Dias (2014)46 | |
| Ceramide inhibits HMGCR activity (cholesterol synthesis) | Subbaiah (2008)62 |
Aβ, amyloid beta; AICD, APP intracellular domain; APP, amyloid precursor protein; HMGCR, HMG‐CoA reductase (3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase); LRP1, Low density lipoprotein receptor‐related protein 1; OHC, hydroxycholesterol; S1P, sphingosine‐1‐phosphate; SM, Sphingomyelin; SphK, Sphingosine kinase; SPT, Serine palmitoyltransferase.
Modeling pharmacological interventions
We linked the physiology model with a model for pharmacological interventions and compared model outcomes with the observed clinical and preclinical data, as described below. Two different pharmacological interventions were considered for model validation, namely, (i) BACE1 inhibitor GNE‐629 and (ii) simvastatin. Similarly, we linked the physiology model to a model for S1PR5 agonist A‐971432 in order to predict the response to this new therapy. The modeling approach used for all pharmacological interventions was to implement the pharmacokinetic (PK) model for each compound and couple the effect of changes in free concentration in blood or brain to specific mechanisms included in the physiology model.
BACE1 inhibitor GNE‐629
The PK model described by Liu et al.23 was used to evaluate the effect of GNE‐629. For model validation, it was assumed that similar changes in Aβ levels should be obtained in monkeys and humans when dosing this particular BACE1 inhibitor. The pharmacodynamic (PD) effect was introduced in the brain physiology model in a similar way as described by Liu et al.23 This effect is modeled with a maximum effect (Emax) function affecting the processing of APP through the β‐secretase cleavage. Table S7 summarizes all PK/PD parameters used.23
Simvastatin
To implement the effect of statins on the synthesis of cholesterol through the inhibition of HmgCoA, a PK model published by Kim et al.24 was used. The PD effect was applied directly on the HmgCoA for the synthesis of mevalonate in the brain compartment. The assumption is that the free concentration of simvastatin acid (the metabolite of simvastatin) will have the main inhibitory effect on HmgCoA reductase, and increasing the free concentration of the drug will decrease the production of cholesterol. Table S8 summarizes all PK/PD parameters used.24
S1PR5 agonist A‐971432
To describe the PK of A‐971432, we extrapolated PK data obtained in the rat (Figure S1). Specifically, the brain PK in the rat was described by a one‐compartment model with volume 11.2 L/kg, clearance 0.57 L/hour/kg, and absorption rate into the brain of 0.32/hour. All parameters are well‐determined with coefficient of variation of 45% or less. We assumed that the human PK can be described by scaling the clearance parameters by body weight (human body weight = 70 kg). We assumed the free fraction of the compound in the brain drives the pharmacological effect, and, therefore, multiplied the total concentration of compound in the brain by the free fraction unbound, brain = 0.005. As A‐971432 is not a substrate of the ABCB1 (P‐glycoprotein) or ABCG23 (breast cancer resistance protein) efflux transporters (data not shown), the brain penetration is likely to be mainly driven by passive diffusion, hence, not subjected to large species differences. For other S1PR5 agonists, we observed in several nonclinical studies that gene expression of transporter molecules, specifically ABCA1 (efflux), but also ABCG1 and ABCA7 (influx) were modulated (data not shown). Specifically, ABCA1 expression was increased by 20–60%. Consequently, in the model, we assumed that the efflux of Cer, S1P, SM, and 24‐OHC from brain via efflux ABC transporters is increased after dosing of A‐971432, making the assumption that the gene expression change translates into a functional change. We assumed an Emax model depending on the free brain concentration of A‐971432 with maximum change in transport via ABC transporter of 25%. The half‐maximal effective concentration was assumed to be 4.1 nM based on an in vitro assay measuring forskolin‐induced cAMP inhibition assay in S1PR5‐transfected CHO cells.20
Calibration of the brain physiology model for AD and healthy case
We aimed at describing specific quantitative differences between the brains of a 70‐year‐old healthy person and a patients with AD (of the same age), to generate specific models to simulate each respective case. We kept one model structure for both cases, and attributed the differences between healthy and AD brain to differences in specific parameter values in the brain physiology model. Some of the changes were based on literature‐reported evidence, and additional changes were introduced by calibration to available data.
Figure 3 summarizes the quantitative differences in specific mechanisms and interactions for the AD case compared to the healthy case used in this work. Changes in AD compared to healthy are related to: (i) perturbations in the sphingolipid balance increasing the production of Cer, (ii) accumulation of Aβ, and (iii) increased levels of cholesterol in the brain compartment.
Figure 3.
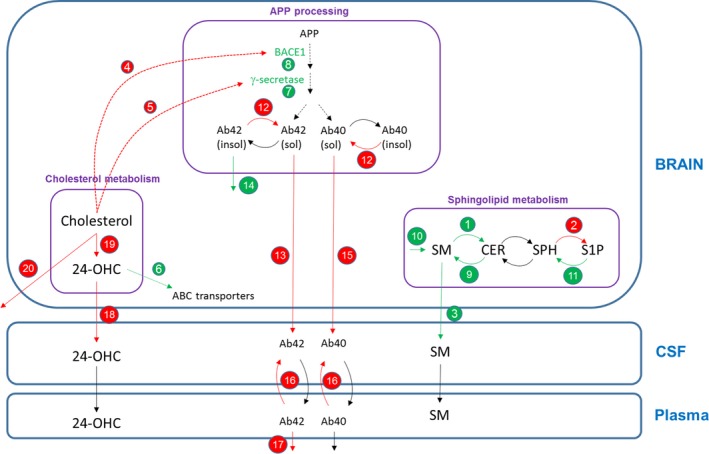
Brain physiology model diagram showing the specific mechanisms and parameter values changing in the Alzheimer disease (AD) case compared to the healthy case. (†) Changes according to evidence found in literature; (‡) changes in amyloid precursor protein (APP) processing model to match published data (as described in the next section); (¥) changes in the cholesterol model to match 24‐OHC and cholesterol levels seen in in‐house preclinical data. Red arrows/text = parameter value decreases for AD case; green arrows/text = parameter value increases for AD case. Quantitative changes in the AD case: (1) production of ceramide (CER) from SM (†),8 (2) production of S1P from SPH (†),8 (3) transfer of SM from the brain to cerebrospinal fluid (CSF) (†),8 (4) half‐maximal effective concentration (EC)50 value for the effect of brain cholesterol on BACE1 (†),30 (5) EC 50 value for the effect of brain cholesterol on γ‐secretase (†),30 (6) effect of 24‐OHC on ABC transporters (†),31 (7) γ‐secretase expression (†),30 (8) BACE1 expression (†),30 (9) production of SM from CER (†), (10) synthesis of SM (†), (11) production of SPH from sphingosine‐1‐phosphate (S1P) (†), (12) reversible reaction from insoluble to soluble Aβ (‡), (13) transfer rate of amyloid beta (Aβ)42 from the brain to CSF (‡), (14) degradation of insoluble Aβ (‡), (15) transfer rate of Aβ40 from the brain to CSF (‡), (16) transfer rate of Aβ from plasma to CSF (‡), (17) degradation of Aβ42 in plasma (‡), (18) efflux of 24‐OHC (from the brain to CSF) (¥), (19) synthesis of 24‐OHC in brain (¥), and (20) degradation of brain cholesterol (¥). SPH, Sphingosine; SM, Sphingomyelin.
Calibration of the brain physiology model
The physiology model was calibrated using baseline data of several biomarkers, predominantly lipids, and Aβ in the brain and CSF, observed in AD and healthy subjects from in‐house‐generated data and literature (Figure 4). Interestingly, for the biomarkers where we had human and nonclinical data (Figure S2), we find that similar changes in baseline values occur in AD vs. healthy subjects as in aged vs. young rodents, indicating that our model – although developed for human physiology – may allow for translational predictions. In particular, qualitatively, either elevation or suppression in a particular biomarker was typically observed consistently in humans and rodents, with the exception of SPH, which was reported slightly elevated in humans, however, we found it suppressed in aged vs. young animals in‐house. Modulation in S1P, Cer, 24‐OHC, and cholesterol in the brain were quantitatively similar between human (AD vs. healthy) and rodents (aged vs. young animals).
Figure 4.
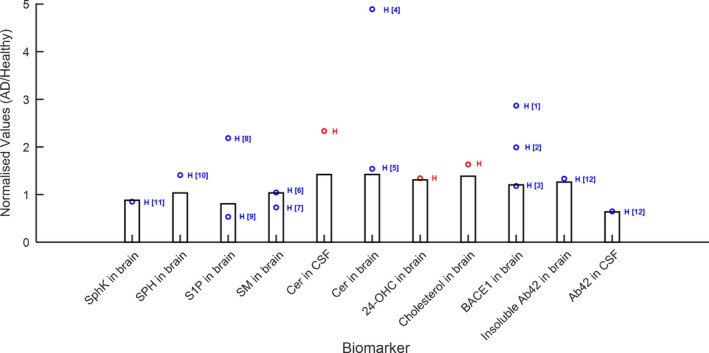
Comparison of quantitative systems pharmacology model simulations (bars represent simulation results at age = 70 years) with data from literature and in‐house studies. (a) Comparison of baseline values in various biomarkers in patients with Alzheimer disease (AD): red points: Abbvie's data; blue points: literature data; H = human data; R = rat data; M = mouse data. References: [1] and [2] = Yang 2003,32 [3] = Xiong 2008,30 [4], [8] = de Wit 2017,33 [5], [6] = Couttas 2016,34 [7], [9], [10] = He 2010,35 [11] = Tagasugi 2011,36 [12] = Mattsson 2014.37 Cer, ceramide; CSF, cerebrospinal fluid; S1P, sphingosine‐1‐phosphate; SPH, Sphingosine.
Model validation: Pharmacological interventions
In order to validate the QSP model, we implemented pharmacological interventions, as described above, and compared simulated outcomes from our calibrated model to clinical responses reported in the literature, without refitting any parameters of the physiology model. Schematic illustrations of PK models and pharmalogical interventions are shown in Figure 5a.
Figure 5.
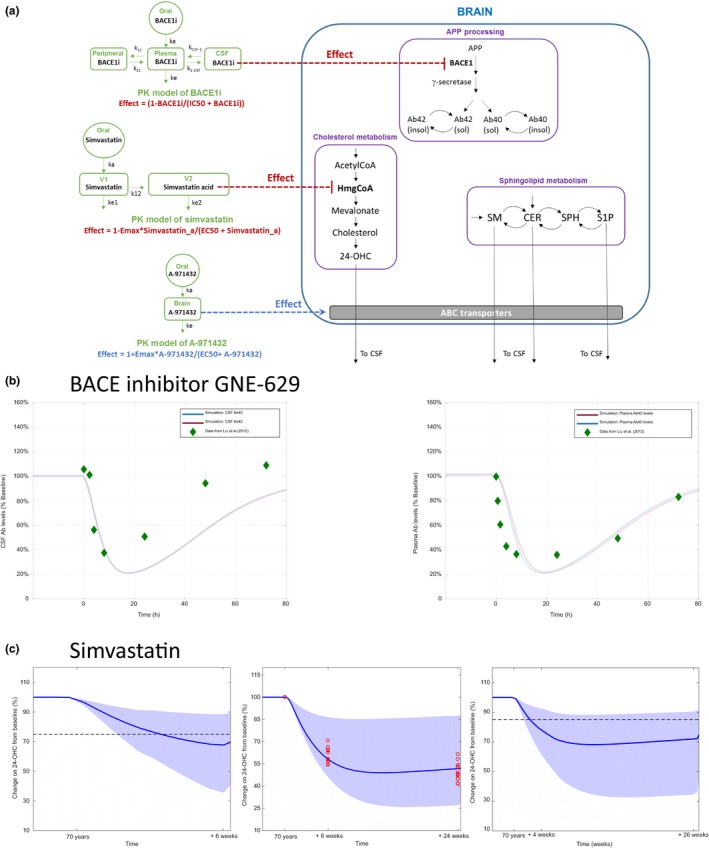
Validation of the quantitative systems pharmacology (QSP) model using published data for therapies. (a) Schematic of the implementation of pharmacokinetic (PK) models for BACEi and simvastatin and their pharmacodynamic effect on BACE1 and HmgCoA in the brain compartment. The model for BACEi is based on the model proposed by ref. 23 and the model for simvastatin is based on the model proposed by ref. 24. (b) Bace inhibitor GNE‐629 (left) changes (% from baseline) in cerebrospinal fluid (CSF) amyloid beta (Aβ40) and Aβ42 levels from QSP model simulation and comparison with data reported by ref. 23. (Right) Changes (% from baseline) in plasma Aβ40 and Aβ42 levels from QSP model simulation and comparison with data reported by ref. 23. (c) Simvastatin. (Left) Comparison of simulations results with change in plasma 24‐OHC reported by ref. 25 after 6 weeks of treatment with a dose of 40 mg q.d. The mean response of the QSP model is just under 70% of the baseline value, which is close to the mean response reported (20–25% decrease of plasma levels of 24‐OHC from baseline). (Middle) Comparison of simulations results with change in plasma 24‐OHC reported by ref. 26 after 6 and 24 weeks of treatment (red points) with a dose of 80 mg q.d. of simvastatin. The mean response of the QSP model is around 35% decrease in 24‐OHC, in good agreement with the reported clinical dynamics of plasma 24‐OHC 26 (40–60% decrease of plasma 24‐OHC compared to baseline). (Right) Comparison of simulations results with change in CSF 24‐OHC reported by ref. 27 after 4 weeks of a dose of 40 mg q.d., followed by a dose of 80 mg q.d. for 22 weeks (red points). The mean response of the QSP model is around 30% decrease in CSF 24‐OHC levels, compared to a reported slight decrease of 8–15% in CSF 24‐OHC levels.27 APP, amyloid precursor protein; Cer, ceramide; EC50, half‐maximal effective concentration; Emax, maximum effect; IC50, half‐maximal inhibitory concentration; OHC, hydroxycholesterol; S1P, sphingosine‐1‐phosphate; SM, Sphingomyelin; SPH, Sphingosine.
BACE1 inhibitor
Model simulations show that treatment with BACE1 inhibitor GNE‐629 results in very similar responses in our model for different classes of Aβ (i.e., Aβ40 and Aβ42); a good quantitative agreement of our simulated outcome is observed compared to the reported data in CSF and plasma23 (Figure 5 b).
Simvastatin
To validate the model further, three different studies, including the following published dose administration regimes were simulated: (i) 40 mg of simvastatin q.d. for 6 weeks,25 (ii) 80 mg of simvastatin q.d. for 24 weeks,26 and (iii) 40 mg of simvastatin q.d. for 4 weeks, followed by 80 mg of simvastatin q.d for 22 weeks.27 In this case, given the high variability observed in the different datasets and the fact that the data were coming from different sources, a population‐based approach for PK was used. To generate the population‐based simulations a virtual population of 100 individuals was created using the variability in PK parameters of simvastatin reported by Kim et al.24 (see Table S8). As shown in Figure 5 c, the model outcome is in good agreement with all three datasets, considering the variability in the population‐based simulations.
Model predictions of sphingolipid and Aβ modulation following treatment with S1PR5 agonists
After validating that the QSP model appropriately captures published literature data for relevant therapies, we used the model to predict pharmacological dynamics due to S1PR5 treatment with an S1PR5 agonist A‐971432.20 The model predicts a dose‐dependent reduction in Cer, as well as insoluble and soluble Aβ42 levels in the brain. The highest dose (3 mg/kg q.d.) is predicted to reduce Cer levels slightly below baseline of healthy subjects (Figure 6 a). In contrast, S1P is predicted to be unaffected by treatment with S1PR5 agonist. Interestingly, the predicted Aβ reduction in the brain is accompanied by the reduction of soluble Aβ levels in the CSF. This indicates that changes in CSF Aβ could be a clinically relevant biomarker to monitor the treatment response to an S1PR5 agonist. Changes due to S1PR5 treatment is predicted to be reversible upon cessation of treatment (data not shown).
Figure 6.
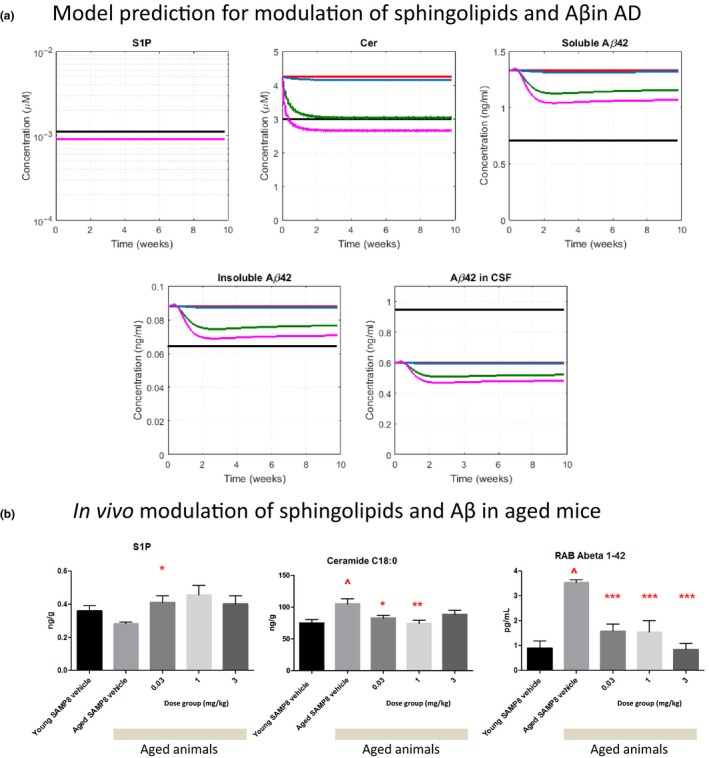
Predictions of Alzheimer disease (AD)‐relevant modulations for sphingosine‐1‐phosphate 5 receptor S1 (PR5) agonist A‐971432 using the quantitative systems pharmacology (QSP) model, and in modulation observed in vivo in an aged mouse model. (a) Predicted modulation of sphingosine‐1‐phosphate (S1P), ceramide (Cer), soluble and insoluble amyloid beta (Aβ)1‐42 in the brain, as well as soluble Aβ1‐42 in cerebrospinal fluid (CSF), after once daily dosing for 10 weeks (Color code: black: healthy BL, red: AD baseline, blue: 0.03 mg/kg, green: 1 mg/kg, magenta: 3 mg/kg). Note that for S1P all simulation curves for AD lie on top of each other. (b) Results for S1P, Cer, and soluble Aβ1‐42 (RAB fraction) in the brains of young and aged SAMP8 mice treated with vehicle or different oral doses of A‐971432 once daily for 10 weeks. Bars represent means and error bars indicate the standard errors of mean. Significant differences are indicated: ^ P < 0.05 in comparison with young vehicle treated animals; * P < 0.05, ** P < 0.01, in comparison with aged vehicle treated animals.
Observed in vivo modulation of sphingolipids and Aβ following treatment with S1PR5 agonists in aged mice
As shown in Figure S2, the modulation in various sphingolipids and cholesterol observed in aged compared with young rodents are qualitatively similar to those observed in patients with AD compared with healthy controls. This may indicate that treatment responses translate across different species. Hence, we aimed to test whether our S1PR5 agonist A‐971432 would modulate sphingolipids in the brain, as well as Aβ, in an aged mouse model. We performed an in vivo study treating aged SAMP8 mice, which have been proposed as a nonclinical animal model for AD28 with A‐971432, as described above. Comparing aged vs. young vehicle‐treated animals, we observed that Cer and Aβ42 levels in the brain were significantly increased in aged mice, whereas S1P levels were not significantly different (Figure 6 b). When treating with A‐971432 (0.03–3 mg/kg q.d., 10 weeks), these age‐related changes in Cer and Aβ42 were reversed in aged animals toward baseline values of young animals. Overall, these preclinical data are in good agreement with predictions of our QSP model for patients with AD and give additional experimental evidence supporting a relevant treatment effect of S1PR5 agonism.
Discussion
In this article, we describe a QSP modeling approach that integrates several interconnected biological pathways, and their dysregulation in AD. Specifically, we include mechanisms related to (i) APP processing and Aβ aggregation, (ii) cholesterol metabolism, and (iii) sphingolipid metabolism. These three submodels were interconnected based on described molecular interactions in order to create a holistic approach for the relevant biology considered in the QSP model (Figure 2). By embedding the description of brain pathobiology into a physiological context, our model enables an enquiry into drug dose and regimen for AD. Specifically, we were interested in the response of various dysregulated lipids in the brain, as well as Aβ as a relevant disease marker in AD, to sphingosine receptor agonists. We used published quantitative models for specific biological pathways from the literature as the basis for our model development.7, 8, 9, 10 The model structure was then carefully altered and amended for additional interactions between different pathways to reflect our understanding of the pathobiology. Parameters of the complete model were recalibrated where necessary to reflect baseline values in a multitude of biomarkers in healthy subjects and patients with AD.
A key assumption of our modeling approach is that healthy individuals and patients with AD can be described by the same model structure, and that the differences between both can be ascribed to changes in parameter values (Figure 3) and the model captures baseline data in healthy and patients with AD in a range of biomarkers (Figure 4). The model was able to accurately predict the time course of response of plasma and CSF Aβ concentration to BACEi (GNE‐629) and simvastatin (Figure 5). Due to the way clinical data is collected there is a limitation to the degree of validation possible, in particular for the interactions between processes described by the submodels. Typically, data collected in clinical trials is related to one of the individual submodels (i.e., in the “molecular vicinity” of the proposed mechanism of action). For instance, the available BACEi data can be used to validate the outputs of the APP processing model as the observed molecular species are close to the pharmacological intervention. Similarly, simvastatin data can be used to predominantly validate the outputs of the cholesterol metabolism model (e.g., 24‐OHC levels). However, typically, data has not been collected for BACEi or simvastatin, which would enable evaluating their effect on other processes not directly related to APP processing or cholesterol metabolism, respectively. It would be beneficial to collect new pharmacological data (e.g., PK/PD), where the effect of a given compound is explored in different submodels, in order to further validate the proposed interactions considered in this QSP approach.
Nevertheless, we are confident that we validated the model as best as possible based on the available published and in‐house data. Therefore, we used the model to evaluate the predicted impact of S1PR5 agonists as potential novel treatment for AD. The hypothesis was that S1PR5 agonists would reverse the lipid imbalance associated with neurodegeneration. Our model predicts that a meaningful reduction of Cer in the brain to the baseline levels of healthy subjects can be achieved via this mechanism with drug candidates with appropriate pharmacological and PK properties (Figure 6). Furthermore, our model predicts that insoluble Aβ in the brain is reduced in patients with AD after treatment with S1PR5 agonists, and that those changes can be monitored by associated changes in CSF Aβ (Figure 6).
Based on the similar modulation of baseline concentrations of several lipids in aged vs. young rodents and patients with AD vs. healthy subjects, the relevant biology to test S1PR5 agonism on lipid dysregulation may be conserved across species. In support, similar to our predictions for the AD case, an in vivo nonclinical study with aged mice showed that Cer can be controlled by S1PR5 activation. Furthermore, modulation of Aβ in the brain was observed following treatment with A‐971432, giving additional evidence for a relevant treatment effect in response to S1PR5 agonism.
In order to identify key controlling species and parameters within the model, we carried out a local sensitivity analysis of the model (Supplementary Information S1). We found that the brain Cer concentration was most dependent upon Cer synthase activity. Hence, this may be another target of interest to control Cer in the brain. Key controlling species and parameters for insoluble Aβ42 in the brain are not surprisingly mainly related to APP processing. However, several key processes modulating cholesterol metabolism, S1P and Cer, are predicted to have significant impact on insoluble Aβ42, with more pronounced effects in AD compared with healthy subjects. These may constitute further potential drug targets against lipid dysregulation in AD.
Here, we constrained the model scope according to our questions, as well as available knowledge and data for lipid metabolism and its relation to Aβ. For example, transport processes are expressed as saturating steady state equations to simplify parameter estimation. However, it may be that further granularity is needed to fully describe the behavior most appropriately using reactant concentrations and rate constants. In addition, further biology, which may be of importance to AD, or which may be connected to our current queries in an unknown way is not yet included. However, our model presents a framework that is easily extendable by other biological mechanisms relevant to AD and aging, such as transient binding kinetic information, tau (patho)biology, physiological changes in brain volume and neuroinflammation, or new interactions as data become available.
Funding
No funding was received for this work.
Conflict of Interest
D.C., A.L.R., J.v.B., L.L., and M.N. are employees of AbbVie. E.v.d.K. was an AbbVie employee at the time when the work reported in the manuscript was performed. C.P.‐A. and N.B. are employees of Certara QSP. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Author Contributions
D.C., C.P.‐A., E.vdK., L.L., N.B., and M.N. wrote the manuscript. D.C., C.P.‐A., A.L.R., J.vB., E.vdK., L.L., N.B., and M.N. designed the research. D.C., C.P.‐.A., A.L.R., J.vB., E.vdK., and N.B. performed the research. D.C., C.P.‐A., and N.B. analyzed the data.
Supporting information
Supplementary Information S1. Tables and Figures, as well as additional information on model components and analyses.
Acknowledgments
AbbVie's rodent tissue (brain, plasma, and CSF) was prepared by Juliane Stein and Susanne Jausel in Ana Lucia Relo's group. AbbVie's lipid data (rodent and human) was generated by Khader Awwad and Michael Schulz. Rat PK data for A‐971432 was generated by Mario Mezler's laboratory. The in vivo experiment in SAMP8 mice was performed by St. Louis University.
References
- 1. Doody, R.S. et al Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N. Engl. J. Med. 370, 311–321 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Prince, M. et al World Alzheimer Report 2015: The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Costs and Trends (Alzheimer's Disease International, London, 2015). [Google Scholar]
- 3. Benson, N. et al Reducing systems biology to practice in pharmaceutical company research; selected case studies. Adv. Exp. Med. Biol. 736, 607–615 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Benson, N. & van der Graaf, P.H. The rise of systems pharmacology in drug discovery and development. Future Med. Chem. 6, 1731–1734 (2014). [DOI] [PubMed] [Google Scholar]
- 5. van der Graaf, P.H. & Benson, N. Systems pharmacology: bridging systems biology and pharmacokinetics‐pharmacodynamics (PKPD) in drug discovery and development. Pharm. Res. 28, 1460–1464 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Mizuno, S. et al AlzPathway: a comprehensive map of signaling pathways of Alzheimer's disease. BMC Syst. Biol. 6, 52 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kyrtsos, C . Of Mice and Math: A Systems Biology Model for Alzheimer's Disease (University of Maryland, College Park, MD, 2011). [Google Scholar]
- 8. Wronowska, W. et al Computational modeling of sphingolipid metabolism. BMC Syst. Biol. 9, 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Potter, R. et al Increased in vivo amyloid‐beta42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 5, 189ra77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ortega, F. et al Interplay between alpha‐, beta‐, and gamma‐secretases determines biphasic amyloid‐beta protein level in the presence of a gamma‐secretase inhibitor. J. Biol. Chem. 288, 785–792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rogers, J.A. et al Combining patient‐level and summary‐level data for Alzheimer's disease modeling and simulation: a beta regression meta‐analysis. J. Pharmacokinet. Pharmacodyn. 39, 479–498 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Delor, I. et al Modeling Alzheimer's disease progression using disease onset time and disease trajectory concepts applied to CDR‐SOB scores from ADNI. CPT Pharmacometrics Syst. Pharmacol. 2, e78 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grimm, M.O. et al The impact of cholesterol, DHA, and sphingolipids on Alzheimer's disease. Biomed. Res. Int. 2013, 814390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosicek, M. & Hecimovic, S. Phospholipids and Alzheimer's disease: alterations, mechanisms and potential biomarkers. Int. J. Mol. Sci. 14, 1310–1322 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aikawa, T. , Holm, M.L. & Kanekiyo, T. ABCA7 and pathogenic pathways of Alzheimer's disease. Brain Sci. 8, 2–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katzov, H. et al Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta‐amyloid metabolism. Hum. Mutat. 23, 358–367 (2004). [DOI] [PubMed] [Google Scholar]
- 17. Kirchgessner, T.G. et al Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. 24, 223–233 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Shepardson, N.E. , Shankar, G.M. & Selkoe, D.J. Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch. Neurol. 68, 1385–1392 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith, K.B. et al The effect of statins on rate of cognitive decline in mild cognitive impairment. Alzheimers Dement. (NY) 3, 149–156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hobson, A.D. et al Discovery of A‐971432, an orally bioavailable selective sphingosine‐1‐phosphate receptor 5 (S1P5) agonist for the potential treatment of neurodegenerative disorders. J. Med. Chem. 58, 9154–9170 (2015). [DOI] [PubMed] [Google Scholar]
- 21. Lutjohann, D. et al Cholesterol homeostasis in human brain: evidence for an age‐dependent flux of 24S‐hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 93, 9799–9804 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Liempd, S. et al Development and validation of a higher‐throughput equilibrium dialysis assay for plasma protein binding. J. Lab. Autom. 16, 56–67 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Liu, X. et al Mechanistic pharmacokinetic‐pharmacodynamic modeling of BACE1 inhibition in monkeys: development of a predictive model for amyloid precursor protein processing. Drug Metab. Dispos. 41, 1319–1328 (2013). [DOI] [PubMed] [Google Scholar]
- 24. Kim, J. et al A population pharmacokinetic‐pharmacodynamic model for simvastatin that predicts low‐density lipoprotein‐cholesterol reduction in patients with primary hyperlipidaemia: simvastatin pharmacokinetic‐pharmacodynamics. Basic Clin. Pharmacol. Toxicol. 109, 156–163 (2011). [DOI] [PubMed] [Google Scholar]
- 25. Vega, G.L. et al Reduction in levels of 24S‐hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch. Neurol. 60, 510 (2003). [DOI] [PubMed] [Google Scholar]
- 26. Locatelli, S. et al Reduction of plasma 24S‐hydroxycholesterol (cerebrosterol) levels using high‐dosage simvastatin in patients with hypercholesterolemia: evidence that simvastatin affects cholesterol metabolism in the human brain. Arch. Neurol. 59, 213–216 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Simons, M. et al Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26‐week randomized, placebo‐controlled, double‐blind trial: simvastatin in Alzheimer's. Ann. Neurol. 52, 346–350 (2002). [DOI] [PubMed] [Google Scholar]
- 28. Akiguchi, I. et al SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda's legacy and future directions. Neuropathology 37, 293–305 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Russell, D.W. et al Cholesterol 24‐hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78, 1017–1040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong, H. et al Cholesterol retention in Alzheimer's brain is responsible for high beta‐ and gamma‐secretase activities and Abeta production. Neurobiol. Dis. 29, 422–437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prasanthi, J.R. et al Differential effects of 24‐hydroxycholesterol and 27‐hydroxycholesterol on beta‐amyloid precursor protein levels and processing in human neuroblastoma SH‐SY5Y cells. Mol. Neurodegener. 4, 1 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang, L.B. et al Elevated beta‐secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4 (2003). [DOI] [PubMed] [Google Scholar]
- 33. de Wit, N.M. et al Altered sphingolipid balance in capillary cerebral amyloid angiopathy. J. Alzheimers Dis. 60, 795–807 (2017). [DOI] [PubMed] [Google Scholar]
- 34. Couttas, T.A. et al Loss of ceramide synthase 2 activity, necessary for myelin biosynthesis, precedes tau pathology in the cortical pathogenesis of Alzheimer's disease. Neurobiol. Aging 43, 89–100 (2016). [DOI] [PubMed] [Google Scholar]
- 35. He, X. et al Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol. Aging 31, 398–408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takasugi, N. et al BACE1 activity is modulated by cell‐associated sphingosine‐1‐phosphate. J. Neurosci. 31, 6850–6857 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mattsson, N. et al Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer's disease. Ann. Clin. Transl. Neurol. 1, 534–543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanabe, F. , Nakajima, T. & Ito, M. The thiol proteinase inhibitor E‐64‐d ameliorates amyloid‐β‐induced reduction of sAPPα secretion by reversing ceramide‐induced protein kinase C down‐regulation in SH‐SY5Y neuroblastoma cells. Biochem. Biophys. Res. Comm. 441, 256–261 (2013). [DOI] [PubMed] [Google Scholar]
- 39. Geekiyanage, H. & Chan, C. MicroRNA‐137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer's disease. J. Neurosci. 31, 14820–14830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grimm, M.O.W. , et al Regulation of cholesterol and sphingomyelin metabolism by amyloid‐beta and presenilin. Nat. Cell Biol. 7, 1118–1123 (2005). [DOI] [PubMed] [Google Scholar]
- 41. Puglielli, L. , et al Ceramide Stabilizes β‐Site Amyloid Precursor Protein‐cleaving Enzyme 1 and Promotes Amyloid β‐Peptide Biogenesis. J. Biol. Chem. 278, 19777–19783 (2003). [DOI] [PubMed] [Google Scholar]
- 42. Jesko, H. , et al Sphingosine kinases modulate the secretion of amyloid β precursor protein from SH‐SY5Y neuroblastoma cells: the role of α‐synuclein. Folia Neuropathol. 52, 70–78 (2014). [DOI] [PubMed] [Google Scholar]
- 43. Grimm, M.O.W. , et al Intracellular APP Domain Regulates Serine‐Palmitoyl‐CoA Transferase Expression and Is Affected in Alzheimer's Disease. Int. J. Alzheimer's Dis. 2011, 695413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gassowska, M. , et al Sphingosine kinases/sphingosine‐1‐phosphate and death Signalling in APP‐transfected cells. Neurochem. Res. 39, 645–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Malaplate‐Armand, C. , et al Soluble oligomers of amyloid‐beta peptide induce neuronal apoptosis by activating a cPLA2‐dependent sphingomyelinase‐ceramide pathway. Neurobiol. Dis. 23, 178–189 (2006). [DOI] [PubMed] [Google Scholar]
- 46. Dias, I.H.K. , et al Oxidized LDL lipids increase β‐amyloid production by SH‐SY5Y cells through glutathione depletion and lipid raft formation. Free Radic. Biol. Med. 75, 48–59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marwarha, G. , et al Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27‐hydroxycholesterol in rabbit organotypic slices. J. Alzheimer's Dis. 19, 1007–1019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marwarha, G. , et al Gadd153 and NF‐κB crosstalk regulates 27‐hydroxycholesterol‐induced increase in BACE1 and β‐amyloid production in human neuroblastoma SH‐SY5Y cells. PLoS One 8, e70773 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49. Prasanthi, J.R.P. , et al Deferiprone reduces amyloid‐β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol‐enriched diet. J. Alzheimer's Dis. 30, 167–182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Famer, D. , et al Regulation of alpha‐ and beta‐secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha‐secretase pathway. Biochem. Biophys. Res. Comm. 359, 46–50 (2007). [DOI] [PubMed] [Google Scholar]
- 51. Grimm, M.O.W. , et al Independent inhibition of Alzheimer disease beta‐ and gamma‐secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 283, 11302–11311 (2008). [DOI] [PubMed] [Google Scholar]
- 52. von Arnim, C.A.F. , et al Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem. Biophys. Res. Comm. 370, 207–212 (2008). [DOI] [PubMed] [Google Scholar]
- 53. Zhu, F. , et al Decrease in the production of β‐amyloid by berberine inhibition of the expression of β‐secretase in HEK293 cells. BMC Neurosci. 12, 125 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malnar, M. , et al Cholesterol‐depletion corrects APP and BACE1 misstrafficking in NPC1‐deficient cells. Biochem. Biophys. Acta. 1822, 1270–1283 (2012). [DOI] [PubMed] [Google Scholar]
- 55. Thirumangalakudi, L. , et al High cholesterol‐induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 106, 475–485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cui, W. , et al Activation of liver X receptor decreases BACE1 expression and activity by reducing membrane cholesterol levels. Neurochem. Res. 36, 1910–1921 (2011). [DOI] [PubMed] [Google Scholar]
- 57. Cole, S.L. , et al Statins cause intracellular accumulation of amyloid precursor protein, beta‐secretase‐cleaved fragments, and amyloid beta‐peptide via an isoprenoid‐dependent mechanism. J. Biol. Chem. 280, 18755–18770 (2005). [DOI] [PubMed] [Google Scholar]
- 58. Fewlass, D.C. , et al Obesity‐related leptin regulates Alzheimer's Abeta. FASEB J. 18, 1870–1878 (2004). [DOI] [PubMed] [Google Scholar]
- 59. Sharman, M.J. , et al The Guinea Pig as a Model for Sporadic Alzheimer's Disease (AD): The Impact of Cholesterol Intake on Expression of AD‐Related Genes. PLoS ONE 8, e66235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu, Q. , et al Amyloid Precursor Protein Regulates Brain Apolipoprotein E and Cholesterol Metabolism through Lipoprotein Receptor LRP1. Neuron 56, 66–78 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davis, W. The ATP‐binding cassette transporter‐2 (ABCA2) regulates esterification of plasma membrane cholesterol by modulation of sphingolipid metabolism. Biochem. Biophys. Acta. 1841, 168–179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Subbaiah, P.V. , Sowa, J.M. & Singh, D.K. Sphingolipids and cellular cholesterol homeostasis. Effect of ceramide on cholesterol trafficking and HMG CoA reductase activity. Arch. Biochem. Biophys. 474, 32–38 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information S1. Tables and Figures, as well as additional information on model components and analyses.