

Abstract
Diazepam is labeled for status epilepticus (SE) in children, but there are limited data characterizing its disposition in pediatric patients. We developed a population pharmacokinetic (PK) model of i.v. diazepam in children with SE. We evaluated relationships between PK parameters and both safety and efficacy, and simulated exposures using dosing regimens from the product label and clinical practice. The model was developed using prospective data from a pediatric clinical trial comparing diazepam to lorazepam for treatment of SE. Altogether, 87 patients aged ≥ 3 months to < 18 years contributed 162 diazepam concentrations. Diazepam PKs were well characterized by a two‐compartment model scaled by body size. No significant or clinically important relationships were observed between diazepam PKs and safety or efficacy. Simulations demonstrated that, compared with label dosing, the study dose (0.2 mg/kg i.v., maximum 8 mg) resulted in greater frequency in rapidly achieving the target therapeutic range of 200–600 ng/mL.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Diazepam is approved to treat status epilepticus in children, but data on its disposition in pediatric patients are limited.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study aimed to characterize diazepam PKs, safety, and efficacy and to evaluate different dosing regimens in children with SE.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The PKs of diazepam was well characterized by a two‐compartment model scaling for body size using weight. Simulations demonstrated that doses recommended by expert opinion achieved therapeutic concentrations more frequently than doses recommended by the product label after a single i.v. dose.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
An initial weight‐based i.v. diazepam dose may be preferred over the product label recommended fixed dose in order to achieve adequate exposure when treating children with SE.
Status epilepticus (SE) is a medical emergency characterized by prolonged or repeated seizures.1 In developed countries, 17–23 per 100,000 children per year will experience an episode of SE.2 Mortality from uncontrolled SE can be as high as 8%, morbidity as high as 34%, and among survivors long‐term consequences include both epilepsy as well as permanent motor deficits, learning difficulties, and behavior problems.3 Because prolonged SE is associated with greater risk for neuronal injury and difficulty in controlling future seizure activity, timely and effective management is critical.4
One of the most widely used antiepileptic drugs for the acute management of SE in pediatric patients is the benzodiazepine diazepam.1 Although diazepam is efficacious in the treatment of pediatric SE, there are limited data characterizing the pharmacokinetics (PKs) of diazepam in pediatric patients.5, 6 Identifying the optimal dose of diazepam for pediatric SE is crucial as rapid reversal of seizures is essential, and benzodiazepines may cause serious side effects, most importantly respiratory depression and respiratory failure.1, 7 Diazepam undergoes cytochrome P450 (CYP)‐dependent hepatic metabolism where it is converted by demethylation and hydroxylation to its active metabolite N‐desmethyldiazepam.8, 9, 10 Diazepam's metabolism is predominantly dependent on the enzymes CYP3A4 and CYP2C19, both of which have low expression at birth and gradually increase with age.8, 11, 12 Additionally, infants demonstrate reduced clearance of diazepam compared with adults, which is attributed to their decreased capacity to hydroxylate diazepam.8, 13 Due to these potential differences in diazepam metabolism between pediatric and adult patients, clinical PK studies characterizing the disposition of diazepam in pediatric patients with SE may result in optimized dosing and improved outcomes of pediatric SE.
Currently, diazepam is approved for pediatric use by the US Food and Drug Administration (FDA) for the treatment of SE in children older than 1 month.7 However, the FDA‐approved product label recommends a range of flat doses varying by age, which contrasts with weight‐based dosing that is recommended by expert opinion in published guidelines and used commonly in clinical practice.7, 14 To date, there have been no published studies comparing diazepam exposure resulting from product label recommended doses to standard of care dosing regimens in pediatrics subjects with SE. We leveraged previously unpublished PK data from a clinical trial of pediatric SE to characterize the PKs of diazepam in children.6 The objectives of this study were as follows: (i) develop a population PK model for diazepam in infants and children ages ≥ 3 months to < 18 years of age; (ii) evaluate the exposure‐response and exposure‐toxicity relationships of i.v. diazepam in children treated with weight‐based dosing with SE; and (iii) simulate and compare exposures resulting from product label flat age‐based dosing vs. clinical practice weight‐based dosing regimens.
Methods
Study design
PK samples providing diazepam exposure data for this analysis were obtained prospectively in a multicenter, double‐blind, randomized clinical trial comparing the efficacy and safety of i.v. lorazepam to i.v. diazepam in children treated in the emergency department (ED) for generalized convulsive SE (NCT00621478, IND #79,010), described previously.6 Children ages ≥ 3 months to < 18 years from 11 large academic pediatric hospitals in the United States were eligible if they presented to the ED with generalized tonic‐clonic SE. Patients were excluded for the following reasons: known pregnancy; hypotensive shock; significant cardiac dysrhythmia; need for emergent surgery and general anesthesia; known contraindication to benzodiazepine use; or benzodiazepine use within 7 days of presentation.
Patients were enrolled using the Exception from Informed Consent for Emergency Research, 21 CFR 50.24, which allows emergency research without prior consent under limited conditions given additional protections for maximizing the well‐being of patients.15 After patients were stabilized, written informed consent was obtained for continued participation. This study design was approved by the FDA, the study sponsor (Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)), an external expert ethics advisory panel, and was conducted in accordance with the ethical standards of the institutional review boards of all participating hospitals and with the Helsinki Declaration.6
Patients randomized in the clinical trial to receive diazepam received an initial i.v. diazepam dose of 0.2 mg/kg (maximum 8 mg) by slow push over 1 minute. Patients who continued to have convulsions received a second dose of 0.1 mg/kg (maximum 4 mg). Rescue treatment with phenytoin, fosphenytoin, or phenobarbital was permitted at 12 minutes in patients with refractory SE. Maintenance anticonvulsant medications were started at 15 minutes and 20 minutes after the first study dose for patients who had cessation of convulsions after the first and second study doses, respectively.6 This study used sparse PK sampling, which included up to 3 samples within 48 hours after the first diazepam dose. Further information regarding the PK sampling and bioanalytical assay for diazepam is included in Supplementary Information S1.
Population PK analysis
PK data were analyzed with a nonlinear mixed effects modeling approach using the software NONMEM version 7.2 (Icon Development Solutions, Ellicott City, MD). Run management was performed using Pirana version 2.8.1.16 Visual predictive checks (VPCs) and bootstrap methods were performed with Perl‐speaks‐NONMEM version 3.6.2.17 Data manipulation and visualization were performed using Stata 13.1 (StataCorp LP, College Station, TX), R version 3.0.2 (R Foundation for Statistical Computing, Vienna, Austria), and RStudio version 0.97.551 (RStudio, Boston, MA), with the packages lattice, Xpose, and ggplot2 used for data visualization.18, 19, 20 The first‐order conditional estimation method with interaction was used for all model runs.
Both one‐compartment and two‐compartment structural PK models were evaluated, and interindividual variability (IIV) was assessed for PK parameters using an exponential model. Proportional and combined (proportional plus additive) residual error models were evaluated. An allometric scale based on total body weight (WT) was used, and a single exponential value was assumed for all clearance terms and a different single exponential value was assumed for all volume terms. The effect of fixing these exponents to standard literature values as well as estimating them was evaluated.21, 22
The potential effects of clinical covariates on PK parameters were evaluated if a relationship was suggested by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the ETA plots of the IIVs for the PK parameters against the following covariates: age, blood urea nitrogen, serum creatinine (SCR), alanine transaminase (ALT), aspartate transaminase (AST), and hematocrit, sex, race, and ethnicity. Continuous covariates, except for WT, were normalized to the population median and evaluated using both linear and power relationships. Covariate testing was performed using a forward inclusion with backward elimination approach. During the forward inclusion process, covariates that reduced the objective function value by > 3.84 (P < 0.05) were retained for the subsequent multivariable analyses. Retention of the covariate in the final model occurred only if removal of the covariate reduced the objective function value by > 6.63 (P < 0.01). Missing covariate values were imputed using the last value carried forward. When no values were available, missing covariate values were imputed using the study population median value.
Model evaluation
Successful minimization, goodness‐of‐fit plots, precision of parameter estimates, bootstrap procedures, and VPCs were used to evaluate model appropriateness. Nonparametric bootstrapping was used to evaluate the precision of final PK parameter estimates using 1,000 replicates to generate the 95% confidence intervals. A VPC was performed using the final model by generating 1,000 Monte Carlo simulation replicates and quantifying the number of observed study concentrations outside the 90% prediction interval.
Assessment of relationships between PK parameters and efficacy or safety end points
Both Wilcoxon rank‐sum tests and multivariable logistic regression was performed to assess the relationship between PK parameters and both efficacy and safety. Further information regarding these comparisons is provided in Supplementary Information S1.
Assessment of dose‐exposure relationship
The final model was used to explore the dose‐exposure relationship. A target therapeutic range of 200–600 ng/mL was used as the surrogate pharmacodynamic end points based on literature‐suggested therapeutic range for diazepam.11, 23, 24, 25 The individual empirical Bayesian estimates generated using the post hoc subroutine from the final model and doses used in the study were applied to predict diazepam concentrations at 10 minutes after a single dose of 0.2 mg/kg (maximum 8 mg). Dose‐exposure relationships were also evaluated using the product labeled dosing, which recommends fixed i.v. doses of (i) 0.2–0.5 mg every 2–5 minutes up to a maximum of 5 mg in infants over 30 days of age and children under 5 years, and (ii) 1 mg every 2–5 minutes up to a maximum of 10 mg in children 5 years or older.
Monte Carlo simulations were used to simulate diazepam concentrations for virtual patients using identical demographics data from all patients in the original clinical trial, including patients originally randomized to the lorazepam study arm. A total of 306 virtual patients were simulated to match the exact number of subjects from the clinical trial. For simulation of the study dosing regimen, a single i.v. dose of 0.2 mg/kg (maximum 8 mg) was simulated. For the product label dosing, simulations of the product label dosing regimens were performed for single i.v. doses of both minimum and maximum amounts recommended per dose. The percentage of simulated patients with concentrations between 200 and 600 ng/mL at 10 minutes after a single dose were calculated for both dosing regimens. Additionally, a multiple‐dose evaluation was performed by simulating repeated doses for the study, product label high, and product label low dosing regimens. In these simulations, repeated doses were simulated for each subject until either the simulated maximum concentration (Cmax) ≥ 200 ng/mL or a time of 10 minutes after the first dose was reached. A timeframe of 10 minutes was chosen as most clinicians would switch to a rescue antiepileptic beyond this window, and also to align with the primary efficacy end point from the original clinical trial.6 The percent of subjects with predicted Cmax ≥ 200 ng/mL, number of predicted doses per subject, and cumulative predicted absolute diazepam dose were calculated and compared across regimens.
Results
Study population and PK specimens
Of 162 patients receiving diazepam, 75 were excluded from the population PK analysis: 60 patients did not have any PK samples collected, 14 patients were treated with diazepam prior to arrival in the ED, and 1 patient did not have any measurable diazepam concentrations. In the remaining 87 patients, the median (range) patient age was 3.9 years (0.4–17.8). The majority of patients had values (median (range)) within the normal range for SCR (0.4 mg/dL (0.2–6.0)), ALT (26 U/L (6–429)), and AST (42 U/L (17–1063)). Of 183 measured concentrations from these 87 patients, 15 (8%) were below the quantifiable limit and were excluded. An additional 6 (3%) concentrations were excluded based on visual inspection suggesting error in sampling collection and/or timing of sample collection. Therefore, the final population PK analysis included 87 patients with 162 concentrations (Table 1). The median (range) number of samples per patient was 2 (1–4). The median (range) first diazepam dose was 0.20 mg/kg (0.09–0.41); a total of 24 patients (28%) received a second dose, and the median (range) second diazepam dose was 0.10 mg/kg (0.083–0.50). The lower limit on this range is a result of a single obese subject who received the maximum absolute dose of diazepam. Two subjects received higher weight‐based doses than the study protocol called for, and the reason for this is unknown to us.
Table 1.
Baseline clinical data for children treated with diazepam (N = 87)
| Age (years) | 3.9 (0.4–17.8) |
| Weight (kg) | 15 (5–89) |
| BUN (mg/dL) | 10 (3–51) |
| SCR (mg/dL) | 0.4 (0.2–6.0) |
| ALT (U/L) | 26 (6–429) |
| AST (U/L) | 42 (17–1063) |
| Hematocrit (%) | 36.4 (29.4–47.2) |
| Age group | – |
| 3 months to < 3 years | 39 (45) |
| 3 years to < 13 years | 38 (44) |
| 13–18 years | 10 (11) |
| Male | 45 (52) |
| White race | 49 (57) |
| Hispanic ethnicity | 33 (38) |
Values are medians (range) for continuous variables and number (%) for categorical variables calculated based on values at the time of first study dose.
ALT, alanine aminotransferase; AST aspartate aminotransferase; BUN, blood urea nitrogen; SCR, serum creatinine.
Population PK model development
A two‐compartment model and a proportional residual error model achieved an improved fit of observed concentrations compared to a one‐compartment model (difference in objective function value (ΔOFV = −32)). Scaling of central compartment clearance (CL), central compartment volume of distribution (V1), intercompartmental clearance (Q), and peripheral compartment volume of distribution (V2) parameters by WT using a fixed exponent allometric relationship (CL, Q = 0.75; V1, V2 = 1) significantly improved model fit (ΔOFV = −81). Estimation of the exponents for the allometric scale of both clearance and volume parameter was also performed; however, this resulted in a significant increase in the degree of shrinkage in the IIV of clearance (from 28.1% to 99.7%), less reliable random effect estimates, and no improvement in the diagnostics plots or parameter estimates. Therefore, the remainder of the model development used these fixed exponents, which is in line with previous literature.26
After scaling for body size based on a 70 kg standardized adult weight, the univariate model‐building process showed that SCR, ALT, and AST on CL and ALT and AST on V1 produced significant reductions in the OFV. However, these changes in OFV were driven by three patients with outlier SCR, ALT, and AST values. Furthermore, the magnitude of each of these covariates was relatively low (estimate for linear covariate relationship < 0.2), and although these produced a statistically significant reduction in the OFV, inclusion of these covariates led to a minimal reduction in the IIV of model parameters and residual variability (< 10% coefficient of variation). Therefore, three outlier patients were excluded from the covariate analysis. When these patients were excluded, the reductions in OFV no longer reached significance (Table S1). The base model with inclusion of WT on all parameters was selected as the final model (Table 2).
Table 2.
Final model parameter estimates and bootstrap results
| Parameter | Final model | Bootstrap (n = 1,000)a | ||||
|---|---|---|---|---|---|---|
| Estimate | RSE (%) | Shrinkage (%) | 2.5th percentile | Median | 97.5th percentile | |
| Structural modelb | ||||||
| CL70KG (L/hour) | 2.36 | 8 | — | 1.84 | 2.26 | 2.69 |
| V170KG (L) | 42 | 19 | — | 22.5 | 41.9 | 66.1 |
| Q70KG (L/hour) | 22.6 | 24 | — | 15.8 | 23.8 | 39.8 |
| V270KG (L) | 56.5 | 12 | — | 41.2 | 57.2 | 72.1 |
| Variance modelc | ||||||
| IIV (CL) | 0.249 | 51 | 28.1 | 0.0266 | 0.206 | 0.431 |
| IIV (V1) | 1.31 | 30 | 32.7 | 0.0594 | 1.21 | 2.20 |
| IIV (Q) | 0 (FIX) | — | — | — | — | — |
| IIV (V2) | 0 (FIX) | — | — | — | — | — |
| Proportional error | 0.132 | 32 | 19.0 | 0.0735 | 0.136 | 0.255 |
CL70KG, population clearance estimate scaled to a 70‐kg adult; IIV, interindividual variability; Q70KG, population intercompartmental clearance estimate scaled to a 70‐kg adult; V170KG, population central volume of distribution estimate scaled to a 70‐kg adult; V270KG, population peripheral volume of distribution estimate scaled to a 70‐kg adult; RSE, relative standard error.aA total of 862 (86.2%) runs successfully minimized and 706 (70.6%) runs completed the covariance step. bFor the final model, the typical values for CL, V1, Q, and V2 could be expressed as the following equations: CL = 2.36*(WT/70) 0.75; V1 = 42*(weight (WT)/70); Q = 22.6*(WT/70) 0.75; and V2 = 56.5*(WT/70).cIIV terms are shown as variance; IIV (CL), interindividual variability in drug clearance; IIV (V1), interindividual variability in central volume of distribution; IIV (Q), interindividual variability in intercompartmental drug clearance; IIV (V2), interindividual variability in peripheral volume of distribution.
A proportional residual error model was used to estimate the unexplained residual error. A combined proportional plus additive residual error model did not improve model fit. Estimation of the IIV for Q and V2 resulted in high shrinkage values (> 50%) for CL, V1, and Q parameter estimates. Therefore, IIV estimates of Q and V2 were fixed to 0. Correlation between the IIV estimates for CL and V1 using a block structure was evaluated, but was unable to produce convergence and, thus, was removed. The final model PK code is provided in Code S1.
Model evaluation
The final two‐compartment PK model well characterized the study data. This is demonstrated by the agreement seen between observations and model predictions (Figure 1 a,b), the normal distribution of residuals (Figure 1 c,d), as well as the low relative standard errors in final model PK parameter estimates, which ranged from 8–24% (Table 2). Additionally, all parameter estimates fell near the median and within the 95% confidence interval of the bootstrap results (Table 2). Goodness‐of‐fit plots showed no obvious trends or model misspecification (Figure 1). A VPC performed using the final model indicated agreement between observed and predicted diazepam concentrations, with 11.1% (18/162) of observed concentrations falling outside the 90% prediction interval (Figure 2). A VPC stratified by age is provided in Figure S1.
Figure 1.
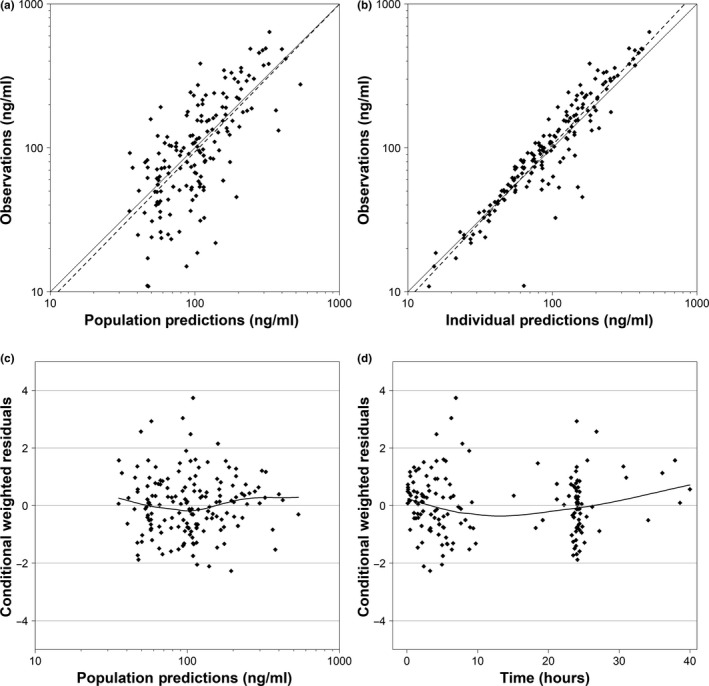
Diagnostic plots of final population pharmacokinetic model. Goodness‐of‐fit plots for the final model. (a) Observed vs. population. (b) Observed vs. individual predictions. (c) Conditional weighted residuals vs. population predictions. (d) Conditional weighted residuals vs. time in hours after first dose. For plots a and b, axes with concentration values are log scaled, the solid black line represents the line of unity, and the dotted black line represents a regression line. For plots c and d, the solid black line represents the locally weighted scatterplot smoothing curve, and reference lines of y = 0 and ± 2, and ± 4 are provided.
Figure 2.
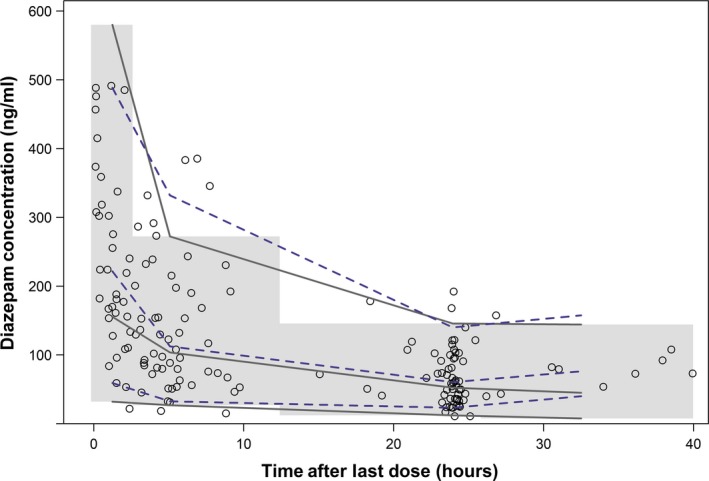
Visual predictive check for the final population pharmacokinetic model based on 1,000 simulations. The open circles represent the observed data, the dashed and solid lines represent the 5th, 50th, and 95th percentiles for the observed and simulated data, respectively. The shaded region represents the 90% prediction interval based on 1,000 simulations.
Exposure‐response relationship
Of the 87 patients, 79 were included in the primary efficacy analysis and 87 patients were included in the safety analysis. Among the included patients, 73% (58/79) achieved the primary efficacy end point of cessation of SE for 10 minutes without recurrence within 30 minutes of initial dose administration. Additionally, 14% (12/87) experienced life‐threatening respiratory depression requiring assisted ventilation. The median (25th and 75th percentiles) first dose in participants who achieved and failed the primary efficacy end point was 0.20 mg/kg (0.19–0.21) and 0.20 mg/kg (0.19–0.21), respectively (P = 0.51). Twenty‐eight percent (24/87) of subjects received a second dose. The median (25th and 75th percentiles) second dose in participants who achieved and failed the primary efficacy end point after the first dose of study drug was 0.10 mg/kg (0.10–0.11) and 0.10 mg/kg (0.10–0.11), respectively (P = 0.15). There were no statistically significant differences in PK parameters (CL, V1, V2, steady‐state volume of distribution (VSS), Cmax, area under the concentration vs. time curve from zero to infinity (AUC0–∞)) between study participants who achieved and failed the primary efficacy and safety end points. Multivariable logistic regression was performed for both the primary efficacy and safety end points after the first and second diazepam doses.
Simulations
Diazepam exposure for various single and multiple dose regimens was simulated for rich sampling in the 306 participants enrolled in the original trial. Overall, higher diazepam concentrations at 10 minutes after dosing were predicted by the study dosing regimen compared with the product labeled dosing regimen (Figure 3). At 10 minutes after simulating a single i.v. diazepam dose of 0.2 mg/kg (maximum 8 mg), 184 patients (60%) had concentrations > 200 ng/mL and 47 patients (15%) had concentrations > 600 ng/mL. At 10 minutes after simulating a single i.v. maximum product labeled dose, 15 patients (5%) had concentrations > 200 ng/mL. At 10 minutes after a single i.v. minimum product labeled dose was simulated, 7 (2%) had concentrations > 200 ng/mL. Neither of the product labeled dose simulations resulted in patients with concentrations > 600 ng/mL at 10 minutes after dosing.
Figure 3.
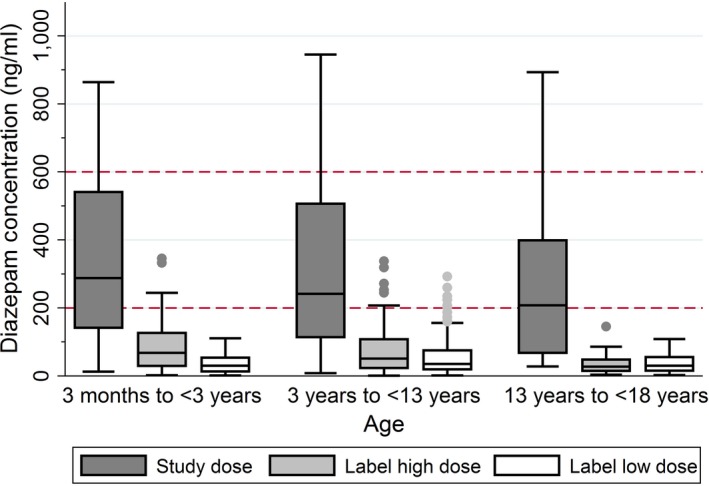
Results from Monte Carlo simulations of a single diazepam dose. Predicted diazepam concentrations at 10 minutes after a single i.v. dose in simulated patients. Study dose: 0.2 mg/kg (maximum 8 mg); product label high dose: 0.5 mg in children 31 days to < 5 years old and 1 mg in children ≥ 5 years; product label low dose: 0.2 mg in children 31 days to < 5 years old and 1 mg in children ≥ 5 years. Horizontal dotted lines indicate the commonly accepted target therapeutic range of 200–600 ng/mL.
The multiple dose simulation results are shown in Table 3. These results demonstrate that the product label high dose resulted in a similar percentage of subjects with concentrations > 200 ng/mL as the study dose (71% and 66%, respectively). However, the median number of doses given per subject until diazepam concentrations exceeded 200 ng/mL or maximum dose was received was 6 in the product label high regimen, compared with only 1 in the weight‐based study dose regimen. Additionally, these results reveal that both the weight‐based and the product label high regimens significantly outperformed the product label low dosing regimen, which only resulted in 28% of subjects with concentrations > 200 ng/mL.
Table 3.
Simulation results comparing study vs. product label dosing for first 10 minutes of seizure treatment
| Dosing regimen | N | Subjects with Cmax > 200 ng/mL, n (%) | Subjects with Cmax > 600 ng/mL, n (%) | Doses per subjecta | Cumulative absolute dose, mga | Cumulative WT‐normalized dose, mg/kga |
|---|---|---|---|---|---|---|
| Studyb | 306 | 216 (71) | 47 (15) | 1 (1–2) | 3.5 (2.2–5.5) | 0.20 (0.20–0.30) |
| < 3 years | 148 | 109 (74) | 23 (16) | 1 (1–2) | 2.2 (1.8–3.0) | 0.20 (0.20–0.30) |
| 3–13 years | 130 | 92 (71) | 20 (15) | 1 (1–2) | 4.8 (3.8–6.6) | 0.20 (0.20–0.30) |
| 13–18 years | 28 | 15 (54) | 4 (14) | 1 (1–2) | 8.0 (7.9–12) | 0.20 (0.16–0.20) |
| Label highc | 306 | 202 (66) | 36 (12) | 6 (5–6) | 3.0 (2.5–6.0) | 0.21 (0.15–0.28) |
| < 3 years | 148 | 106 (72) | 24 (16) | 6 (4–6) | 3.0 (2.0–3.0) | 0.25 (0.20–0.30) |
| 3–13 years | 130 | 84 (65) | 12 (9) | 6 (5–6) | 3.0 (3.0–6.0) | 0.19 (0.13–0.24) |
| 13–18 years | 28 | 12 (43) | 0 (0) | 6 (6–6) | 6.0 (6.0–6.0) | 0.10 (0.08–0.15) |
| Label lowd | 306 | 86 (28) | 6 (2) | 3 (3–3) | 0.6 (0.6–3.0) | 0.06 (0.05–0.09) |
| < 3 years | 148 | 37 (25) | 0 (0) | 3 (3–3) | 0.6 (0.6–0.6) | 0.06 (0.05–0.07) |
| 3–13 years | 130 | 44 (34) | 5 (4) | 3 (3–3) | 3.0 (0.6–3.0) | 0.08 (0.04–0.12) |
| 13–18 years | 28 | 5 (18) | 1 (4) | 3 (3–3) | 3.0 (3.0–3.0) | 0.05 (0.04–0.08) |
Cmax, maximum simulated concentration; WT, body weight.
aReported as median (25th and 75th percentile). bSubjects were dosed at 0.2 mg/kg (maximum 8 mg) i.v. push; if at 5 minutes after initial dose their simulated Cmax was < 200 ng/mL, subjects received a second dose of 0.1 mg/kg i.v. push (maximum 4 mg). cSubjects ages 30 days–5 years of age were dosed 0.5 mg every 2 minutes until Cmax was > 200 ng/mL; subjects aged > 5 years of age were dosed 1 mg every 2 minutes. dSubjects ages 30 days–5 years of age were dosed 0.2 mg every 5 minutes until Cmax was > 200 ng/mL; subjects aged ≥ 5 years of age were dosed 1 mg every 5 minutes until Cmax was > 200 ng/mL.
Discussion
Pediatric SE is a challenging clinical scenario requiring rapid seizure control to avoid permanent and potentially life‐threatening neuronal damage.27 Current available literature describing PKs of diazepam in pediatric SE is scant, and therefore optimal dosing of diazepam for the treatment for pediatric SE remains unclear.1, 14 The lack of PK analyses of diazepam in pediatric SE is likely due to the emergent nature of this condition, which makes obtaining informed consent and performing blood sampling extremely difficult. Our study was made possible by leveraging PK data obtained from a clinical trial evaluating the efficacy of diazepam for the treatment of pediatric SE in the ED.6 This clinical trial took advantage of the Exception from Informed Consent for Emergency Research, 21 CFR 50.24, which significantly enhanced recruitment. The use of this protocol enabled PK sampling to be performed during the time of active convulsions, which strengthened our ability to evaluate the relationships between diazepam PKs and safety or efficacy end points in children with SE. Additionally, we applied a population modeling approach, which allowed us to utilize the limited PK sampling.
To date, this is the largest PK study of diazepam in pediatric subjects with SE, and the first study to describe a population PK model of diazepam in this patient population. Diazepam PKs in 87 patients aged ≥ 3 months to < 18 years were well characterized by a two‐compartment model scaled by body size. The final model IIV estimates for both CL and V1 were somewhat high with coefficients of variation of 49.9% and 115%, respectively. This may be explained by the wide range of weights of subjects enrolled in the clinical trial (5–89 kg), or by the sampling scheme that resulted in PK samples 3 clusters, rather than uniformly over 48 hours. The simulation results of the VPC demonstrate that the model captures the observed variability in the data well (Figure 2).
Our results are similar to a previous analysis of diazepam that used more traditional PK parameter estimation methods. In an adult study of nine patients with epilepsy with a mean weight of 66 kg, a two−compartment model was chosen to characterize the PKs of diazepam.28 The mean CL estimate in this previously published study was 3.10 L/hour (0.0469 L/hour/kg), which is similar to that from our final model adult‐scaled estimate for CL of 2.36 L/hour (0.034 L/hour/kg).28 The mean VSS from this previously published study was 61.1 L (0.916 L/kg), which is also comparable to our final model estimate for VSS (the sum of V1 and V2) of 98.5 L (1.41 L/kg).29 Additionally, our final model population estimates are also comparable to those reported in a noncompartmental PK analysis of 11 children ages 6 months to 13 years with severe malaria given i.v. diazepam for seizures. Investigators from this analysis reported a mean (95% confidence interval) clearance of 0.044 L/hour/kg (0.024–0.062) and a mean (95% confidence interval) VSS of 3.5 L/kg (1.3–5.7), both of which are similar to our clearance estimates of 0.034 L/hour/kg and our VSS of 1.41 L/kg.23
Our analysis did not identify any significant impact of organ function or age on diazepam CL. In regard to organ function, the vast majority of patients had normal liver and kidney functions suggested by SCR, ALT, and AST values within the normal range. Therefore, our dataset lacked a robust distribution of these covariates needed for accurate modeling of their effects on the model parameters. When these covariate effects were estimated, the statistical significance was driven by 3.4% (3/87) of our subjects, and the magnitude of the effect was weak, likely due to the limited number of subjects with organ dysfunction. Regarding patient age, it has been estimated that adult levels of CYP3A4 expression are approached by 6–12 months of age, and CYP2C19 expression are approached by 2 years of age.12, 30, 31 Because our analysis includes only 2.3% (2/87) of subjects < 6 months of age, it is likely that the vast majority of patients in this study would have similar biotransformation capacity for hepatic metabolism of diazepam as in adults.
There were no statistically significant relationships noted between diazepam PK parameters or Cmax and the primary efficacy or safety end points, which are in line with previous literature.11, 23, 32 The reason for the lack of a relationship between diazepam PKs and efficacy end points remains unclear. One possible explanation may be that current diazepam dosing recommendations achieve diazepam concentrations at the top of the exposure‐response relationship. Therefore, given the exposures achieved by the subjects in our study, it is possible that other external factors, such as primary diagnosis or comorbidities, may drive efficacy and safety end points rather than exposure. A second explanation might be related to the active metabolites of diazepam, which may correlate with safety and efficacy measures, but were not quantified in our analysis.8, 9, 10 However, previous research investigating the effect of diazepam's major metabolite desmethyldiazepam suggested that due to the slow rate of formation, diazepam's metabolites are unlikely to contribute significantly to the pharmacologic effect of diazepam during acute treatment.33 Additionally, given that only 12 subjects experienced the primary safety outcomes, it is likely that our analysis was underpowered to detect a relationship between diazepam PKs and respiratory depression.
Monte Carlo simulations showed that compared to either product label recommended dose, the study dose of 0.2 mg/kg (8 mg maximum) resulted in a greater percentage of simulated patients exceeding the minimal therapeutic concentration of 200 ng/mL at 10 minutes after a single dose. Importantly, no simulated concentrations based on the study dose group were predicted to exceed 2,000 ng/mL, which is considered to be the toxic range in adults.34 Therefore, it is possible that an initial weight‐based dosing of 0.2 mg/kg may improve seizure control without compromising safety. This may be beneficial in older and heavier children whose product label recommended absolute doses are less likely to achieve exposures of > 200 ng/mL. Our multiple‐dose simulation results support this, as the percent of subjects ≥ 13 years of age with predicted concentrations ≥ 200 ng/mL was higher for the weight‐based study dose (54%) compared with the product label high dose (43%) and the product label low dose (18%). This is consistent with the findings from the diazepam arm of the original clinical trial, which demonstrated a high rate of primary efficacy of 69.2% (9/13) in patients ≥ 13 years of age, which was similar to the overall rate of primary efficacy of 72.1% (101/140). However, the implementation of weight‐based dosing of diazepam should be done with caution, given that the high‐stress environment of the ED where SE is treated may increase the risk of medication errors.35
Although this is the first population PK model of diazepam in children with SE, our analysis has several limitations. As mentioned previously, the patient population evaluated in this study did not include a large percentage of children with renal insufficiency, liver impairment, or children < 6 months old. Therefore, application of our final population PK model in these patients is an extrapolation, and may not be appropriate. Furthermore, our simulations were performed in a virtual population of subjects with identical demographics of children in our study. Due to the higher incidence of pediatric SE in children < 5 years of age, the distribution of ages and weights are skewed toward younger individuals where SE is much more common (Figure S2). Therefore, our results are more representative of the true population of pediatric subjects presenting with SE, rather than a uniform distribution of age and weights. Second, the half‐life of diazepam has previously been estimated as 13–18 hours in similar pediatric populations.13, 28 Given the limited number of concentrations drawn beyond 24 hours, our sampling scheme may not be ideal for characterizing the PKs of diazepam, and thus, the model may not predict accumulation with repeated administration with high accuracy. Further PK sampling was not feasible in this setting as most subjects are discharged from the hospital after 1–2 days of observation. A third limitation pertains to the bioanalysis of diazepam, which is described in Supplementary Information S1. Plasma diazepam concentrations were quantified after samples were stored at −70°C for up to 4 years; however, long‐term stability data have only been established up to 1,044 days (unpublished data). Despite this, the diazepam concentrations we observed in this study are very similar to those of previously published literature in both children and adults receiving similar weight‐based dosing.23, 28 Furthermore, neither unbound diazepam nor its metabolites were quantified in our analysis, and the therapeutic range chosen has not been prospectively evaluated in children, both of which may limit the pharmacodynamic assessment. Other limitations of this study were that the underlying etiology of SE for each patient was not reported, and clinical criteria were used to determine SE termination, which is consistent with methods from previous studies.6, 36, 37, 38, 39
In summary, a two‐compartment population PK model accounting for size‐based differences using allometrically scaled WT characterized diazepam disposition in children with SE, and no significant relationships were noted between diazepam PK parameters and the primary efficacy or safety end points. Monte Carlo simulations demonstrated that, compared with the product label recommended diazepam dose, the study dose of 0.2 mg/kg (maximum 8 mg) resulted in a higher percentage of simulated patients exceeding the commonly accepted therapeutic range of 200–600 ng/mL at 10 minutes after dosing. Future clinical pharmacology studies of diazepam should be performed to better characterize the therapeutic range of diazepam for safe and effective treatment of pediatric SE. Additionally, these studies should include both neonates as well as children with organ dysfunction to better inform the effects of organ maturation and dysfunction on diazepam PKs.
Source of Funding
This work was funded under National Institute of Child Health and Human Development (NICHD) contract HHSN275201000003I for the Pediatric Trials Network (Principal Investigator Daniel K. Benjamin, Jr.). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under award number UL1TR001117. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflicts of Interest
L.C.K. received support from NIH grants T32GM086330, 5T32HD043029, and 4K12HD043494. R.J.B. is supported by the National Institute of General Medical Sciences (NIGMS) of the NIH under award T32GM086330. C.P.H. receives salary support for research from NICHD (K23HD090239), the US government for his work in pediatric and neonatal clinical pharmacology (Government Contract HHSN267200700051C, PI: Benjamin, under the Best Pharmaceuticals for Children Act), and industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). M.C‐W. receives support for research from the NIH (1R01‐HD076676‐01A1), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the National Institute of Allergy and Infectious Disease (NIAID; HHSN272201500006I and HHSN272201300017I), NICHD (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201300009C), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). J.T.G. receives support for research from the National Institute of Neurological Disorders and Stroke (K23NS085049; HHSN27100001), The Patient‐Centered Outcomes Research Institute, and industry sponsors (www.dcri.duke.edu/research/coi.jsp). D.G. receives support for research from NICHD (K23HD083465). The remaining authors have no funding to disclose.
Author Contributions
L.C.K., R.J.B., and D.G. wrote manuscript. L.C.K., C.P.H., M.C‐W., J.M.C., and D.G. designed the research. L.C.K., C.P.H., J.M.C., J.T.G., B.H., E.V.C., K.M., R.A., M.C‐W., and D.G. performed the research. L.C.K., R.J.B., and D.G. analyzed the data.
Supporting information
Figure S1. Visual predictive check results stratified by age from final population PK model.
Figure S2. Histograms of age and weight of the subjects use in dosing simulations.
Table S1. Covariate model‐testing process.
Code S1. NONMEM code for final population PK model describing diazepam in children with SE.
Supplementary Information S1. PK sample collection and bioanalytical assay methods description.
Acknowledgments
The assay measuring diazepam concentrations was developed by Robert Wurm and performed by Michael O'Mara at OpAns Laboratory (Durham, NC).
The Best Pharmaceuticals for Children Act – Pediatric Trials Network Publication Committee: Gary Furda, Duke Clinical Research Institute, Durham, NC; Danny Benjamin, Duke Clinical Research Institute, Durham, NC; Edmund Capparelli, University of California San Diego, San Diego, CA; Gregory L. Kearns, Arkansas Children's Hospital Research Institute, Little Rock, AR; Ian M. Paul, Penn State College of Medicine, Hershey, PA; Jan Sullivan, University of Louisville, Louisville, KY; Christoph P. Hornik, Duke Clinical Research Institute, Durham, NC; Kelly Wade, Children's Hospital of Philadelphia, Philadelphia, PA. The Eunice Kennedy Shriver National Institute of Child Health and Human Development: David Siegel, Perdita Taylor‐Zapata, Anne Zajicek, Zhaoxia Ren, Ekaterini Tsilou, and Alice Pagan. The EMMES Corporation (Data Coordinating Center): Ravinder Anand and Gina Simone. Pediatric Trials Network Diazepam Study Team, Principal Investigators, and Study Coordinators: Duke Clinical Research Institute: Lawrence Ku, Christoph P. Hornik, Barrie Harper, Mary Mills, Jeffrey T. Guptill, Kevin Watt, and Michael Cohen‐Wolkowiez; The University of North Carolina at Chapel Hill: Daniel Gonzalez; University of California, San Diego: Edmund V. Capparelli.
References
- 1. Sofou, K. , Kristjánsdóttir, R. , Papachatzakis, N.E. , Ahmadzadeh, A. & Uvebrant, P. Management of prolonged seizures and status epilepticus in childhood: a systematic review. J. Child Neurol. 24, 918–926 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Neville, B.G.R. , Chin, R.F.M. & Scott, R.C. Childhood convulsive status epilepticus: epidemiology, management and outcome. Acta Neurol. Scand. Suppl. 186, 21–24 (2007). [PubMed] [Google Scholar]
- 3. Raspall‐Chaure, M. , Chin, R.F. , Neville, B.G. & Scott, R.C. Outcome of paediatric convulsive status epilepticus: a systematic review. Lancet Neurol. 5, 769–779 (2006). [DOI] [PubMed] [Google Scholar]
- 4. Lowenstein, D.H. & Alldredge, B.K. Status epilepticus at an urban public hospital in the 1980s. Neurology 43, 483–488 (1993). [DOI] [PubMed] [Google Scholar]
- 5. De Negri, M. & Baglietto, M.G. Treatment of status epilepticus in children. Paediatr. Drugs 3, 411–420 (2001). [DOI] [PubMed] [Google Scholar]
- 6. Chamberlain, J.M. et al Lorazepam vs diazepam for pediatric status epilepticus. JAMA 311, 1652–1660 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Rebel Distributors Corp . DIAZEPAM ‐ diazepam injection, solution. <http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=d1b5762f-307c-4c99-977d-d91d7dd22141>. Accessed 8 November 2017.
- 8. Riss, J. , Cloyd, J. , Gates, J. & Collins, S. Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol. Scand. 118, 69–86 (2008). [DOI] [PubMed] [Google Scholar]
- 9. Kaplan, S.A. , Jack, M.L. , Alexander, K. & Weinfeld, R.E. Pharmacokinetic profile of diazepam in man following single intravenous and oral and chronic oral administrations. J. Pharm. Sci. 62, 1789–1796 (1973). [DOI] [PubMed] [Google Scholar]
- 10. Greenblatt, D.J. , Divoll, M.K. , Soong, M.H. , Boxenbaum, H.G. , Harmatz, J.S. & Shader, R.I. Desmethyldiazepam pharmacokinetics: studies following intravenous and oral desmethyldiazepam, oral clorazepate, and intravenous diazepam. J. Clin. Pharmacol. 28, 853–859 (1988). [DOI] [PubMed] [Google Scholar]
- 11. Anderson, M. Benzodiazepines for prolonged seizures. Arch. Dis. Child Educ. Pract. Ed. 95, 183–189 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Stevens, J.C. et al Developmental expression of the major human hepatic CYP3A enzymes. J. Pharmacol. Exp. Ther. 307, 573–582 (2003). [DOI] [PubMed] [Google Scholar]
- 13. Morselli, P.L. et al Diazepam elimination in premature and full term infants, and children. J. Perinat. Med. 1, 133–141 (1973). [DOI] [PubMed] [Google Scholar]
- 14. Brophy, G.M. et al Guidelines for the evaluation and management of status epilepticus. Neurocrit. Care 17, 3–23 (2012). [DOI] [PubMed] [Google Scholar]
- 15. Food and Drug Administration . Guidance for institutional review boards, clinical investigators, and sponsors: exception from informed consent requirements for emergency research. <https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM249673.pdf>. Published 2013. Accessed 8 November 2017.
- 16. Keizer, R.J. , van Benten, M. , Beijnen, J.H. , Schellens, J.H. & Huitema, A.D. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput. Methods Programs Biomed. 101, 72–79 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 18. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 19. Sarkar, D. Lattice: Multivariate Data Visualization with R (Springer, New York, 2008). [Google Scholar]
- 20. Wickham, H. Ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2009). [Google Scholar]
- 21. Holford, N. , Heo, Y.A. & Anderson, B. A pharmacokinetic standard for babies and adults. J. Pharm. Sci. 102, 2941–2952 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Anderson, B.J. & Holford, N.H. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 24, 25–36 (2009). [DOI] [PubMed] [Google Scholar]
- 23. Ogutu, B.R. et al Pharmacokinetics and anticonvulsant effects of diazepam in children with severe falciparum malaria and convulsions. Br. J. Clin. Pharmacol. 53, 49–57 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferngren, H.G. Diazepam treatment for acute convulsions in children. A report of 41 patients, three with plasma levels. Epilepsia 15, 27–37 (1974). [DOI] [PubMed] [Google Scholar]
- 25. Remy, C. , Jourdil, N. , Villemain, D. , Favel, P. & Genton, P. Intrarectal diazepam in epileptic adults. Epilepsia 33, 353–358 (1992). [DOI] [PubMed] [Google Scholar]
- 26. Anderson, B. & Holford, N. Mechanism‐based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 48, 303–332 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Gathwala, G. , Goel, M. , Singh, J. & Mittal, K. Intravenous diazepam, midazolam and lorazepam in acute seizure control. Indian J. Pediatr. 79, 327–332 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Dhillon, S. & Richens, A. Pharmacokinetics of diazepam in epileptic patients and normal volunteers following intravenous administartion. Br. J. Clin. Pharmacol. 12, 841–844 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibaldi, M. & Perrier, D. Pharmacokinetics 2nd edn (Marcel Dekker Inc, New York, 1982). [Google Scholar]
- 30. Kearns, G.L. , Abdel‐Rahman, S.M. , Alander, S.W. , Blowey, D.L. , Leeder, J.S. & Kauffman, R.E. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349, 1157–1167 (2003). [DOI] [PubMed] [Google Scholar]
- 31. Ku, L.C. & Smith, P.B. Dosing in neonates: special considerations in physiology and trial design. Pediatr. Res. 77, 2–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scott, R.C. , Besag, F.M. & Neville, B.G. Buccal midazolam and rectal diazepam for treatment of prolonged seizures in childhood and adolescence: a randomised trial. Lancet 353, 623–626 (1999). [DOI] [PubMed] [Google Scholar]
- 33. Sunzel, M. , Paalzow, L. , Berggren, L. & Eriksson, I. Respiratory and cardiovascular effects in relation to plasma levels of midazolam and diazepam. Br. J. Clin. Pharmacol. 25, 561–569 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. North Carolina Office of the Medical Examiner . Toxicology. <http://www.ocme.dhhs.nc.gov/toxicology/>. Published 2009. Accessed 8 November 2017.
- 35. McDowell, S.E. , Ferner, H.S. & Ferner, R.E. The pathophysiology of medication errors: how and where they arise. Br. J. Clin. Pharmacol. 67, 605–613 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leppik, I.E. , Derivan, A.T. , Homan, R.W. , Walker, J. , Ramsay, R.E. & Patrick, B. Double‐blind study of lorazepam and diazepam in status epilepticus. JAMA 249, 1452–1454 (1983). [PubMed] [Google Scholar]
- 37. Chiulli, D.A. , Terndrup, T.E. & Kanter, R.K. The influence of diazepam or lorazepam on the frequency of endotracheal intubation in childhood status epilepticus. J. Emerg. Med. 9, 13–17 (1991). [DOI] [PubMed] [Google Scholar]
- 38. Giang, D.W. & McBride, M.C. Lorazepam versus diazepam for the treatment of status epilepticus. Pediatr. Neurol. 4, 358–361 (1988). [DOI] [PubMed] [Google Scholar]
- 39. Alldredge, B.K. et al A comparison of lorazepam, diazepam, and placebo for the treatment of out‐of‐hospital status epilepticus. N. Engl. J. Med. 345, 631–637 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Visual predictive check results stratified by age from final population PK model.
Figure S2. Histograms of age and weight of the subjects use in dosing simulations.
Table S1. Covariate model‐testing process.
Code S1. NONMEM code for final population PK model describing diazepam in children with SE.
Supplementary Information S1. PK sample collection and bioanalytical assay methods description.