

Abstract
This analysis describes the population pharmacokinetics (PPK) of apixaban in nonvalvular atrial fibrillation (NVAF) subjects, and quantifies the impact of intrinsic and extrinsic factors on exposure. The PPK model was developed using data from phase I–III studies. Apixaban exposure was characterized by a two‐compartment PPK model with first‐order absorption and elimination. Predictive covariates on apparent clearance included age, sex, Asian race, renal function, and concomitant strong/moderate cytochrome P450 (CYP)3A4/P‐glycoprotein (P‐gp) inhibitors. Individual covariate effects generally resulted in < 25% change in apixaban exposure vs. the reference NVAF subject (non‐Asian, male, aged 65 years, weighing 70 kg without concomitant CYP3A4/P‐gp inhibitors), except for severe renal impairment, which resulted in 55% higher exposure than the reference subject. The dose‐reduction algorithm resulted in a ~27% lower median exposure, with a large overlap between the 2.5‐mg and 5‐mg groups. The impact of Asian race on apixaban exposure was < 15% and not considered clinically significant.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Previous clinical pharmacology studies indicate that individual intrinsic and extrinsic factors have limited effect on apixaban exposure. However, the impact of a combination of factors on exposure was not known.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study developed a comprehensive PPK model of apixaban in subjects with NVAF in order to quantify the impact of intrinsic and extrinsic factors.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Apixaban exposure was adequately characterized by a two‐compartment PPK model with first‐order absorption and first‐order elimination. With the exception of severe renal impairment, changes in apixaban exposure due to individual demographic factors were < 25% relative to the reference subject.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
These analyses evaluating the impact of factors, such as Asian race, body weight, renal function, and age, support the use of the fixed‐dose regimen and dose‐modification scheme used in the global phase III clinical trials.
Apixaban is a novel, orally active, selective, direct, reversible inhibitor of the coagulation factor Xa and is approved in a number of countries for multiple indications, including thromboprophylaxis after knee or hip replacement surgery, treatment of deep vein thrombosis (DVT) and PE, prevention of recurrent DVT and PE, and reduction of the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (NVAF).1, 2, 3, 4, 5, 6, 7
Apixaban exhibits a pharmacokinetic (PK) profile with the following characteristics: oral bioavailability of ~50%, dose‐proportional increase in exposure throughout the therapeutic dose range (2.5–10 mg), and no time dependency. Apixaban has a total clearance of 3.3 L/hour, a steady‐state volume of distribution of ~21 liters, and a half‐life of ~12 hours following oral administration.8, 9 Multiple pathways are involved in the plasma elimination of apixaban, including metabolism primarily by cytochrome P450 3A4 (CYP3A4), and biliary, renal, and direct intestinal excretion.8, 10, 11, 12 Renal clearance following intravenous administration, accounts for ~27% of total systemic clearance in healthy subjects.9, 13 Apixaban is a substrate for P‐glycoprotein (P‐gp) and breast cancer resistance protein.14, 15
Two NVAF phase III trials evaluating apixaban have demonstrated a significantly improved benefit–risk profile compared to the standard of care.3, 4 The selected apixaban dose of 5 mg b.i.d. was based primarily on the results and analysis of the phase II dose‐ranging study in venous thromboembolism prevention.16, 17 Additionally, the phase III studies3, 4 implemented a dose‐modification strategy (which was not evaluated in phase II) based on advanced age, low body weight, and reduced renal function to minimize the potential for higher exposures in a population that may be at an inherently higher bleeding risk than the typical subject.18, 19 Although results of the clinical pharmacology studies indicated that these factors individually have limited effect on apixaban exposure, a combination of more than one intrinsic factor may increase apixaban exposure to a greater extent than a given factor alone.20, 21, 22 In addition, advanced age and poor renal function have been associated with an inherent increased risk of bleeding. Thus, in the phase III trials, a reduced apixaban dose of 2.5 mg b.i.d. was used in subjects meeting two or more of the following criteria: an age of ≥ 80 years, a body weight of ≤ 60 kg, or a serum creatinine level of ≥ 1.5 mg/dL (133 μmol/L), whereas all other subjects received the standard 5‐mg b.i.d. dose.
The aim of this analysis was to describe the population pharmacokinetics (PPK) of apixaban in subjects with NVAF, to quantify the impact of intrinsic and extrinsic factors on apixaban PK, and to predict apixaban exposure for the approved dosing regimens in patients with NVAF. To support global development, an ad hoc analysis was performed to estimate the impact of Japanese, Korean, and other Asian backgrounds on apixaban PK relative to non‐Asian patients with NVAF.
METHODS
Study populations and data
The PPK analysis included intensive and sparse apixaban concentration–time data collected from 12 clinical studies: 8 phase I studies, a phase II study in Japanese subjects with acute coronary syndrome (ACS; Japan ACS phase II), a global phase II study in subjects with ACS (APPRAISE‐1), a phase II study in Japanese subjects with NVAF (Japan NVAF phase II), and a subset of subjects from one global phase III NVAF clinical trial, ARISTOTLE (Table 1 ).4, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 This analysis utilized a two‐stage approach so the majority of PK model development could be performed with data from the phase I and phase II studies (stage 1). Once data from the phase III study became available, the stage 1 models were re‐estimated (updated stage 1) and then redefined with additional covariate testing (stage 2). Two ACS phase II studies, including one in Japanese subjects, were included in the stage 1 analysis to provide apixaban concentration data in a non‐NVAF population with similar demographic characteristics, enabling a better evaluation of the effects of race and patients with NVAF status on apixaban PK.
Table 1.
Summary of data sources
| Study type | Target population | Apixaban dose and regimen | Nominal PK sampling schedule |
|---|---|---|---|
| Phase I studiesa | |||
| Multiple doses22 | Healthy adult male subjects (N = 36) |
2.5–25 mg b.i.d. for 7 days 10–25 mg q.d. for 7 days |
Days 1, 4, and 7: predose (0 hour); postdose (hour), b.i.d. and q.d.: 1, 2, 3, 4, 6, 9, 12, and 24; b.i.d. only: 13, 14, 15, 16, 18, and 21 day 3, b.i.d. and q.d.: predose (72 hours) postdose day 7, b.i.d. and q.d.: 48 (day 9) and 72 hours (day 10) |
| Race23 | Healthy white and Japanese subjects (N = 24) | 2.5, 10, 25, and 50‐mg single dose |
Predose (0 hour) postdose (hour): 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48, and 72 |
| Renal impairment19 | Healthy and renally impaired subjects (N = 32) | 10‐mg single dose |
Predose (0 hour) postdose (hour): 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48, 72, and 96 |
| Age and sex20 | Healthy young (≤ 45 years) and elderly (≥ 65 years) male and female subjects (N = 79) | 20‐mg single dose |
Predose (0 hour) postdose (hour): 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 18, 24, 36, 48, 72, and 96 |
| Race24 | Healthy Japanese subjects (N = 18) | 2.5, 5, and 10 mg b.i.d. for 7 days |
Days 1 and 7: predose (0 hour); postdose (hour): 1, 2, 3, 4, 5, 6, 8, 12, 13, 14, 15, 16, and 17; days 2, 3, 5, 6, 8, and 9: predose (0 hour) or corresponding timea
day 4: predose (0 hour), 12 hours |
| Race25 | Healthy Chinese subjects (N = 12) | 10‐mg single dose, then 10 mg b.i.d. for 6 days |
Day 1: predose (0 hour); postdose (hour): 0.5, 1, 2, 3, 4, 6, 9, 12, 18, 24, 36, 48, 60, and 72 days 7 and 8: predose (0 hour) day 9: predose (0 hour); postdose (hour): 0.5, 1, 2, 3, 4, 6, 9, 12, 18, 24, 36, 48, 60, and 72a |
| Body weight21 | Healthy subjects of high (≥ 120 kg), normal (65–85 kg, and low (≤ 50 kg) body weight (N = 55) | 10‐mg single dose | Day 1: predose (0 hour); postdose (hour): 0.5, 1, 2, 3, 4, 6, 9, 12, 18, 24, 36, 48, 60, and 72 |
| Multiple doses | Healthy subjects (N = 14) | 2.5 mg b.i.d. for 4 days |
Day 1: predose (0 hour) day 4: predose (0 hour); postdose (hour): 0.5, 1, 2, 3, 4, 6, 8, 12, 12.5, 13, 14, 15, 16, 18, 20, 24, 36, 48, and 60 |
| Phase II studya | |||
| APPRAISE 126 | ACS treatment subjects (N = 951) |
2.5 and 10 mg b.i.d. 10 and 20 mg q.d. for 26 weeks |
Weeks 3 and 26: random sample |
| JAPAN ACS phase II27 | Japanese subjects with ACS (N = 93) | 2.5 and 5 mg b.i.d. for 24 weeks | Weeks 1, 4, and 12: predose (0 hour); postdose: 1–3 hours and 3–5 hours |
| Japan NVAF phase II28 | Japanese subjects with NVAF (N = 139) | 2.5 and 5 mg b.i.d. for 12 weeks | Weeks 1 and 8: predose (0 hour); postdose: 1–3 hours and 3–5 hours |
| Phase 3 study | |||
| ARISTOTLE4 | Adult subjects with NVAF (N = 2932) | 5 mg b.i.d. (2.5 mg b.i.d. in select subjects) for 6 months | Month 2, single random sample |
ACS, acute coronary syndrome; NVAF, nonvalvular atrial fibrillation; PK, pharmacokinetic.
Dose‐reduction algorithm was not evaluated in phase II studies including Japan NVAF phase II.
This investigation primarily focused on the estimation of effects of covariates, including, but not restricted to, sex, age, renal function, race, body weight, time of administration (diurnal variation), and concomitant medications on the PK of the compound. These factors were selected based on the known clinical pharmacology profile of apixaban at the time, results of previous PPK analyses, and factors considered to be pertinent to the NVAF patient population.
All study protocols, their amendments, and informed‐consent documentation for studies included in the analyses were reviewed and approved by institutional review boards, and all studies were conducted in accordance with Good Clinical Practice, the Declaration of Helsinki, and local regulations.
Pharmacokinetic assay
Apixaban concentrations were determined using a validated liquid chromatography assay coupled with atmospheric pressure ionization mass spectrometry method (Intertek Pharmaceutical Service, El Dorado Hills, CA), with a lower limit of quantification of 1 ng/mL.30
Stage 1 population pharmacokinetic analysis
Stage 1 base model
The structural component of the base model was taken from a previously developed31 linear two‐compartment PPK model, parameterized in terms of apparent oral clearance (CL/F; L/hour), apparent intercompartmental clearance (Q/F; L/hour), apparent volume of distribution (central (Vc/F), and peripheral (Vp/F); L), and first‐order absorption rate constant (ka; hour−1). Total apixaban clearance was split into renal and nonrenal elimination components. The effect of renal function (calculated creatinine clearance (cCrCL): calculated with the Cockcroft‐Gault equation32) on CL/F was incorporated into the base model using a power function. To model the effect of cCrCL on the apparent renal clearance (CLR/F) and avoid the limitations of the Cockcroft‐Gault formula32 for extremes of body weight, a breakpoint of 150 mL/minute was included as follows (Eq. 1):
| (1) |
where CLi/F is apixaban apparent plasma clearance for individual i, CLNR/F is the nonrenal component of CLi/F, CLR/F is the renal component of CLi/F, cCrCLi is the calculated creatinine clearance for individual i, cCrCLref is the reference value of 80 mL/minute, θ1 and θ2 are the power coefficients for the relationship between cCrCLi and CLi/F, FLAG is an indicator variable that is 1 when cCrCLi is > 150 mL/minute and zero otherwise, and ηi is the random effect for individual i.
Based on the inclusion of data from dose‐ranging studies, and observation of less than proportional exposure with doses > 10 mg due to decreased dissolution at higher doses,8 a reduction in relative bioavailability (Frel) was incorporated into the model. The Frel was fixed to 1 for the lowest dose group (2.5 mg) and estimated as a parameter for doses > 2.5 mg using Eq. 2:
| (2) |
where F rel is the relative bioavailability of a given dose > 2.5 mg, IMAX is the maximum reduction in relative bioavailability, ED50 is the dose at which half of the maximal reduction in Frel is achieved, and γ is the shape parameter that controls the relationship between dose and Frel.
Interindividual variance (IIV) terms were described by an exponential error model, with the random variability distributed with a mean of zero and a variance of ω2. The residual variability was described by a log‐normal error model (implemented as an additive error model using a log transform‐both‐sides approach). All modeling and simulations were conducted using First Order Conditional Estimation with Interaction with NONMEM (version 7.1).33 Postprocessing of NONMEM output and creation of derived datasets and figures was completed using SAS (version 9) or S‐plus (version 8.2) software.
Stage 1 full and final models
The following covariates were incorporated into the base model simultaneously to form the stage 1 full PPK model.
Apparent renal clearance (CLR/F): race (white, black, Asian, other), subject status (healthy, ACS, NVAF), and dosing time (morning (12:01am to 11:00am), afternoon (11:01am to 5:00pm), and evening (5:01pm to 12:00am).
The apparent nonrenal clearance (CLNR/F): age, sex, baseline body weight, race, subject status, and dosing time.
Apparent volume of central compartment (VC/F): baseline body weight and subject status.
Absorption rate (ka): dosing time.
Continuous covariates were normalized to a typical reference value and were included in the model using a power function. Categorical covariate effects were parameterized as a fractional change. The covariate parameterizations are described by Eq. 3:
| (3) |
where TVP is the typical value of a model parameter, covmi is the individual continuous covariate, refm is the reference continuous covariate value, covpi is the individual categorical (0–1) covariate, θn is an estimated parameter describing the typical PK parameter value for an individual with covariates equal to the reference covariate values (coνmi= refm, covpi= 0), and θ( m + n ) and θ( p + m + n ) are estimated parameters describing the magnitude of the covariate–parameter relationships.
Wald's Approximation Method (WAM) was used to identify a subset of reduced PK models relative to the stage 1 full model.34 The WAM procedure ranks all 2k possible submodels derived from the presence or absence of the k covariate parameters in the full model. The WAM algorithm approximates the log‐likelihood surface by a quadratic equation in the covariate effects based on the estimates and asymptotic variance–covariance matrix of the estimates from the stage 1 full model fit. Maximization of Schwarz's Bayesian Criterion (SBC), calculated using the approximation, was used to rank all 2k possible models. The top 15 ranked models were fit using NONMEM to calculate the actual SBC.
The stage 1 final parsimonious model (Supplemental File S1) was selected to be the model with the maximum value of NONMEM‐based SBC among the top 15 ranked models determined by the WAM procedure.
Stage 2 population pharmacokinetic analysis
Stage 1 base, full, and final model parameter estimates were updated to include the apixaban concentrations from the NVAF phase III study and refitted to ensure that the stage 1 final model remained parsimonious (referred to as the updated stage 1 final model; Table 3). The updated stage 1 final model was then adapted to include concomitant medications known to produce changes in apixaban PK. The influence of strong and moderate CYP3A4/P‐gp inhibitors as well as strong CYP3A4/P‐gp inducers were evaluated as time‐varying covariates on total CL/F after initially being tested on both CLR/F and CLNR/F (Table 3). Model reduction was then performed using a WAM procedure as described for the stage 1 analysis to determine the stage 2 final model (Table 3 and Supplemental File S2).
Model assessment
The adequacy of the model to describe the data was assessed through evaluation of objective function values (OFVs), standard diagnostic and graphical assessments, as well as precision of parameter estimates. Additionally, a posterior predictive check (PPC) method was performed to determine if the model was capable of simulating data that was consistent with the observed apixaban concentrations.35 The time course of the mean and 90% prediction intervals were plotted stratified by study and dose, and the corresponding observed PPC statistics were overlaid.
Prediction of steady‐state exposure and evaluation of apixaban exposure obtained with dose‐adjustment algorithm
The stage 2 final PPK model was used to predict the steady‐state concentration–time profiles in subjects with NVAF who received 5 mg b.i.d. and in those who met the criteria to receive the reduced dose of 2.5 mg b.i.d. (subjects with at least two of the three following factors: age ≥ 80 years, body weight ≤ 60 kg, or serum creatinine ≥ 1.5 mg/dL). In order to generate predictions of apixaban, the multivariate normal distribution was used as an approximate asymptotic posterior distribution to generate 500 sets of population parameter values from which 500 simulated datasets were generated. For each of 500 simulated parameter datasets, 1,000 patients were simulated each for the dose‐modification group receiving 2.5 mg b.i.d. apixaban and the reference group receiving 5 mg b.i.d. apixaban. Simulated total daily apixaban exposures were generated for each group based on covariates that were generated using a nonparametric bootstrap (sampling with replacement) of the observed PK covariates within each group to preserve the correlation structure among the covariates.
Ad hoc analysis of stratification of Asian race into Japanese, Korean, and other Asians
The stage 2 final PPK model that included Asian race as a covariate on the total CL/F of apixaban was subsequently refined to include separate covariate effects for Japanese, Korean, and other Asian races. As the Japan NVAF phase II trial randomized 2.5‐mg and 5‐mg apixaban dose levels, and did not evaluate the dose‐reduction algorithm, apixaban concentrations were generated for the overall Japanese population by dose and compared with observed concentrations according to whether or not the phase III NVAF dose‐reduction algorithm criteria would have been met.
RESULTS
The stage 2 final PPK analysis dataset consisted of 11,968 apixaban concentrations from 4,385 subjects. The stage 1 analysis included 9,036 apixaban concentrations from 270 phase I study subjects and 1,183 phase II subjects with NVAF or ACS. Stage 2 included an additional 2,932 apixaban concentrations from 2,932 NVAF subjects from the phase III ARISTOTLE study. Table 2 provides summary statistics of the baseline demographic covariates and concomitant medication usage, stratified by indication. The overall analysis population had a mean age of 65 years, an average body weight of 83.4 kg, and a mean cCrCL of 85.2 mL/minute. NVAF subjects comprised 70% of the subjects in the PPK analysis dataset. Asians accounted for ~15% of the analysis dataset (9.1% Japanese, 1.6% Korean, and 4.2% other Asians). Approximately 5% of the NVAF population enrolled in the phase III trial met the criteria for the 2.5‐mg dose. For graphical displays and comparison across populations, a typical reference NVAF subject was defined as a man, non‐Asian subject aged 65 years, with a body weight of 70 kg and a cCrCL of 80 mL/minute.
Table 2.
Summary of baseline demographic covariates for PPK analysis
| Covariate | Phase I subjects | NVAF | ACS | Total |
|---|---|---|---|---|
| Age (years) | ||||
| n | 270 | 3,071 | 1,044 | 4,385 |
| Mean (SD) | 38.89 (18.2) | 69.33 (9.2) | 60.84 (11.4) | 65.44 (13.0) |
| Median (min–max) | 33 (18–85) | 70 (26–94) | 61 (28–89) | 68 (18–94) |
| Baseline body weight (kg) | ||||
| n | 270 | 3,071 | 1,044 | 4,385 |
| Mean (SD) | 75.97 (21.9) | 84.74 (20.4) | 81.52 (15.7) | 83.44 (19.6) |
| Median (min–max) | 71.2 (37.7–175.1) | 83.0 (32–198.2) | 80.0 (44–175.5) | 81.4 (32–198.2) |
| Creatinine clearance (mL/minute) | ||||
| n | 270 | 3,071 | 1,044 | 4,385 |
| Mean (SD) | 114.32 (47.2) | 79.87 (31.5) | 93.35 (33.5) | 85.2 (34.5) |
| Median (min–max) | 112.8 (15–317.5) | 74.35 (18.8–279.8) | 89.27 (11.9–319.7) | 79.3 (11.9–319.7) |
| Sex, n (%) | ||||
| Male | 181 (67.04) | 2,079 (67.70) | 820 (78.54) | 3,080 (70.24) |
| Female | 89 (32.96) | 992 (32.30) | 224 (21.46) | 1,305 (29.76) |
| Race, n (%) | ||||
| White | 181 (67.04) | 2,531 (82.42) | 937 (89.75) | 3,649 (83.22) |
| Asian | 49 (18.15) | 509 (16.57) | 94 (9.00) | 652 (14.87) |
| Japanese | 30 (11.11) | 278 (9.05) | 93 (8.91) | 401 (9.14) |
| Korean | 0 (0) | 69 (2.25) | 0 (0) | 69 (1.57) |
| Other Asian | 19 (7.04) | 162 (5.28) | 1 (0.10) | 182 (4.15) |
| Black/African American | 30 (11.11) | 25 (0.81) | 6 (0.57) | 61 (1.39) |
| Other | 10 (3.70) | 6 (0.20) | 7 (0.67) | 23 (0.52) |
| Strong CYP3A4/P‐gp inhibitors,a n (%) | ||||
| No | 270 (100) | 3,068 (99.9) | 1,044 (100) | 4,382 (99.93) |
| Yes | 0 (0) | 3 (0.1) | 0 (0) | 3 (0.07) |
| Moderate CYP3A4/P‐gp inhibitors,a n (%) | ||||
| No | 270 (100) | 2,399 (78.12) | 998 (95.59) | 3,667 (83.63) |
| Yes | 0 (0) | 672 (21.88) | 46 (4.41) | 718 (16.37) |
| Strong CYP3A4/P‐gp inducers,a n (%) | ||||
| No | 270 (100) | 3,017 (98.24) | 1,030 (98.66) | 4,317 (98.45) |
| Yes | 0 (0) | 54 (1.76) | 14 (1.34) | 68 (1.55) |
ACS, acute coronary syndrome; CYP, cytochrome P450; NVAF, nonvalvular atrial fibrillation; P‐gp, P‐glycoprotein; PPK, population pharmacokinetic.
Concomitant medication usage was designated as “Yes” if a subject was on a pertinent concomitant medication when at least one pharmacokinetic sample was collected.
PPK model development
Stage 1 population pharmacokinetic analysis
Parameter estimates and precision for the stage 1 base, full, and final models are shown in Table S1 . Initial attempts to fit a full model to the apixaban concentration–time data did not result in successful convergence. A series of modifications were made, including separate residual variability for healthy subjects vs. patients and estimation of off‐diagonal elements of the variance–covariance matrix of IIV. Parameterization resulted in an estimate of ~1 for the correlation between IIV‐CL/F and IIV‐Vc/F, indicating a perfect correlation for these random effects. It was hypothesized that the high correlation could be reduced by reparameterizing the base model so that subject‐specific random effects were estimated on the microconstants (k10, k21, and k12). This model converged successfully, but the covariance step failed with a singular R‐matrix. Thus, the model was further simplified by assuming that changes in CLR/F were proportional to changes in cCrCL, forming the final stage 1 base PPK model. Additionally, the covariate effects for race, subject status, and dosing time were evaluated on total CL/F instead of each component separately in the stage 1 full PPK model.
Nine covariate effects were incorporated into this model, including dosing time on ka; baseline body weight, NVAF subject status, and ACS subject status on Vc/F; age and sex on CLNR/F; and Asian race, NVAF subject status, and ACS subject status on CL/F. This model accounted for 96.4% of the reduction in OFV relative to the full model. Based on these results, the highest ranked model was parsimonious and selected as the stage 1 final model (Table S1 ). Overall, the stage 1 final model provides an adequate fit to the apixaban plasma concentration vs. time data in healthy subjects and subjects with NVAF or ACS.
Stage 2 population pharmacokinetic analysis
Although successful convergence was achieved when phase III data were added and the stage 1 models refitted, some model instability was noted. The models estimated with the updated dataset were more susceptible to numerical problems and demonstrated higher correlations (ρ > 0.95) for the parameters describing the relationship between Frel and dose (i.e., γ, logit IMAX, and ED50). Therefore, the parameterization for Frel was simplified using a power model to stabilize the updated stage 1 model:
| (4) |
where Frel is the relative bioavailability of a given dose > 2.5 mg, I50 is the decrease in relative bioavailability for a 50‐mg apixaban dose, and γ is the shape parameter that controls the relationship between dose and Frel.
A total of 10 intrinsic and extrinsic covariate effects were included in the stage 2 final model (Table 3 and Table S2 ). The WAM procedure was not implemented in stage 2 model development because only two covariate parameters (strong or moderate CYP3A4/P‐gp inhibitors and strong CYP3A4/P‐gp inducers) were added to the model. The concomitant medication categories for strong and moderate CYP3A4/P‐gp inhibitors were collapsed because only three subjects contributed a PK sample while on a concomitant strong inhibitor (Table 2 ). The effects of concomitant medications on apixaban CL/F were estimated to be relatively small in magnitude; the effect of strong or moderate CYP3A4/P‐gp inhibitors resulted in only a 14.6% decrease in CL/F. The effect of strong inducers on apixaban CL/F, included in the full stage 2 model, was very small and poorly estimated, which suggested that the data do not contain much information to describe this relationship. Thus, the effect was not included in the final stage 2 model.
Table 3.
Updated stage 1 final, stage 2 final, and stage 2 final ad hoc model parameter estimates
| Fixed effects parameters | Estimate ± SE updated stage 1 final model | Estimate ± SE stage 2 final model | Estimate ± SE stage 2 final ad hoc d model |
|---|---|---|---|
| OFV | −5,878.214 | −5,888.403 | −5,900.112 |
| ka (θ1) (1/hour) | 0.471 ± 0.0204 | 0.473 ± 0.0208 | 0.471 ± 0.0218 |
| Evening dosing (am/pm = 2) (θ10) | −0.434 ± 0.0234 | −0.433 ± 0.0236 | −0.436 ± 0.0237 |
| Afternoon dosing (am/pm = 3) (θ27) | NE | NE | NE |
| CLR/F (θ2) (L/hour) | 1.57 ± 0.118 | 1.57 ± 0.117 | 1.60 ± 0.126 |
| cCrCL (θ7) | 1 FIXED | 1 FIXED | 1 FIXED |
| CLNR/F (θ6) (L/hour) | 2.02 ± 0.11 | 2.02 ± 0.108 | 2.02 ± 0.113 |
| D_WTB (θ26) | NE | NE | NE |
| Age (θ14) | −0.429 ± 0.0681 | −0.429 ± 0.0678 | −0.414 ± 0.0727 |
| Female (θ15) | −0.215 ± 0.0263 | −0.216 ± 0.0263 | −0.227 ± 0.0269 |
| CL/F | 3.59a | 3.59a | 3.62a |
| Black (θ28) | NE | NE | NE |
| Asian (θ16) | −0.12 ± 0.0184 | −0.119 ± 0.0183 | |
| Japanese (θ24) | NE | NE | −0.151 ± 0.0202 |
| Korean (θ25) | NE | NE | 0.0325 ± 0.0753 |
| Other Asians (θ16) | NE | NE | −0.0427 ± 0.0403 |
| NVAF (θ17) | −0.149 ± 0.0298 | −0.139 ± 0.0305 | −0.145 ± 0.0326 |
| ACS (θ18) | −0.217 ± 0.0284 | −0.215 ± 0.0286 | −0.212 ± 0.0304 |
| Strong/moderate inhibitors (θ19) | NE | −0.146 ± 0.0421 | −0.140 ± 0.0419 |
| Strong/moderate inducers (θ20) | NE | NE | NE |
| Vc/F (θ3) (L) | 29.8 ± 0.981 | 30.0 ± 0.971 | 30.0 ± 1.04 |
| D_WTB (θ11) | 0.806 ± 0.0649 | 0.790 ± 0.0645 | 0.792 ± 0.0648 |
| NVAF subjects (θ12) | −0.0217 ± 0.0501 | −0.0405 ± 0.0491 | −0.0530 ± 0.0508 |
| ACS subjects (θ13) | −0.177 ± 0.0449 | −0.180 ± 0.0446 | −0.184 ± 0.0463 |
| Q/F (θ4) (L/hour) | 1.89 ± 0.151 | 1.91 ± 0.163 | 1.92 ± 0.202 |
| Vp/F (θ5) (L) | 26.7 ± 1.89 | 27.0 ± 2.03 | 27.3 ± 2.78 |
| Shape parameter for Frel (γ) (θ8) | 0.853 ± 0.0693 | 0.857 ± 0.0684 | 0.875 ± 0.0779 |
| Logit for reduction in Frel at 50 mg (I50) (θ9) | −0.321 ± 0.0512 | −0.322 ± 0.0512 | −0.327 ± 0.0519 |
| IIV components | Estimate ± SE (%CVb) | Estimate ± SE (%CVb) | Estimate ± SE (%CVb) |
|---|---|---|---|
| ω2‐ka | 0.271 ± 0.0319 (52.1) | 0.263 ± 0.0311 (51.3) | 0.264 ± 0.0312 (51.4) |
| ω2‐k | 0.101 ± 0.00989 (31.8) | 0.0954 ± 0.00964 (30.9) | 0.0961 ± 0.00987 (31.0) |
| ω2‐Vc/F | 0.0287 ± 0.0055 (16.9) | 0.0294 ± 0.00553 (17.1) | 0.0290 ± 0.00550 (17.0) |
| ω2‐k21 | 0.243 ± 0.0412 (49.3) | 0.240 ± 0.0404 (49.0) | 0.241 ± 0.0409 (49.1) |
| ω2‐k12 | 1.49 ± 0.163 (122) | 1.55 ± 0.169 (124) | 1.55 ± 0.186 (124.5) |
| Residual variance components | Estimate ± SE (%CVc) | Estimate ± SE (%CVc) | Estimate ± SE (%CVc) |
|---|---|---|---|
| σ HV and Studies Japan NVAF phase II28, Japan ACS phase II27 (θ21) | 0.31 ± 0.00282 (31) | 0.31 ± 0.00282 (31) | 0.31 ± 0.00284 (31) |
| σ Study APPRAISE 126 (θ22) | 0.666 ± 0.0182 (66.6) | 0.668 ± 0.0183 (66.8) | 0.667 ± 0.0187 (66.7) |
| σ Study ARISTOTLE4 (θ23) | 0.451 ± 0.0169 (45.1) | 0.460 ± 0.0163 (46.0) | 0.457 ± 0.0165 (45.7) |
%CV, percentage of coefficient of variation; ACS, acute coronary syndrome; am/pm, dose time; cCrCL, calculated creatinine clearance; CL/F, apparent total clearance; CLNR/F, apparent nonrenal clearance; CLR/F, apparent renal clearance; Frel, relative bioavailability; HV, healthy volunteer; decrease in relative bioavailability for 50‐mg dose; IIV, interindividual variability; k, k12, and k21, first‐order rate constants; ka, absorption rate constant; NE, not evaluable; NVAF, nonvalvular atrial fibrillation; OFV, objective function value; Q/F, apparent intercompartmental clearance; Vc/F, apparent volume of distribution of the central compartment; Vp/F, apparent volume of distribution of the peripheral compartment; WTB, baseline body weight.
aCL/F values were calculated as the simple sum of the population parameters estimates for CLR/F and CLNR/F and do not represent model‐estimated parameters. bApproximate %CV reported as 100·ω. cApproximate %CV reported as 100·σ. dParameter estimates associated with the ad hoc analysis of the stratification of the Asian race into Japanese, Korean, and other Asian.
Administration of apixaban in the evening resulted in a 43% decrease in ka relative to administration in the morning or afternoon. Baseline body weight and subject status were found to influence Vc/F, with the effect estimated as a 23.3% reduction for a 50‐kg subject and a 22% increase for a 90‐kg subject relative to the typical 70‐kg NVAF subject. In addition, female subjects had a 21.6% reduction in CLNR/F relative to male subjects. Finally, Asian race, NVAF, ACS, or strong or moderate CYP3A4/P‐gp inhibitors resulted in reduced CL/F of 11.9%, 13.9%, 21.5%, and 14.6%, respectively, compared with non‐Asian subjects, healthy subjects, or subjects who did not receive concurrent administration of a CYP3A4/P‐gp inhibitor (Figure 1 ).
Figure 1.
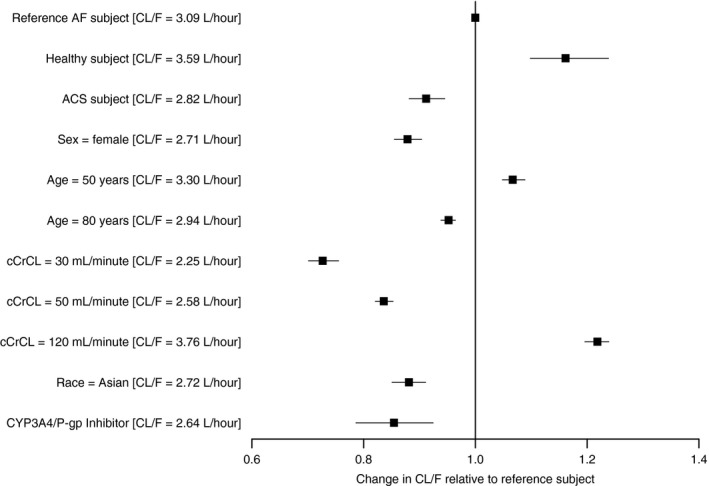
Illustration of covariate effects on total apparent clearance (CL/F). Solid squares represent the ratio of the typical predicted CL/F relative to the reference subject. The black line represents the 90% confidence interval of the ratio. The reference subject is a 65‐year‐old, non‐Asian, male subject with nonvalvular atrial fibrillation (NVAF) that has a calculated creatinine clearance (cCrCL) with the Cockcroft‐Gault equation of 80 mL/minute and did not receive a concomitant strong or moderate cytochrome P450 (CYP)3A4/P‐glycoprotein (P‐gp) inhibitor. ACS, acute coronary syndrome; AF, atrial fibrillation.
Model assessment
The final stage 2 model parameters were well estimated (Table 2 ) and no trends for model misspecification were observed in the diagnostic plots (data on file; Bristol‐Myers Squibb). A PPC was conducted to determine if the stage 2 final model was capable of simulating data that were consistent with the observed PK measurements in NVAF subjects (Figure S1 ); PPCs for the NVAF population for healthy and ACS subjects are shown in Figures S2 and S3 , respectively. Overall, the model captures the general shape of the apixaban concentration–time profile in both healthy subjects and patients, despite some instances where the observed data are not contained within the 90% prediction interval. Although there seemed to be a modest underprediction of the 5‐mg b.i.d. data, no systematic trends for model misspecification were observed in the NVAF subject data.
Predicted apixaban exposure in NVAF subjects
The distribution of the predicted total daily exposure (area under the curve at steady‐state (AUCss)) stratified by dose for subjects enrolled in the ARISTOTLE study4 is shown in Figure 2 . The median exposure predicted by the PPK model is lower for the dose‐modification group than the reference group assigned to the 5‐mg b.i.d. dose (Figure 2 ). Table 4 provides a summary of the predicted daily steady‐state apixaban exposure in NVAF subjects upon treatment with the 5‐mg or 2.5‐mg b.i.d. dose. These results show that NVAF subjects who require dose adjustment to 2.5 mg b.i.d. would be expected to have ~27% lower AUC compared with subjects receiving 5 mg b.i.d. Table S3 shows the predicted exposures for NVAF subjects who met dose‐modification criteria.
Figure 2.
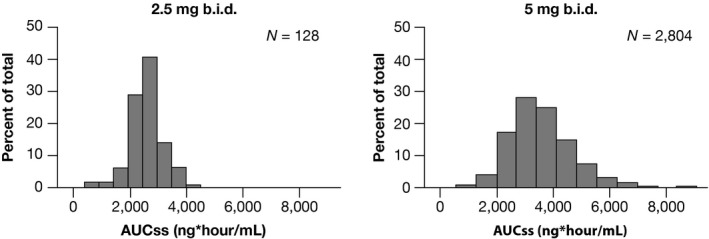
Distribution of predicted daily area under the curve at steady state (AUCss) for the ARISTOTLE Study Stratified by Dose Group. The 2.5‐mg dose group is the modified dose group and includes subjects meeting at least 2 of the 3 dose‐reduction criteria.
Table 4.
Predicted apixaban steady‐state exposure in patients with NVAF
| Steady‐state parameter (units)a | 5 mg b.i.d. | 2.5 mg b.i.d.b | ||||
|---|---|---|---|---|---|---|
| Median (90% CI) | 5th percentile | 95th percentile | Median (90% CI) | 5th percentile | 95th percentile | |
| Daily AUC (ng*hour/mL) | 3,280 (3,170–3,390) | 1,600 | 6,590 | 2,410 (2,320–2,510) | 1,250 | 4,560 |
| Cmax (ng/mL) | 171 (167–177) | 91 | 321 | 123 (119–128) | 68.5 | 221 |
| Cmin (ng/mL) | 103 (98.7–107) | 40.9 | 230 | 79.2 (75.2–83.2) | 34.4 | 162 |
| Tmax (hour) | 2.89 (2.81–2.99) | 1.63 | 4.13 | 2.86 (2.75–2.96) | 1.55 | 4.14 |
AUC, area under the concentration time curve; CI, confidence interval; Cmax, peak plasma concentration; Cmin, trough plasma concentration; NVAF, nonvalvular atrial fibrillation; Tmax, time of maximum plasma concentration.
Ad hoc analysis of stratification of Asian race into Japanese, Korean, and other Asian subgroups
Stratification of the effects of Asian race on CL/F resulted in −15.1%, + 3.3%, and −4.3% change in population mean total CL/F for Japanese, Korean, and other Asian races, respectively, relative to non‐Asian typical patients with NVAF (men, 65 years, 70 kg, and cCrCL of 80 mL/minute), corresponding to + 17.7%, −3.2%, and + 4.5% changes in daily AUCss, respectively (Table 3 and Table S2 ).
The plots of the observed and predicted apixaban plasma concentrations in Japanese subjects with NVAF at steady state are shown in Figure 3 . The predicted apixaban plasma concentrations for 2.5 and 5 mg b.i.d. showed a general dose‐proportionality. Observed concentrations after administration of apixaban 2.5 mg b.i.d. in Japanese subjects not meeting the phase III dose‐reduction criteria were generally lower than the apixaban 2.5‐mg b.i.d. observed data in subjects who met the phase III dose‐reduction criteria. Additionally, observed concentrations after administration of apixaban 5 mg b.i.d. in Japanese subjects meeting the phase III dose‐reduction criteria seemed to be distributed around the 5‐mg b.i.d. predicted line in typical Japanese subjects and were within the variability of apixaban 5‐mg b.i.d. observed data in Japanese subjects who did not meet the dose‐reduction criteria.
Figure 3.
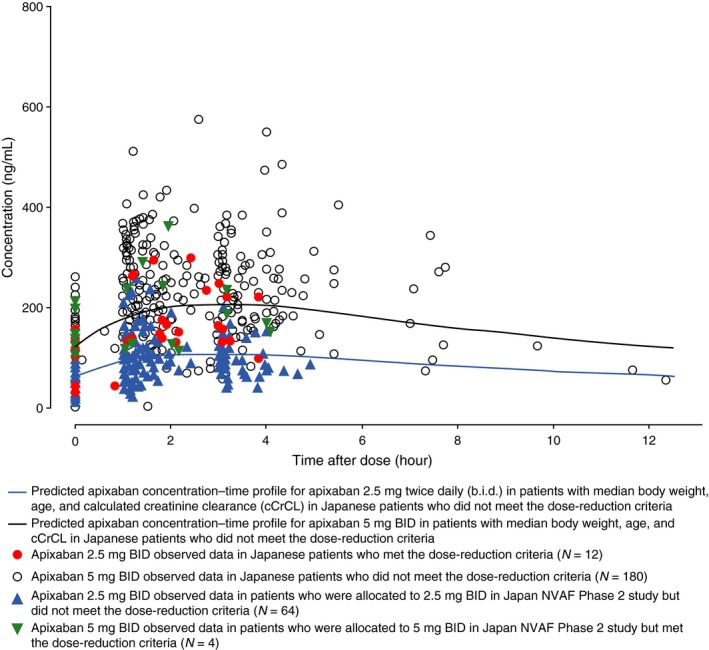
Observed and predicted apixaban plasma concentrations in Japanese patients with nonvalvular atrial fibrillation (NVAF) at steady state. Median values for body weight, age, and calculated creatinine clearance used to calculate typical Japanese predictions were 64.1 kg, 70 years, and 65.1 mL/minute, respectively.
Discussion
Apixaban exposure in NVAF subjects was adequately described with a two‐compartment model with first‐order absorption and elimination. The finding of a lower ka in the evening does not seem to be of clinical relevance given the b.i.d. dosing of apixaban and robust safety and efficacy findings from the phase III NVAF clinical trials.3, 4 Overall, the effects of renal function, race, patient type, and strong/moderate CYP3A4/P‐gp inhibitors were found to be significant predictors of apixaban clearance. The model allowed for empirical separation of the total plasma CL/F into renal and nonrenal components with predicted increases in AUCss of 9%, 28%, and 55% for patients with NVAF with mild (cCrCL = 65 mL/minute), moderate (40 mL/minute), and severe (15 mL/minute) renal impairment, respectively, compared with those with normal renal function. The results are generally consistent with the estimates from phase I studies20, 22 in which mild, moderate, and severe renal impairment were associated with an ~16%, 29%, and 44% higher estimated apixaban AUCINF than those with normal renal funtion.20 Several factors identified in previous phase I clinical trials, including age, sex, body weight, and mild‐to‐moderate renal impairment, were shown to modestly influence apixaban PK and these factors alone do not require any clinical dose adjustment.
Considering the elimination pathways of apixaban and the results of human drug–drug interaction studies, strong/moderate inhibitors of both CYP3A4 and P‐gp were suspected to be an influential covariate. Although they were evaluated in this analysis, the overall results estimated a small change (~17% increase) in exposure. A 99% and 40% increase in AUC was observed when apixaban was co‐administered with ketoconazole, a strong CYP3A4/P‐gp inhibitor, and diltiazem, a moderate CYP3A4/P‐gp inhibitor, respectively.8, 36 The effect of strong CYP3A4/P‐gp inducers on clearance was also evaluated in this analysis. Given the sparse sampling strategy and limited data available, the effect was very small and poorly estimated, and, thus, this covariate was removed from the final model. Given the limitation of the PPK data with respect to these variables, greater emphasis should be placed on the phase I results for the purposes of clinical considerations regarding the potential impact of CYP3A4 and P‐gp inhibitors.
The phase III NVAF studies included a dose modification for subjects who met at least two of the following criteria: age ≥ 80 years, body weight ≤ 60 kg, or serum creatinine ≥ 1.5 mg/dL.3, 4 Individually, these factors have been shown to have limited effects on exposure,20, 21, 22 both within the analyses described herein as well as in individual trials. In addition, each of these factors on their own was not expected to result in clinically meaningful change in the benefit–risk profile of apixaban, as confirmed by a subgroup analysis performed in subjects who met only one of the dose‐reduction criteria.37 However, an inherently higher risk of bleeding may be associated with the population of subjects exhibiting at least two of these criteria.38 Coupled with the knowledge that the combination of at least two of these factors may increase apixaban exposure to a greater extent than a given factor alone, the dosing algorithm for NVAF was designed to address these concerns while maintaining efficacy.38 The dose‐reduction algorithm resulted in a ~27% lower median exposure for the 2.5‐mg dose group compared to the 5‐mg dose group, with a large overlap between the groups. Most importantly, primary study outcomes (stroke or systemic embolism, International Society of Thrombosis and Haemostasis (ISTH) major bleeding) vs. warfarin were similar in those receiving 2.5 mg b.i.d. or 5 mg b.i.d.37
The overall impact of Asian race on apixaban exposure was small. The Asian population represented 15% of the overall population in the analysis dataset, with 9% being Japanese subjects. The CL/F for Asian subjects was 11.9% lower compared to non‐Asian subjects. An ad hoc analysis of further stratification of Asian race to Japanese, Korean, and other Asians showed that Asian race effects on CL/F of apixaban were 15.1% lower, 3.3% higher, and 4.3% lower in the Japanese, Korean, and other Asian subjects, respectively, relative to non‐Asian subjects. The reasons for the differences in the CL/F between Asian and non‐Asian subjects are unclear. In spite of no dose‐reduction criteria being implemented in the Japan NVAF phase II study, the apixaban PK data in Japanese subjects from the study showed that the global dose‐reduction criteria can be used in Japanese NVAF subjects.
A comprehensive PPK model for apixaban in the NVAF population was developed. Apixaban exposure was adequately characterized by a two‐compartment PPK model with first‐order absorption and first‐order elimination. Demographic factors that may affect apixaban PK were identified, however, none resulted in more than a 25% change in apixaban exposure relative to the reference subject, except for severe renal impairment. The effect of Japanese race was small and consistent with the effect of Asian race,24 and exposure was not meaningfully different from that in the non‐Asian population. In summary, these analyses evaluating the impact of factors, such as Asian race, body weight, renal function, and age, support the use of the fixed‐dose regimen and dose‐modification scheme used in the global phase III clinical trials.
Funding
This analysis was funded by Bristol‐Myers Squibb and Pfizer.
Conflict of interest
A.R., N.T., X.W., T.L., and C.F. are employees of Bristol‐Myers Squibb. B.C. and F.L. were employees of Bristol‐Myers Squibb at the time of the research. W.B. and R.B. are employees of Pfizer. T.U. is an employee of Bristol Myers Squibb K.K. M.O. is an employee of Pfizer Japan. K.K. and J.N. were paid consultants to Bristol‐Myers Squibb.
Author Contributions
B.C., K.K., J.N., A.R., N.T., W.B., R.B., X.W., T.L., F.L., T.U., M.O., and C.F. wrote the manuscript. X.W., F.L., and C.F. designed the research. R.B., X.W., and C.F. performed the research. B.C., K.K., J.N., A.R., N.T., W.B., R.B., X.W., T.L., F.L., T.U., M.O., and C.F. analyzed the data.
Supporting information
Figure S1. Posterior predictive check for atrial fibrillation patients following apixaban doses at steady‐state.
Figure S2. (a) Posterior predictive check for healthy subjects following apixaban dose on day 1. (b) Posterior predictive check for healthy subjects following apixaban doses at steady‐state.
Figure S3. Posterior predictive check for acute coronary syndrome patients following apixaban dose at steady‐state.
Table S1. Stage 1 base, full, and final model parameter estimates.
Table S2. Covariate parameters of the final model including separate race effects for Japanese, Koreans, and other Asians.
Table S3. Predicted apixaban steady‐state exposure in patients with NVAF in dose modification subgroups receiving 2.5 mg b.i.d. apixban.
Supplemental File S1. NONMEM code – apixaban stage 1 final model
Supplemental File S2. NONMEM code – apixaban stage 2 final model – Asian race
Acknowledgments
The authors would like to thank the participants, investigators, and study personnel who contributed to making the study possible. Editorial assistance was provided by Sandi Lusk and Dana Fox, PhD, at Caudex.
References
- 1. Agnelli, G. et al Oral apixaban for the treatment of acute venous thromboembolism. N. Engl. J. Med. 369, 799–808 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Agnelli, G. et al Apixaban for extended treatment of venous thromboembolism. N. Engl. J. Med. 368, 699–708 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Connolly, S.J. et al Apixaban in patients with atrial fibrillation. N. Engl. J. Med. 364, 806–817 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Granger, C.B. et al Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 365, 981–992 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Lassen, M.R. , Raskob, G.E. , Gallus, A. , Pineo, G. , Chen, D. & Portman, R.J. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N. Engl. J. Med. 361, 594–604 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Lassen, M.R. , Raskob, G.E. , Gallus, A. , Pineo, G. , Chen, D. & Hornick, P. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE‐2): a randomised double‐blind trial. Lancet 375, 807–815 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Lassen, M.R. , Gallus, A. , Raskob, G.E. , Pineo, G. , Chen, D. & Ramirez, L.M. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N. Engl. J. Med. 363, 2487–2498 (2010). [DOI] [PubMed] [Google Scholar]
- 8. Bristol‐Myers Squibb Company . P.I. Eliquis (apixaban) prescribing information. Revised 6/2018 (2016).
- 9. Vakkalagadda, B. et al Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am. J. Cardiovasc. Drugs 16, 119–127 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Raghavan, N. et al Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab. Dispos. 37, 74–81 (2009). [DOI] [PubMed] [Google Scholar]
- 11. Wang, X. et al Effect of activated charcoal on the pharmacokinetics of apixaban in healthy subjects [abstract PI‐90]. Clin. Pharmacol. Ther. 91(suppl. 1), S41 (2012). [Google Scholar]
- 12. Zhang, D. et al Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: administration of activated charcoal to bile duct‐cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab. Dispos. 41, 906–915 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Frost, C. et al Apixaban, a direct factor Xa inhibitor: single‐dose pharmacokinetics and pharmacodynamics of an intravenous formulation [abstract 148]. J. Clin. Pharmacol. 48, 1132 (2008). [Google Scholar]
- 14. Wang, L. et al In vitro assessment of metabolic drug‐drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab. Dispos. 38, 448–458 (2010). [DOI] [PubMed] [Google Scholar]
- 15. Zhang, D. et al Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab. Dispos. 41, 827–835 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Leil, T.A. , Feng, Y. , Zhang, L. , Paccaly, A. , Mohan, P. & Pfister, M. Quantification of apixaban's therapeutic utility in prevention of venous thromboembolism: selection of phase III trial dose. Clin. Pharmacol. Ther. 88, 375–382 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Levine, M.N. et al A randomized phase II trial of apixaban for the prevention of thromboembolism in patients with metastatic cancer. J. Thromb. Haemost. 10, 807–814 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Geldhof, V. , Vandenbriele, C. , Verhamme, P. & Vanassche, T. Venous thromboembolism in the elderly: efficacy and safety of non‐VKA oral anticoagulants. Thromb. J. 12, 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jun, M. et al The association between kidney function and major bleeding in older adults with atrial fibrillation starting warfarin treatment: population based observational study. BMJ 350, h246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang, M. et al Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J. Clin. Pharmacol. 56, 637–645 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Frost, C.E. et al Effects of age and sex on the single‐dose pharmacokinetics and pharmacodynamics of apixaban. Clin. Pharmacokinet. 54, 651–662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Upreti, V.V. et al Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br. J. Clin. Pharmacol. 76, 908–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frost, C. et al Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 76, 776–786 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu, Z. , Nepal, S. , Bragat, A. , Shenker, A. & Frost, C. Single dose apixaban pharmacokinetics and pharmacodynamics in healthy male Japanese and Caucasian subjects (A647). Can. J. Clin. Pharmacol. 15, e420–e781 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamahira, N. et al Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int. J. Clin. Pharmacol. Ther. 52, 564–573 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Cui, Y. et al Single‐ and multiple‐dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin. Pharmacol. 5, 177–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander, J.H. et al Apixaban, an oral, direct, selective factor Xa inhibitor, in combination with antiplatelet therapy after acute coronary syndrome: results of the Apixaban for Prevention of Acute Ischemic and Safety Events (APPRAISE) trial. Circulation 119, 2877–2885 (2009). [DOI] [PubMed] [Google Scholar]
- 28. Ogawa, H. , Goto, S. , Matsuzaki, M. , Hiro, S. & Shima, D. Randomized, double‐blind trial to evaluate the safety of apixaban with antiplatelet therapy after acute coronary syndrome in Japanese patients (APPRAISE‐J). Circ. J. 77, 2341–2348 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Ogawa, S. , Shinohara, Y. & Kanmuri, K. Safety and efficacy of the oral direct factor Xa inhibitor apixaban in Japanese patients with non‐valvular atrial fibrillation. The ARISTOTLE‐J study. Circ. J. 75, 1852–1859 (2011). [DOI] [PubMed] [Google Scholar]
- 30. Pursley, J. et al LC‐MS/MS determination of apixaban (BMS‐562247) and its major metabolite in human plasma: an application of polarity switching and monolithic HPLC column. Bioanalysis 6, 2071–2082 (2014). [DOI] [PubMed] [Google Scholar]
- 31. Leil, T.A. , Frost, C. , Wang, X. , Pfister, M. & LaCreta, F. Model‐based exposure‐response analysis of apixaban to quantify bleeding risk in special populations of subjects undergoing orthopedic surgery. CPT Pharmacometrics Syst. Pharmacol. 3, e136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cockcroft, D.W. & Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 16, 31–41 (1976). [DOI] [PubMed] [Google Scholar]
- 33. Beal, S.L. & Sheiner, L.B. NONMEM User's Guides (NONMEM Project Group, University of California, San Francisco, CA, 1992). [Google Scholar]
- 34. Kowalski, K.G. & Hutmacher, M.M. Efficient screening of covariates in population models using Wald's approximation to the likelihood ratio test. J. Pharmacokinet. Pharmacodyn. 28, 253–275 (2001). [DOI] [PubMed] [Google Scholar]
- 35. Yano, Y. , Beal, S.L. & Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28, 171–192 (2001). [DOI] [PubMed] [Google Scholar]
- 36. Frost, C.E. et al Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br. J. Clin. Pharmacol. 79, 838–846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alexander, J.H. et al Apixaban 5 mg twice daily and clinical outcomes in patients with atrial fibrillation and advanced age, low body weight, or high creatinine: a secondary analysis of a randomized clinical trial. JAMA Cardiol., 1, 673–681 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Lip, G.Y. , Frison, L. , Halperin, J.L. & Lane, D.A. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS‐BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J. Am. Coll. Cardiol. 57, 173–180 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Posterior predictive check for atrial fibrillation patients following apixaban doses at steady‐state.
Figure S2. (a) Posterior predictive check for healthy subjects following apixaban dose on day 1. (b) Posterior predictive check for healthy subjects following apixaban doses at steady‐state.
Figure S3. Posterior predictive check for acute coronary syndrome patients following apixaban dose at steady‐state.
Table S1. Stage 1 base, full, and final model parameter estimates.
Table S2. Covariate parameters of the final model including separate race effects for Japanese, Koreans, and other Asians.
Table S3. Predicted apixaban steady‐state exposure in patients with NVAF in dose modification subgroups receiving 2.5 mg b.i.d. apixban.
Supplemental File S1. NONMEM code – apixaban stage 1 final model
Supplemental File S2. NONMEM code – apixaban stage 2 final model – Asian race