

Abstract
The aim of the present study was to establish a physiologically based pharmacokinetic (PBPK) model for coproporphyrin I (CP‐I), a biomarker supporting the prediction of drug‐drug interactions (DDIs) involving hepatic organic anion transporting polypeptide 1B (OATP1B), using clinical DDI data with an OATP1B inhibitor rifampicin (300 and 600 mg, orally). The in vivo inhibition constants of rifampicin used as initial input parameters for OATP1Bs (K i,u,OATP1Bs) and multidrug resistance‐associated protein two‐mediated biliary excretion were estimated as 0.23 and 0.87 μM, respectively, from previous reports. Sensitivity analysis demonstrated that the K i,u,OATP1Bs and biosynthesis rate of CP‐I affected the magnitude of the interaction. K i,u,OATP1Bs values optimized by nonlinear least‐squares fitting were ~0.5‐fold of the initial value. It was determined that the blood concentration‐time profiles of four statins were well‐predicted using corrected individual K i,u,OATP1B values (ratio of in vitro K i,u(statin)/in vitro K i,u(CP‐I)). In conclusion, PBPK modeling of CP‐I supports dynamic prediction of OATP1B‐mediated DDIs.
Study Highlights.
What is the current knowledge on the topic?
CP‐I has been identified as an endogenous substrate for hepatic OATP1Bs and MRP2, and accumulates following a single dose of rifampicin, an OATP1Bs/MRP2 inhibitor, thereby presenting as a surrogate probe for DDI assessment.
What question did this study address?
Does a PBPK model for CP‐I, in which hepatic uptake and efflux processes are incorporated, enable us to describe its pharmacokinetics in the absence and presence of a perpetrator drug such as rifampicin?
What does this study add to our knowledge?
The presented PBPK modeling approach enables a greater understanding of the mechanisms of CP‐I pharmacokinetics and evaluation of the dose‐dependent inhibitory effects of rifampicin.
How might this change drug discovery, development, and/or therapeutics?
A useful PBPK model for an important OATP1Bs/MRP2 biomarker (CP‐I) is described. The model can support the translation of the effect of a new chemical entity on CP‐I pharmacokinetics to that on clinically used drugs (e.g., statins). In so doing, the model can complement existing OATP1B‐mediated DDI risk assessment approaches based on agency guidelines.
It has become widely accepted that the combinatorial use of drugs can give rise to clinically relevant drug‐drug interactions (DDIs), especially when the efficacy and safety profile of the victim drug is impacted. Therefore, DDI screening, modeling, and prediction has become standard practice. Such efforts now include the organic anion transporting polypeptides 1B (OATP1B1 and OATP1B3), which are multispecific transporters mediating the sinusoidal uptake of various drugs into hepatocytes. The inhibition of OATP1Bs is an important DDI mechanism that often presents as a significant increase in the systemic exposure of OATP1Bs substrates (e.g., statins, sulfonylureas and meglitinides, and antihepatitis C virus drugs) following the concomitant administration of OATP1B inhibitors, such as cyclosporine A, rifampicin (RIF), and gemfibrozil.1, 2, 3, 4 Typically, OATP1B in vitro inhibition data support DDI risk assessment after following agency guidelines. Such an approach, however, based on a target threshold (e.g., R > 1.1; where R = 1 + [(f u,p·I)/half‐maximal inhibitory concentration (IC50)]) that if triggered necessitates clinical follow‐up, is known to yield relatively high false‐positive rates.5 Therefore, various endogenous compounds have been proposed as surrogate probes to facilitate early clinical assessment of pharmacokinetic DDIs, which could reduce the need for unnecessary DDI studies using established probe drugs.5
Probe candidates for hepatic OATP1Bs have been reported, such as bilirubin and its glucuronides,6 bile acids, especially glucuronide and sulfate conjugates,7, 8 glucuronide and sulfate conjugates of steroids,9 fatty acid dicarboxylates,10 and coproporphyrin I and III (CP‐I and CP‐III). Of these, CP‐I and III, both the byproducts of heme synthesis, can support the evaluation of OATP1B‐mediated DDIs because of their specificity for OATP1B1 and OATP1B3,11, 12 sensitivity,12 and the good association of the area under the plasma concentration‐time curve (AUC) between CP‐I and statins (pitavastatin, atorvastatin, and fluvastatin).13 Importantly, both plasma CP forms present negligible diurnal variation, which is a desirable characteristic of a biomarker.12, 13
Mechanistic physiologically based pharmacokinetic (PBPK) modeling has increasingly been used as a tool to predict DDIs in recent years by combining the PBPK models of both victim and perpetrator. In this study, we aimed to establish a PBPK model for CP‐I in which hepatic uptake and efflux processes are considered. Followed by hepatic uptake, CP‐I undergoes biliary excretion14 via multidrug resistance‐associated protein 2 (MRP2).15 Clinical data, obtained with the positron emission tomography probe [11C]‐TIC‐Me, has shown that MRP2‐mediated biliary excretion16 can be significantly inhibited by RIF at a clinically relevant dose (600 mg). Such data suggest the DDI potential of RIF against MRP2,17 but in vitro inhibition data can be problematic because of the high variability in the reported values of in vitro inhibition constants (K i) for MRP2 (7.9–83 μM, as listed in the University of Washington Metabolism and Transport Drug Interaction Database). Overall, it was considered important to account for the involvement of both hepatic OATP1Bs and MRP2 in the interaction between CP‐I and RIF. CP‐I is also excreted into the urine, and its renal clearance is not significantly altered after the administration of RIF (600 mg).12
We previously presented a stepwise protocol to analyze multiple clinical cases of DDI involving drug transporters using a standardized PBPK model structure and unified model parameters for each victim or perpetrator drug.18 Using this standardized protocol and a previously established model for RIF,19 we constructed a PBPK model for CP‐I and optimized parameters, including K i for OATP1Bs, which could explain our clinical data showing a dose response with oral RIF at 300 and 600 mg.13 Subsequently, we evaluated the predictability of the DDIs between statins and RIF in the same clinical dataset,13 taking into account the difference between the in vitro K i of RIF for statins and CP‐I. Accounting for intersubstrate differences in K i is consistent with the recent report of Barnett et al.,20 who considered the IC50 values of RIF for both CP‐I and rosuvastatin in vitro and conducted mixed model analysis of clinical interaction data. Such considerations enhance the value of a PBPK model used to translate CP‐I interaction data to other OATP1B substrates.
Methods
Overview of PBPK modeling and simulation strategy
We previously reported a framework for constructing PBPK models.18 A PBPK model for CP‐I was constructed by introducing a parameter for CP‐I biosynthesis (v syn) into our previously proposed PBPK model (Supplementary Text S1). The v syn was incorporated in the liver compartment; CP‐I and CP‐III are spontaneously generated from coproporphyrinogen I and III, which are synthesized from uroporphyrinogen I and III by uroporphyrinogen decarboxylase expressed in both liver and erythrocytes. In patients of porphyria cutanea tarda caused by a deficient hepatic activity of uroporphyrinogen decarboxylase due to hepatitis,21 the urinary ratio of CPs to uroporphyrins (spontaneously generated from uroporphyrinogens) was decreased to 0.28, whereas the ratio was 4.2 in healthy volunteers,22 suggesting > 90% of CPs in humans in normal condition is synthesized in the liver. Thus, we regarded the liver as the major organ for CP‐I biosynthesis. The enterohepatic circulation (EHC) of CP‐I was incorporated in the model according to a previous report of kinetic model analyses using the plasma concentration data and urinary and fecal excretion data of 14C‐labeled CP‐I and CP‐III.23 Using the PBPK model and parameters for RIF reported by Asaumi et al.,19 RIF‐mediated inhibition of hepatic OATP1B and MRP2 was incorporated. Differential equations and other equation settings describing the PBPK model are presented in Supplementary Text S1. Finally, (i) simulation of blood concentration‐time profiles of CP‐I in the absence and presence of RIF, (ii) sensitivity analyses to examine the impact of change in each parameter on the simulation outcome, and (iii) parameter optimization by nonlinear least‐squares fitting were performed.
Simulation designs for our clinical study13 (volunteers, drug administration, time course, and sampling) and model parameter settings are described in Supplementary Text S1. Details for uptake and efflux experiments using plated human hepatocytes (Figure S1) for the determination of hepatic membrane permeation parameters for CP‐I are also described in Supplementary Text S1.
Software
The nonlinear least‐squares fitting software, Napp version 2.31,24 was used in all the optimization and simulation processes applied in the present study. The weight for the nonlinear least‐squares calculation was set as the square root of the original value. The weighted sum of squares (WSS) and the Akaike's information criteria (AIC) shown in Eqs. (1) and (2), respectively, were used to evaluate the fitting results.
| (1) |
y i: ith observed value, : ith predicted value.
| (2) |
Model predictability
To quantitatively evaluate model predictability for blood concentration‐time profiles, AUC ratio, and maximum blood concentration (Cmax) ratio, commonly used accuracy test criteria, the average fold errors (AFEs), were calculated, as shown in Eq. (3):
| (3) |
where n is the number of observations.
Results
Simulation of blood and liver concentration‐time profiles of RIF using the reported PBPK model
As shown in Figure 1, the observed blood concentration‐time profiles of RIF administered orally at 300 and 600 mg13 were reproduced by the simulation with the comprehensive PBPK model and parameters reported previously.19 The unbound blood concentration of RIF reached maximum values of 0.43 and 0.94 μM with 300 mg (Figure 1 a) and 600 mg (Figure 1 b) administration, respectively. The predicted intrahepatic unbound concentration reached maximum values of 1.0 and 1.5 μM following 300 and 600 mg administration, respectively.
Figure 1.
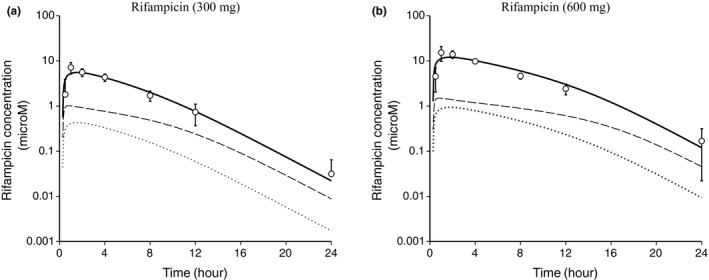
Simulation of the blood and liver concentration‐time profiles of rifampicin (RIF). The circles represent the observed blood concentrations of RIF administered at (a) 300 mg and (b) 600 mg, orally (mean ± SD, n = 8).13 The solid line represents a simulated blood concentration‐time profile using the physiologically based pharmacokinetic model and parameters reported previously.19 The dotted line represents a simulated blood unbound concentration‐time profile. The broken line represents a simulated intrahepatic unbound concentration‐time profile (the first hepatocyte compartment shown in Supplementary Text S1).
Setting of in vivo inhibition constants for OATP1Bs and MRP2 by RIF
To simulate the dynamic change in the blood concentration of CP‐I caused by RIF, the in vivo K i,u,OATP1Bs (0.23 μM)18 was set initially. Our in vitro experiments using OATP1B‐expressing cells (Figure S2) suggested that the calculated K i for OATP1B1 (0.78 μM) and that for OATP1B3 (0.18 μM) using CP‐I as a substrate tends to be lower than those using statins as substrates (1.1–2.8 μM for OATP1B1, 0.30–0.86 μM for OATP1B3; Table S2). Thus, we assumed some intersubstrate differences in the inhibitory characteristics by RIF, as reported previously.25 As part of the PBPK‐enabled modeling exercise, we considered a range of in vivo K i,u,OATP1Bs values (between 0.33‐fold and 3‐fold of the initial value) to support parameter sensitivity (Figure S3) and optimization by fitting (Figure 2). In addition, the in vivo K i,u,MRP2 (0.87 μM), calculated based on the change in the biliary clearance of [11C]‐TIC‐Me,17 was used (see details in Supplementary Text S1).
Figure 2.
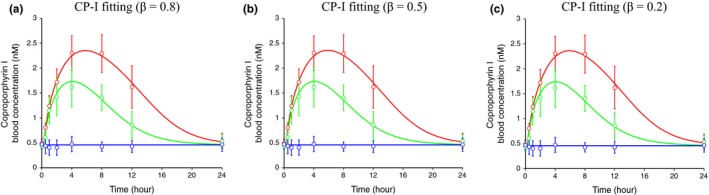
Simultaneously fitted blood concentration‐time profiles of coproporphyrin (CP)‐I in the absence and presence of rifampicin (RIF) after parameter optimization using the physiologically based pharmacokinetic (PBPK) model incorporating the inhibition of organic anion‐transporting polypeptide (OATP)1Bs and multidrug resistance‐associated protein 2 (MRP)2. The blue (control), green (300 mg RIF, orally), and red (600 mg RIF, orally) circles represent the observed blood concentrations of CP‐I (mean ± SD, n = 8).13 The lines represent simultaneously fitted blood concentration–time profiles after optimization of parameters in the PBPK model, in which the inhibition of hepatic OATP1Bs and MRP2 by RIF are incorporated. The fixed and optimized parameters are shown in Tables 2 and S1 A,B. The β values are set at 0.8 (a), 0.5 (b), and 0.2 (c).
Sensitivity analyses of model parameters to demonstrate the effects on the blood concentration‐time profiles of CP‐I
The blood concentration‐time profile of CP‐I following RIF administration was simulated (Figure S3) using the constructed PBPK model and parameters shown in Table 1 and Table S1. Sensitivity analyses of the hepatic overall intrinsic clearance, CLint,all (and correspondingly altered v syn according to the theoretical equation shown in Supplementary Text S1) showed that these parameters could change the turnover of CP‐I and the magnitude of the interaction with RIF. As the CLint,all and v syn are increased, the RIF‐mediated interaction is increased regardless of the different preset β values of 0.8 (Figure S3 a), 0.5 (Figure S3 b), and 0.2 (Figure S3 c). In addition, our sensitivity analyses also showed that the OATP1B inhibitor potency, reflected by K i,u,OATP1Bs (Figure S3 d–f), was a determinant of the interaction between RIF and CP‐I.
Table 1.
Parameters used in the simulation of the blood concentration‐time profiles of CP‐I in the absence and presence of RIF (Figure S3)
| ID | CLint,all a | v syn b | βc | R dif d | γd | f bile e | k transit e | k a e | F a F g e | K i,u,OATP1Bs f | K i,u,MRP2 g |
|---|---|---|---|---|---|---|---|---|---|---|---|
| l/h/kg | nmol/h/kg | – | – | – | – | h−1 | h−1 | – | μM | μM | |
| A | 33.3 | 0.153 | 0.8 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.66 | 0.23 | 0.87 |
| B | 33.3 | 0.125 | 0.5 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.66 | 0.23 | 0.87 |
| C | 33.3 | 0.105 | 0.2 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.66 | 0.23 | 0.87 |
CP‐I, coproporphyrin; RIF, rifampicin.
aCalculated from the reported CLint,all of 14C‐CP‐III administered intravenously to humans.23 bCalculated using the parameters including CLint,all and F a F g according to Eqs. S7–S10. See details in METHODS. cFixed to three different values indicating different conditions of the major rate‐determining processes in the hepatic intrinsic clearance. dCalculated according to the in vitro data using plated human hepatocytes. See details in METHODS. eDerived from the previous multicompartment analyses using 14C‐labeled coproporphyrins in humans.23 fThe in vivo K i,u,OATP1Bs estimated from physiologically based pharmacokinetic analyses of the drug‐drug interaction between pitavastatin and rifampicin.18 gCalculated based on the change in the in vivo biliary excretion clearance reported in the previous clinical study using a positron emission tomography probe (11C‐TIC‐Me).17
To evaluate the effects of K i,u,MRP2 and other parameters relating to EHC (F a F g, k a, and k transit), whose initial values were derived from the previous report,23 further sensitivity analyses were performed (Figure S3 g–l). The K i,u,MRP2 slightly affected the CP‐I blood concentration when the β value was set at 0.5 and 0.2 (Figure S3 h,i). As the F a F g is decreased, RIF‐mediated interaction is increased (Figure S3 j–l). Little change in the magnitude of the interaction was observed in sensitivity analyses of k a and k transit (data not shown).
Previous evidences suggested that > 90% of CPs in humans in normal condition is synthesized in the liver.22 We performed sensitivity analyses setting the fraction of CP‐I biosynthesis in the liver (f syn) as 1, 0.9, and 0.8 (Figure S3 m–o). Little change in the magnitude of the interaction was observed with these different f syn. Thus, we used f syn = 1 in the following analyses.
Optimization of model parameters by fitting of the blood concentration‐time profiles of CP‐I
Because CLint,all (v syn), K i,u,OATP1Bs, and F a F g markedly affected the magnitude of interaction between CP‐I and RIF (Figure S3 a–f,j–l), these parameters were optimized by nonlinear least‐square fitting to the CP‐I blood concentration‐time profiles in the absence and presence of RIF13 simultaneously. These parameters were moved freely between 0.33‐fold and 3‐fold in the initial values described in Table 1. We could obtain parameter sets that can explain the observed data (Figure 2 a–c). The optimized CLint,all, v syn, and F a F g were 35.1–43.1 l/h/kg, 0.212–0.442 nmol/h/kg, and 0.256–0.318, respectively, depending on the setting of β values (Table 2). The optimized values of K i,u,OATP1Bs ranged between 0.0824 and 0.106 μM, which were 0.36‐fold to 0.46‐fold of the initial value (0.23 μM).
Table 2.
Parameters fixed and optimized in the fitting of the blood concentration‐time profiles of CP‐I in the absence and presence of RIF (Figure 2)
| ID | CLint,all | v syn a | β | R dif | γ | f bile | k transit | k a | F a F g | K i,u,OATP1Bs | K i,u,MRP2 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| l/h/kg | nmol/h/kg | – | – | – | – | h−1 | h−1 | – | μM | μM | |||
| Fixed | Fixed | Fixed | Fixed | Fixed | Fixed | Fixed | SS | AIC | |||||
| A | 35.1 ± 2.7 | 0.442 (0.345–0.567) | 0.8 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.256 ± 0.067 | 0.0824 ± 0.0105 | 0.87 | 1.73 | 19.1 |
| B | 38.3 ± 2.4 | 0.264 (0.219–0.315) | 0.5 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.309 ± 0.079 | 0.0965 ± 0.0123 | 0.87 | 1.75 | 19.5 |
| C | 43.1 ± 2.3 | 0.212 (0.182–0.245) | 0.2 | 0.035 | 0.020 | 0.84 | 5.2 | 3.0 | 0.318 ± 0.087 | 0.106 ± 0.013 | 0.87 | 1.81 | 20.2 |
AIC, Akaike information criterion; CP‐I, coproporphyrin; RIF, rifampicin.
Calculated using the parameters including CLint,all and F a F g according to Eqs. S7–S10. In the parentheses, the minimum and maximum values calculated using SDs of CLint,all and F a F g are described.
Simulation of blood concentration‐time profiles of CP‐I using its optimized parameters and PBPK model incorporating the inhibition of OATP1Bs alone
To compare the contribution of OATP1Bs and MRP2 to the magnitude of the CP‐I‐RIF interaction, a simulation was performed using a PBPK model for CP‐I, in which only the inhibition of hepatic OATP1Bs by RIF was incorporated. Using the parameters described in Table 2, with exclusion of K i,u,MRP2, the simulated CP‐I blood concentrations (Figure S4 a–c) were lower than those using the original PBPK model incorporating dual (OATP1Bs/MRP2) inhibition (Figure 2 a–c). Such a model‐dependent difference was more significant when the β value was set lower.
The observed and the simulated AUC0–24 h and Cmax of CP‐I in the presence of RIF are summarized in Table 3. The simulated AUC and Cmax using the PBPK model with dual OATP1Bs/MRP2‐inhibition were around 96% and 102–108% of the observed values, respectively. In comparison, the simulated AUC and Cmax using the PBPK model with OATP1B inhibition only was 75–85% and 79–89% vs. the observed values, respectively.
Table 3.
The blood AUC and Cmax of CP‐I estimated by PBPK modeling compared to the observation
| AUC0–24h (nM h) | Cmax (nM) | ||||
|---|---|---|---|---|---|
| β | RIF 300 mg | RIF 600 mg | RIF 300 mg | RIF 600 mg | |
| Observationa | – | 23.9 ± 4.8 | 36.3 ± 6.7 | 1.61 ± 0.38 | 2.30 ± 0.35 |
| Inhibition of OATP1Bs and MRP2 (Figure 2) | 0.8 | 23.0 (96.2) | 34.8 (95.9) | 1.73 (108) | 2.35 (102) |
| 0.5 | 22.9 (95.8) | 34.8 (95.9) | 1.73 (108) | 2.36 (103) | |
| 0.2 | 22.9 (95.8) | 34.7 (95.6) | 1.74 (108) | 2.36 (103) | |
| Inhibition of OATP1Bs (Figure S4) | 0.8 | 20.3 (84.9) | 29.0 (79.9) | 1.43 (88.8) | 1.87 (81.3) |
| 0.5 | 19.3 (80.8) | 27.5 (75.8) | 1.35 (83.9) | 1.79 (77.8) | |
| 0.2 | 19.0 (79.5) | 27.3 (75.2) | 1.35 (83.9) | 1.80 (78.3) | |
In the parentheses, the estimation/observation ratios (%) are shown.
AUC, area under the plasma concentration‐time curve; Cmax, peak plasma concentration; CP‐I, coproporphyrin I; MRP2, multidrug resistance‐associated protein 2; OATP, organic anion‐transporting polypeptide; PBPK, physiologically based pharmacokinetic; RIF, rifampicin.
Data from the previous study.13
Prediction of the effect of RIF on blood concentration‐time profiles of statins using their PBPK models and parameters including substrate‐dependent K i,u,OATP1Bs
We evaluated the predictability of DDIs between four statins and RIF using our clinical data,13 as well as the K i,u,OATP1Bs for CP‐I optimized by PBPK modeling (Table 2) and the ratio of in vitro K i for each statin to CP‐I (Table S2). Previously reported PBPK models for pitavastatin, fluvastatin,18 rosuvastatin,26 and a newly constructed PBPK model for atorvastatin, whose structure is the same as pitavastatin and fluvastatin, leveraged their basic physiological and pharmacokinetic parameters (Table S1 A,C). After obtaining some parameters by fitting to the control data alone (Table S1 D), the blood concentration‐time profile of each statin in the presence of RIF was simulated (Figure 3 a–d) using substrate‐dependent K i,u,OATP1Bs, shown in Table S1 D, calculated by multiplying the K i,u,OATP1Bs for CP‐I by the ratio of in vitro K i for OATP1B1 (statin/CP‐I). AFEs for concentration‐time predictions in the presence of 300 and 600 mg RIF were calculated to be 1.71/1.97, 1.87/2.42, 1.50/1.56, and 1.43/1.73 for pitavastatin, rosuvastatin, atorvastatin, and fluvastatin, respectively. The predicted AUC and Cmax of statins were well‐correlated with their observed AUC and Cmax (Figure 3 e,f).
Figure 3.
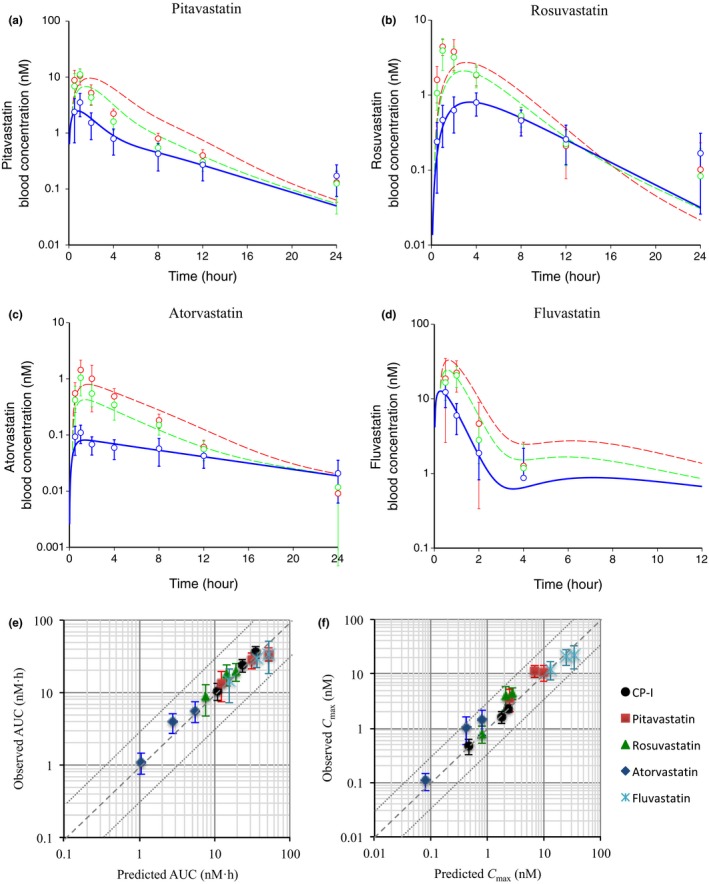
Prediction of the effect of rifampicin (RIF) on blood concentration‐time profiles of statins using their physiologically based pharmacokinetic (PBPK) models and parameters, including substrate‐dependent K i,u,OATP1Bs. (a–d) The blue (control), green (300 mg RIF, orally), and red (600 mg RIF, orally) circles represent the observed blood concentrations of statins (mean ± SD, n = 8).13 The blue solid lines (control) represent fitted blood concentration‐time profiles of statins after optimization of parameters in their PBPK models. The green and red broken lines (+RIF) represent predicted blood concentration‐time profiles of statins. (a) pitavastatin, (b) rosuvastatin, (c) atorvastatin, and (d) fluvastatin. The used parameters, including substrate‐dependent K i,u,OATP1Bs values are shown in Table S1 A,C,D. (e, f) Comparison of the predicted and observed area under the plasma concentration‐time curve (AUC) (e) and peak plasma concentration (Cmax) (f) of statins in the absence and presence of RIF. The AUC and Cmax of coproporphyrin (CP)‐I are also shown. Dashed and dotted lines denote unity and threefold boundaries, respectively.
On the other hand, when the prediction was performed using the in vivo K i,u,OATP1Bs for CP‐I (0.1 μM) without any correction (Figure S5 a–d), AFEs were calculated to be 2.11/2.54, 1.92/2.53, 1.61/2.34, and 2.63/2.98 for pitavastatin, rosuvastatin, atorvastatin, and fluvastatin with 300 and 600 mg RIF, respectively. Therefore, the inhibitory effect of RIF was overestimated, except for rosuvastatin whose in vitro RIF K i was not so different from CP‐I. The predicted (without correcting K i,u,OATP1Bs) vs. observed AUC and Cmax are shown in Figure S5 e,f.
Finally, the AUC ratio and Cmax ratio were calculated, as shown in Figure S5 g–j. In this instance, with correction of the in vivo K i based on differences of in vitro K i, the AFE for predicted vs. observed AUC ratio was 1.20, and that for the Cmax ratio was 1.36 (Figure S5 g,h). Without correction of the in vivo K i based on differences of in vitro K i, AFE values for the AUC ratio and Cmax ratio were higher (1.74 and 1.39, respectively; Figure S5 i,j).
Discussion
In this study, a PBPK model for CP‐I was constructed to provide mechanistic insight regarding the dose‐dependent inhibition of OATP1B and MRP2 by RIF. Sensitivity analyses demonstrated that model parameters including the in vivo K i,u,OATP1Bs, v syn, and CLint,all, could affect CP‐I blood concentration‐time profiles following RIF. After optimization of these parameters by nonlinear least‐squares fitting, blood concentration‐time profiles of statins affected by RIF were well‐predicted using substrate‐dependent K i values.
The initial value in our present analyses was based on the in vivo K i,u,OATP1Bs values of RIF, which were estimated to be 0.23 μM (vs. pitavastatin) and 0.19 μM (vs. pravastatin) in our previous PBPK‐DDI analyses.18 Parameter optimization yielded 0.37‐fold to 0.48‐fold of the initial K i,u,OATP1Bs (0.082–0.11 μM in Table 2) to explain the CP‐I blood concentration‐time profiles with RIF (Figure 2). Some substrate‐dependent differences in the sensitivity to OATP1B inhibitors in vitro, including RIF, have been reported.25 In order to support the intersubstrate difference suggested by the model‐based analysis in this study, we compared the inhibition profiles of RIF between CP‐I and the statins, by performing uptake experiments using OATP1B1/OATP1B3‐expressing HEK293 cells (Figure S2). The calculated K i values for OATP1B1 and OATP1B3 were 1.5‐fold to 3.6‐fold and 1.6‐fold to 4.8‐fold lower using CP‐I as a substrate than using statins (Table S2), which might support the lower estimated in vivo K i,u,OATP1Bs for CP‐I (Table 2) than for statins.18
By comparing the simulation results using the model incorporating dual OATP1B/MRP2 inhibition (Figure 2), to that incorporating OATP1B inhibition only (Figure S4), 75–85% of the observed AUC and 78–89% of the observed Cmax were explained by inhibition of OATP1Bs (Table 3), and the remaining by inhibition of MRP2. Thus, OATP1Bs had a major contribution to the magnitude of the CP‐I‐RIF interaction; however, the magnitude suggests that the influence of MRP2 inhibition on CP‐I pharmacokinetics should not be ignored during the assessment of DDIs.
Using the first model with an initial parameter setting (Figure S3), sensitivity analysis of K i,u,MRP2 from 0.33 to 3‐fold of the initial value affected blood concentrations in the presence of RIF (especially when β = 0.8) less significantly than K i,u,OATP1Bs. Actually, the contribution of RIF‐mediated inhibition of MRP2 was estimated to be ~10% of the AUC increase at most (β = 0.5 and 0.2). The reduced impact of K i,u,MRP2 on CP‐I blood concentrations is expected because of (i) increased sinusoidal efflux of CP‐I is offset by (ii) the decreased EHC (with F a F g of 0.66) of CP‐I, both due to the inhibition of MRP2‐mediated biliary excretion. On the other hand, the effect of MRP2‐inhibition was more clearly observed when using the final model with an optimized parameter setting (Figure 2), because (I) a larger v syn setting led to the more significant effect of RIF, and (II) a smaller F a F g setting (0.26–0.32) led to less contribution of EHC.
For some time it has been known that the urinary CP‐I/(CP‐I + CP‐III) ratio is associated with ABCC2 mutations and it has been suggested that the ratio can serve as a MRP2 trait measure.27 The increase in CP‐I/(CP‐I + CP‐III) ratio associated with Dubin–Johnson syndrome28, 29 might result from increased plasma concentration and urinary excretion of CP‐I due to the diminished MRP2‐mediated biliary excretion of CP‐I. Although MRP2 is also expressed on human kidney proximal tubule epithelia,30 inhibition of renal MRP2 was not incorporated in our PBPK model because the renal clearance of CP‐I is not affected by RIF.12
Yoshida et al.31 summarized the effects of Rotor Syndrome (RS) on the urinary excretion of CP‐I from six previous articles. It was concluded that the ratio of urinary excretion in healthy control (non‐RS) subjects vs. patients with RS (non‐RS/RS) was comparable across the various studies, ranging from 5–20% with the geometric mean of 9.8%. We simulated CP‐I blood concentrations in RS subjects using our PBPK model, assuming that (i) PSact,inf mediated by OATP1B1 and OATP1B3 is decreased to zero and (ii) v syn is unchanged in RS (Supplementary Text S1). The resultant simulated steady‐state CP‐I blood concentration for RS subjects was approximately sevenfold higher than that reported for reference (non‐RS) subjects (3.2 nM vs. 0.46 nM, as shown in Table S1 B). Assuming that renal clearance is unchanged in patients with RS, the ratio of urinary excretion (non‐RS/RS) should be 14%, which is within the range that has been reported.
We predicted the blood concentration‐time profiles of four statins in the presence of RIF (Figure 3 a–d), using substrate‐dependent K i,u,OATP1Bs values (Table S1 D) calculated by multiplying the K i,u,OATP1Bs for CP‐I (Table 2) by the ratio of in vitro K i,u for each statin to CP‐I (Table S2). The prediction accuracy (AFE) in the blood concentrations of statins in our clinical data13 was within 2, except for one case (600 mg RIF with rosuvastatin). In addition, the predicted AUC and Cmax of statins was well‐correlated with the observed AUC and Cmax (Figure 3 e,f). Furthermore, the AFE for predicted and observed AUC ratio is 1.20, and that for Cmax ratio is 1.36 (Figure S5 g,h). The differences between predicted and observed AUC ratios for pitavastatin and fluvastatin were larger than those for other compounds (overestimation). This might be caused by nonobserved dose‐dependence of RIF on the pharmacokinetics pitavastatin and fluvastatin in the clinical study by Takehara et al.13 For rosuvastatin and atorvastatin, the differences between the predicted and observed Cmax ratio were large (underestimation). The considerable reason for this is that RIF might affect the absorption process of these compounds by unknown mechanisms (e.g., increase in k a by intestinal efflux transporters, such as breast cancer resistance protein, MRP2, and P‐glycoprotein). Overall, the results of the present study support the predictive performance of our method for practical use in drug development.
Barnett et al.20 estimated in vivo K i,u for the RIF‐mediated inhibition of hepatic elimination clearance using empirical models for CP‐I and rosuvastatin. They showed the difference of in vivo RIF K i,u for the hepatic elimination of CP‐I (0.13 μM) and rosuvastatin (0.25 μM). Yoshida et al.31 have also adopted an empirical model for CP‐I. After estimating an in vivo RIF K i,u for the hepatic elimination (0.02 μM), the clinical DDI between pravastatin and RIF was well predicted. Thus, in vivo K i,u values estimated in these empirical models were sufficient for explanation of RIF‐mediated change in the blood concentration‐time profile of CP‐I. However, we should keep in mind that the obtained in vivo K i,u value is much lower compared with in vitro K i,u and IC50 values for CP‐I (0.18 and 0.78 μM in Table S2; 0.25 and 3.3 μM from the previous reports32, 33) and those for statins as other OATP1B substrates (0.30–2.8 μM in Table S2; 0.30–5.7 μM in the Drug Interaction Database). In vivo K i,u values estimated using the empirical models should not be applied to other probes directly, because (i) the hepatic clearance used in the empirical models is a hybrid parameter containing the hepatic blood flow and intrinsic clearances (uptake, basolateral efflux, metabolism, and biliary excretion) for CP‐I. By not considering the site of inhibition by RIF, one could greatly impact the estimation of in vivo K i,u; and (ii) the former studies did not consider the site of biosynthesis for CP‐I. Actually, the liver is the major organ responsible for the biosynthesis of CP‐I (see METHODS). Therefore, hepatic MRP2 function as well as OATP1Bs should be important determinants for CP‐I blood concentration. This should also affect the estimation of in vivo K i,u.
Thus, our PBPK modeling approach for CP‐I has the advantage and advancement from the previous empirical modeling approach in terms of consideration of the site of biosynthesis, rate‐determining process in the hepatic elimination, and the inhibition mechanisms, which enables the prediction of complex DDI involving a new chemical entity (NCE) via hepatic transport processes (OATP1Bs and MRP2) based on in vitro information, including substrate‐dependent K i, as we propose a workflow in Figure 4. (I) Concentration‐time profiles of given NCE obtained in an exploratory, first‐in‐human study (phase‐0) or in a phase‐1 study (dose escalation study) would provide an opportunity to construct a PBPK model, in which several parameters determined in vitro and in silico are incorporated according to our previous standardized method.18 Using the NCE's PBPK model and our PBPK model for CP‐I, in vivo K i,OATP1Bs for CP‐I can be obtained by analyzing its concentration‐time profiles in the absence and presence of the NCE. (II) In vitro K i,OATP1B values for CP‐I and for probe substrate drugs (e.g., statins) would be needed to calculate in vivo K i,OATP1Bs for the drugs according to the equation shown in Figure 4. (III) Finally, using the calculated in vivo K i,OATP1Bs for the probe substrate drugs, PBPK modeling and simulation would support the prediction of changes in substrate concentration‐time profiles, AUC, and Cmax caused by the NCE. Although some problems ascribed to experimental techniques in vitro remain to be solved (i.e., difficulty in obtaining reliable in vitro K i,u of highly lipophilic compounds due to nonspecific binding), translation of the effect of an NCE on CP‐I pharmacokinetics to effects on clinically used drugs is gradually becoming more accepted.5, 7, 12, 20, 34
Figure 4.

Scheme of the workflow for predicting drug‐drug interactions using coproporphyrin (CP)‐I as an endogenous biomarker. (I) Concentration‐time profiles of a new chemical entity (NCE) obtained in an exploratory, first‐in‐human study (phase‐0) or in a phase‐1 study (dose escalation study) will provide an opportunity to construct its physiologically based pharmacokinetic (PBPK) model, in which several parameters determined in vitro and in silico are incorporated according to our previous standardized method.18 Using PBPK models for the NCE and CP‐I, in vivo K i,OATP1Bs for CP‐I can be obtained by analyzing its concentration‐time profiles in the absence and presence of the NCE. (II) In vitro K i,OATP1Bs values for CP‐I and for probe substrate drugs (e.g., statins) will be needed to calculate in vivo K i,OATP1Bs for the drugs. (III) Using the calculated in vivo K i,OATP1Bs for the probe substrate drugs, PBPK modeling and simulation will enable us to predict changes in their concentration‐time profiles, area under the plasma concentration‐time curve (AUC) and peak plasma concentration (Cmax) caused by the NCE.
In conclusion, the PBPK modeling approach presented herein provides a deeper understanding of the mechanisms governing CP‐I pharmacokinetics and enables complex analyses of the dose‐dependent inhibitory effects of RIF on the hepatic OATP1Bs/MRP2‐mediated transport of CP‐I. Based on the in vivo K i for CP‐I optimized by PBPK modeling, and the ratio of in vitro K i for each statin to CP‐I, we showed that it is possible to accurately predict the blood concentration‐time profiles of OATP1B probe drugs affected by RIF at two dose levels. Presently, the vision is to expand the use the model to include additional OATP1Bs inhibitors beyond RIF.
Funding.
This study was financially supported by Grant‐in‐Aid for Scientific Research (S) (Grant 24229002) and Grant‐in‐Aid for Young Scientists (B) (Grant 17K15536) from the Ministry of Education, Culture, Sports, Science, and Technology in Japan.
Conflict of Interest
The authors declared no conflict of interest.
Author Contributions
T.Y., K.T., K.M., H.K., E.K., A.D.R., K.C., and Y.S. wrote the manuscript. T.Y., K.M., H.K., and Y.S. designed the research. T.Y., K.T., and Y.S. performed the research. T.Y. and Y.S. analyzed the data.
Supporting information
Figure S1. Uptake and efflux profiles of CP‐I in plated human hepatocytes.
Figure S2. Saturation of CP‐I uptake by OATP1B1, and inhibitory effects of RIF on OATP1B1‐mediated and OATP1B3‐mediated uptakes of CP‐I and statins in vitro.
Figure S3. Sensitivity analyses of model parameters to examine effects on the blood concentration‐time profiles of CP‐I in the absence and presence of RIF using the PBPK model incorporating the inhibitions of OATP1Bs and MRP2.
Figure S4. Simulation of blood concentration–time profiles of CP‐I in the absence and presence of RIF using parameters optimized as shown in Figure 2 and the PBPK model incorporating the inhibition of OATP1Bs alone.
Figure S5. Prediction of the effect of RIF on blood concentration–time profiles of statins using their PBPK models and parameters including substrate‐dependent K i,u,OATP1Bs (without correction of the in vivo K i based on differences of in vitro K i).
Table S1. Parameters used in the PBPK model for CP‐I and statins.
Table S2. IC50 values by RIF determined from the uptake experiments using OATP1B1‐ and OATP1B3‐expressing HEK293 cells, and calculated K i values.
Supplementary Text S1. Legends for the supplementary figures. Simulation design. PBPK model structure, parameters and equations. Material and methods for in vitro experiments.
References
- 1. Morrissey, K.M. , Wen, C.C. , Johns, S.J. , Zhang, L. , Huang, S.M. & Giacomini, K.M. The UCSF‐FDA TransPortal: a public drug transporter database. Clin. Pharmacol. Ther. 92, 545–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshida, K. , Maeda, K. & Sugiyama, Y. Transporter‐mediated drug–drug interactions involving OATP substrates: predictions based on in vitro inhibition studies. Clin. Pharmacol. Ther. 91, 1053–1064 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Elsby, R. , Hilgendorf, C. & Fenner, K. Understanding the critical disposition pathways of statins to assess drug‐drug interaction risk during drug development: it's not just about OATP1B1. Clin. Pharmacol. Ther. 92, 584–598 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Snoeys, J. , Beumont, M. , Monshouwer, M. & Ouwerkerk‐Mahadevan, S. Mechanistic understanding of the nonlinear pharmacokinetics and intersubject variability of simeprevir: a PBPK‐guided drug development approach. Clin. Pharmacol. Ther. 99, 224–234 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Rodrigues, A.D. , Taskar, K.S. , Kusuhara, H. & Sugiyama, Y. Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther. 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Yang, K. et al Systems pharmacology modeling of drug‐induced hyperbilirubinemia: differentiating hepatotoxicity and inhibition of enzymes/transporters. Clin. Pharmacol. Ther. 101, 501–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takehara, I. et al Investigation of glycochenodeoxycholate sulfate and chenodeoxycholate glucuronide as surrogate endogenous probes for drug interaction studies of OATP1B1 and OATP1B3 in healthy Japanese volunteers. Pharm. Res. 34, 1601–1614 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Thakare, R. et al Leveraging of rifampicin‐dosed cynomolgus monkeys to identify bile acid 3‐O‐sulfate conjugates as potential novel biomarkers for organic anion‐transporting polypeptides. Drug Metab. Dispos. 45, 721–733 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Watanabe, M. , Watanabe, T. , Yabuki, M. & Tamai, I. Dehydroepiandrosterone sulfate, a useful endogenous probe for evaluation of drug‐drug interaction on hepatic organic anion transporting polypeptide (OATP) in cynomolgus monkeys. Drug Metab. Pharmacokinet. 30, 198–204 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Yee, S.W. et al Metabolomic and genome‐wide association studies reveal potential endogenous biomarkers for OATP1B1. Clin. Pharmacol. Ther. 100, 524–536 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen, H. et al Coproporphyrins I and III as functional markers of OATP1B activity: in vitro and in vivo evaluation in preclinical species. J. Pharmacol. Exp. Ther. 357, 382–393 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Lai, Y. et al Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J. Pharmacol. Exp. Ther. 358, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Takehara, I. et al Comparative study of the dose‐dependence of OATP1B inhibition by rifampicin using probe drugs and endogenous substrates in healthy volunteers. Pharm. Res. 35, 138 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Beukeveld, G.J. , Meerman, L. , Huizenga, J.R. , Venekamp‐Hoolsema, E.E. , Gips, C.H. & Wolthers, B.G. Determination of porphyrins in bile using high performance liquid chromatography and some clinical applications. Eur. J. Clin. Chem. Clin. Biochem. 32, 153–159 (1994). [DOI] [PubMed] [Google Scholar]
- 15. Gilibili, R.R. et al Coproporphyrin‐I: a fluorescent, endogenous optimal probe substrate for ABCC2 (MRP2) suitable for vesicle‐based MRP2 inhibition assay. Drug Metab. Dispos. 45, 604–611 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Takashima, T. et al Positron emission tomography studies using (15R)‐16‐m‐[11C]tolyl‐17,18,19,20‐tetranorisocarbacyclin methyl ester for the evaluation of hepatobiliary transport. J. Pharmacol. Exp. Ther. 335, 314–323 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Takashima, T. et al PET imaging‐based evaluation of hepatobiliary transport in humans with (15R)‐11C‐TIC‐Me. J. Nucl. Med. 53, 741–748 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Yoshikado, T. et al Quantitative analyses of hepatic OATP‐mediated interactions between statins and inhibitors using PBPK modeling with a parameter optimization method. Clin. Pharmacol. Ther. 100, 513–523 (2016). [DOI] [PubMed] [Google Scholar]
- 19. Asaumi, R. et al Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug‐drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacometrics Syst. Pharmacol. 7, 186–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barnett, S. et al Gaining mechanistic insight into coproporphyrin I as endogenous biomarker for OATP1B‐mediated drug‐drug interactions using population pharmacokinetic modeling and simulation. Clin. Pharmacol. Ther. 104, 564–574 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kushner, J.P. , Barbuto, A.J. & Lee, G.R. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J. Clin. Invest. 58, 1089–1097 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moran, M.J. et al Hepatic uroporphyrinogen decarboxylase activity in porphyria cutanea tarda patients: the influence of virus C infection. Hepatology 27, 584–589 (1998). [DOI] [PubMed] [Google Scholar]
- 23. Koskelo, P. & Kekki, M. Multicompartment analysis of 14C‐labelled coproporphyrin and uroporphyrin kinetics in human beings. Ann. Clin. Res. 8(suppl. 17), 198–202 (1976). [PubMed] [Google Scholar]
- 24. Hisaka, A. & Sugiyama, Y. Analysis of nonlinear and nonsteady state hepatic extraction with the dispersion model using the finite difference method. J. Pharmacokinet. Biopharm. 26, 495–519 (1998). [DOI] [PubMed] [Google Scholar]
- 25. Izumi, S. et al Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug‐drug interactions. Drug Metab. Dispos. 43, 235–247 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Futatsugi, A. , Toshimoto, K. , Yoshikado, T. , Sugiyama, Y. & Kato, Y. Evaluation of alteration in hepatic and intestinal BCRP function in vivo from ABCG2 c.421C>A polymorphism based on PBPK analysis of rosuvastatin. Drug Metab. Dispos. 46, 749–757 (2018). [DOI] [PubMed] [Google Scholar]
- 27. Benz‐de Bretagne, I. et al Urinary elimination of coproporphyrins is dependent on ABCC2 polymorphisms and represents a potential biomarker of MRP2 activity in humans. J. Biomed. Biotechnol. 2011, 498757 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frank, M. , Doss, M. & de Carvalho, D.G. Diagnostic and pathogenetic implications of urinary coproporphyrin excretion in the Dubin‐Johnson syndrome. Hepatogastroenterology 37, 147–151 (1990). [PubMed] [Google Scholar]
- 29. Toh, S. et al Genomic structure of the canalicular multispecific organic anion‐transporter gene (MRP2/cMOAT) and mutations in the ATP‐binding‐cassette region in Dubin‐Johnson syndrome. Am. J. Hum. Genet. 64, 739–746 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaub, T.P. et al Expression of the MRP2 gene‐encoded conjugate export pump in human kidney proximal tubules and in renal cell carcinoma. J. Am. Soc. Nephrol. 10, 1159–1169 (1999). [DOI] [PubMed] [Google Scholar]
- 31. Yoshida, K. , Guo, C. & Sane, R. Quantitative prediction of OATP‐mediated drug‐drug interactions with model‐based analysis of endogenous biomarker kinetics. CPT Pharmacometrics Syst. Pharmacol. 7, 517–524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen, H. et al Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab. Dispos. 45, 908–919 (2017). [DOI] [PubMed] [Google Scholar]
- 33. Bednarczyk, D. & Boiselle, C. Organic anion transporting polypeptide (OATP)‐mediated transport of coproporphyrins I and III. Xenobiotica 46, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Kunze, A. , Ediage, E.N. , Dillen, L. , Monshouwer, M. & Snoeys, J. Clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B‐mediated drug‐drug interactions. Clin. Pharmacokinet; e‐pub ahead of print 16 April 2018. doi: 10.1007/s40262-018-0648-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Uptake and efflux profiles of CP‐I in plated human hepatocytes.
Figure S2. Saturation of CP‐I uptake by OATP1B1, and inhibitory effects of RIF on OATP1B1‐mediated and OATP1B3‐mediated uptakes of CP‐I and statins in vitro.
Figure S3. Sensitivity analyses of model parameters to examine effects on the blood concentration‐time profiles of CP‐I in the absence and presence of RIF using the PBPK model incorporating the inhibitions of OATP1Bs and MRP2.
Figure S4. Simulation of blood concentration–time profiles of CP‐I in the absence and presence of RIF using parameters optimized as shown in Figure 2 and the PBPK model incorporating the inhibition of OATP1Bs alone.
Figure S5. Prediction of the effect of RIF on blood concentration–time profiles of statins using their PBPK models and parameters including substrate‐dependent K i,u,OATP1Bs (without correction of the in vivo K i based on differences of in vitro K i).
Table S1. Parameters used in the PBPK model for CP‐I and statins.
Table S2. IC50 values by RIF determined from the uptake experiments using OATP1B1‐ and OATP1B3‐expressing HEK293 cells, and calculated K i values.
Supplementary Text S1. Legends for the supplementary figures. Simulation design. PBPK model structure, parameters and equations. Material and methods for in vitro experiments.