

Abstract
Fluorescence resonance energy transfer (FRET) technology is a useful tool to monitor protein interactions as well as protease activity. We have recently reported a biochemical assay utilizing a FRET reporter peptide to monitor the activity of the 20S catalytic particle (20S CP) of the proteasome. This assay is designed specifically to have increased sensitivity to identify stimulators of the 20S CP, which may hold therapeutic potential to treat protein accumulation diseases. The protocol described here details the necessary steps in synthesizing the FRET reporter peptide and performing the FRET assay with the 20S CP.
Keywords: Proteasome, Stimulation, Screening, Peptide
INTRODUCTION
The 20S catalytic particle (20S CP) of the proteasome plays many vital roles to maintain healthy cell function. When the 20S CP is capped by the 19S regulatory particle (19S RP) it can perform ubiquitin-mediated protein hydrolysis (Bhattacharyya, 2014). If the 20S CP is not associated with the 19S RP it can only degrade damaged or intrinsically disordered proteins in a ubiquitin-independent manner (Ben-Nissan, 2014). In diseases, such as Parkinson’s or Alzheimer’s, these types of proteins accumulate and lead to apoptosis (Irvine, 2008). It has been hypothesized that the 20S CP could be stimulated as a therapeutic target to decrease the amount of stress associated with protein accumulation, limiting or potentially reversing any protein accumulation related toxicity. Unfortunately, the methods commonly used to monitor proteasome activity do not possess the appropriate sensitivity to identify a broad range of small molecule stimulators of the 20S CP. We have therefore developed a FRET reporter to be used in a biochemical assay to screen for stimulators of the 20S CP (Coleman, 2018).
Our reporter is 11 amino acids in length, seven amino acids longer than the commonly utilized coumarin reporters. To generate a FRET reporter, it must include a fluorescent donor moiety, which absorbs and emits lights, and an acceptor moiety, which absorbs light in a range similar to the emission wavelength of the donor moiety. When the donor and acceptor are in close proximity, FRET occurs, reducing the measurable signal that could be produced by the FRET donor. Utilizing a FRET donor and acceptor in a reporter peptide allows one to monitor protease activity by recording the emission wavelength intensity of the FRET donor. Once the protease has cleaved the peptide between the FRET donor and acceptor, the proximity between these two moieties increases, preventing FRET from occurring and leading to an increase in signal at the donor emission wavelength (Figure 1A). Inspired by a published FRET reporter to monitor HIV protease activity (Jin, 2011), we chose the FRET pair EDANS (donor; ex. 335, em. 495) and Dabcyl (acceptor; ex. 473) to use for our FRET peptide (Figure 1B).
Figure 1.
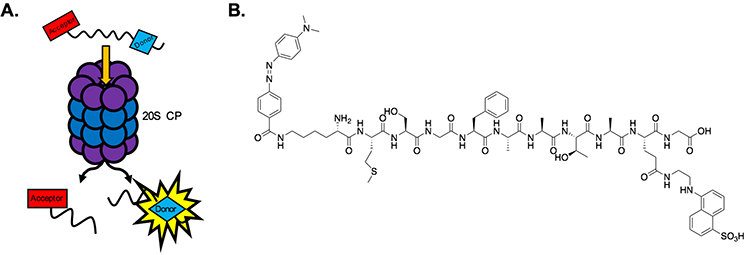
A) Hydrolysis of the FRET reporter peptide by the 20S CP. Once the peptide has been cleaved, a measurable signal is produced by the FRET donor moiety. B) Structure of the FRET reporter peptide: NH2-Lys(Dabcyl)-Met-Ser-Gly-Phe-Ala-Ala-Thr-Ala-Glu(EDANS)-Gly-OH.
We hypothesized that a longer peptide would be harder for the 20S CP to accept without any stimulation, leading to a significant increase in the dynamic range of molecules that could be detected during a screening campaign. The length of our FRET reporter peptide was therefore chosen in order to achieve the longest peptide possible while still maintaining the appropriate distance between the donor and acceptor to maintain FRET. The Forster radius, or the distance at which the energy transfer between the FRET donor and acceptor is 50% efficient, for our chosen FRET pair is 30 angstroms. For our FRET peptide, the distance between EDANS and Dabcyl is approximately 30–35 angstroms, maintaining a high degree of FRET efficiency. This is important to achieve a high signal-to-noise ratio.
BASIC PROTOCOL 1
FRET PEPTIDE SYNTHESIS
Described below is the general procedure for synthesizing the FRET reporter peptide. The FRET reporter is synthesized using solid-phase peptide synthesis (SPPS) procedures. We chose to utilize Fmoc-Gly-Wang resin to obtain a carboxylic acid at the C-terminus, which slightly improves solubility. In addition, using a preloaded resin makes coupling the first amino acid easier, especially when trying to couple an unnatural amino acid containing a fluorophore, by creating a greater distance from the resin to decrease steric hindrance. In accordance with SPPS procedures, the peptide is synthesized in the C- to N-terminus direction. The amino acids between the FRET pair were chosen for cost effectiveness, ease of coupling, and to increase solubility. To increase the efficiency of each coupling, the reaction is heated at 60 °C with agitation. This is usually sufficient to push all coupling reactions to completion within 1 hr. However, any reaction that is unsuccessful as determined by the Kaiser test, is repeated. After all of the coupling reactions are completed successfully, the peptide is deprotected and cleaved from the resin using a strong acid before HPLC purification and lyophilization. The lyophilized FRET peptide powder is then used to make the FRET solution for the 20S CP activity assay.
Materials
Fmoc-Gly-Wang Resin (Cat. #01908; Chem-Impex Int’l INC.)
Dichloromethane (DCM)
N-N-Dimethylformamide (DMF)
Piperidine
Kaiser reagents:
Solution 1: 5 g ninhydrin in 100 mL ethanol
Solution 2: 80 g phenol in 20 mL ethanol
Solution 3: 2 mL 1 mM aqueous KCN in 98 mL pyridine
COMU coupling reagent (CAS #1075198–30-9)
Fmoc-protected natural amino acids: Fmoc-Ala-OH, Fmoc-Thr(tBu)-OH, Fmoc-Phe-OH, Fmoc-Gly-OH, Fmoc-Ser(tBu)-OH, Fmoc-Met-OH
Fmoc-protected unnatural amino acids containing FRET donor and acceptor: Fmoc-Glu(EDANS)-OH and Fmoc-Lys(Dabcyl)-OH (purchased from Chem-Impex Int’l INC.)
Diisopropylethylamine (DIPEA)
Trifluoroacetic acid (TFA)
Triisoproylsilane (TIPS)
Fritted syringe (reaction vessel)
Safety Note: All reagents should be dispensed in a chemical fume hood. All proper PPE should be worn, including chemical resistant gloves. Always use extreme caution when using TFA and be careful when opening resin vessel as it can become pressurized during peptide cleavage.
FRET Peptide Synthesis Protocol
Weigh the appropriate amount of Fmoc-Gly-loaded Wang Resin into a fritted syringe.
Swell the resin in a 1:1 ratio of DCM:DMF for 30 min at room temperature (RT) with shaking.
Deprotect the resin (remove the Fmoc) in 20% piperidine in DMF at RT with shaking for 20 min. Drain the solution and add fresh 20% piperidine in DMF, allowing the syringe to shake at RT for another 20 min. Drain the solution.
After draining the solution, wash the resin first with DMF 3x, followed by 3 washes with DCM.
-
Perform Kaiser test:
- Carefully remove a few beads of resin from the syringe and place into a small tube.
- Add 2 drops of each Kaiser reagent.
- Heat the mixture at 110–120 °C for 2 min.
- Analyze the color of the beads: blue/purple (the resin is deprotected), yellow/clear (the resin is protected)
The Kaiser test is used to determine the state of the resin by indicating the presence of free primary amines.
Interpreting results: Blue/purple indicates there is a free amine present, i.e. that the deprotection was successful, or that the coupling was unsuccessful. Yellow/clear indicates that the deprotection was unsuccessful, or that the coupling was successful as there is no free amine present.
-
Following a successful deprotection, mix the coupling reagents as follows:
- Weigh out the appropriate amount of amino acid (4 molar equivalents) and COMU (4 molar equivalents).
- Dissolve the solids in as little DMF as possible.
- Add 8 molar equivalents of DIPEA.
- Agitate for 2 minutes.
Ex. If you started with 50 mg of the Fmoc-Gly-loaded Wang resin with a loading capacity of 0.658 meq/gram:
(0.05 g) × ( 0.658 meq/g) = 3.29 × 10−2 meq
To determine the number of moles of reagents you need:
Amino acids: (3.29 × 10−2 meq) × 4 = 0.132 mmol
COMU: (3.29 × 10−2 meq) × 4 = 0.132 mmol
DIPEA: (3.29 × 10−2 meq) × 8 = 0.263 mmol
Converting moles to grams:
Using Fmoc-Glu(EDANS)-OH: 617.68 g/mol
(1.32 × 10−4 mol) × (617.68 g/mol) = 81.5 mg
COMU: 428.27 g/mol
(1.32 × 10−4 mol) × (428.27 g/mol) = 56.5 mg
DIPEA: 129.25 g/mol
(2.63 × 10−4 mol) × (129.25 g/mol) = 34.0 mg
Converting grams to liters:
DIPEA: density - 0.742 g/mL Add the coupling reagent mixture to the resin. Heat the reaction vessel at 60 °C for 1 hr while shaking.
-
After the 1 hr reaction period, repeat steps 4 and 5 to determine whether the reaction was successful.
If the coupling was unsuccessful, repeat the reaction with fresh reagents until it is deemed successful by the Kaiser test.
-
Following a successful coupling, repeat steps 3–8 until all of the amino acids have been successfully coupled.
Coupling Fmoc-Lys(Dabcyl)-OH may stain the resin and make it difficult to determine the results of the Kaiser test. It is helpful to save the results of the previous Kaiser test for comparison.
- Cleave the peptide from the resin following the successful deprotection of the last amino acid.
- Create a solution of 95% TFA, 2.5% TIPS, and 2.5% DCM – be extremely careful when working with TFA. TFA should only be used in a chemical hood and by a trained user.
- Add the solution to the resin.
- Shake the reaction at RT for 2 hr. Open vessel in hood and take extra care as pressure could have built in the resin vessel.
Collect the solution from the reaction vessel, as this will contain the peptide. Rinse the resin with DCM three times and collect the rinses.
Using argon or nitrogen gas, blow off the TFA and DCM until you have a small residue at the bottom of the tube of 100 μL or less.
-
Add 5 mL diethyl ether to the residue and agitate for 5 min.
The peptide is insoluble in ether; this step is used to crash the peptide out of solution to separate it from other impurities that may be soluble.
Centrifuge the solution at 4700 × g to collect the solid peptide at the bottom of the tube. Decant the ether.
Repeat steps 13 and 14 two more times.
Following the last decantation, insert a small hole in the container using a needle, and place the tube in a vacuum sealed container overnight. This is performed to remove all remaining ether from the solid.
-
Dissolve the solid and purify the peptide by high-pressure liquid chromatography (HPLC) or by other methods available to you.
You may need to first dissolve the solid in as little DMSO as possible before diluting with HPLC solvents, such as water, acetonitrile, or methanol. Be careful the peptide does not crash out of solution.
If you do use HPLC for purification, set up the method as follows:
Begin with an isocratic elution of 95% Solution A (100% Water) for 2 min pumping at 15 mL/min.
Followed by a linear gradient of 5–95% Solution B (100% Acetronitrile) over a 16 min period.
Perform an isocratic elution of 95% Solution B for 2 min.
Re-equilibrate with 95% Solution A for 2 min.
Broadly collect fractions for the first run. Once you know where the peptide will elute, collect fractions in that region. The samples containing the peptide will be pink to red in appearance. This method works with an Agilent Preparatory 1200 system with a Zorbax Eclipse XDB-C18 9.4 × 250 mm, 5 μm particle size.
-
Verify the identity of the peptide by mass spectrometry.
Liquid chromatography mass spectrometry (LC-MS) is the preferred method and will also give insight into the purity of the peptide.
-
Once the FRET peptide has been purified, lyophilize the fractions to obtain the peptide as a solid powder.
It is helpful to weigh the container before collecting the fractions to be lyophilized. After obtaining the powder, weigh the container again and calculate the difference in order to attain the total mass of your peptide. Notably, the powder will be red in appearance.
Basic Protocol 2
20S CP FRET ASSAY
In this section, we detail the necessary steps to set up and perform the FRET assay, using the FRET reporter peptide. This biochemical assay is best suited to screen for stimulators of the 20S CP; however, it can also be used to look at inhibition of the proteasome. Before beginning, ensure you have access to a plate reader that can read fluorescence intensity in the excitation and emission region of the fluorophore, EDANS (ex. 335 nm, em. 493 nm). The steps involved in setting up a method on the plate reader to perform the kinetic assay are detailed below. In addition, we will instruct how to prepare your samples: the FRET solution, test samples, and controls; and how to prepare the plate before placing it into the plate reader for analysis. At the end of this section will be a discussion of how to analyze the data and interpret the results.
Materials
Plate reader that can heat to 37 °C (Synergy plate reader, Tecan plate reader, etc.)
Tris-HCl (50 mM, pH 7.6)
Dimethylsulfoxide for molecular biology (DMSO)
Small molecules for screening
Lyophilized FRET peptide
20 μM FRET solution
Tris-HCl (50 mM, pH 7.6)
Control solution
Small molecule solutions
90 nM 20S CP, diluted in Tris-HCl (purchased or purified)
96-well plate (black) or 384-well plate (black)
Creating a Plate Reader Method
Set the method to read fluorescence intensity.
-
Assign the excitation and emission wavelengths as 335 nm and 493 nm, respectively, corresponding to the excitation and emission wavelengths of Edans.
If you are unable to specify an exact wavelength, ensure that the wavelengths above are within the ranges listed as the excitation and emission wavelengths for your specific plate reader.
Ensure that the method is set up as a kinetic assay. This will allow for fluorescent readings over time.
Set the method to read the fluorescence intensity every 2 min over a 1 hr period. The 2 min interval ensures that a sufficient number of readings will be taken to achieve a linear slope for even the most potent of stimulators.
Finally, set the plate reader to have an internal temperature of 37 °C, which should be reached prior to adding the prepared plate.
The 20S CP functions best at physiological temperature. Setting the temperature to 37 °C also provides a better indication of how the small molecule might affect the 20S CP in cells.
Preparing small molecule solutions and control solution
-
6.
Determine the solvent most appropriate to dissolve the small molecules you wish to test.
Oftentimes, these small molecules are not water soluble. However, if the most suitable solvent is water, use the Tris-HCl buffer (50 mM, pH 7.6). The solvent most often used is DMSO.
If you do use DMSO, make the concentration at least 50x the concentration you wish to test in the assay. This is to ensure that the concentration of DMSO in the assay is at most 2%. If you must use more DMSO, be careful that the final concentration of DMSO in the assay is not greater than 5% as a higher concentration will dramatically affect the activity of the 20S CP.
Ex. Testing AM-404, which is often used as a positive control at 25 μM.
Calculating the number of moles of AM-404:
From the number of moles, we can calculate how much DMSO we need to obtain a 1250 μM stock solution (testing at 25 μM):You can make a more concentrated solution to dilute later if necessary.
-
7.
Dilute the stock solution to 10x that you wish to test in the assay.
If you use a solvent other than Tris-HCl to dissolve the small molecule(s), ensure your 10x solution is a maximum of 50% of that solvent. Many solvents, such as DMSO and ethanol, are structurally destructive to the proteasome and can interfere with results.
Ex. Diluting AM-404 to 250 μM in 20% DMSO.
Stock solution: 1250 μM in 100% DMSO
We want 500 μL of 250 μM AM-404 in 20% DMSO100 μL of 1250 μM AM-404 should be diluted in 400 μL Tris-HCl to produce a 250 μM solution of AM-404 that is 20% DMSO.
-
8.
Make a control solution like that described in step two, lacking the small molecule.
This is especially important if you use a solvent like DMSO. You want to ensure that any effect on the activity of the proteasome is due to the small molecule and not a result of the solvent used. This solution is used to calculate the basal level of activity of the 20S CP.
Preparation of FRET solution
-
9.
Using the purified, lyophiized FRET peptide, make a 10 mM stock solution in DMSO.
Ex. If you have 1 mg of FRET peptide, you need to know the number of moles in 1 mg:
From the number of moles, we can calculate how much DMSO we need to obtain a 10 mM stock solution:This solution can be aliquoted and stored at −80 °C for later use. It’s important to avoid multiple freeze-thaw cycles, as this can damage the peptide and affect the quality of the assay.
-
10.
Dilute the 10 mM FRET peptide solution in Tris-HCl (50 mM, pH 7.6) to obtain a 20 μM solution.
The amount of solution you need for your assay will depend on the number of samples you have and the size plate you are using.
Ex. If you have three samples you would like to test in triplicate, you will be using 15 wells of your 96- or 384-well plate (6 of these wells are to run your 2 controls in triplicate). To ensure you will have enough of the solution for all of your wells, you should use the x+1 rule. In this case, × refers to the number of individual samples you have, including your controls, and would be equal to 5 total samples. Following the x+1 rule, you should make enough of the 20 μM FRET solution to run 6 sets of triplicates, or 18 wells of your plate.
96-well plate:
For this sized plate, you will be adding 40 μL of the 20 μM FRET solution to each of the wells.
To calculate the total amount of solution you need:
18 wells × (40 μL/well) = 720 μL
To calculate the amount of 10 mM (or 10,000 μM) stock solution you need to dilute:384-well plate:
For this sized plate, you will be adding 20 μL of the 20 μM FRET solution to each of the wells.
To calculate the total amount of solution you need:
18 wells × (20 μL/well) = 360 μL
To calculate the amount of 10 mM (or 10,000 μM) stock solution you need to dilute:
Preparing the plate and plate reader set Up
-
11.
Add 40 μL (96-well plate) or 20 μL (384-well plate) of your 20 μM FRET solution to each of the wells.
If possible, try not to use the wells around edges of the plate. This should help reduce any interference that may occur from evaporation. To keep your samples in order, it’s easiest to use a different column or row for each sample (Fig. 2).
-
12.
To your first control, containing “No 20S CP,” add 5 μL (96-well plate) or 2.5 μL (384-well plate) Tris-HCl.
-
13.
To both of your control samples (“No 20S CP” and “Basal Level”), add 5 μL (96-well plate) or 2.5 μL (384-well plate) of the control solution.
-
14.
Ensure the plate reader has reached 37 °C.
-
15.
To the “Basal Level” and “Test Sample” wells, add 5 μL (96-well plate) or 2.5 μL (384-well plate) of the 90 nM 20S CP solution, resulting in a final concentration of 9 nM 20S CP in those wells. After the addition of the 20S CP, ensure that all the liquid in the wells is at the bottom of the plate.
This is most efficiently done using a plate spinner; however, if a plate spinner is unavailable, gently tap the edges of the plate to try to get all the liquid to the bottom.
This should be done fairly quickly and immediately prior to analyzing the plate with the plate reader.
-
16.
Place the plate in the plate reader and begin analyzing the fluorescent signal, using the method described previously.
Figure 2.
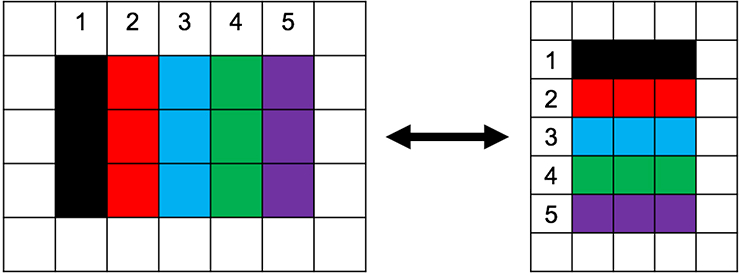
Triplicate sample orientation to limit potential error. Avoiding the edges of the plate helps prevent any interference that might occur from evaporation of the sample. In the example shown above, the samples are as follows: #1, No 20S CP; #2, Basal Level; #3, Test Sample 1; #4, Test Sample 2; #5, Test Sample 3.
Data Analysis
-
17.
Plot the relative fluorescence units (RFU) against time using a program such as GraphPad Prism or Microsoft Excel.
If using Microsoft excel, take the average and standard deviation of all the triplicates for each sample. GraphPad Prism will allow you to input each of the triplicates and will calculate the average and standard deviations for each time point for you.
-
18.
Perform a linear derivation of the data points.
Note that the curves produced by the data points may not be a line. In this case, reduce the number of data points to the linear region. This usually approximates the time range to 40 min, rather than 60 min. This must be performed for all data sets for appropriate comparison.
-
19.
Obtain the slopes for each of the lines.
The slope for the “No 20S CP” samples should be around zero. If the slope is greatly positive, that might indicate that the FRET peptide has destabilized, and a fresh solution should be used. Generally, the slope is approximated to the level of activity of the 20S CP, with the exception of the “No 20S CP” control samples. For example, the slope obtained for the “Basal Level” control samples will be considered the basal level of activity to which you will compare your test samples. Talk more here about the controls. Especially the basal level.
-
20.
Calculate the change in activity produced by your test samples compared to the “Basal Level” control.
For example, if the slope you obtain for the “Basal Level” is 5.6 RFU/min and that you obtain for the Test Sample is 20.3 RFU/min:
COMMENTARY
Background Information
The ubiquitin-proteasome system (UPS) is the prominent protein degradation pathway in cells and its activity is necessary for cells to maintain proper protein homeostasis, or proteostasis (Bhattacharyya, 2014). In cells, proteins can become damaged due to oxidative stress, disease, errors in transcription and/or translation, and many other mechanisms, which makes the proteins ineffective. These damaged or non-functional proteins must be degraded by the UPS or through the autophagy pathway. (Dunn Jr., 1994). In the UPS, proteins are tagged with a poly-ubiquitin chain, which targets them for degradation by the 26S proteasome (Hochstrasser, 1995). The 26S proteasome is comprised of one 19S regulatory particle (19S RP) and one 20S catalytic particle (20S CP). The 19S RP is responsible for recognizing and removing the ubiquitin tag, unfolding the protein, and shuttling it into the opened gate of the 20S CP. The 20S CP houses the catalytically active subunits that hydrolyze the protein substrates into small peptides (Nandi, 2006). The 20S CP is comprised of four heptameric protein rings of the form α7β7β7α7, forming a barrel-like structure. The β-subunits are responsible for structural integrity and contain the catalytic sites. Specifically, the β1-, β2-, β5-subunits perform caspase-like, trypsin-like, and chymotrypsin-like activities, respectively (Orlowski, 2000). The α-subunits form the gate of the 20S CP, which is in a closed state with an opening of only 13 angstroms when the 20S CP is not capped by the 19S RP or another gate-opening moiety (Groll, 2000).
In addition to its role in the UPS, the 20S CP also has an important function in disease states. In a number of diseases including Parkinson’s and in aging cell models, the expression of the 19S RP is greatly diminished, which limits the amount of ubiquitin-dependent degradation that can occur (Husom, 2004; McNaught, 2001). These disease states are also characterized by an accumulation of oxidatively damaged proteins and intrinsically disordered proteins, such as α-synuclein, which form cytotoxic aggregates, resulting in cell death (Spillantini, 1997). The 20S CP, without a regulatory cap, is capable of degrading oxidatively damaged and intrinsically disordered proteins (Ben-Nissan, 2014), through the ubiquitin-independent proteasome system (UIPS, Opoku-Nsiah, 2018). It has been hypothesized that a small molecule could enhance the activity of the 20S CP by performing a similar gate-opening mechanism as the 19S RP and therefore increase the degradation of accumulated proteins. A few small molecules capable of modulating the activity of the 20S CP have been discovered and have shown success in enhancing the degradation of proteins, such as α-synuclein and tau (Trader, 2017; Coleman, 2018, Jones, 2017). Unfortunately, current methods of monitoring proteasome-mediated hydrolysis are not well-suited for screening for gate-opening small molecules, leading to the development of the FRET reporter peptide described here.
The protocol for the FRET assay to analyze proteasome-mediated hydrolysis described above is similar to that used with the 3- to 4-amino acid coumarin reporters that have been used for decades (Kisselev, 2005). These coumarin reporters are each specific to one activity of the 20S CP and easily enter the closed gate of the 20S CP to be hydrolyzed, resulting in fluorescence. These reporters are efficient at analyzing the various activities of the 20S CP and at identifying inhibitors of the 20S CP; however, using these reporters to screen for gate-opening stimulators of the 20S CP can result in many false negatives by failing to identify weak modulators, which hold therapeutic value. Therefore, we decided to create a larger reporter more specifically geared to screening for gate-opening stimulators of the 20S CP. This reporter is more than three-times more sensitive to these types of stimulators than the coumarin reporters previously used (Fig. 3).
Figure 3.
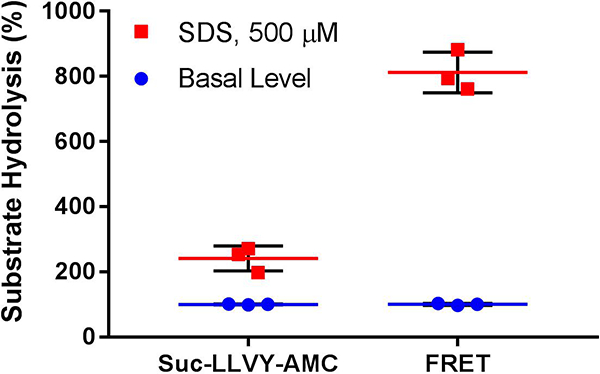
In triplicate, a coumarin reporter (Suc-LLVY-AMC) of proteasome activity was used to determine the stimulating ability of 500 μM sodium dodecyl sulfate (SDS). For comparison, this was also performed using the FRET reporter. The basal level of activity determined by each reporter was normalized to 100%. Using the coumarin reporter, SDS stimulated the hydrolysis activity 2.4x, whereas FRET reporter hydrolysis was stimulated 8.3x. Therefore, the FRET reporter increases the dynamic range of measurable stimulated activity.
Critical Parameters
FRET peptide purity
It is important for the FRET reporter to be > 95% pure. Other products that can result from the synthesis can interfere with the results of the assay by interfering with the FRET of the peptide. It is also possible that smaller FRET peptides could be produced, which would have less sensitivity to stimulation and generate skewed results.
Black 96- or 384-well plate
The plate used for this assay should be solid black to prevent any interference from reflection or light outside of the plate. In addition, the plate used should be made of polystyrene or a similar material and be untreated. There have been reports of plates affecting results, therefore once a plate type is selected we recommend using that plate brand for all further assays.
Small molecule screening
In order to achieve accurate results, it is important that the small molecules being screened do not absorb or emit light in the ranges involved in the FRET between the donor and acceptor on the FRET peptide. It is recommended that an initial scan is performed to remove any molecules that show fluorescent signal in wavelengths associated with the FRET peptide. This is done by performing the assay using the small molecules at the intended testing concentration in Tris-HCl. No FRET reporter or 20S CP should be used in this assay. Any small molecule that displays a high degree of fluorescent signal or a change in the fluorescent signal over time cannot be accurately assessed using the FRET assay and should be removed from the screen.
Troubleshooting
| Problem | Solution |
|---|---|
| The control samples containing “No 20S CP” are showing an increase in signal over time. |
Discard current FRET reporter solution and make more from stored aliquots. |
| The signal produced by the sample with the small molecule is lower/higher than that in the control samples but still increases over time. |
Perform assay looking at intrinsic signal of small molecule and effect the small molecule has on the FRET reporter. Background: Tris-HCl and small molecule at screening concentration Effect on reporter: 20 μM FRET solution and small molecule at screening concentration If the fluorescence increases over time in either sample, this small molecule cannot be screened by this assay. |
| The signal produced by the sample with the small molecule is higher than that in the control samples and decreases over time. |
This small molecule cannot be screened in this assay. |
| There is no increase in signal in the control “Basal Level” samples, but there is in the positive control samples. |
Increase the gain in the settings for the method. |
| There is no increase in signal in any of the samples. |
Double check that the emission wavelength is set to 493 nm (or a range around 493 nm). Try increasing the gain. If the emission wavelength is set correctly and increasing the gain does not fix the problem, the 20S CP may not be active. This could be due to deterioration of the sample over time, in which case fresh 20S CP will need to be used. |
Understanding Results
Synthesizing the FRET peptide is straightforward. The successful synthesis should be verified by mass spectrometry, and the purity should be analyzed by HPLC.
There can be several results of the FRET assay, and all the possibilities are detailed below:
20S CP Stimulation:
In this scenario, the slope obtained by the sample containing the small molecule is greater than that obtained for the basal level control. It is also important that the initial fluorescence readings for the test sample and the controls are in the same range (i.e. y-intercepts are similar). This is to ensure that there is no intrinsic fluorescence associated with the small molecule being tested. An example of the results for 20S CP stimulation is shown in Fig. 4.
Figure 4.

Simulated example data of 20S CP stimulation. The small molecule being tested is able to stimulate the 20S CP well over the basal level of activity. The y-intercepts of all of the samples are similar, indicating there is no intrinsic fluorescence associated with the small molecule.
20S CP Inhibition:
Inhibition of the 20S CP will result in a smaller slope associated with the sample containing the small molecule compared to that of the basal level control. Similarly, the y-intercepts of all of the samples should be around the same range. An example of 20S CP inhibition is shown in Fig. 5.
Figure 5.

Simulated example data of 20S CP inhibition. The slope corresponding to the small molecule being tested is less than that of the basal level of activity, indicating inhibition.
No effect on the 20S CP:
If the small molecule being tested has no effect on the 20S CP, the slope obtained for this test sample will not be significantly different from that obtained for the basal level control, Fig. 6.
Figure 6.
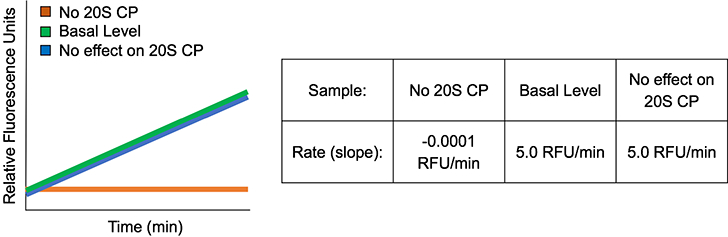
Simulated example data of a small molecule that has no effect on the 20S CP. The rate of hydrolysis of the FRET peptide does not change in the presence of the small molecule.
Inconclusive/Small molecule cannot be analyzed in this assay:
Small molecules that cannot be analyzed using the assay are those that possess intrinsic fluorescence in the range of the emission wavelength of EDANS (493 nm) or have properties that interfere with the FRET of the peptide. A molecule that has intrinsic fluorescence will have a y-intercept greatly different from that of the controls, Fig. 7. This fluorescence intensity will likely change over time by either decaying with successive readings or increasing with each excitation. Small molecules that interfere with the FRET may display erratic fluorescence readings or have y-intercepts much lower than that of the controls.
Figure 7.
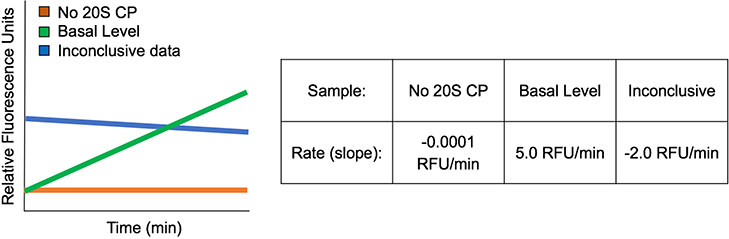
Simulated example data of a small molecule that cannot be tested using the FRET assay. The y-intercept corresponding to the sample containing the small molecule is much greater than the control samples, indicating the small molecule is intrinsically fluorescent. The fluorescent signal decays over time, leading to an inconclusive effect on the 20S CP.
Time Considerations
Synthesizing the FRET reporter peptide, from beginning synthesis to obtaining a purified, lyophilized powder, takes approximately 1 week.
The FRET assay requires 2 hours of time: 1 hour for preparing the plate and 1 hour for running the assay in the plate reader. Analysis of the data is not cumbersome.
SIGNIFICANCE.
The 20S catalytic particle (20S CP) of the proteasome has been implicated as a target for stimulation to combat diseases characterized by protein accumulation. The tools currently available to identify small molecules that modulate proteasome activity lack the sensitivity to recognize weak stimulators of the 20S CP, which may still hold therapeutic value, resulting in a high rate of false negatives during a high throughput screening campaign. To limit the number of false negatives in a screen, we have designed a FRET reporter peptide to broaden the dynamic range of stimulated proteasome activity, promoting the discovery of small molecules with a variety of stimulating capabilities.
ACKNOWLEDGMENT
The work described here was supported by a start-up package from Purdue University School of Pharmacy, NIH grant P30 CA023168 (Purdue University Center for Cancer Research), and a grant from the Ralph W. and Grace M. Showalter Research Trust.
LITERATURE CITED
- Ben-Nissan G, & Sharon M (2014). Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules, 4, 862–884. https://doi.org/10.3390/biom4030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Yu H, Mim C, & Matouschek A (2014). Regulated protein turnover: snapshots of the proteasome in action. Nature Reviews Molecular Cell Biology, 15, 122–133. doi: 10.1038/nrm3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R, & Trader D (2018). Development and Application of a Sensitive Peptide Reporter to Discover 20S Proteasome Stimulators. ACS Combinatorial Science, 20, 269–276. doi: 10.1021/acscombsci.7b00193. [DOI] [PubMed] [Google Scholar]
- Dunn WA (1994). Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends in Cell Biology, 4, 139–143. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, … Finley D (2000). A gated channel into the proteasome core particle. Nature Structural Biology, 7, 1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M (1995). Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Current Opinion in Cell Biology, 7, 215–223. doi: 10.1016/0955-0674(95)80031-X. [DOI] [PubMed] [Google Scholar]
- Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, & Ferrington DA (2004). Altered proteasome function and subunit composition in aged muscle. Archives of Biochemistry and Biophysics, 421, 67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Irvine GB, El-Agnaf OM, Shankar GM, & Walsh DM (2008). Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Molecular Medicine (Cambridge, Mass.), 14, 451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Njomen E, Sjogren B, Dexheimer TS, Tepe JJ (2017) Small molecule enhancement of 20S proteasome activity targets intrinsically disordered proteins. ACS Chemical Biology, 12, 2240–2247. doi: 10.1021/acschembio.7b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Ellis E, Veetil JV, Yao H, & Ye K (2011). Visualization of human immunodeficiency virus protease inhibition using a novel Förster resonance energy transfer molecular probe. Biotechnology Progress, 27, 1107–1114. doi: 10.1002/btpr.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KS, & Jenner P (2001). Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neuroscience Letters, 297, 191–194. doi: 10.1016/S0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- Nandi D, Tahiliani P, Kumar A, & Chandu D (2006). The ubiquitin-proteasome system. Journal of Biosciences, 31, 137–155. doi: 10.031/01/0137-0155. [DOI] [PubMed] [Google Scholar]
- Opoku-Nsiah KA, & Gestwicki JE (2018) Aim for the core: suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Translational Research, In Press. doi: 10.1016/j.trsl.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski M, & Wilk S (2000). Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Archives of Biochemistry and Biophysics, 383, 1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, & Goedert M (1997). Alpha-synuclein in Lewy bodies. Nature, 388, 839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Trader D, Simanski S, & Kodadek T (2017). Establishment of A Suite of Assays That Support the Discovery of Proteasome Agonists. Biochimica et Biophysica Acta, 1861, 892–899. doi: 10.1016/j.bbagen.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]