

Abstract
In amyotrophic lateral sclerosis (ALS), mitochondrial dysfunction and oxidative stress form a vicious cycle that promotes neurodegeneration and muscle wasting. To quantify the diseasestage-dependent changes of mitochondrial function and their relationship to the generation of reactive oxygen species (ROS), we generated double transgenic mice (G93A/cpYFP) that carry human ALS mutation SOD1G93A and mt-cpYFP transgenes, in which mt-cpYFP detects dynamic changes of ROS-related mitoflash events at individual mitochondria level. Compared with wild type mice, mitoflash activity in the SOD1G93A (G93A) mouse muscle showed an increased flashing frequency prior to the onset of ALS symptom (at the age of 2 months), whereas the onset of ALS symptoms (at the age of 4 months) is associated with drastic changes in the kinetics property of mitoflash signal with prolonged full duration at half maximum (FDHM). Elevated levels of cytosolic ROS in skeletal muscle derived from the SOD1G93A mice were confirmed with fluorescent probes, MitoSOX™ Red and ROS Brite™ 570. Immunoblotting analysis of subcellular mitochondrial fractionation of G93A muscle revealed an increased expression level of cyclophilin D (CypD), a regulatory component of the mitochondrial permeability transition pore (mPTP), at the age of 4 months but not at the age of 2 months. Transient overexpressing of SOD1G93A in skeletal muscle of wild type mice directly promoted mitochondrial ROS production with an enhanced mitoflash activity in the absence of motor neuron axonal withdrawal. Remarkably, the SOD1G93A-induced mitoflash activity was attenuated by the application of cyclosporine A (CsA), an inhibitor of CypD. Similar to the observation with the SOD1G93A transgenic mice, an increased expression level of CypD was also detected in skeletal muscle following transient overexpression of SOD1G93A. Overall, this study reveals a disease-stage-dependent change in mitochondrial function that is associated with CypD-dependent mPTP opening; and the ALS mutation SOD1G93A directly contributes to mitochondrial dysfunction in the absence of motor neuron axonal withdrawal.
Keywords: ALS, skeletal muscle, mitochondria
Graphical abstract
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive fatal neuromuscular disease characterized by motor neuron death and severe skeletal muscle degeneration. Currently there is no effective treatment. 95% of ALS patients die within 5 years after disease onset 1. Most ALS cases are sporadic (SALS), with about 10% being familial (FALS). While multiple factors could contribute to SALS, both SALS and FALS manifest similar pathological and clinical phenotypes of neuromuscular degeneration2, indicating a common downstream pathological mechanism underlying the disease progression. Despite intensive research efforts, the pathogenic mechanism underlying progressive neuromuscular degeneration in ALS remains largely obscure. Although the death of motor neuron is a pathological hallmark of ALS, defects in other cell types or organs may also actively contribute to ALS disease progression3–10. Skeletal muscle is substantially affected in ALS, as ALS patients experience progressive and severe muscle wasting during the disease progression.
The progressive and severe muscle wasting in ALS certainly contributes to the disease progression and impacts the life quality of patients. Individual muscle fibers communicate with motor neuron at the neuromuscular junction (NMJ), whereas retrograde signals are also conducted from muscle back to motor neuron11. While ALS is generally considered as a “dying-back” process of motor neurons12–14, studies from us5, 8, 9 and other research groups 4, 6 support that muscle appears to be a primary target of ALS, in addition to being a victim of motor neuron axonal withdrawal. Thus, understanding the pathological mechanism of muscle degeneration would provide opportunities to develop potential interventions to sustain muscle function that could constitute an alternative means to alleviate the disease progression and improve the life quality of ALS patients.
Spinal cord and muscle autopsy/biopsy samples derived from both SALS and FALS patients show remarkable mitochondrial defects 15–20. Mitochondrial dysfunction is a major player in neuronal degeneration during ALS progression 21–23. Mouse models expressing human ALS mutations (e.g., SOD1G93A) recapitulate many features of the human disease 24, and have been widely used to investigate pathogenic mechanisms and preclinical therapies for ALS 21, 25–27. Our previous study using the ALS mouse model G93A established a role for abnormal mitochondrial Ca2+ signaling in mediating the crosstalk between muscle and neurons during ALS progression 8, 9. In addition, we have found that skeletal muscle of G93A mice show abnormal mitochondrial network and reduced dynamics that occur early before the onset of ALS neuromuscular symptoms 5. Dysfunctional mitochondria are a major source of excessive reactive oxygen species (ROS) production 28–30. Oxidative stress is an essential feature of ALS muscle pathology (reviewed by Loeffler et al, 2016) 31. Oxidative stress caused by excessive mitochondrial ROS production can further exacerbate mitochondrial dysfunction 23, which could form a vicious cycle promoting muscle wasting in the course of ALS. In the current study, we examined the ROS-related mitochondria dysfunction and the underlying potential molecular mechanism in skeletal muscle derived from the ALS mouse model G93A at different disease stages.
Methods
Animal models
There are three types of mouse models used in the current study. The ALS transgenic mouse model (SOD1G93A) (G93A) was provided by Dr. Han-Xiang Deng (Northwestern University) 24. The cpYFP transgenic mouse model with same genetic background as G93A mice (B6SJL) was produced in our laboratory previously 30 using the same transgene (mtcpYFP/pUCCAGGS) originally developed by Dr. Heping Cheng’s research group 32. Through cross-breeding G93A and cpYFP mice, we generated double transgenic mouse model G93A/cpYFP that carries both SOD1G93A ALS mutation and mitochondria targeted biosensor mt-cpYFP, which enables recording of the dynamic change of mitoflash activity. The double transgenic mouse model G93A/cp-YFP does not show significant difference in the disease onset and life span, compared with the original ALS G93A mouse model. All experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols on the usage of mice were approved by the Institutional Animal Care and Use Committee of Rush University, University of Missouri at Kansas City and Kansas City University of Medicine and Bioscience.
Muscle fiber preparation
Experimental mice were euthanized by CO2 inhalation followed by cervical dislocation, and the flexor digitorum brevis (FDB) muscles were removed for functional or biochemical studies. Individual FDB muscle fibers were isolated using a collagenase-digestion method described previously 8, 9, 30. Briefly, FDB muscles were digested in modified Krebs solution (0 Ca2+) containing 0.2% type I Collagenase (Sigma), for 1 hour at 37 °C. After digestion, muscles were kept in collagenase-free Krebs solution (with 2 mM Ca2+ and 10 mM glucose) at 4 °C, and used for studies within 6 hours.
Electroporation and gene expression in FDB muscle of adult mice
The procedure was modified from our previous studies 5, 8. The anesthetized mice were first injected with 10 μl of 2 mg/ml hyaluronidase in sterile saline at the ventral side of both hind paws through a 29-gauge needle. One hour later, 10–15 μg of plasmid DNA of mt-SOD1G93A-mCherry, 4mt-SOD1G93A-GFP, mt-SOD1-mCherry or 4mt-SOD1-GFP 33 in 10μl of sterile saline were injected at the same site. Fifteen minutes later, two electrodes (gold-plated stainless steel acupuncture needles) were placed at the starting lines of paw and toes, with about 9 mm in between. Twenty pulses of 100 V/cm with 20 ms duration were applied at 1 Hz (ECM 830 Electro Square Porator, BTX Harvard Apparatus). Seven days later, the mouse was euthanized and FDB muscles were removed for functional studies.
ROS level evaluation on live FDB muscle fibers
The commercially available fluorescent dyes MitoSOX™ Red (M36008, Invitrogen) and ROS Brite™ 570 (16000, AAT) were used to evaluate mitochondrial superoxide production level and total cellular ROS production level in live FDA muscle fibers respectively. For detecting mitochondrial superoxide production level, FDB muscle fibers were incubated with 1μM MitoSOX™ Red in the Krebs solution for 10 min at 37°C. For detecting total cellular ROS production level, FDB muscle fibers were incubated with 500X diluted ROS Brite™ 570 in Krebs solution for 30 min at 37°C. The fluorescence intensity of MitoSOX™ Red and ROS Brite™ 570 was evaluated on Leica TCS SP8 confocal microscope (Leica Microsystems Inc., Germany). MitoSOX™ Red was excited at 514 nm with the fluorescent images collected at 570–600 nm, and ROS Brite™ 570 was excited at 561 nm with the fluorescent images collected at 570–600 nm. To avoid the artificial biases induced during the recording of images, the conditions for imaging colletion were kept the same between control and experimental groups, including power of the laser used, pin hole and the gain for fluorescence measurement.
Confocal imaging of mitoflash (cpYFP) signals and image analysis
Both Zeiss LSM 510 Live confocal microscope (Zeiss, Germany) and Leica TCS SP8 confocal microscope were used for imaging cpYFP fluorescence. Images were captured with a 40X, 1.2 NA water immersion objective at a sampling rate of 1sec/frame. The excitation of mt-cpYFP was achieved by alternating excitation at 488 nm, and the emission was collected at >505 nm. Experiments were performed at room temperature (23°C). Time-lapse confocal images were analyzed using custom-developed algorisms written in Interactive Data language (IDL) and Image J (NIH) 30, 34.
Isolation and purification of mitochondrial fraction from skeletal muscle
Crude mitochondrial fractionations from skeletal muscle were first obtained using a modified method previously reported 35, 36 with modifications 30. Briefly, the FDB muscle was removed from the experimental mouse and placed in ice-cold phosphate-buffered saline (PBS) containing 10 mM EDTA. The muscle was suspended in 10 ml/gram weight ice-cold homogenization buffer (100 mM sucrose, 10 mM EDTA, 100 mM Tris-HCl, 46 mM KCl, pH 7.4 with 5 mg/ml BSA and proteinase inhibitor (Thermo Fisher)), minced into small pieces and homogenized on ice. The homogenate was centrifuged at 800g 4°C for 10 min. The supernatant was transferred to a centrifuge tube and centrifuged at 10,000g 4°C for 10 min. The resulting pellets were collected as the crude mitochondrial fraction. The supernatant was centrifuged at 100,000g 4°C for 60 min for collection of the cytosolic fraction. The crude mitochondrial fraction was suspended in 1.5 ml 25% nycodenz buffer, layered onto 1.25 ml 30% nycodenz buffer, and overlaid with 1.25 ml 23% nycodenz buffer containing 5 mM Tris, 3 mM KCl, 0.3 mM EDTA, pH7.5. The sample was centrifuged at 52,000 for 90 min at 4°C in a swinging bucket rotor (BECKMAN, SW60 Ti). The mitochondrial fraction was collected from the 25%/30% interface, and suspended in equal volume of homogenization buffer and centrifuged at 10,000 g at 4°C for 10 min. The same step was repeated three times and the final pellet was collected as the pure mitochondrial fraction used for the immunoblotting assay.
Immunoblotting assay
Protein concentrations were determined by BCA protein assay (Thermo Scientific). Equal mass protein samples (10 μg) were subjected to 10% SDS-polyacrylamide gel electrophoresis, transferred to PVDF membrane (MILIPORE), and immunoblotted with primary antibodies. Antibodies used were anti-Cyclophilin D (Abcam, ab110324), 1:1000 dilution; anti-COX-IV (Cell Signaling, 4844S), 1:5000 dilution and anti-GAPDH (Cell signaling, 5174S), 1:10000 dilution. Results were visualized with ECL reagents (Thermo Scientific). Densitometry evaluation was conducted using ImageJ software (NIH, Bethesda, MD).
Statistics
Numeric data were presented as mean ± S.E. of the independent determinations. Statistical comparisons were done using students t-test for single mean or ANOVA test for multiple means when appropriate. All graphs were plotted in Sigmaplot (Systat Software Inc.) and p<0.05 was considered as significant.
Results
Elevated ROS production in skeletal muscle of G93A mice
Mitochondria are a main source of ROS production that can lead to oxidative stress in skeletal muscle. We conducted experiments to measure ROS levels in live skeletal muscle derived from G93A mice at two ages: 2-month old before disease onset and 4-month old after disease onset 24. Individual flexor digitorum brevis (FDB) muscle fibers isolated from G93A mice and wild type (WT) littermates were first incubated with ROS Brite™ 570 for evaluation of total cytosolic ROS levels. ROS Brite™ 570 is a non-fluorescent cell-permeable reagent and produces bright orange fluorescence upon ROS oxidation. Because this reaction is non- reversible and non-ratiometric, to obtain reliable fluorescent intensity that could be used to compare the total cellular ROS production level among different muscle fibers, we kept the same experimental conditions such as the muscle digestion condition, dye loading and the confocal microscope setting for all experimental groups. As shown in Figure 1A and 1B, the relative fluorescent intensity of ROS Brite™ 570 in muscle fibers derived from 2-months old and 4 months old G93A mice are significantly increased when compared to the age matched wild type control (2-month G93A: 104.7.0±6.1, n=26 fibers, N=3 mice. p<0.01 vs WT: 69.8±5.0, n= 28 fibers, N=3 mice; and 4-month G93A: 70.9±0.91, n=20 fibers, N=3 mice. p<0.01 vs WT: 42.2±0.69, n=24 fibers, N=3 mice).
Figure 1. Increased ROS level detected in live muscle fibers derived from G93A mice before and after ALS disease onset.

Representative confocal images of live FDB muscle fibers loaded with ROS Brite™ 570 (A) and MitoSOX™ Red (C). Note that both fluorescent intensity of ROS Brite™ 570 (B) and MitoSOX™ Red (D) in muscle fibers from G93A mice at 2-months or 4-months old show significant increase when compared to the muscle fibers of age-matched WT mice. 2M: 2-month old mice; 4M: 4-month old mice. (n=20–54 fibers; N=3 mice/experimental group; ** p<0.01).
We further examined whether the increased cellular ROS level in G93A muscle fibers was related to increased mitochondrial ROS production. MitoSOX™ Red is a fluorogenic dye for highly selective detection of superoxide level in mitochondria. As shown in Figure 1C and 1D, the relative fluorescent intensity of MitoSOX™ Red in muscle fibers of G93A mice is significantly higher when compared with the age controlled WT muscle fibers (2-month G93A: 96.5±2.6, n=54 fibers, N=3 mice; p<0.01 vs WT: 51.3±7.1, n=45 fibers, N=3 mice; 4-month G93A: 58.7±19.5, n=34 fibers, N=3 mice; p<0.01 vs WT: 30.9±7.7, n=27 fibers, N=3 mice). Our data suggest that skeletal muscle of G93A mice shows elevated level of ROS production. This elevated ROS production occurs early in young G93A mice before disease onset, and mitochondria are likely a major source for this elevated total ROS level in ALS muscle.
Disease stage-dependent changes in mitoflash activity in G93A muscle
Although application of ROS Brite™ 570 and MitoSOX™ Red detected an elevated level of ROS production in skeletal muscle derived from G93A mice at the age before and after the ALS disease onset, both indicators do not provide quantitative evaluation of the ROS production due to their nature as non-ratiometric dyes. Thus the readout of ROS Brite™ 570 and MitoSOX™ Red did not reveal whether there is a correlation between mitochondrial ROS production and the ALS disease progression. In addition, the signal detected by ROS Brite™ 570 or MitoSOX™ Red is not reversible and will not distinguish dynamic changes of mitochondrial ROS production in live muscle cells.
The mt-cpYFP transgenic mouse model provides a unique opportunity to examine the dynamic changes of ROS-related mitochondrial function, defined as mitoflash in live muscle cells 29, 30, 32, 37, 38. The mt-cpYFP transgenic mouse model was previously generated in our laboratory 30. We crossbred the mt-cpYFP mice with the G93A mice and generated a double transgenic mouse model (G93A/cpYFP). The double transgenic mouse model enabled us to evaluate the dynamic changes of mitoflash signals in live skeletal muscle fibers at different ALS disease stages. Individual FDB muscle fibers were isolated from G93A/cpYFP mice (before and after disease onset) and age controlled WT mice for recording of mitoflash activity. A standard protocol was established to record the mitoflash events in FDB muscle fibers, in which 100 images were taken continuously at a speed of 1 image/sec 30. The property of mitoflash signal was analyzed using the established software, FlashSniper 34, to obtain kinetics parameters such as the full duration at half maximum (FDHM), the rising and decay time, and the amplitude of the signal. In addition, the fiber area giving mitoflash signal during the 100-sequential-imaging time period was summed as Total Flash Area. The ratio of Total Flash Area over the whole fiber area named Total Flash Area/Fiber Area was then calculated. Total flash Area/Fiber area provides quantification of the number of mitochondria in a single muscle fiber that are involved in generating mitoflash events, as well as the frequency of the mitoflash events 30.
Remarkably, the parameters of the mitoflash signal in muscle fibers show disease stagedependent changes in G93A muscle. The G93A mice start to show neuromuscular symptoms around the age of 100 days. Thus, we examined the mitoflash signal in skeletal muscle of G93A mice at the age of 2-month old (before the disease onset) and 4-month old (after the disease on set). As demonstrated in Fig 2A1/2, 2B1/2 and Supplementary data 1 movie 1 and 2), the FDB muscle fibers derived from 2-month old G93A/cpYFP mice showed an increased mitochondrial mitoflash activity, which was manifested by a significant increase in the mitoflash frequency demonstrated by the ratio of Total Flash Area/Fiber Area as shown in Fig 2C (2 month old G93A/cpYFP: 2.3±0.29, n=80 fibers, N=5 mice; vs WT/cpYFP: 1.2±0.18, n=74 fibers, N=4 mice, p<0.01), while there was no significant change in the kinetic parameters of the mitoflash signal at this stage. After disease onset, the Total Flash Area/Fiber Area was significantly decreased in the skeletal muscle of 4-month old G93A/cpYFP mice (0.69±0.11, n=36 fibers, N=4 mice) p<0.01 when compared with 2-month old G93A/cpYFP mice (Fig 2C). Remarkably, the kinetics of the mitoflash signals were significantly changed in muscle fibers after disease onset. Specifically, the full duration at half maximum (FDHM) elongated almost twice compared to the 2-month old G93A/cpYFP mice (4-month old G93A/cpYFP: 33.37±5.33, vs 2-month old G93A/cpYFP: 18.28±2.07, p<0.01; and vs WT/cpYFP: 16.76±1.72, p<0.01) (Fig 2D). There are no significant changes in parameters of mitoflash in WT/cpYFP mice at the age from 2 to 7 months (data not shown). Our data suggest that change in ROS-related mitochondrial function is disease stage-dependent in ALS skeletal muscle.
Figure 2. Disease-stage-dependent changes in mitoflash activity in G93A muscle.

Representative peak intensity map of all mitoflash events detected in 100 sequentially recorded confocal images (1 image/sec) of muscle fibers isolated from the FDB muscles with WT cpYFP (A1), G93A/cpYFP (2-month old) (B1) and G93A/cpYFP (4-month old) (C1). (A2, B2, C2) are the 3D surface plots of (A1, B1, C1). The quantification analysis of the mitoflash for three conditions are illustrated in (D) Total Flash Area/Fiber Area (in 100-sequential-images). (E) FDHM. Note that Total Flash Area/Fiber Area for 2-month old G93A/cpYFP mice significantly increased compared with the WT cpYFP mice, however there was a significant decrease in 4month old G93A/cpYFP mice. While there was no change in FDHM between WT cpYFP and 2-month old G93A/cpYFP mice, there was a drastic increase in FDHM in 4-month old G93A/cpYFP mice. (n=36–80 fibers; N=3–5 mice/experimental group; ** p<0.01).
Disease stage-dependent changes in expression levels of CypD in mitochondria of G93A muscle
Mitochondrial permeability transition pore (mPTP) is a high conductance channel in the inner membrane of mitochondria (IMM) 39, 40. Uncontrolled opening of mPTP is a key step to promote mitochondrial ROS production 41. Opening of mPTP is accompanied by the loss of mitochondrial membrane potential and proton gradient across the IMM 42. It has been shown that mitoflash signal is associated with a transient opening of mPTP in both cardiac and skeletal muscles, as the mitoflash signal is simultaneously coupled with the depolarization of mitochondrial inner membrane 29, 43, 44.
While the molecular composition of mPTP is still incompletely understood 42, 45, studies have shown that Cyclophilin D (CypD) promotes the opening of mPTP 46, 47. Thus, we examined whether CypD expression level in mitochondria of skeletal muscle was altered during ALS disease progression by conducting immunoblotting assay using an anti-CypD antibody. Mitochondria were fractionated and purified from skeletal muscle of G93A mice before (2-month old) and after (4-month old) disease onset, and used for examining the expression level of CypD. We first demonstrated the purity of the mitochondrial fractionations derived from skeletal muscle of the mice. As shown in Figure 3A, the mitochondrial fractionations isolated from three different mice show positive to mitochondrial protein COX-IV antibody without detectable contamination of cytosolic proteins examined by the cytosolic protein antibody Argo2, while the cytosol portions show positive to Argo2 antibody without detectable contamination of mitochondria. Then the protein expression level of CypD was examined in G93A muscle before and after ALS disease onset. As shown in Figure 3B and 3C, the relative level of CypD in mitochondria in the skeletal muscle of 2-month old mice is not changed compared to age-matched WT mice (WT: 2.07±0.21; G93A: 1.69±0.11; N=4 mice, p=0.148). However, there is a significant increase in mitochondrial CypD level in the skeletal muscle of 4-month old mice compared with age-matched WT (WT: 0.63±0.06; G93A: 1.29±0.14; N=3 mice, p<0.01) (Figure 3D and 3E). The original images of the immunoblotting analysis were presented in Supplementary data 4 and 5. Together, our data suggest that both CypD expression level in mitochondria and the mitoflash activity in G93A skeletal muscle show disease stage-dependent changes, suggesting that CypD may play an important role in regulating mitochondrial mPTP opening in muscle wasting during ALS disease progression.
Figure 3. Disease-stage-dependent changes of the CypD expression level in muscle mitochondria of G93A mice.

(A) Immunoblotting analysis revealed the purified mitochondria without contamination of cytosolic compartment. (B, C) There is no detected difference in the CypD expression level in muscle mitochondria before disease onset. (D, E) After ALS disease onset, the CypD expression level significantly increased in muscle mitochondria of G93A mice compared with the age-matched WT mice (**p<0.01).
Overexpression of ALS mutation SOD1G93A in normal skeletal muscle enhanced mitochondrial ROS production
During ALS disease progression, skeletal muscle experiences motor neuron axonal withdrawal. Previously we have shown that denervation leads to increased ROS production with enhanced mitoflash activity in skeletal muscle 30. The observed increase in ROS production in ALS G93A skeletal muscle could be a response of mitochondria to denervation. However, it is not known whether expression of ALS mutation directly promotes mitochondrial ROS production. Accumulation of mutant SOD1 in mitochondria is linked to motor neuron degeneration in ALS cellular and animal models 48, 49. Our previous study also showed that ALS mutation SOD1G93A formed protein aggregates inside mitochondria when SOD1G93A was overexpressed in skeletal muscle of normal mice 5. We reasoned that the ALS mutation SOD1G93A might promote mitochondrial ROS production in skeletal muscle fibers similar to the nerve cells.
Mitochondria targeted ALS mutation mt-SOD1G93A-GFP was transfected to the FDB muscle of a normal mouse in one hind limb through the electroporation procedure. The wild type SOD1 (mt-SOD1-GFP) was transfected in the FDB muscle of the same mouse in another hind limb as a control to rule out potential unexpected effects of overexpression of SOD1 and GFP inside mitochondria on the intracellular ROS production. Seven days after electroporation, the FDB muscle was isolated for digestion to obtain individual single muscle fibers for evaluation of the ROS level. Expression of the green fluorescent protein GFP allows identification of the positively transfected muscle fibers. The fibers with similar GFP expression level were selected for the ROS level evaluation. A standard was set as that the expression level of SOD1-GFP should not be less than the expression level of SOD1G93A-GFP, as illustrated in the representative images of Fig 4A and 4C.
Figure 4. Overexpression of ALS mutation SOD1G93A in normal skeletal muscle leads to enhanced mitochondrial ROS production.

Individual FDB muscle fibers with overexpression of mt-SOD1-GFP or mt-SOD1G93A-GFP were loaded with ROS Brite™ 570 (A) or MitoSOX™ Red (C). mt-SOD1G93A-GFP overexpression significantly increased the total ROS production level (B) and the mitochondrial superoxide production level (D) when compared with those muscle fibers with WT mt-SOD1-GFP overexpression. (n=27–31 fibers; N=3 mice; **p<0.01).
The total cytosolic ROS production level in transfected muscle fibers was evaluated using ROS Brite™ 570 (Fig 4A and 4B), and the mitochondrial superoxide production level was evaluated using MitoSOX™ Red (Figure 4C and 4D). FDB muscle fibers with overexpression of mt-SOD1G93A-GFP showed elevated fluorescence intensity of ROS Brite™ 570 (4mtSOD1G93A-GFP: 70.4±4.6, n=27 fibers, N=3 mice vs. mt-SOD1-GFP: 45.3±3.8, n=27 fibers, N=3 mice; p<0.01) and MitoSOX™ Red (mt-SOD1G93A-GFP: 75.8±9.5, n=31 fibers, N=3 mice vs. mt-SOD1-GFP: 30.7±5.9, n=20 fibers, N=3 mice; p<0.01). Our data suggest that overexpression of ALS mutation in skeletal muscle leads to enhanced intracellular ROS production, and mitochondria are likely the target of ALS mutation and become a major source for generating excessive ROS.
Overexpression of ALS mutation SOD1G93A in normal skeletal muscle promoted mitoflash activity that was attenuated by the application of cyclosporine A (CsA)
To examine whether ALS mutation SOD1G93A has a direct impact on the mitoflash activity, mitochondria targeted ALS mutation mt-SOD1G93A-mCherry was transfected in one of the FDB muscles of the cp-YFP transgenic mouse, while mt-SOD1-mCherry was transfected in the another FDB muscle of the same mouse as a control. The emission wavelength of mCherry has limited overlap with that of cpYFP. In addition, the red fluorescence of mCherry allowed identification of positively transfected FDB muscle fibers (see Figure 6C). Seven days after electroporation, FDB muscles were isolated for preparation of individual muscle fibers. The muscle fibers identified with mCherry overexpression were used for mitoflash recording. As shown in the representative images in Figure 5A (a1, a2) and Supplemental Data 2 (movie 3), more regions in the muscle fiber with mt-SOD1G93A-mCherry overexpression showed mitoflash signal during the 100 sec recording time period when compared with fibers with mtSOD1-mCherry overexpression (Figure 5B (b1, b2) and Supplemental Data 2 (movie 4)), indicating an increased frequency of mitoflash activity in muscle fibers with overexpression of the ALS mutation. Indeed, the Total Flash Area/Fiber Area is enhanced in the fibers with mtSOD1G93A-mCherry overexpression (Figure 5C, mt-SOD1G93A-mCherry: 5.28±1.19, n=14 fibers, N=3 mice vs. mt-SOD1-mCherry: 0.12±0.03, n=14 fibers, N=3 mice; p<0.01). In addition, the full duration of half maximal (FDHM) was also significantly elongated in fibers with mtSOD1G93A-mCherry overexpression (Figure 5D, mt-SOD1G93A-mCherry: 10.88±2.18, n=14 fibers, N=3 mice vs. mt-SOD1-mCherry: 6.72±0.99, n=14 fibers, N=3 mice; p<0.05).
Figure 6. Immunoblotting analysis of individual muscle fibers with overexpression of mt-SOD1G93A-mCherry or mt-SOD1-mcherry.

(A) Anti-CypD immunoblotting analysis of three independent experiments with 20 transfected individual FDB muscle fibers collected from both conditions (7 days after the electroporation). (B) Quantitative analyses show that overexpression of ALS mutation mt-SOD1G93A-mCherry significantly increased CypD expression level when compared with muscle fibers with overexpression of mt-SOD1-mCherry. (C) Representative image shows individual FDB muscle fibers having mCherry expression (red). (n=3 independent experiments, *p<0.05).
Figure 5. Overexpression of ALS mutation SOD1G93A in normal skeletal muscle promoted mitoflash activity that could be inhibited by CsA.

Representative peak intensity map of all mitoflash events detected in 100 recorded confocal images and the related surface plot of FDB muscle fibers with overexpression of mt-SOD1G93A-mCherry without (a1, a2) and with (a3, a4) CsA treatment; mt-SOD1-mCherry without (b1, b2) and with (b3, b4) CsA treatment. The quantification analysis of mitoflash events for those four conditions are illustrated in (C) Total Flash Area/Fiber Area (in 100-sequential-images); and (D) Full Duration of the Half Maximum (FDHM). Note that the Total Flash Area/Fiber Area and FDHM of the mt-SOD1G93A-mCherry muscle fibers significantly increased compared with that of the mt-SOD1-mCherry control fibers. The application of 1μM CsA drastically attenuated the Total Flash Area of the mitoflash activity of the mt-SOD1G93A-mCherry muscle fibers, while the FDHM showed a tendency of reduction without statistical significance (n=14, *p<0.05; **p<0.01). No significant differences were detected in fibers with overexpression of WT control mt-SOD1-mCherry in the presence or absence of CsA application.
Mitoflash signal is associated with a transient opening of mPTP in both cardiac and skeletal muscles 29, 30, 43. We previously have shown that the enhanced mitoflash activity in denervated muscle fibers can be attenuated by the application of cyclosporin (CsA) 30, an established inhibitor of mitochondrial transition pore (mPTP) opening via inhibition of cyclophilin D (CypD) 46, 47, 50–52,53. Here we examined whether ALS mutation-induced mitoflash signal could be inhibited by CsA. mt-cpYFP muscle fibers with overexpression of mt-SOD1G93A-mCherry or mtSOD1-mCherry were incubated with 1 μM CsA for 30 minutes prior to imaging. Supplemental data 3 (Movie 5) and Figure 5A (a3, a4) show representative images of SOD1G93A-mCherry muscle fiber treated with CsA. The mt-SOD1-mCherry control fiber treated with CsA was shown in Supplemental data 3 (Movie 6) and Figure 5A (b3, b4). The application of CsA reduced the Total Flash Area/Fiber Area in muscle fibers with mt-SOD1G93A-mCherry overexpression (5.28±1.19, n=14 fibers without CsA vs 0.21±0.09, n=11 fibers with CsA, p<0.01) (Fig 5 C). In addition, the mean signal duration FDHM in G93A-mCherry transfected fibers was also shortened by CsA application (10.88±2.18, n=14 fibers without CsA vs 6.99±1.76, n=11 fibers with CsA), although it is not statistically significant (Fig 5D). No significant differences were detected in fibers with overexpression of WT control mt-SOD1-mCherry in the presence or absence of CsA application (Fig 5C and 5D).
Transient overexpression of ALS mutation SOD1G93A led to increased CypD expression in skeletal muscle
We further examined whether overexpression of ALS mutation SOD1G93A in normal muscle fibers could alter the expression level of CypD. For each experimental mouse, one side of the FDB muscle was transfected with mt-SOD1G93A-mCherry, and another side was transfected with mt-SOD1-mCherry as a control through electroporation. Because of the highly variable transfection efficiency between individual FDB muscles, we examined the CypD expression level by collecting individual live FDB fibers with positive expression of ALS mutation (mtSOD1G93A-mCherry) or WT SOD1 (mt-SOD1-mCherry) from digested FDB muscles, in order to avoid the potential biases caused by the inconsistent transfection efficiency on the readout of the immunoblotting analysis. The expression of red fluorescence of mCherry allowed the selection of positively transfected fibers (Figure 6C). Because it is not possible to collect mitochondrial fractionation from such small amount of muscle fibers, we detected the total expression level of CypD in 20 transfected fibers for both conditions at 7 days after transfection. Thus, GAPDH was used as the reference protein. Interestingly, in the two of three independent experiments, the amount of reference protein GAPDH significantly reduced in muscle fibers with overexpression of the ALS mutation (Fig 6A, (2) and (3)). This indicates a possible muscle atrophy induced by overexpression of ALS mutation SOD1G93A. Nevertheless, the relative expression level of CypD/GAPDH in muscle fibers with ALS mutation overexpression is significantly increased (mt-SOD1-mCherry: 1.37±0.30 vs. mt-SOD1G93A-mCherry: 2.74±0.43; N=3 mice, p<0.05) (Fig 6A, 6B). The original images of the immunoblotting analysis were presented in Supplementary data 6. These data suggest that SOD1G93A mutation directly leads to CypD-dependent mPTP opening in skeletal muscle fibers, which is associated with the enhanced mitochondrial ROS production and oxidative stress in the ALS muscle.
Discussion
To examine the disease-stage-dependent changes of mitochondrial function in ALS skeletal muscle, we generated double transgenic mouse model (G93A/cpYFP) that carries human ALS mutation SOD1G93A and mt-cpYFP transgenes, in which mt-cpYFP detects the dynamic changes of ROS-related mitoflash events at individual mitochondrion level. Through examining live skeletal muscle fibers derived from the ALS mouse model G93A/cpYFP, we have detected changes in ROS-related mitochondrial function, which is characterized by the different kinetics of mitoflash signals at different disease stages. The altered mitoflash activity is also associated with a changed expression level of CypD in mitochondria in the course of ALS progression, indicating an involvement of mitochondrial mPTP opening in promoting ROS production in ALS muscle. Remarkably, through overexpression of ALS mutation SOD1G93A in skeletal muscle of normal mice, we found that ALS mutation could directly lead to an enhanced mitochondrial ROS production and mPTP-related mitoflash activity in the absence of motor neuron withdrawal.
Oxidative stress is an essential feature of ALS muscle pathology 31, while dysfunctional mitochondria are a major source of excessive ROS production 28–30. The biochemical study from Halter et al, 2010 has shown an increased amount of ROS in skeletal muscle of SOD1 mutant mice even before motor impairment 54. In the current study, we detected an increased ROS production level in skeletal muscle of ALS mouse model G93A by applying fluorescent dyes (ROS Brite™ and MitoSOX™ Red) to detecting total cellular ROS and mitochondrial superoxide levels in freshly isolated live muscle fibers. Although both commercially available dyes provided consistent results of elevated ROS level in ALS skeletal muscle, they could not distinguish whether there is any difference in the ROS production during ALS disease progression due to the non-ratiometric measurement of the signal. In order to quantify the disease-stage-dependent changes of mitochondrial function, we generated double transgenic mice (G93A/cpYFP) that carry human ALS mutation SOD1G93A and mt-cpYFP transgenes. mtcpYFP was first developed by Wang et al (2008) as a mitochondrial-targeted fluorescent protein with its signal associated with ROS production and energy metabolism of individual mitochondrion in live cells 29, 32, 37. The signal of mt-cpYFP was revealed as transient waves at the level of individual mitochondria, a phenomenon defined as the mitoflash 32. Using the same transgene we produced mt-cpYFP transgenic mice 30 and crossbred this mouse line with the ALS mouse model G93A. Remarkably, the mitoflash signal reported by cpYFP in muscle fibers derived from those double transgenic mice (G93A/cpYFP) showed disease-stage-dependent features. Before ALS disease onset (2-month old), the total numbers of mitoflash events has doubled compared to the wild type control muscle, indicating a higher frequency of individual mitochondrion to give mitoflash signals. Other kinetic parameters of the mitoflash signal remain the same as the control, indicating there is no change in the kinetics of the mitoflash signal at early stage. However, after disease onset, the duration of mitoflash signal is prolonged. It is very well established that mitoflash activity is dynamically coupled with the mPTP opening in both cardiac and skeletal muscle, because the transient increase in mt-cpYFP fluorescence was always coupled with the depolarization of mitochondrial membrane potential at the same location measured by TMRE during a real time dual recording 29, 43, 44. The elongated FDHM of mitoflash signal suggests an altered mPTP functional status in skeletal muscle when ALS disease progresses. Published studies have shown that mPTP has two opening states: a low conductance state and a high conductance state 55. In the low-conductance state, the mPTP has a MW cut-off below 300Da and only allows passage of small ions including H+ and Ca2+. Under this condition, mPTP opens transiently without mitochondrial swelling 55, 56 and may have a role in regulation of mitochondrial Ca2+ level 57. In the high conductance state, the mPTP displays a much higher cut-off (below 1500 Da) with longer opening, resulting in sustained mitochondrial membrane potential depolarization, mitochondrial swelling/rupture and cell death 55, 56. It is possible, at early stage, the increased frequency of mitoflash events is only associated with the low-conductance state of the mPTP, while at later stage of ALS, the elongated duration of mitoflash is coupled with the high conductance state of the mPTP. Ca2+ overload in mitochondria was found in motor neuron 58. Our previous study also identified mitochondrial Ca2+ overload in skeletal muscle of SOD1G93A transgenic mice 8, 9. At early stage of ALS, the increased frequency of transient mPTP opening could be an adaptive response to help alleviate mitochondrial Ca2+ overload59, which may have some beneficial roles in protection of mitochondria. As the disease progresses to a later stage, the FDHM almost doubled, which suggests an elongated mPTP opening. Thus, functional defects of mitochondria might progress to a state that can no longer be rescued, causing an irreversible opening of the mPTP and cell death in skeletal muscle of G93A mice at later stage of ALS.
It has been shown that CypD promotes mPTP opening 46, 47, 53. Published studies have shown a strong connection between CypD-related mPTP opening and the mitoflash signal29, 32, 44, 60. While no significant change of CypD expression level was detected in the spinal cord of SOD1G93A transgenic mice 61, we observed disease-stage-dependent changes of CypD expression in skeletal muscle of SOD1G93A transgenic mice. G93A skeletal muscle experiences double insults during ALS progression, including the motor neuron axonal withdrawal and a direct effect of the ALS mutation on skeletal muscle. We previously showed that denervation promoted mitoflash activity in skeletal muscle, which was blocked by the application of CsA30. In the current study, we found that ALS mutation SOD1G93A directly promoted mitoflash activity when the mutated isoform was overexpressed in skeletal muscle of normal mice. This SOD1G93A-induced mitoflash activity could also be blocked by the application of CsA. Together, our data suggest that an altered mitoflash activity in G93A muscle is likely CypD-related. Indeed, there is an increased CypD expression level in mitochondrial fraction of skeletal muscle derived from G93A mice at later stage of the disease. This disease-stage-dependent change of CypD expression level in mitochondria is in line with the disease-stage-dependent change of mitoflash activity. We speculate that increased CypD level in mitochondria of SOD1G93A muscle may contribute to the elongated and irreversible opening of mPTP at later stage of ALS. Interestingly, there were no detectable changes in CypD level before the disease onset in the G93A skeletal muscle, although the frequency of the mitoflash signal significantly increased at early stage. We have already shown that both denervation- and ALS mutation-induced mitoflash activity is CypD related. It is not clear how CypD is linked to increased mitoflash frequency without changes in its expression level at early stage of the disease. One possibility could be a functional change in CypD protein, such as a changed phosphorylation status 62.
Studies have shown that ALS-linked mutant SOD1 is recruited to spinal cord mitochondria in ALS murine models 63–67. Accumulation of mutant SOD1 in mitochondria was found to be associated with increased oxidative damage and decreased mitochondrial respiratory activity in the spinal cord, although the exact mechanism remains obscure 48, 49. Previously, we have found that mitochondria in G93A skeletal muscle show similar abnormal morphology as detected in motor neuron 9, and ALS mutation SOD1G93A forms aggregates inside muscle mitochondria5. Here, we further discovered that ALS-linked mutation SOD1G93A directly promoted mitochondrial ROS production in skeletal muscle in the absence of motor neuron axonal withdrawal. Through overexpression of mitochondria targeted SOD1G93A in skeletal muscle of normal cpYFP transgenic mice, we found that SOD1G93A directly caused an increased mitoflash activity, which was inhibited by CsA. Remarkably, overexpression of SOD1G93A in skeletal muscle of normal mice directly increased CypD expression level, indicating that accumulation of protein aggregates of SOD1G93A inside mitochondria could be a potential trigger for the changes in CypD expression and function. Autophagy targets misfolded proteins and damaged intracellular organelles including mitochondria for lysosomal degradation. We previously demonstrated that SOD1G93A formed aggregates in mitochondria of skeletal muscle, and this aggregation led to mitochondrial depolarization, disruption of the mitochondrial network and dynamics in skeletal muscle5. Remarkably, in an additional study, we found that skeletal muscle of the ALS G93A mice showed a disease-stage-dependent reduction in autophagy capacity 68 that could also contribute to the accumulation of damaged mitochondria and the enhanced CypD level in the later stage of ALS.
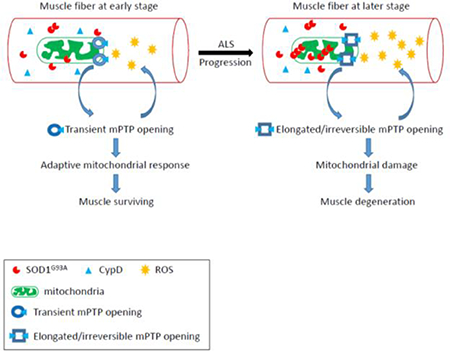
Based on past and present results, a potential disease-stage-dependent pathologic sequence of muscle degeneration in ALS G93A mouse model is illustrated in Fig 7. At earlier stage, there is less aggregation of mutant SOD1 mutant protein in mitochondria. The increased frequency of mitoflash events may be an early adaptive response of mitochondria, and may have some beneficial role in preserving mitochondrial function. In contrast, after disease onset, the mitoflash activity shows a significant reduction in signal frequency, but with much longer signal duration. This may imply that increased mutant SOD1 aggregation inside mitochondria promotes CypD expression that favorites an irreversible opening status of the mPTP. Thus, mPTP with increased CypD binding may lose the adaptive ability to respond to Ca2+ overload, and promote more ROS production that form vicious cycles leading to permanent mitochondrial damage and muscle cell degeneration at later stage of the disease.
Figure 7. Proposed pathologic sequence of muscle degeneration in the ALS mouse model.

Before the ALS disease onset, the skeletal muscle of ALS G93A mice starts to have enhanced mitochondrial ROS production, which could be associated with a transient opening of mPTP. This transient opening of mPTP could be an adaptive response at the early stage of ALS to preserve mitochondrial function for muscle survival. At later stage of ALS, the aggregation of ALS mutation SOD1G93A inside mitochondria further promotes mitochondrial damage and ROS production that form a vicious cycle. The increased CypD expression level in muscle mitochondria at later stage may play a role in changing the mPTP to an elongated and irreversible status, which leads to irreversible mitochondrial damage and muscle degeneration.
CypD inactivation has shown benefits in protection of mitochondrial function in myopathies and multiple sclerosis 69–71. Baines CP et al (2005) generated the CypD (Ppif) null mice that were resistant to mitochondrial swelling and permeability transition46. Martin et al (2009) crossed the CypD null mice with ALS G93A mice and found that deletion of CypD delayed the disease onset and extended survival61, while Kim et al (2012) found that CypD ablation completely abolished the phenotypic advantage of G93A females, with no effect on the disease course of G93A males72. A more comprehensive study from Parone et al (2013) found that elimination of CypD expression in three different SOD1 mutation mouse lines had no beneficial effect on the disease progression and survival, although mitochondrial morphology and function were preserved in the motor neuron of those mice73. As discussed in Parone et al (2013), the divergent outcome of those ALS mice with CypD ablation may be attributable to confounding genetic changes in the CypD-gene-disrupted mouse lines73. In addition, as a naturally existing protein, CypD may also play a physiological role in regulating mitochondrial function. Complete ablation of CypD may not necessarily be beneficial for the integrity of mitochondrial or cellular function. In the currently study, we discovered a diseasestage-dependent change in the CypD expression level that was associated with the diseasestage-dependent change in ROS-related mitoflash activity in skeletal muscle mitochondria of ALS G93A mice. Our result provides additional clues that CypD is likely involved in mitochondrial dysfunction in skeletal muscle during ALS disease progression, and could be a potential therapeutic target for alleviating muscle mitochondrial dysfunction.
Although mt-cpYFP has been characterized and used as a biomarker of mitochondrial ROS generation32, 38, 74, studies have suggested that mitoflash may also report ATP 75 or pH changes of mitochondria76, 77. cpYFP likely reports both ROS and pH signal of mitochondria, and is thus a robust indicator for detecting the dynamic status of mitochondrial function 78, 79. A study by Ding et al indicates that mitoflash represents a dynamic signal of mitochondrial ROS production and energy metabolism37. In our recent published study, mitoflash signal served as a unique biosensor that allowed us to discover the early response of mitochondria to denervation, and the role of physiological Ca2+ transients in maintaining the functional integrity of mitochondria in skeletal muscle30. In addition, we used commercially available dyes, MitoSOX™ Red and ROS Brite™ 570, as additional fluorescent indicators for monitoring mitochondrial superoxide and total cellular ROS levels. Although, MitoSOX™ Red and ROS Brite™ 570 further confirmed the enhanced ROS production in skeletal muscle of ALS mice, they were not able to detect the dynamic changes of mitochondrial ROS production. Thus, cpYFP is a valuable biosensor allowing us to explore the disease-stage-dependent changes of ROS-related mitochondrial function in skeletal muscle of the ALS mouse model. The G93A/cpYFP double transgenic mouse model could be a unique animal model for testing potential means for ALS treatment, because it can test whether a potential treatment reverses mitochondrial functional status from the later stage to an earlier stage.
Conclusion
In summary, we detected increased mitochondrial ROS levels in live skeletal muscle fibers derived from ALS SOD1G93A transgenic mice. Through exploring the mitoflash activity in G93A/cpYFP double transgenic mice, we demonstrated a disease-stage-dependent change in mPTP-related mitochondrial dysfunction in ALS skeletal muscle. The cpYFP mitoflash signal provides a quantitative biomarker for evaluating ROS-related mitochondrial function in skeletal muscle during ALS disease progression, and the double transgenic mouse model G93A/cpYFP could provide a unique means for testing the efficacy of potential interventions for treating ALS in future studies. Through overexpression of ALS mutation SOD1G93A in skeletal muscle of normal cpYFP mice, we determined that SOD1G93A alone could promote mitochondrial ROS production, which is associated with CypD-related mitochondrial mPTP opening. Future studies are required to explore the molecular mechanism of the underlying effect of ALS mutation on mitochondria through regulating the expression and function of CypD and other possible signaling pathways involved.
Supplementary Material
Acknowledgments
Funding
This work was fully supported by grants from NIAMS/NIH R01 AR057404, Bank of American Victor E. Speas Foundation and ALS Association (16-IIP-288) to JZ, and partially supported by NIAMS/NIH R01-AR070752 to JM. Zhou laboratory was also supported by the McCown Gordon Gala Research Gift. CK was a recipient of a postdoctoral fellowship from NIH/NHLBI T32 training grant HL 07692-(21-25). LZ was a recipient of a scholarship from Zunyi Medical University.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- CsA
Cyclosporine A
- CypD
cyclophilin D
- FDB
flexor digitorum brevis
- G93A
transgenic mice with overexpression of ALS-associated mutation SOD1G93A
- Mptp
mitochondrial transition pore
- ROS
reactive oxygen species
- mt-cpYFP
mitochondrial targeted biosensor
- FDHM
full duration of half maximum
- WB
Western Blotting
Footnotes
Competing interests
The authors declare that they have no competing interests.
Ethics approval
All animal experiments were performed according to the procedures approved by Institutional Animal Care and Use Committee of Rush University, Kansas City University and University of Missouri at Kansas City.
List of chemical compounds
Cyclosporine A
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alonso A, Logroscino G, Jick SS and Hernan MA. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2009;16:745–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasinelli P and Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature reviews Neuroscience. 2006;7:710–23. [DOI] [PubMed] [Google Scholar]
- 3.Boillee S, Vande Velde C and Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. [DOI] [PubMed] [Google Scholar]
- 4.Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fano G, Sandri M and Musaro A. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–36. [DOI] [PubMed] [Google Scholar]
- 5.Luo G, Yi J, Ma C, Xiao Y, Yi F, Yu T and Zhou J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PloS one. 2013;8:e82112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong M and Martin LJ. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Human molecular genetics. 2010;19:2284–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu S, Yi J, Zhang YG, Zhou J and Sun J. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol Rep. 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi J, Ma C, Li Y, Weisleder N, Rios E, Ma J and Zhou J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J Biol Chem. 2011;286:32436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Yi J, Fu R, Liu E, Siddique T, Rios E and Deng HX. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu K, Yi J, Xiao Y, Lai Y, Song P, Zheng W, Jiao H, Fan J, Wu C, Chen D, Zhou J and Xiao G. Impaired bone homeostasis in amyotrophic lateral sclerosis mice with muscle atrophy. J Biol Chem. 2015;290:8081–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen QT, Son YJ, Sanes JR and Lichtman JW. Nerve terminals form but fail to mature when postsynaptic differentiation is blocked: in vivo analysis using mammalian nerve-muscle chimeras. J Neurosci. 2000;20:6077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dadon-Nachum M, Melamed E and Offen D. The “dying-back” phenomenon of motor neurons in ALS. Journal of molecular neuroscience : MN. 2011;43:470–7. [DOI] [PubMed] [Google Scholar]
- 13.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA and Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:23240. [DOI] [PubMed] [Google Scholar]
- 14.Frey D, Schneider C, Xu L, Borg J, Spooren W and Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Echaniz-Laguna A, Zoll J, Ponsot E, N’Guessan B, Tranchant C, Loeffler JP and Lampert E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp Neurol. 2006;198:25–30. [DOI] [PubMed] [Google Scholar]
- 16.Napoli L, Crugnola V, Lamperti C, Silani V, Di Mauro S, Bresolin N and Moggio M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Archives of neurology. 2011;68:1612–3. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki S and Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Journal of neuropathology and experimental neurology. 2007;66:10–6. [DOI] [PubMed] [Google Scholar]
- 18.Siciliano G, Pastorini E, Pasquali L, Manca ML, Iudice A and Murri L. Impaired oxidative metabolism in exercising muscle from ALS patients. Journal of the neurological sciences. 2001;191:61–5. [DOI] [PubMed] [Google Scholar]
- 19.Soraru G, Vergani L, Fedrizzi L, D’Ascenzo C, Polo A, Bernazzi B and Angelini C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropathol Appl Neurobiol. 2007;33:204–11. [DOI] [PubMed] [Google Scholar]
- 20.Wiedemann FR, Manfredi G, Mawrin C, Beal MF and Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. Journal of neurochemistry. 2002;80:616–25. [DOI] [PubMed] [Google Scholar]
- 21.Carri MT and Cozzolino M. SOD1 and mitochondria in ALS: a dangerous liaison. Journal of bioenergetics and biomembranes. 2011;43:593–9. [DOI] [PubMed] [Google Scholar]
- 22.Cozzolino M, Ferri A, Valle C and Carri MT. Mitochondria and ALS: implications from novel genes and pathways. Molecular and cellular neurosciences. 2013;55:44–9. [DOI] [PubMed] [Google Scholar]
- 23.Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN and Radi E. Mitochondria, oxidative stress and neurodegeneration. Journal of the neurological sciences. 2012;322:254–62. [DOI] [PubMed] [Google Scholar]
- 24.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX and et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–5. [DOI] [PubMed] [Google Scholar]
- 25.Carri MT, D’Ambrosi N and Cozzolino M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochemical and biophysical research communications. 2016. [DOI] [PubMed] [Google Scholar]
- 26.Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ and Eisenberg DS. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGoldrick P, Joyce PI, Fisher EM and Greensmith L. Rodent models of amyotrophic lateral sclerosis. Biochimica et biophysica acta. 2013;1832:1421–36. [DOI] [PubMed] [Google Scholar]
- 28.Brookes PS, Yoon Y, Robotham JL, Anders MW and Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology Cell physiology. 2004;287:C817–33. [DOI] [PubMed] [Google Scholar]
- 29.Fang H, Chen M, Ding Y, Shang W, Xu J, Zhang X, Zhang W, Li K, Xiao Y, Gao F, Shang S, Li JC, Tian XL, Wang SQ, Zhou J, Weisleder N, Ma J, Ouyang K, Chen J, Wang X, Zheng M, Wang W and Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell research. 2011;21:1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karam C, Yi J, Xiao Y, Dhakal K, Zhang L, Li X, Manno C, Xu J, Li K, Cheng H, Ma J and Zhou J. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skeletal muscle. 2017;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loeffler JP, Picchiarelli G, Dupuis L and Gonzalez De Aguilar JL. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016;26:227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT and Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Yi J, Li X, Xiao Y, Dhakal K and Zhou J. ALS-associated mutation SOD1(G93A) leads to abnormal mitochondrial dynamics in osteocytes. Bone. 2018;106:126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li K, Zhang W, Fang H, Xie W, Liu J, Zheng M, Wang X, Wang W, Tan W and Cheng H. Superoxide flashes reveal novel properties of mitochondrial reactive oxygen species excitability in cardiomyocytes. Biophysical journal. 2012;102:1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong M, Gertz B, Chestnut BA and Martin LJ. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Frontiers in cellular neuroscience. 2013;7:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frezza C, Cipolat S and Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–95. [DOI] [PubMed] [Google Scholar]
- 37.Ding Y, Fang H, Shang W, Xiao Y, Sun T, Hou N, Pan L, Sun X, Ma Q, Zhou J, Wang X, Zhang X and Cheng H. Mitoflash altered by metabolic stress in insulin-resistant skeletal muscle. Journal of molecular medicine. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei L, Salahura G, Boncompagni S, Kasischke KA, Protasi F, Sheu SS and Dirksen RT. Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:3068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasola A, Sciacovelli M, Pantic B and Bernardi P. Signal transduction to the permeability transition pore. FEBS Lett. 2010;584:1989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javadov S and Kuznetsov A. Mitochondrial permeability transition and cell death: the role of cyclophilin d. Front Physiol. 2013;4:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zorov DB, Juhaszova M and Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological reviews. 2014;94:909–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernardi P The mitochondrial permeability transition pore: a mystery solved? Frontiers in physiology. 2013;4:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Zhang H and Cheng H. Mitochondrial flashes: From indicator characterization to in vivo imaging. Methods. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei L and Dirksen RT. Perspectives on: SGP symposium on mitochondrial physiology and medicine: mitochondrial superoxide flashes: from discovery to new controversies. The Journal of general physiology. 2012;139:425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernardi P and Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. Journal of molecular and cellular cardiology. 2015;78:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J and Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T and Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. [DOI] [PubMed] [Google Scholar]
- 48.Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS and Carri MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13860–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF and Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–33. [DOI] [PubMed] [Google Scholar]
- 50.Halestrap AP and Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. The Biochemical journal. 1990;268:153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hausenloy DJ, Boston-Griffiths EA and Yellon DM. Cyclosporin A and cardioprotection: from investigative tool to therapeutic agent. British journal of pharmacology. 2012;165:1235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM and Crompton M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. European journal of biochemistry / FEBS. 1996;238:166–72. [DOI] [PubMed] [Google Scholar]
- 53.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA and Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. [DOI] [PubMed] [Google Scholar]
- 54.Halter B, Gonzalez de Aguilar JL, Rene F, Petri S, Fricker B, Echaniz-Laguna A, Dupuis L, Larmet Y and Loeffler JP. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free radical biology & medicine. 2010;48:915–23. [DOI] [PubMed] [Google Scholar]
- 55.Ichas F and Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et biophysica acta. 1998;1366:33–50. [DOI] [PubMed] [Google Scholar]
- 56.Brenner C and Moulin M. Physiological roles of the permeability transition pore. Circulation research. 2012;111:1237–47. [DOI] [PubMed] [Google Scholar]
- 57.Korge P, Yang L, Yang JH, Wang Y, Qu Z and Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. The Journal of biological chemistry. 2011;286:34851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guatteo E, Carunchio I, Pieri M, Albo F, Canu N, Mercuri NB and Zona C. Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels’ activation in a genetic model of amyotrophic lateral sclerosis. Neurobiology of disease. 2007;28:90–100. [DOI] [PubMed] [Google Scholar]
- 59.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G and Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W, Gong G, Wang X, Wei-LaPierre L, Cheng H, Dirksen R and Sheu SS. Mitochondrial Flash: Integrative Reactive Oxygen Species and pH Signals in Cell and Organelle Biology. Antioxidants & redox signaling. 2016;25:534–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD and Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218:333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS and Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Rehnmark A and Marklund SL. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci. 2006;26:4147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV and Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS and Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. [DOI] [PubMed] [Google Scholar]
- 66.Vande Velde C, Miller TM, Cashman NR and Cleveland DW. Selective association of misfolded ALSlinked mutant SOD1 with the cytoplasmic face of mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vijayvergiya C, Beal MF, Buck J and Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiao Y, Ma C, Yi J, Wu S, Luo G, Xu X, Lin PH, Sun J and Zhou J. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol Rep. 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, Mattioli E, Gualandi F, Ferlini A, Merlini L, Maraldi NM, Bonaldo P and Bernardi P. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P and Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Merlini L, Angelin A, Tiepolo T, Braghetta P, Sabatelli P, Zamparelli A, Ferlini A, Maraldi NM, Bonaldo P and Bernardi P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim HJ, Magrane J, Starkov AA and Manfredi G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain : a journal of neurology. 2012;135:2865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E and Cleveland DW. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33:4657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pouvreau S Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PloS one. 2010;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muller FL. A critical evaluation of cpYFP as a probe for superoxide. Free radical biology & medicine. 2009;47:1779–80. [DOI] [PubMed] [Google Scholar]
- 76.Schwarzlander M, Logan DC, Fricker MD and Sweetlove LJ. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. The Biochemical journal. 2011;437:381–7. [DOI] [PubMed] [Google Scholar]
- 77.Schwarzlander M, Wagner S, Ermakova YG, Belousov VV, Radi R, Beckman JS, Buettner GR, Demaurex N, Duchen MR, Forman HJ, Fricker MD, Gems D, Halestrap AP, Halliwell B, Jakob U, Johnston IG, Jones NS, Logan DC, Morgan B, Muller FL, Nicholls DG, Remington SJ, Schumacker PT, Winterbourn CC, Sweetlove LJ, Meyer AJ, Dick TP and Murphy MP. The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature. 2014;514:E12–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei-LaPierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S, Wang X, Sheu SS, Cheng H, Dirksen RT and Wang W. Respective contribution of mitochondrial superoxide and pH to mitochondriatargeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J Biol Chem. 2013;288:10567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang M, Sun T, Jian C, Lei L, Han P, Lv Q, Yang R, Zhou X, Xu J, Hu Y, Men Y, Huang Y, Zhang C, Zhu X, Wang X, Cheng H and Xiong JW. Remodeling of Mitochondrial Flashes in Muscular Development and Dystrophy in Zebrafish. PloS one. 2015;10:e0132567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.