

Abstract
Although a pre-pregnancy dietary intervention is believed to be able to prevent offspring obesity, research evidence is absent. We hypothesize that a long period of pre-pregnancy maternal diet transition from a high fat (HF)-diet to a normal fat (NF)-diet effectively prevents offspring obesity, and this preventive effect is independent of maternal body weight change. In our study, female mice were either continued on a NF diet (NF-group) or a HF diet (HF-group) until weaning; or switched from a HF to a NF for 1-week (H1N-group), 5-week (H5N-group) or 9- week (H9N-group) before pregnancy. After weaning, the offspring were given the HF diet for 12 weeks to promote obesity. The mothers, regardless of which group, did not display maternal body weight change and glucose intolerance either before pregnancy or after weaning. Compared to the HF group, the H1N and H5N, but not the H9N offspring, developed glucose intolerance earlier, with more severely imbalanced glucose homeostasis. These offspring also displayed hepatocyte degeneration and significant adipocyte hypertrophy associated with higher expression of lipogenesis genes. The molecular mechanistic study showed blunted insulin signaling, overactivated adipocyte Akt signaling and hepatic AMPK signaling with enhanced lipogenesis genes in the H1N and H5N versus the NF offspring. However, maternal H9N diets normalized glucose and lipid metabolism of the offspring, via re-sensitized insulin signaling and normalized Akt and AMPK signaling. In summary, we showed that a long-term maternal diet intervention effectively released the inter-generational obesogenic effect of maternal HF diet, independent of maternal weight management.
Keywords: maternal diet intervention, high-fat diet, offspring obesity, insulin signaling, Akt signaling, AMPK signaling
INTRODUCTION
Over the past two decades, obesity and its associated diseases have become a global problem on human health and health-care resources. In the United States, 68.5% of adults were either overweight or obese, 34.9% were obese, and 6.4% were extremely obese in 2011-2012[1]. It is noticed that 58.5% of women at childbearing age (20-39 y) and 31.9% of youth (2-19 y) were either overweight or obese[1]. Authorities from the American Heart Association Council on Epidemiology and Prevention recently states that obesity among girls and women has generated a vicious cycle that may also contributes to the obesity epidemic for the past two decades [2].
Recent studies have put more efforts on investigating maternal over-nutrition by high fat diet, which reflects the dietary habits of the Western society. In humans, babies exposed to over-nutrition during gestation have increased risks of obesity, diabetes and other complications [3-6]. In animal models, offspring of mothers exposed to over-nutrition have common phenotypes that include catch-up growth, increased adiposity, impaired glucose tolerance, impaired insulin sensitivity and liver dysfunction [7-11]. It is noteworthy that maternal overnutrition is responsible for promoting offspring obesity and the early onset of glucose intolerance, even while maternal obesity may not have developed [12]. Thus, it is suggested that effective prevention of obesity and its related diseases via maternal lifestyle intervention may need to begin even before pregnancy [13-20].
A maternal healthy diet intervention is thought to be beneficial to both pregnant mothers and babies. As the gestational weight gain (GWG) guidelines by the Institute of Medicine (IOM) recommended a weight gain of only 25 to 35 pounds for normal women and only 11 to 20 pounds for all classes of obesity during pregnancy [21], women must cultivate and maintain a healthy dietary style before pregnancy. Because previous studies evaluating pre-conception lifestyles and interventions were not focused on offspring health, the results of the studies only showed a reduced risk of gestational diabetes and preeclampsia [22-24]. Follow-up studies of the babies, however, are lacking. Hence, no evidence-based strategy monitoring women’s diets prior to and during pregnancy is currently available; the development of which would be of ultimate benefit to the mother and the fetus [25].
In an effort to devise such a strategy, our group used a murine model to study whether switching from a high fat (HF) diet to a non-fat (NF) diet 1 week before pregnancy (H1N group) and maintaining the NF diet through weaning could reprogram offspring obesity [26]. We reported that the obesity phenotype of offspring upon maternal HF or H1N diet are gender dependent. Our study focused on female offspring reported that a short-term switch in maternal diet prior to pregnancy is not necessarily beneficial and may even be harmful to offspring health.
However, maternal diet intervention might take longer for the mother to readjust its body metabolism and therefore it would be beneficial for the pregnancy and the offspring. We hypothesized that the transition from a HF to a NF diet before pregnancy could reduce the risk for offspring obesity; however, a longer transition term may be required. To test this hypothesis, we evaluated strategies regarding different transition durations (1, 5 or 9-week) from a HF- to a NF- diet before pregnancy and maintaining this NF diet until weaning. We evaluated whether different durations of diet transition would differentially impact the offspring obesity and whether a long-term diet transition would be beneficial to offspring health. In addition, we investigated whether the effect of maternal diet intervention was independent of certain maternal factors, such as maternal body weight change and glucose intolerance.
EXPERIMENTAL METHODS
Study design and animal models (Summarized in Table 1)
Table 1.
Study Design
| Mother’s Diet | Offspring’s Diet | |||
|---|---|---|---|---|
| Pre-pregnancy | Pregnancy/Lactation | After Weaning | ||
| Pre-transition | Transition | |||
| REF | NF | - | NF | NF |
| NF | NF | - | NF | HF |
| HF | HF | - | HF | HF |
| H1N | HF | NF (1Wk) | NF | HF |
| H5N | HF | NF (5 Wks) | NF | HF |
| H9N | HF | NF (9 Wks) | NF | HF |
Four-week-old female mice of mixed background (B6/129/SvEv) were fed with either the control diet (NF, 10% fat) or the HF diet (60% fat) for 12 weeks. The breeding pairs given the HF diet either continued this diet throughout gestation and lactation (the HF group) or were switched to a NF diet 1 week (the H1N group), 5 weeks (the H5N group) or 9 weeks (the H9N group) before conception and continued this diet throughout gestation and lactation. After weaning, all offspring were given the identical HF diet for 12 weeks to promote weight gains and then sacrificed. At least five litters per treatment group were used in this study (n=5-7 per group) From each litter, three female offspring mice were randomly selected for 12 weeks of post- weaning monitoring. Another group of offspring, 7 mice from 3 litters, which was fed with the NF diet and was born by breeding pairs on NF diet (the REF group), served as the reference group to show the normal ranges for a given characteristic. Mouse experiments were completed according to a protocol reviewed and approved by the Institutional Animal Care and Use Committee of the University of North Dakota and Texas A&M University, in compliance with the USA Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Animal Diet
Diet was purchased from Research Diets, LLC (New Brunswick, NJ). The control diet (Cat#D12450B) had an energy density of 3.771 kcal/g (10% fat energy, 70% carbohydrate energy, and 20% protein energy). The HF diet (Cat#D12492) had an energy density of 5.157 kcal/g (60% fat energy, 20% carbohydrate energy, and 20% protein energy). The fat source is composed of 92% of lard and 8% of soybean oil.
Antibodies and Reagents
Primary antibody: Rabbit anti-mouse Glut4 antibody was purchased from Abcam (CAT#ab654, Cambridge, MA); Rabbit anti-mouse IRS1 (#CAT2390S), Ser612-IRS1 (CAT#3203S), Ser636/639-IRS1 (CAT#2388S), AKT (CAT#9272S), Thr308-AKT (CAT#13038), Ser-473- AKT (CAT#4060S) antibodies and the AMPK and ACC antibody sampler kit (CAT#9957) were obtained from Cell Signaling Technology (Danvers, MA); Rabbit anti-mouse Glut2 (CAT#sc-518022), IRS2 (CAT#sc-390761) and Actin antibodies (CAT#sc-58673) were purchased from Santa Cruz (Stockton, CA). Goat anti-rabbit IgG-HRP (CAT#sc-2004) was also obtained from Santa Cruz (Stockton, CA).
Intraperitoneal injected glucose tolerance test (IPTGG)
IPTGG was performed as described as previously described [26].
Immunohistochemical staining and adipocyte cell sizing
Visceral white adipose tissue and liver tissue were fixed in 10% formalin/PBS. The tissue was embedded in paraffin blocks after processing and was cut into 5-μm-thick slices using a TC-2 tissue sectioner (Sorvall Instruments). Adipocyte diameter was measured using Image J software. Two hundred cells were randomly counted in each gonadal adipose sample. The adipocytes of five animals from each group were counted. An average diameter was recorded for each animal.
Measurement of Hepatic Glucose, Glycogen and Triglyceride
Hepatic glucose level was measured using an Amplex red glucose assay kit (Thermo Fisher Scientific Inc, #A22189). Hepatic glycogen level was measured using a glycogen assay kit (Thermo Fisher Scientific Inc, #MAK016-1KT). Hepatic Triglyceride content was measured using an adipogenesis detection kit, which quantifies triglyceride accumulation in cells and tissues (Abcam’s ab102513). They were performed as previously described [26].
Realtime-PCR
The total mRNA of gonadal adipocytes and hepatocytes were extracted using Invitrogen Trizol methods. cDNA were synthesized using Sigma Readyscript cDNA Synthesis Mix. Real-time PCR was performed with a Bio-Rad IQ5 realtime-PCR system. Statistical analysis were performed using the ΔCT method. All primers are listed in Table 2.
Table 2.
Primers used for mRNA quantification by real-time PCR.
| Gene | Forward primer 5’−3’ | Reverse primer 5’−3’ |
|---|---|---|
| Pklr | CACTCAGCTACAGACCCGTG | CCACACTGTCTTTGCATCGC |
| Pfkfb1 | CTCTACGAAGCTCACACGCT | AACTGGCTCAAGACGCTGAA |
| Pfkl | TTTTGGAGGTGATGGGACGG | TCCATGTTGTCTGGGCGAAA |
| G6pc | GCTGGAGTCTTGTCAGGCAT | ATCCAAGCGCGAAACCAAAC |
| Phkal | GGGAACGTGGCGATAAGACA | GCCCACCTTTCACACCAAAC |
| Fbp | GTCGCTCTTTCCACAGGACA | TTGGCATAGCCCTCGTTGAG |
| Pepck | ATGAAAGGCCGCACCATGTA | GCACAGATATGCCCATCCGA |
| Gsk3b | CGAGACACACCTGCACTCTT | TCCGAGCATGTGGAGGGATA |
| Phkγl | TTGACGCCTATGCTTTCCGT | AAGTCCTCCTCCTCAGCCAA |
| Phkb | GATGCTCACAACCGCAACAG | CAAGGCAAACGCAGGGTAAC |
| Phkg1 | TTGACGCCTATGCTTTCCGT | AAGTCCTCCTCCTCAGCCAA |
| Pygl | CCAGCTTGGGCCACTTACTT | GGAGGGACCCAGCTTACTC |
| Srebf1 | AGCAGTCACCAGCTTCAGTC | GGTCATGTTGGAAACCACGC |
| Pparg | TGTAATGGAAGGGCAAAAGG | TGGCTTCCAGTGCATAAGTT |
| Fasn | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| Ppargclα | CAAAGCTGAAGCCCTCTTGC | CCTTTCTTGGTGGAGTGGCT |
| Cebpb | TGGACAAGAACAGCAACGAG | GCCATGGCCTTGACCAAGGAG |
| Acaca | AGATGTGCTGGGTCATGTGG | ATCCAGGCCATGTTGAGACG |
| B2m* | TTCTGGTGCTTGTCTCACTGA | CAGTATGTTCGGCTTCCCATTC |
B2m was selected as the internal control. Regarding to the criteria for selecting B2m as the internal control please see the supplementary figure 1
Statistical analysis
At least five litters per treatment group were included in this study (n=5-7). Differences among the REF, NF, HF and H1N, H5N and H9N groups at the single time points, were analyzed by Fishers’ least significant difference test in order to take into account the multiple comparisons. Fisher’s least significant difference test was performed by first carrying out one-way analysis of variance (ANOVA) for all treatment groups, and was then followed by two-group t tests if ANOVA test was statistically significant. For the longitudinal data such as body weight and food consumption, a linear mixed model was used for the analysis of repeated measures with each individual mouse as a random effect. For all the statistical tests, the results were considered significant if the p value is less than 0.05. All analyses were carried out by using SAS JMP software (SAS Institute Inc., Cary, NC, USA) and R statistical programming language.
RESULTS
The maternal diet interventions with different transition periods did not result in maternal body weight changes and glucose intolerance of the mothers.
Our previous study provided evidence that a maternal HF diet for 12 weeks before pregnancy promoted offspring obesity and glucose intolerance is not dependent on the presence of maternal obesity or overweight status [12], suggesting the maternal HF diet was an independent factor to program offspring obesity. In the current study, a confounding problem has been whether the maternal diet interventions are independent of maternal physical changes, such as maternal weight gain/loss or glucose intolerance development [4, 6, 27], to result in different offspring phenotypes. To address this question, we recorded the body weight of the mother and performed an intraperitoneal glucose tolerance test (IPGTT). We showed that every mother, regardless which group it was randomly assigned to, had similar body weight and were not obese or overweight at the beginning of the experimental period and before the dietary transition (Wk0 and Wk12, Figs.1A and B). It is also clear that all mothers, regardless of diet transition or the duration of diet transition, weighed similarly before pregnancy (Fig.1C, T1). The H1N, H5N and H9N mothers did not significantly gain or lose weight during pregnancy and lactation comparing to HF mothers (Fig.1C, T2 and T3), possibly due to ad libitum feeding during the transition. Additionally, no mothers displayed glucose intolerance before pregnancy or after the lactation period (Figs. 1D and E).
Figure 1. Different maternal interventions led to different phenotypes of offspring obesity was not due to maternal obesity and glucose intolerance.
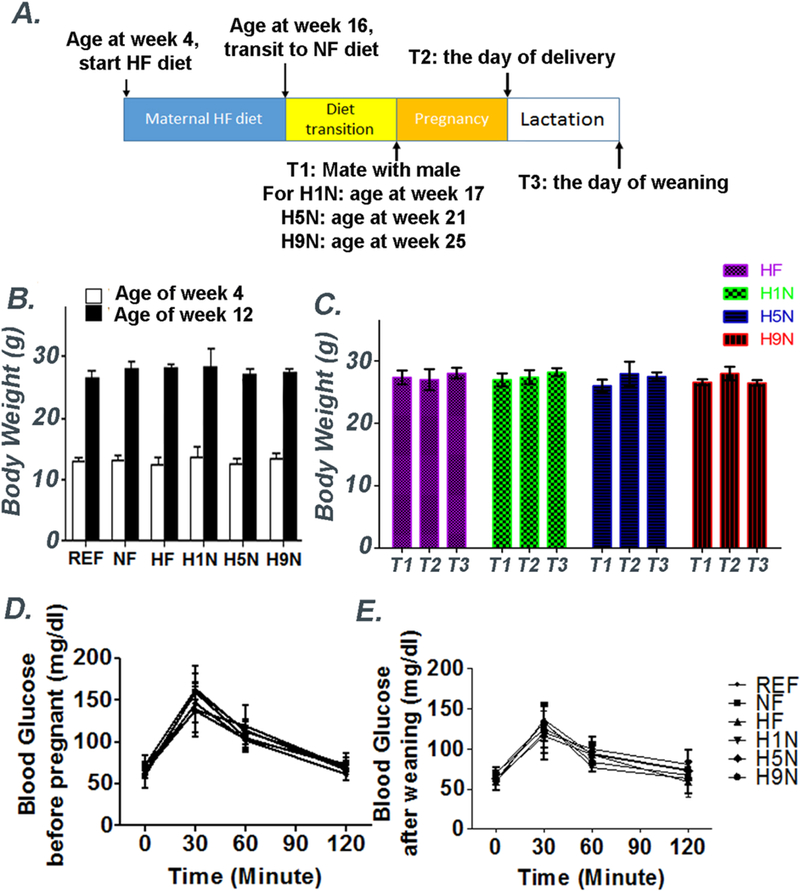
1A. Female mice were treated with HF diet (60% fat) for 12 weeks at the age of week 4 (experimental Week 0). Body weight of each group were recorded at week 1 and week 12. At experimental week 12, the mice either continued this diet throughout gestation and lactation (the HF group) or were switched to a NF diet 1 week (the H1N group), 5 weeks (the H5N group) or 9 weeks (the H9N group) before conception and continued this diet throughout gestation and lactation. The body weight of HF, H1N, H5N and H9N groups were recorded before pregnancy (T1), after delivery (T2) and after lactation period (T3).
1B and C. Mean body weight of the mothers before pregnancy, after delivery and after lactation. The results are presented as Mean ± SE, n=3-7.
1D and E. IPGTT of the mothers before pregnancy and after lactation. The results are presented as Mean ± SE, n=3-7.
The H9N, but not the H1N and H5N diet, prevented offspring obesity and glucose intolerance.
We recorded the food consumption and the body weight of the offspring mice once every week during the experimental period. Not surprisingly, the REF groups consumed a lower level of calories than the NF, HF, H1N and H9N offspring, except for offspring from H5N group (Fig.2A, P<0.01). The H5N offspring has a trend to consume less energy compared to the NF, HF, H1N or H9N offspring, however the difference is not statistically significant during the experimental period (P>0.05).
Figure 2. The H9N, but not the H5N and H1N diets, prevented offspring obesity and glucose intolerance.
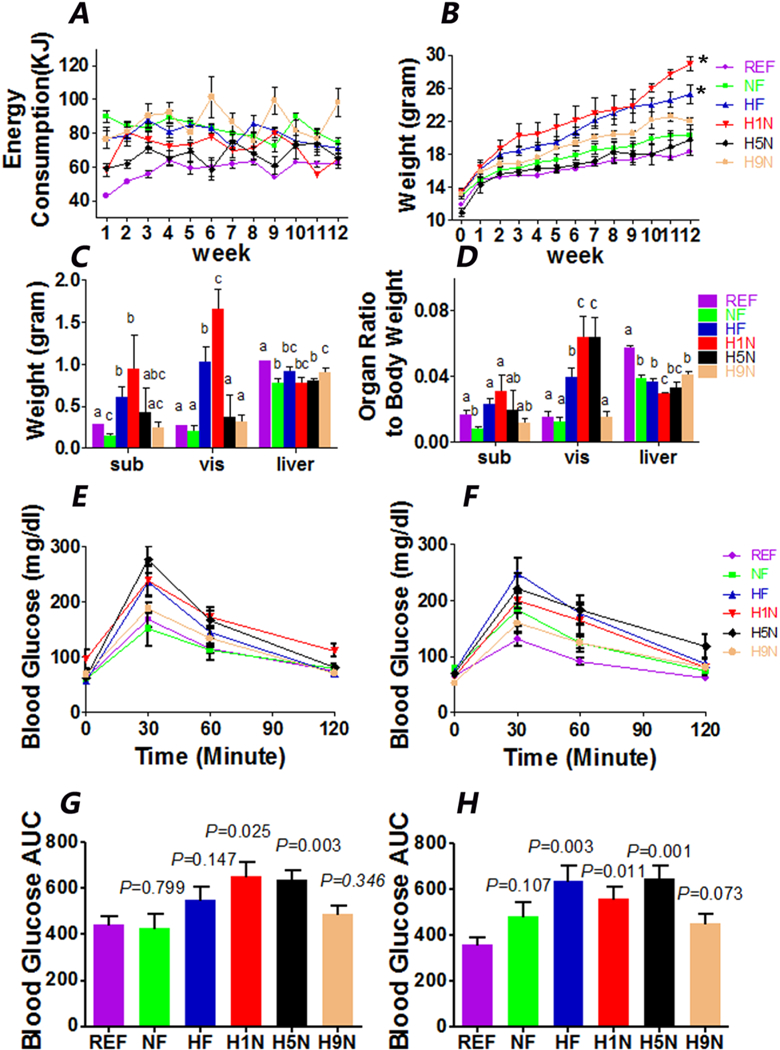
2A. Food consumption of each offspring mouse was calculated as kilocalories from post weaning weeks 1 to 12. The offspring mice were separately housed and the food consumption (kcal) of each cage was calculated as follows: food consumption (kcal) = (weight of input - weight of leftover) x 5.157 for H1N, H5N, H9N, HF, and NF offspring (or x 3.771 for REF offspring). The result are presented as Mean ± SE, n=3-7.
2B. Body weight of female offspring from wean to post-weaning week 12. The statistics for time serial data was analyzed by repeated analysis and significance test was performed to evaluate the difference between the treatment groups (HF, H1N, H5N and H9N groups) versus the NF groups. Result is presented as Mean ± SE, n=3-7. The statistical significance was indicated with “*”.
2C. Tissue (subcutaneous fat, gonadal fat and liver) weight of female offspring at week 12. The result are presented as Mean ± SE, n=3-7. Significance (p < 0.05) is presented by different characters comparing among treatment groups in each tissue.
2D. The relative adipose tissue weight normalized on body weight of female offspring at week 12. The results are presented as Mean ± SE, n=3-7. Significance (p < 0.05) is presented by different characters comparing among treatment groups in each tissue.
2E and F. IPGTT results at week 9 (E) and week 12 (F) on postweaning HF diet. The result are presented as Mean ± SE, n=3-7.
2G and H. AUC of IPGTT results at week 9 and week 12. The result are presented as Mean ± SE, n=3-7. P value is calculated by student t-test to REF group.
At weaning, H5N and REF offspring had lower mean body weight (MBW) compared to any of the other experimental groups of which MBW at weaning were not significantly different (Fig. 2B). At the end of week 12, the MBW among all groups from the highest to the lowest ranked: H1N>HF> (H9N≈NF≈H5N≈REF). HF and H1N offspring had the largest subcutaneous fat and gonadal fat pads measured by the weight among all groups (Fig.2C). Since the MBW at week 12 of each group varies, we measured the relative weight by normalizing the adipose tissue weight on the MBW (Fig.2D). HF diet significantly increased the relative gonadal fat weight compared to NF diet, and the H1N and H5N offspring had the largest relative gonadal adipose tissue. Notably, the gonadal fat accumulation found in the maternal HF diet offspring was not found in the H9N diet offspring.
Next, we evaluated whether these offspring mice were glucose intolerant by IPGTT at week 9 and week 12 of the post-weaning HF diet. Consistent with our previous report, HF offspring displayed glucose intolerance at week 12 (P=0.003, Figs.2F and H) but not week 9 (P=0.147, Figs.2E and G). Starting as early as week 9 and until week 12, H1N and H5N offspring were glucose intolerant. However, H9N offspring was glucose tolerant at both week 9 (P=0.346, Figs.2E and G) and week 12 (P=0.073, Figure 2F and H). Two hours after a glucose challenge, the glucose levels of all experimental groups were able to return to the basal level (Fig.2E and F).
The H9N, but not the H5N and H1N diet, prevented the adipocyte hypertrophy and the hepatocyte degeneration caused by maternal HF diet.
We observed significantly enlarged gonadal adipose tissue in HF, H1N and H5N offspring (Fig.2D), which is consistent with the histology study (Fig.3A). Furthermore, compared to NF offspring, the HF, H1N and H5N offspring had significantly enlarged visceral adipocytes, while visceral adipocytes in H9N offspring were smaller than those of NF offspring (Figs.3A and B).
Figure 3. The H5N and H1N, but not the H9N diet, induced the adipocyte hypertrophy and the hepatocyte degeneration.
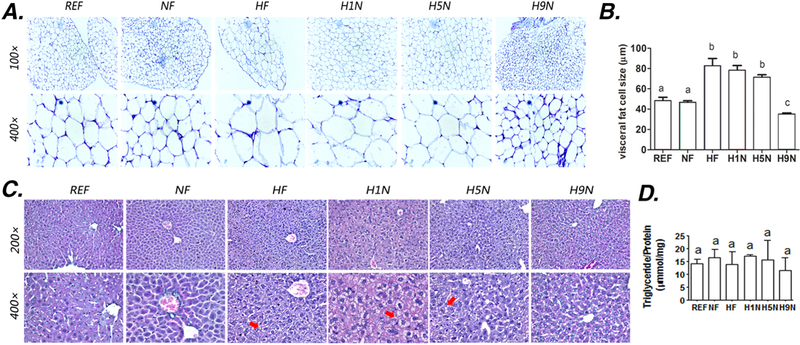
3A. HE staining of gonadal adipose tissue from the female offspring on 12-week postweaning HF diet
3B. Average size of the adipocyte was measured using Image J software as described in Methods section
3C. HE staining of liver samples from the female offspring on 12-week postweaning HF diet
3D. Amount of hepatic triglyceride content after 12-week exposure of postweaning HF diet. Hepatic Triglyceride content was measured using an adipogenesis detection kit (Abcam’s ab102513).
The results are presented as Mean ± SE, n= 3-7. Significance (p < 0.05) is presented by different characters comparing among groups in each tissue.
We further assessed if the maternal diet transition from HF diet to NF diet would impact the hepatic lipid deposition and injury. We did not observe obvious steatosis in any of the six experimental groups (Fig.3C), which is confirmed by the experiment to detect the triglyceride (TG) content that all groups had similar amount of TG content (Fig.3D). However, the microvesicular fatty change of the hepatocytes without steatosis [28], in which the nuclei remain centrally located and appear to be indented by the small fat droplets, was found exclusively in HF and H1N liver and partially in H5N liver, while seldom displayed in H9N offspring liver (Fig.3C).
The H9N, but not the H1N and H5N diet, normalized offspring glucose homeostasis disrupted by maternal HF diet.
To investigate the impacts of maternal diet transitions on the offspring glucose metabolism in liver, we measured the hepatic glucose content. The offspring hepatic glucose contents from the highest to the lowest were: H1N>H5N>HF>H9N>NF>REF (Fig.4A).
Figure 4. The H9N, but not the H5N and H1N diets, normalized offspring glucose homeostasis broken by maternal HF diet.
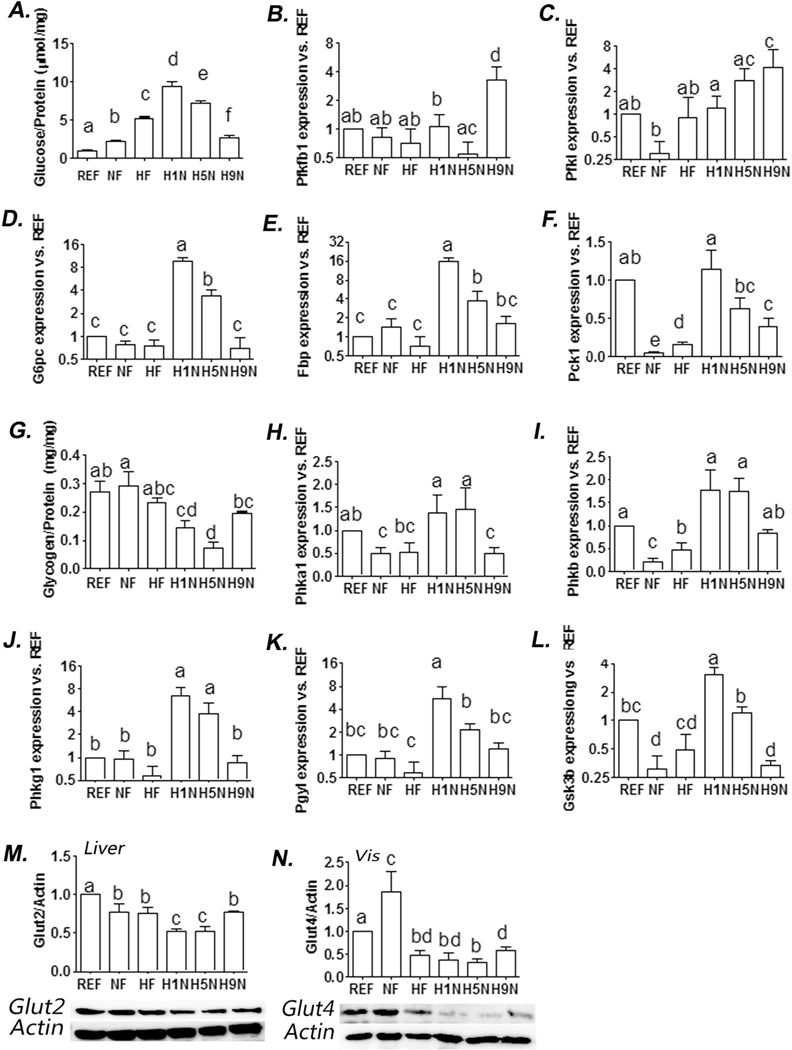
4A. Amount of hepatic glucose content after 12-week exposure of postweaning HF diet. Hepatic glucose level was measured using an Amplex red glucose assay kit (Thermo Fisher Scientific Inc, #A22189). 4B-F. Hepatic expression of genes involved in glycogenesis and gluconeogenesis, and glycolysis was measured by real-time PCR. 4G. Amount of hepatic glycogen content after 12-week exposure of postweaning HF diet. Hepatic glycogen level was measured using a glycogen assay kit (Thermo Fisher Scientific Inc, #MAK016-1KT). 4H-L. Hepatic expression of genes involved in inhibiting glycogenesis was measured by realtime PCR. 4M. Hepatic Glut2 expression was detected by Western blot analysis. Relative amounts of Glut2 were expressed as the ratio Glut2/Actin. 4N. Adipocyte Glut4 expression was detected by Western blot analysis. Relative amounts of Glut4 were expressed as the ratio Glut4/Actin. The results are presented as Mean ± SE, n= 3-7. Significance (p < 0.05) is presented by different characters comparing among groups in each tissue.
We next measured the offspring hepatic levels of genes involved in glucose metabolism. The H9N offspring showed significantly higher expression levels of key genes for glycolysis, Pfkfbl and Pfkl, than either NF or REF offspring, while the NF, HF, H1N and H5N showed no expression alterations (Figs.4 B and C), suggesting over-activation of glycolysis of the H9N hepatocytes.
For hepatic gluconeogenesis involved genes, H1N and H5N diet increased expression of all three key genes we observed (G6pc, Fbp and Pckl) (Figs.4D-F), suggesting over-activation of gluconeogenesis in these groups. However, such upregulations of the gluconeogenesis genes were not observed in H9N hepatocytes (Figs.4D-F).
Unlike the extremely high level of glucose in H1N and H5N offspring, we observed extremely low levels of hepatic glycogen content in these two groups, comparing to either NF or REF offspring (Fig.4G). The glycogen content in H9N offspring recovered to normal levels and was only slightly lower than NF offspring and was not different from REF offspring (Fig.3G). We wonder if the glycogen content changes were negatively associated with the expression levels of glycogenesis inhibitor genes involving Gsk3β, Phkαl, Phkβ, Phkγl and Pygl. Compared to both HF and NF offspring, the H1N and H5N offspring had over-expression of almost all glycogenesis inhibitor genes; while NF, HF, H9N and REF had overall similar levels of glycogenesis inhibitor genes (Figs.4H-L).
We measured if the maternal diet intervention affected the glucose transportation by changing the expression of Glut2 in the liver and Glut4 in the gonadal fat tissue. The data showed significant decrement of hepatic Glut2 and adipose Glut4 in H1N and H5N comparing to NF offspring, while the H9N diet at least partially recovered their expression (Figs.4M and N).
The H9N, but not the H1N and H5N diet, ameliorated the insulin desensitization in both hepatocytes and adipocytes.
We previously showed that H1N diet exacerbated deleterious effects on insulin signaling caused by maternal HF diet [26] and considered whether a longer term of transition from HF diet to NF diet before pregnancy would at least partially recover the insulin sensitivity in liver and adipocytes. As the data showed, both H5N and H1N diet, but not the NF diet, decreased the expression of IRS1 in hepatocytes compared to either REF or NF offspring, with a level even lower than HF offspring (Figs.5A and B). NF diet did not affect the phosphorylation efficiency at Ser612 and Ser636/639, while HF diet enhanced phosphorylation efficiency at Ser612. There were overphosphorylation at both Ser612 and Ser636/639 of H1N offspring comparing to either NF or HF offspring. The H5N and the H9N diets did not affect the phosphorylation at Ser636/639 (Figs.5C and D). In adipocytes, HF, H1N and H5N diets inhibited Irs1 expression to a level lower than REF (Figs. 5E and F) and overphosphorylated IRS1 at Ser612 (Figs. 5E and H), suggesting an overall desensitization of insulin signaling. Unlike H1N and H5N offspring, H9N offspring expressed similar level of IRS1 and p-IRS1 at both Ser612 and Ser636/639 as REF or NF offspring did (Figs.5E, G and H), suggesting an overall recovering of the insulin sensitivity.
Figure 5. The H9N, but not the H1N and H5N diets, ameliorated the insulin desensitization and repressed the hepatic AMPK activation.
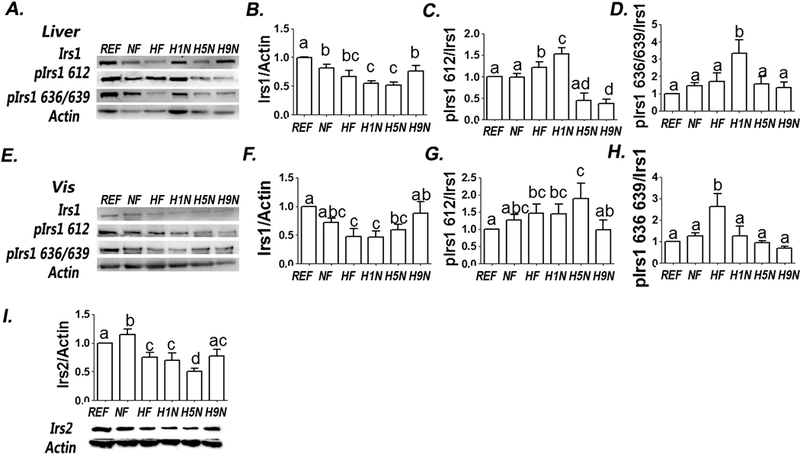
5A. Hepatic IRS1 and phosphorylation of IRS1 were measured by Western Blots.
5B-D. Relative amounts of IRS1 and phosphorylation of IRS1 were expressed as the ratio of IRS1/ Actin (B) and as the ratios of Ser636/639/IRS1 (C) and Ser612/IRS1 (D), respectively.
5E. Adipocyte IRS1 and phosphorylation of IRS1 were measured by Western Blots.
5F-H. Relative amounts of IRS1 and phosphorylation of IRS1 were expressed as the ratio of IRS1/ Actin (F) and as the ratios of Ser636/639/IRS1 (G) and Ser612/IRS1 (H), respectively.
5I. Hepatic IRS2 and phosphorylation of IRS2 were measured by Western Blots. Relative amounts of IRS2 was described as the ratio of IRS2/ Actin.
The results are presented as Mean ± SE, n=3-7 litters/group. Significance (p < 0.05) is presented by different characters comparing among groups in each tissue.
We measured the expression of IRS2 in hepatocytes, considering its important role in regulating insulin signaling in the liver. The NF diet significantly enhanced the expression level of IRS2 in the liver; however, HF diet not only thwarted this effect but also further depressed its expression. The H1N diet had similar effect as HF diet and the H5N diet further inhibited IRS2. However, the IRS2 expression in H9N offspring was not different from in REF, although it was still lower than NF offspring (Fig.5I).
The H9N, but not the H1N and H5N diet, normalized the overactivation of Akt signaling and lipogenesis in adipocytes.
AKT signaling is a key element in the insulin signal transduction network and is well-known to mediate glucose and lipid metabolism. In adipocytes, the AKT expression level from the highest to the lowest was NF≈H5N>H9N≈H1N≈HF≈REF (Figs. 6A). Both the NF and HF diet overphosphorylated AKT at Ser308 and Ser473. With the diet transiting from a HF-diet to a NF- diet, the H1N further enhanced phosphorylation at both sites and the H5N further overphosphorylated AKT at Ser473 (Figs.6A-C). In contrast, the H9N diet brought both expression and phosphorylations of AKT back to the similar level as REF offspring (Figs.6A-C).
Figure 6. The H9N, but not the H5N and H1N diets, prevented the overactivation of the Akt signaling and lipogenesis in adipocytes.
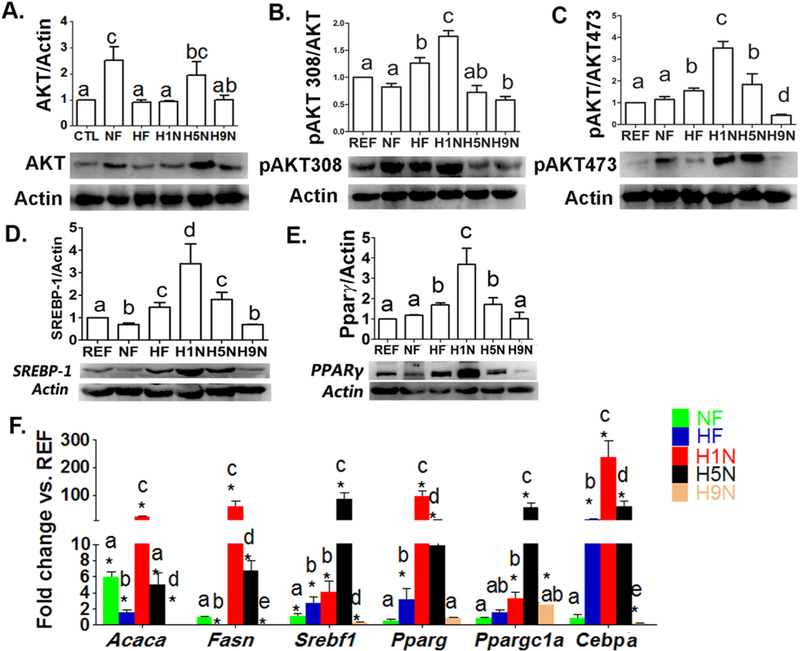
6A-C. Adipocyte AKT (M) and phosphorylation of AKT at Thr308 (N) and Ser473 (O) were measured by Western Blots. Relative amounts of AKT and phosphorylation of AKT were calculated as the ratio of AKT/Actin, pAKT308/AKT and pAKT473/AKT.
6D. Adipocyte Srebpl expression was detected by Western blot analysis. Relative amounts of Srebpl were expressed as the ratio Srebpl/Actin.
6E. Adipocyte Ppar-γ expression was detected by Western blot analysis. Relative amounts of Ppar-y were expressed as the ratio Ppar-γ/Actin.
6F. Adipocyte expression of genes involved in lipogenesis and adipogenesis was measured by real-time PCR.
The results are presented as Mean ± SE, n= 3-7. Significance (p < 0.05) is presented by different characters comparing among groups in each tissue.
The expression of Srebp-1c and Ppar-γ are under the regulation of Akt signaling [29, 30]. They have been well established to regulate adipocyte differentiation and overexpression of Srebp-1c causes adipocyte hypertrophy [31, 32]. Srebp-1c expression in H1N offspring was the highest among all three groups, the HF, H1N and H5N, that upregulated its expression, while H9N diet totally repressed the overexpression of Srebp-1 caused by maternal HF diet in adipocytes (Fig.6D). For Ppar-γ, the H9N diet completely impeded the upregulation of Ppar-γ by maternal HF diets. Unlike H9N diets, the H1N and H5N diets failed to reverse Ppar-γ upregulation; instead, H1N diet further enhanced its expression (Fig.6E). These protein expression level changes were consistent with the mRNA level changes of other important adipogenesis genes (Fig.6F). Overall, H1N and H5N offspring expressed remarkably higher levels of Acacα, Fasn, Pparγ, Ppargclα and Cebpβ than HF or NF offspring. Opposite to the expression in H1N and H5N groups, the expressions of almost all detected adipogenic genes in H9N offspring were either same as (Pparγ and Ppargclα) or significantly lower than those of NF or HF offspring.
The H1N, H5N and H9N diets differentially regulated the AMPK signaling pathway for lipid homeostasis.
To understand the molecular mechanism of how maternal diet intervention regulated the cellular lipid homeostasis in liver, we tested the AMPK pathway activity under different duration of maternal diet shift. We did not observe significant changes in pAMPK-α, pAMPK-β and pACC/ACC/Actin ratio, markers for AMPK signaling activity, in both NF and HF offspring (Figs.7A-F). Surprisingly, transition from a maternal HF to a NF diet affected AMPK signaling associated with different transition periods differently. We observed higher levels of p-AMPK-α, p-AMPK-β1 and p-ACC ratio to ACC of the H1N and H5N offspring compared to either NF or HF offspring, however H9N offspring brought these values back to control levels (Figs 7A-F).
Figure 7. The H1N, H5N and H9N differentially regulate the hepatic AMPK signaling and hepatic lipogenesis for lipid homeostasis.
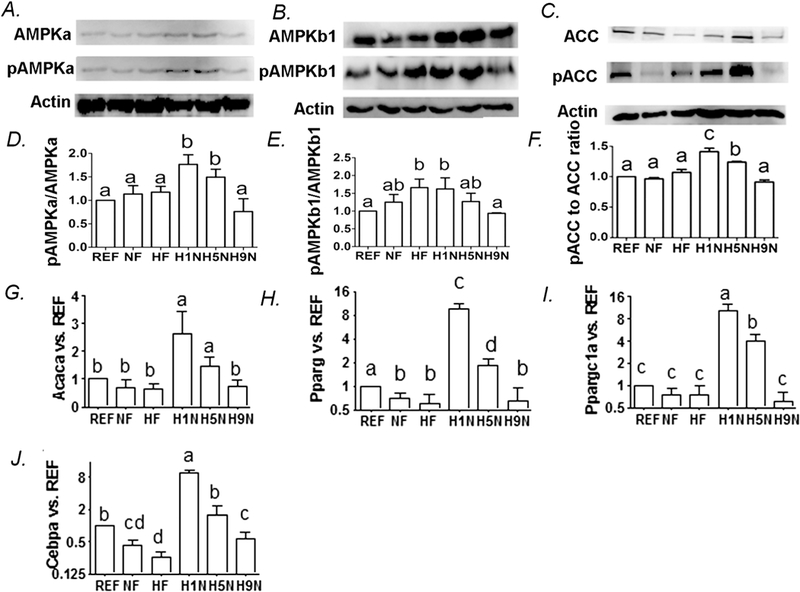
7A-C. Hepatic AMPKα, phosphorylation of AMPKα at Thr172, AMPK β1, phosphorylation of AMPKβ1 at Ser108, ACC and phosphorylation of ACC at Ser79 were measured by Western Blots.
7D-F. Relative amounts of phosphorylation of AMPKα, phosphorylation of AMPKβ1, p-ACC to ACC ratio.
7G-J. Hepatic expression of genes involved in lipogenesis was measured by real-time PCR.
The results are presented as Mean ± SE, n= 3-7. Significance (p < 0.05) is presented by different characters comparing among groups in each tissue.
To understand the lipid homeostasis, we further measured mRNA levels of other key genes for lipogenesis in liver. Similar to the H1N and H5N diet overactivating AMPK signaling, they also enhanced the hepatic expression of the lipogenesis genes, Acaca, Pparγ, Ppargcla and Cebpα (Figs.7G-J). In H9N and NF offspring, these overall gene expression upregulations were not observed (Figs.7G-J).
DISCUSSION
Lifestyle modification is easy to access and economical, and thus is considered the first-line intervention in the treatment and prevention of obesity. Previously, we have reported that transitions from a maternal HF diet to a NF diet one week before pregnancy did not prevent the early onset of diet-induced obesity (DIO) in the next generation [26]. However, Nathanielsz’s group described a preventive effect on obesity of the offspring from the dam whose mother was overweight and was switched from a HF to a NF diet four weeks before pregnancy [12]. These inconsistent results suggest that an optimized maternal strategy needs to be carefully selected to maximize the beneficial effects to both the mother and the baby.
Based on Nathanielsz’s group and our data [12, 26], it is clear that different terms of maternal diet interventions differentially impact the offspring phenotype. By evaluating different phenotypes of the offspring, we noticed that a short- or a medium-term maternal transition was not beneficial to the offspring’s health; rather, the H1N and H5N offspring were more sensitive to postnatal HF diet evidenced by: (1) earlier onset of glucose intolerance with imbalanced glucose homeostasis; (2) more relative gonadal adipose tissue with adipocyte hypertrophy; (3) more extensively presented hepatocyte degeneration. Although H5N offspring did not display a higher MBW at week 12, they had enlargement of visceral adipose tissue which is a well-known risk factor for adult cardiovascular disease [33, 34]. The low MBW of H5N at week 12 is possibly due to its low MBW at weaning, which is another risk factor for adulthood obesity, hypertension and diabetes mellitus [35-38]. On the contrary, we found that the H9N offspring were born with normal body weight and consistently gained weight but was neither obese nor glucose intolerant on 12-week HF diet. Together these observations support that a long transitional term is required to reverse the adverse effects of maternal HF diet on offspring obesity induced by postweaning from the HF diet.
It is also possible that the prevention of the downstream obesity is actually due to the maternal body weight loss before pregnancy. This is supported by the Nathanielsz’s group that the mothers with diet intervention lost 7% of the body weight before getting pregnant [12]. Similarly, maternal weight loss surgery has been shown to effectively reduce offspring obesity up to three times [39, 40]; however, this data was not justified by the lifestyle modifications after the surgery. Therefore, there is still a gap in knowledge as to whether the prevention of downstream obesity is due to the dietary intervention or simply because of maternal weight management. In our study, none of the mothers were obese on 9-week HFD treatment, before the diet intervention initiated. This is consistent with various reports that female mice is not sensitive to HFD and 9-week treatment of HFD is not long enough to induce significant body weight gain [41-43]. In addition, all mothers, regardless how early they were switched from a HF to a NF diet, maintained consistent body weight before pregnancy and after lactation, possibly due to ad libitum feeding. These results provide robust evidence to suggest that maternal diet intervention, independent of maternal obesity or body weight gain/loss, plays an important role in reprogramming the body weight of the offspring. However, the recommended long-term dietary switch from HF to NF diet without maternal body weight loss, while suggested as beneficial for non-obese mothers of prior unhealthy dietary habits, may not be similarly efficient on obese mothers-to-be. Future work will need to focus on this interesting and important question.
Our data described a molecular mechanism, with key signaling pathways, to explain why pre-conceptional diet intervention must be long enough to prevent offspring obesity and glucose intolerance. It is clear that the H1N and H5N diets desensitized insulin signaling and were favored to hepatic glucose anabolism and adipocyte lipogenesis via selective insulin resistance and through activation of AKT signaling in adipocytes [44, 45], the H9N diet was sensitive to insulin signaling and promoted glucose catabolism and reversed lipogenesis overactivation caused by maternal HF diet, via normalizing the AKT signaling in adipocytes.
Our data also suggested an important role of the AMPK signaling pathway under different maternal diet interventions in maintaining hepatic lipid homeostasis of the female offspring. AMPK signaling plays as a metabolic sensor responsible for nutrient deprivation through increased AMP levels and therefore switches on/off energy expenditure and storage [46-50]. Previous studies have reported conflicting data about the AMPK activity in male offspring on maternal HF diet [51-53]. Our study showed that neither maternal nor postnatal HF diet affected AMPK signaling in female hepatocytes. However, H1N and H5N offspring seemed to experience lipid catabolism in liver, evidenced by enhanced pACC/ACC. The enhanced lipid expenditure was balanced by a potential increase in lipid storage, evidenced by the upregulation of lipogenesis genes in the offspring of mothers on H1N and H5N diets. Increase in either lipid expenditure or lipid storage was not found in H9N. Taking this evidence into account, our study, for the first time, suggested a potential mechanism involving an adaptive regulation of AMPK signaling responsible for lipogenesis, which co-contributes to maintaining hepatic lipid homeostasis by maternal diet intervention.
In summary, our work demonstrates the ability of properly timed maternal diet intervention to reduce risk yet effectively illustrates that maternal diet, if poorly timed, can exacerbate negative offspring phenotypes, in some instances more so than without intervention. In our study, the transition periods of 1week, 5 weeks and 9 weeks were selected due to three reasons. First, like human, the mouse has a “metabolic memory” from 1 week up to 5 months according to different treatments and different characteristics being evaluated [54-56]; second, according to Jackson laboratory [57], a mouse at the age of 3 to 6 months is considered as a young and mature adult which is at the human age of 20 to 30 years old; third, during the age of 3 to 6 months, every 4-week is considered as three-year of human [58]. Thus, we selected these transition periods so that the female mouse age “equals to” the human age of 24 yrs (H1N), 27 yrs (H5N) and 30 yrs (H9N), and the transition period is about 1 years (H1N), 4 years (H5N) and 7 years (H9N) in human life. With the female age spans from 20 to 30 years old, the concern about the age-related high-risk pregnancy that affects the offspring health might be ignored. The early onset of symptoms in our mice suffered with short- or medium-term maternal diet intervention, independent of maternal glucose intolerance or obesity, prompted the question as to if and how this maternal diet transition time point may contribute to in utero metabolic programming in human. Targeting the critical window prior to gestation by providing feasible, lifestyle modifications may assist in delaying or reversing the intergenerational cycle of obesity and aberrant metabolic outcomes in both mother and offspring. Although the period for human to adapt for metabolic changes is absolutely not as simple as we describe earlier, our study will help to lay the framework for designing future clinical trials as a feasible and efficient option to enact change and provide guidelines for both mothers and healthcare providers.
Supplementary Material
Highlights.
-
□
Different maternal diet intervention times differentially affect offspring obesity.
-
□
A long-term maternal diet (H9N) intervention effectively prevented offspring obesity.
-
□
H9N diet intervention rebalanced offspring glucose homeostasis.
-
□
H9N normalized the blunted insulin signaling and overactivated Akt signaling in offspring.
-
□
H9N diet normalized hepatic AMPK signaling pathway for lipid homeostasis.
ACKNOWLEDGMENTS
This project is supported by grants from the National Institutes of Health (NIDDK 1R01DK112368-01 to LX and KZ). This work is supported by the USDA National Institute of Food and Agriculture, [Hatch] project [1010406] to LX.
Footnotes
Declarations of interest:
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ogden CL, Carroll MD, Kit BK, Flegal KM: Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH: Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. Journal of molecular endocrinology 2008, 41:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charlton M: Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol 2004, 2:1048–1058. [DOI] [PubMed] [Google Scholar]
- 4.Tenenbaum-Gavish K, Hod M: Impact of maternal obesity on fetal health. Fetal diagnosis and therapy 2013, 34:1–7. [DOI] [PubMed] [Google Scholar]
- 5.Leddy MA, Power ML, Schulkin J: The impact of maternal obesity on maternal and fetal health. Reviews in obstetrics & gynecology 2008, 1:170–178. [PMC free article] [PubMed] [Google Scholar]
- 6.Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH: Predicting obesity in young adulthood from childhood and parental obesity. The New England journal of medicine 1997, 337:869–873. [DOI] [PubMed] [Google Scholar]
- 7.Ainge H, Thompson C, Ozanne SE, Rooney KB: A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int J Obes (Lond) 2011, 35:325–335. [DOI] [PubMed] [Google Scholar]
- 8.Alfaradhi MZ, Ozanne SE: Developmental programming in response to maternal overnutrition. Frontiers in genetics 2011, 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams L, Seki Y, Vuguin PM, Charron MJ: Animal models of in utero exposure to a high fat diet: a review. Biochim Biophys Acta 2014, 1842:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masuyama H, Hiramatsu Y: Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153:2823–2830. [DOI] [PubMed] [Google Scholar]
- 11.Ornellas F, Souza-Mello V, Mandarim-de-Lacerda CA, Aguila MB: Programming of obesity and comorbidities in the progeny: lessons from a model of diet-induced obese parents. PLoS One 2015, 10:e0124737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zambrano E, Martinez-Samayoa PM, Rodriguez-Gonzalez GL, Nathanielsz PW: Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J Physiol 2010, 588:1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stettler N, Kumanyika SK, Katz SH, Zemel BS, Stallings VA: Rapid weight gain during infancy and obesity in young adulthood in a cohort of African Americans. Am J Clin Nutr 2003, 77:1374–1378. [DOI] [PubMed] [Google Scholar]
- 14.Magarey AM, Daniels LA, Boulton TJ, Cockington RA: Predicting obesity in early adulthood from childhood and parental obesity. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2003, 27:505–513. [DOI] [PubMed] [Google Scholar]
- 15.Danielzik S, Czerwinski-Mast M, Langnase K, Dilba B, Muller MJ: Parental overweight, socioeconomic status and high birth weight are the major determinants of overweight and obesity in 5–7 y-old children: baseline data of the Kiel Obesity Prevention Study (KOPS). International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2004, 28:1494–1502. [DOI] [PubMed] [Google Scholar]
- 16.Salsberry PJ, Reagan PB: Dynamics of early childhood overweight. Pediatrics 2005, 116:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravelli GP, Stein ZA, Susser MW: Obesity in young men after famine exposure in utero and early infancy. The New England journal of medicine 1976, 295:349–353. [DOI] [PubMed] [Google Scholar]
- 18.Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP: Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr 1999, 70:811–816. [DOI] [PubMed] [Google Scholar]
- 19.Law CM, Barker DJ, Osmond C, Fall CH, Simmonds SJ: Early growth and abdominal fatness in adult life. Journal of epidemiology and community health 1992, 46:184–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips DI, Walker BR, Reynolds RM, Flanagan DE, Wood PJ, Osmond C, Barker DJ, Whorwood CB: Low birth weight predicts elevated plasma cortisol concentrations in adults from 3 populations. Hypertension 2000, 35:1301–1306. [DOI] [PubMed] [Google Scholar]
- 21.In: Weight Gain During Pregnancy: Reexamining the Guidelines. edn. Edited by Rasmussen KM, Yaktine AL. Washington (DC); 2009. [PubMed] [Google Scholar]
- 22.Schmidt SF, Jorgensen M, Chen Y, Nielsen R, Sandelin A, Mandrup S: Cross species comparison of C/EBPalpha and PPARgamma profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC genomics 2011, 12:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villamor E, Cnattingius S: Interpregnancy weight change and risk of adverse pregnancy outcomes: a population-based study. Lancet 2006, 368:1164–1170. [DOI] [PubMed] [Google Scholar]
- 24.Bogaerts A, Van den Bergh BR, Ameye L, Witters I, Martens E, Timmerman D, Devlieger R: Interpregnancy weight change and risk for adverse perinatal outcome. Obstet Gynecol 2013, 122:999–1009. [DOI] [PubMed] [Google Scholar]
- 25.Shapira N: Prenatal nutrition: a critical window of opportunity for mother and child. Womens Health (Lond Engl) 2008, 4:639–656. [DOI] [PubMed] [Google Scholar]
- 26.Fu Q, Olson P, Rasmussen D, Keith B, Williamson M, Zhang KK, Xie L: A short-term transition from a high-fat diet to a normal-fat diet before pregnancy exacerbates female mouse offspring obesity. Int J Obes (Lond) 2016, 40:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ong ZY, Muhlhausler BS: Maternal “junk-food” feeding of rat dams alters food choices and development of the mesolimbic reward pathway in the offspring. Faseb J 2011, 25:2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh MM, Brunt EM: Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol 2007, 128:837–847. [DOI] [PubMed] [Google Scholar]
- 29.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH et al. : Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 2011, 14:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang HH, Huang J, Duvel K, Boback B, Wu S, Squillace RM, Wu CL, Manning BD: Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 2009, 4:e6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horton JD, Shimomura I, Ikemoto S, Bashmakov Y, Hammer RE: Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J Biol Chem 2003, 278:36652–36660. [DOI] [PubMed] [Google Scholar]
- 32.Kim JB, Spiegelman BM: ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996, 10:1096–1107. [DOI] [PubMed] [Google Scholar]
- 33.Giby VG, Ajith TA: Role of adipokines and peroxisome proliferator-activated receptors in nonalcoholic fatty liver disease. World J Hepatol 2014, 6:570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Souza-Mello V: Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J Hepatol 2015, 7:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferezou-Viala J, Roy AF, Serougne C, Gripois D, Parquet M, Bailleux V, Gertler A, Delplanque B, Djiane J, Riottot M et al. : Long-term consequences of maternal high-fat feeding on hypothalamic leptin sensitivity and diet-induced obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 2007, 293:R1056–1062. [DOI] [PubMed] [Google Scholar]
- 36.Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ: Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation 1996, 94:3246–3250. [DOI] [PubMed] [Google Scholar]
- 37.Ribeiro AM, Lima Mde C, de Lira PI, da Silva GA: [Low birth weight and obesity: causal or casual association?]. Rev Paul Pediatr 2015, 33:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu ZB, Han SP, Zhu GZ, Zhu C, Wang XJ, Cao XG, Guo XR: Birth weight and subsequent risk of obesity: a systematic review and meta-analysis. Obes Rev 2011, 12:525–542. [DOI] [PubMed] [Google Scholar]
- 39.Smith J, Cianflone K, Biron S, Hould FS, Lebel S, Marceau S, Lescelleur O, Biertho L, Simard S, Kral JG et al. : Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab 2009, 94:4275–4283. [DOI] [PubMed] [Google Scholar]
- 40.Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P: Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 2006, 118:e1644–1649. [DOI] [PubMed] [Google Scholar]
- 41.Pettersson US, Walden TB, Carlsson PO, Jansson L, Phillipson M: Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One 2012, 7:e46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS et al. : Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 2010, 18:463–469. [DOI] [PubMed] [Google Scholar]
- 43.Medrikova D, Jilkova ZM, Bardova K, Janovska P, Rossmeisl M, Kopecky J: Sex differences during the course of diet-induced obesity in mice: adipose tissue expandability and glycemic control. Int J Obes (Lond) 2012, 36:262–272. [DOI] [PubMed] [Google Scholar]
- 44.Brown MS, Goldstein JL: Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 2008, 7:95–96. [DOI] [PubMed] [Google Scholar]
- 45.Matsumoto M, Han S, Kitamura T, Accili D: Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest 2006, 116:2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wrighton KH: AMPK moonlights in mitosis. Nat Rev Mol Cell Biol 2012, 13:64. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG, Ross FA, Hawley SA: AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012, 13:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF et al. : Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472:230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA et al. : AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A 2011, 108:16092–16097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM et al. : Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 2010, 11:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shankar K, Kang P, Harrell A, Zhong Y, Marecki JC, Ronis MJ, Badger TM: Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 2010, 151:2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang K, Li X, Zhang L, Yang H: Association between intrauterine mild hyperglycemia and postnatal highfat diet with adiponectin and AMPK pathway genes. Gynecol Endocrinol 2016, 32:110–115. [DOI] [PubMed] [Google Scholar]
- 53.Benatti RO, Melo AM, Borges FO, Ignacio-Souza LM, Simino LA, Milanski M, Velloso LA, Torsoni MA, Torsoni AS: Maternal high-fat diet consumption modulates hepatic lipid metabolism and microRNA-122 (miR-122) and microRNA-370 (miR-370) expression in offspring. Br J Nutr 2014, 111:2112–2122. [DOI] [PubMed] [Google Scholar]
- 54.Intine RV, Sarras MP Jr.: Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Curr Diab Rep 2012, 12:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ceriello A, Ihnat MA, Thorpe JE: Clinical review 2: The “metabolic memory”: is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab 2009, 94:410–415. [DOI] [PubMed] [Google Scholar]
- 56.Cameron KM, Miwa S, Walker C, von Zglinicki T: Male mice retain a metabolic memory of improved glucose tolerance induced during adult onset, short-term dietary restriction. Longev Healthspan 2012, 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jackson Labratory: https://www.journals.elsevier.com/the-journal-of-nutritional-biochemistry/call-for-papers/call-for-papers-special-issue-on-nutritional-modulation.
- 58.Dutta S, Sengupta P: Men and mice: Relating their ages. Life Sci 2016, 152:244–248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.