

Abstract
Background
The highly selective oral tropomyosin receptor kinase (TRK) inhibitor larotrectinib has demonstrated significant activity in adult and pediatric TRK fusion cancers. In the current study, the authors describe the clinical course of children with locally advanced TRK fusion sarcoma who were treated preoperatively with larotrectinib and underwent subsequent surgical resection.
Methods
A total of 24 children were treated on a pediatric phase 1 trial of larotrectinib (ClinicalTrials.gov identifier NCT02637687). Five children who had a documented TRK fusion sarcoma and underwent surgical resection were included in the current analysis. Tumor response (Response Evaluation Criteria In Solid Tumors [RECIST] version 1.1) and surgical outcomes were collected prospectively.
Results
A total of 5 patients (median age, 2 years; range, 0.4‐12 years) had locally advanced infantile fibrosarcoma (3 patients) or soft‐tissue sarcoma (2 patients). Four patients had disease that was refractory to standard therapy. All 5 patients achieved a partial response to larotrectinib by version 1.1 of RECIST and underwent surgical resection after a median of 6 cycles (range, 4‐9 cycles) of treatment. Surgical resections were R0 (negative resection margins with no tumor at the inked resection margin) in 3 patients, R1 (microscopic residual tumor at the resection margin) in 1 patient, and R2 (macroscopic residual tumor at the resection margin) in 1 patient. Three patients achieved complete (2 patients) or near‐complete (>98% treatment effect; 1 patient) pathologic responses. These patients remained in follow‐up and were no longer receiving larotrectinib for a minimum of 7 to 15 months postoperatively. Two patients had viable tumor at the time of surgical resection and positive resection margins and continued to receive adjuvant larotrectinib. No patients experienced postoperative complications or wound healing issues.
Conclusions
Children with locally advanced TRK fusion sarcomas may proceed to surgical resection after treatment with the selective TRK inhibitor larotrectinib, thereby sparing them the potentially significant morbidity noted with current approaches. These results support the evaluation of larotrectinib as presurgical therapy in children with newly diagnosed TRK fusion sarcomas.
Keywords: infantile fibrosarcoma, larotrectinib, local control, neurotrophic receptor tyrosine kinase (NTRK), pediatric, sarcoma, surgery, tropomyosin receptor kinase (TRK) fusion.
Short abstract
Children with locally advanced tropomyosin receptor kinase (TRK) fusion sarcomas may proceed to surgical resection after neoadjuvant treatment with the selective oral TRK inhibitor larotrectinib, sparing them the potentially significant morbidity noted with current approaches. The results of the current study support the further evaluation of larotrectinib as neoadjuvant therapy in children with newly diagnosed TRK fusion sarcomas.
Introduction
The neurotrophin tropomyosin receptor kinases TRKA, TRKB, and TRKC are encoded by the neurotrophic receptor tyrosine kinase 1 (NTRK1), NTRK2, and NTRK3 genes, respectively.1 These receptors play a diverse role in neurobiology, but also are implicated in the pathogenesis of a subset of cancers with oncogenic fusions involving one of the NTRK genes. These fusions include the region encoding the tyrosine kinase domain of the TRK protein and result in constitutive kinase activity. TRK fusions have been described in a diverse range of pediatric malignancies, particularly infantile fibrosarcoma and other sarcomas.2
Larotrectinib is a highly potent and selective inhibitor of TRKA, TRKB, and TRKC. This agent has been shown to have significant activity in adults and children with TRK fusion cancers.3 A pediatric phase 1 trial has completed accrual (ClinicalTrials.gov identifier NCT02637687) and demonstrated that larotrectinib was tolerable in this population.4 Moreover, an objective response rate of 93% (by investigator and independent review) was observed in children with TRK fusion tumors and Response Evaluation Criteria in Solid Tumors (RECIST)‐measurable disease.
The pediatric phase 1 trial of larotrectinib included a group of children with locally advanced TRK fusion sarcomas who underwent surgical resection after treatment with larotrectinib. Given the high response rate with this agent and the importance of local control in the management of sarcoma, the objective of the current study was to provide additional data regarding this group of patients. Key areas of interest included histologic response at the time of surgery, surgical resection margin status, surgical complications, and clinical course after recovery from surgery.
Materials and Methods
Patients
Patients were eligible for inclusion in the current study if they participated in the phase 1 dose escalation portion of the pediatric phase 1/2 trial of larotrectinib (ClinicalTrials.gov identifier NCT02637687) and therefore met the inclusion criteria for that clinical trial as described in the full report from the phase 1 dose escalation portion (total of 24 patients enrolled).4 It is important for the current analysis to note that patients in the phase 1 dose escalation portion of the clinical trial needed to have disease that met one of the following criteria: 1) nonresponsive to standard therapy; 2) recurrent or progressive after standard therapy; 3) no available standard therapy; or 4) locally advanced infantile fibrosarcoma that would necessitate disfiguring surgery or amputation to achieve complete surgical resection. Additional criteria for inclusion in the current study included local determination of a TRK fusion sarcoma that was locally advanced, no known metastatic disease, and any attempted surgical resection of the sarcoma after the initiation of larotrectinib.
The pediatric phase 1/2 clinical trial of larotrectinib was approved by the institutional review boards at each institution and informed consent was obtained from the families/legal guardians of all patients.
Treatment
Patients received larotrectinib orally twice daily on a continuous schedule according to the clinical trial protocol and assigned dose level.4 Required disease evaluations occurred after every other cycle during the first 12 cycles, with an optional evaluation after the first cycle. Surgical resection was permitted on study for patients when, in the opinion of the treating investigator, a patient’s tumor became capable of being resected without mutilating surgery or limb amputation. As per protocol, larotrectinib was withheld for a minimum of 24 hours prior to surgical resection. Larotrectinib could be resumed a minimum of 48 hours after surgery at the discretion of the treating investigator. Patients who discontinued treatment with larotrectinib after surgery (eg, due to an R0 surgical resection or investigator decision) continued to be followed on study and could resume treatment with larotrectinib if they experienced disease recurrence. Patients in follow‐up who were no longer receiving larotrectinib underwent repeat imaging every 3 months.
Endpoints
Response was assessed according to RECIST version 1.15 and centrally reviewed. Adverse events, including operative and postoperative complications, were coded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (version 4.03). All surgical reports were reviewed centrally by an orthopedic oncologist (M.E.A.). Tumor pathologic response and surgical resection margin status were assessed locally following the standard practice at each institution. No immunohistochemistry was performed on surgical resection specimens. The extent of surgical resection was coded as R0 (negative resection margins with no tumor at the inked resection margin), R1 (microscopic residual tumor at the resection margin), or R2 (macroscopic residual tumor at the resection margin) according to the International Union Against Cancer TNM Classification of Malignant Tumours.6 All pathology reports were reviewed centrally by an orthopedic oncologist (M.E.A.) and a pediatric pathologist (J.L.D.).
Results
Patient Characteristics
As of February 19, 2018 (the data cutoff date for this analysis), a total of 5 patients met the criteria for inclusion in the analytical cohort (Table 1). The 5 patients (2 girls and 3 boys; median age at enrollment of 2 years [range, 0.4‐12 years]) had infantile fibrosarcoma (3 patients) or other soft‐tissue sarcoma (2 patients). The patients had disease that either was refractory to prior standard therapy (4 patients) or had no standard therapy option at the time of the initial diagnosis (1 patient). Two patients had undergone previous surgical resections, but experienced disease progression after surgery and prior to treatment with larotrectinib. Barriers to surgical resection prior to treatment with larotrectinib included relationship between the tumor and major neurovascular structures (4 patients) and the extent of acetabulum involvement (1 patient).
Table 1.
Characteristics of the 5 Patients With TRK Fusion Sarcomas Treated With Larotrectinib Followed by Surgical Resection
| Baseline Characteristics | Value |
|---|---|
| Median age at time of initial diagnosis (range) | 9 mo (1 mo‐12 y) |
| Male:female ratio | 3:2 |
| Diagnosis, no. | |
| Infantile fibrosarcoma | 3 |
| Other sarcoma | 2 |
| Extent of disease at initial diagnosis, no. | |
| Locally advanced | 5 |
| Metastatic | 0 |
| Primary tumor site, no. | |
| Lower extremity | 2 |
| Upper extremity/shoulder | 2 |
| Pelvis | 1 |
| TRK fusion | |
| ETV6‐NTRK3 | 2 |
| TPM3‐NTRK1 | 1 |
| PDE4DIP‐NTRK1 | 1 |
| SQSTM1‐NTRK1 | 1 |
| Characteristics at Initiation of Larotrectinib | |
| Median age at enrollment (range) | 2 y (0.4‐12 y) |
| Extent of disease at time of study enrollment, no. | |
| Locally advanced | 5 |
| Metastatic | 0 |
| No. of prior systemic therapies, no. | |
| 0 | 1 |
| 1 | 1 |
| ≥2 | 3 |
| No. of prior surgical resections, no. | |
| 0 | 3 |
| 1 | 1 |
| 2 | 1 |
| Median age at time of first surgical resection prior to larotrectinib (range)a | 15.5 mo (2‐29 mo) |
| Characteristics of First Surgical Resection After Initiating Larotrectinib | |
| Median age at time of first surgical resection after initiating larotrectinib (range) | 35 mo (11‐163 mo) |
| Median no. of neoadjuvant cycles (range) | 6 (4‐9) |
| Median d from last larotrectinib to surgery | 1 (0‐1) |
| Extent of surgical resection, no. | |
| R0 | 3 |
| R1 | 1 |
| R2 | 1 |
| Pathologic response, no. | |
| Complete or near‐complete | 3 |
| Viable tumor seen | 2 |
Abbreviations: ETV6, ETS variant 6; NTRK1, neurotrophic receptor tyrosine kinase 1; NTRK3, neurotrophic receptor tyrosine kinase 3; PDE4DIP, phosphodiesterase 4D‐interacting protein; R0, negative resection margins with no tumor at the inked resection margin; R1, microscopic residual tumor at the resection margin; R2, macroscopic residual tumor at the resection margin; SQSTM1, sequestosome 1; TPM3, tropomyosin 3; TRK, tropomyosin receptor kinase.
Three patients did not undergo surgical resection prior to initiating treatment with larotrectinib.
Summary of Oncologic Outcomes
All 5 patients achieved a partial response with larotrectinib and proceeded to undergo surgical resection after a median of 6 cycles (range, 4‐9 cycles). Surgical resections were R0 in 3 patients, R1 in 1 patient, and R2 in 1 patient. At the time of surgical resection, 3 patients had achieved complete (2 patients) or near‐complete (>98% treatment effect; 1 patient) pathologic responses. These 3 patients remained in follow‐up and no longer receiving larotrectinib for 7 to 15 months postoperatively. Reductions in the maximal tumor diameter as per RECIST in the 3 patients who underwent R0 resections were 52%, 45%, and 31%, respectively (cases 1‐3 detailed below). Two patients had viable tumor at the time of surgical resection and resumed treatment with larotrectinib postoperatively. One of these patients also received adjuvant radiotherapy. No patients experienced postoperative complications or wound healing issues.
Case Vignettes
Additional details of the 5 patients in the analytical cohort are included here.
Case 1
The patient was a girl aged 2 years at the time of enrollment with an ETS variant 6 (ETV6)–neurotrophic receptor tyrosine kinase 3 (NTRK3) fusion, localized, infantile fibrosarcoma arising in the soft tissues posterior to the knee. She received vincristine, actinomycin D, and cyclophosphamide (VAC) for 2 cycles and experienced disease progression. Her only standard oncologic surgical option was deemed to be above‐knee amputation and therefore she was enrolled into the phase 1 larotrectinib trial. After 4 cycles of larotrectinib, the patient achieved a confirmed partial response with a 52% reduction in tumor burden and proceeded to limb‐sparing surgical resection. Her pathology demonstrated microscopic residual viable tumor foci with >98% treatment effect and clear resection margins (R0 resection). She had no intraoperative or postoperative complications. She had no wound healing issues or functional deficits on physical examination. At the time of last follow‐up, the patient was disease free for >15 months after surgery.
Case 2
The patient was a boy aged 5 months at the time of enrollment with an ETV6‐NTRK3 fusion, localized, infantile fibrosarcoma arising in the soft tissues of the forearm. He received vincristine and actinomycin D for 2 cycles and experienced disease progression. His chemotherapy was changed to VAC, but the response remained inadequate to allow a limb‐sparing surgery (Fig. 1A). Therefore, the patient initiated treatment with larotrectinib and achieved a confirmed partial response after 4 cycles with a 45% reduction in tumor burden. He was referred for definitive limb‐sparing surgery after 6 cycles of larotrectinib (Fig. 1B). His pathology demonstrated a complete pathologic response and clear resection margins with scar tissue noted (R0 resection). He had no intraoperative or postoperative complications, and no wound healing issues or functional deficits. At the time of last follow‐up, the patient was disease free at >12 months of follow‐up and was without evidence of disease.
Figure 1.
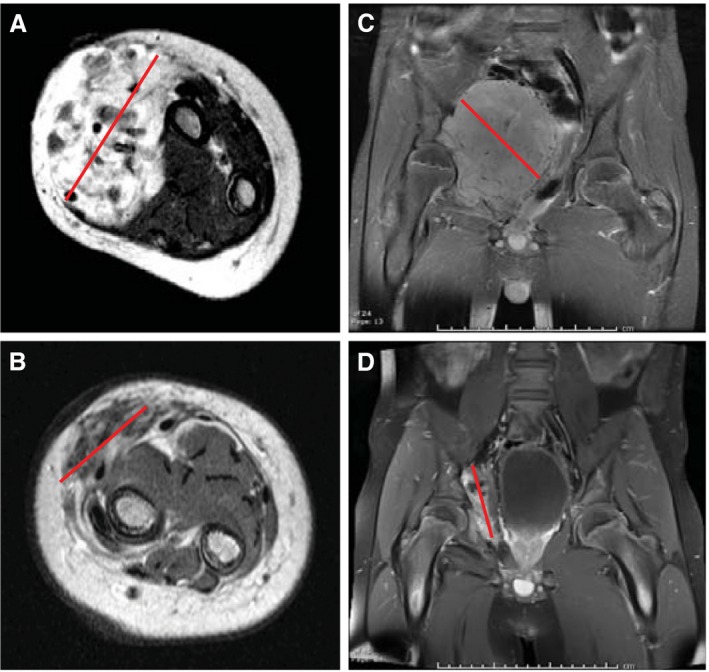
(A) Baseline T1‐weighted magnetic resonance imaging (MRI) with gadolinium obtained prior to treatment with larotrectinib in case 2 with an ETS variant 6 (ETV6)–neurotrophic receptor tyrosine kinase 3 (NTRK3) fusion infantile fibrosarcoma arising in the forearm. (B) Preoperative T1‐weighted MRI with gadolinium obtained after 6 cycles of larotrectinib in case 2. (C) Baseline T1‐weighted MRI with gadolinium obtained prior to treatment with larotrectinib in case 3 with a tropomyosin 3 (TPM3)–NTRK1 fusion spindle cell sarcoma arising in the pelvis. (D) Preoperative T1‐weighted MRI with gadolinium after 9 cycles of larotrectinib in case 3. The red line indicates the maximum dimension in each panel.
Case 3
The patient was a boy aged 12 years at the time of enrollment with a tropomyosin 3 (TPM3)–NTRK1 fusion, localized, poorly classified spindle cell sarcoma arising in the pelvis and involving the acetabulum (Fig. 1C). He did not have a standard medical option available. Surgical resection was anticipated to result in significant functional deficits. Therefore, the patient initiated treatment with larotrectinib as his first form of anticancer therapy. He achieved a confirmed partial response after 8 cycles of larotrectinib with a 31% reduction in tumor burden and was referred for surgical resection after 9 cycles of larotrectinib (Fig. 1D). He underwent an uncomplicated surgical resection and had no viable tumor noted in the surgical resection specimen (R0 resection). No intraoperative or postoperative complications were noted, including no delay in wound healing. Treatment with larotrectinib was not restarted postoperatively and at the time of last follow‐up the patient had remained in follow‐up for >7 months after surgery with no evidence of tumor recurrence.
Case 4
The patient was a boy aged 15 months at the time of enrollment with a phosphodiesterase 4D‐interacting protein (PDE4DIP)–NTRK1 fusion, localized, intramuscular soft‐tissue sarcoma of the right upper thigh. He underwent an initial attempt at surgical resection with tumor regrowth noted 1 to 2 months postoperatively along with new extension into muscle. The patient subsequently was treated with 1 cycle of VAC with evidence of progressive disease despite treatment, followed by 1 cycle of chemotherapy with ifosfamide and doxorubicin. He continued to exhibit no response to systemic therapy and therefore treatment with larotrectinib was initiated, with the patient receiving his first dose approximately 1 month after the last dose of ifosfamide and doxorubicin. He achieved an initial partial response after 2 cycles of larotrectinib with a 37% reduction in tumor burden. After 6 cycles of larotrectinib, he had experienced a 64% reduction in tumor burden and was referred for surgery. He underwent an R2 surgical resection without intraoperative or postoperative complications and with preservation of function of the leg. Approximately 40% to 60% viable tumor was observed, including at multiple surgical resection margins (R2 resection). The tumor was noted to demonstrate significant morphologic treatment‐related changes with alteration from a cellular, round cell morphologic pattern to a marked histologic variability including areas described as myxoid to fibrotic/scar‐like and cytomorphology attributed to treatment effect. No necrosis was identified, although a portion of the tumor bed consisted of nonneoplastic tissue. Given these pathology findings and evidence of short‐interval local recurrence with measurable disease, the patient resumed treatment with larotrectinib 4 weeks after surgery and experienced a second partial response. After an additional 7 cycles of larotrectinib, he underwent a second uncomplicated R2 resection, with viable tumor again noted at the resection margin. He was treated with adjuvant radiotherapy (5400‐centigray proton equivalents) and ongoing adjuvant larotrectinib for 7 cycles and remained in complete response at the time of last follow‐up.
Case 5
The patient was a girl aged 2 years at the time of enrollment with a large sequestosome 1 (SQSTM1)–NTRK1 fusion, localized, infantile fibrosarcoma of the shoulder, extending to the neck, back, and axilla. She received 4 cycles of VAC and achieved stable disease followed by an initial attempt at a complete surgical resection. Her pathology demonstrated resection margins that were microscopically focally positive (R1 resection). She developed a local recurrence approximately 7 months after undergoing complete surgical resection and underwent another surgical resection; microscopic residual disease was noted and the patient received 4 additional cycles of VAC. Approximately 6 months after completing chemotherapy, she experienced another local disease recurrence and underwent interventional radiology ablation. Short‐interval disease progression again was noted. Standard oncologic surgical resection would have required a forequarter amputation and therefore a chemotherapy approach was considered preferential. The patient received 2 cycles of ifosfamide and etoposide. She had an inadequate response and subsequently initiated treatment with larotrectinib. She achieved a confirmed partial response with a 93% reduction in tumor burden and was referred for surgery after 14 cycles of larotrectinib to resect a small residual nodule that was slowly increasing in size. The patient underwent an R1 surgical resection without intraoperative or postoperative complications reported. Viable tumor was noted, with histology including large hyperchromatic cells and scattered multinucleated cells. Tumor was focally present at the surgical resection margin. Given these pathology findings, the patient resumed adjuvant larotrectinib postoperatively. At the time of last follow‐up, she had remained on larotrectinib and without evidence of disease progression after more than a total of 20 cycles completed (7 of which were administered after surgical resection).
Discussion
To our knowledge, the current study is the first to demonstrate the feasibility of using a highly selective therapy targeted toward oncogenic TRK fusions to facilitate surgical resection in children with sarcoma. The results indicate that this approach appears to be tolerable and can render tumors resectable that otherwise would require radical or morbid surgical procedures in these young patients. We observed no wound healing issues associated with the perioperative use of larotrectinib. Moreover, larotrectinib resulted in marked histologic treatment response, including alterations in tumor morphology with some descriptions of increased cellular pleomorphism, decreased tumor burden, increased inflammation, and fibrosis of the tumor bed. Frank necrosis was observed less often. Adjuvant larotrectinib may contribute to disease control in those patients with evidence of viable tumor after neoadjuvant larotrectinib and R1/R2 surgical resections.
The role of surgery in the management of patients with infantile fibrosarcoma has evolved in recent years. Unlike many other sarcomas for which an R0 surgical resection is deemed to be a necessary component of curative therapy, more conservative procedures now are routinely performed for patients with infantile fibrosarcoma.7, 8 These conservative procedures appear to be associated with favorable outcomes in the majority of patients, although the cases highlighted in the current study suggest that a subset of these tumors have a propensity for aggressive local behavior after R1 surgical resections are performed. To the best of our knowledge, it remains unknown whether the use of larotrectinib in the neoadjuvant setting may obviate the need for surgical resection altogether in a subset of patients with infantile fibrosarcoma and a favorable response to targeted therapy.
Pertinent to this last point, it is interesting to note that all 5 patients described herein had dramatic radiographic responses. Regardless of these tumor reductions, 2 patients had extensive residual viable tumor at the time of surgical resection and subsequently were restarted on larotrectinib in an adjuvant fashion. Likewise, 3 patients who achieved partial responses by RECIST nevertheless experienced complete or nearly complete histologic responses. Additional experience will be needed to understand which patients are likely to have a complete or nearly complete histologic response to larotrectinib and which patients are likely to have residual viable tumor. Additional tools, such as functional imaging and/or the assessment of circulating tumor DNA to detect TRK fusions,9 may serve as biomarkers with which to noninvasively assess the degree of necrosis after neoadjuvant larotrectinib. It is possible that the reliable preoperative assessment of histologic response ultimately may enable the surgeon to tailor the extent of surgical resection, with more aggressive resections planned for patients with expected residual viable tumor after achieving a maximum response to neoadjuvant larotrectinib.
We acknowledge certain limitations to the current study. The cohort included only 5 patients in total, 2 of whom had soft‐tissue sarcomas and 3 of whom had infantile fibrosarcoma. We noted that the phase 2 portion of the parent phase 1/2 clinical trial currently is ongoing and provides a similar allowance for surgical resection as well as the use of adjuvant larotrectinib depending on the specific clinical scenario for each patient. Because the phase 2 portion of the trial includes a cohort of children with TRK fusion infantile fibrosarcoma and a separate cohort of children with other TRK fusion solid tumors, we expect that we will obtain additional data regarding this approach. The follow‐up period for this cohort remains limited and we continue to track their outcomes. Although radiographic responses to neoadjuvant larotrectinib, surgical reports, and pathology reports were reviewed centrally, surgical resection margin status and pathologic response were not. The assessment of surgical resection margin status may be particularly challenging in these tumors, in which treated tumor may resemble scar tissue, and therefore it is possible that some tumors classified as R0 surgical resections could have represented R1 surgical resections. Finally, adverse events were captured according to routine reporting requirements standard to clinical trials of oncology agents, but a dedicated case report form for capturing operative or postoperative adverse events was not included in the phase 1 portion.
We plan to continue to evaluate surgical outcomes in children with locally advanced TRK fusion sarcoma in the ongoing phase 2 portion of the current clinical trial. The promising initial results with this approach argue for an evaluation of the role of larotrectinib earlier in the course of the disease in children with TRK fusion sarcoma. We noted that all 3 patients with infantile fibrosarcoma in the current study cohort received alkylator‐based chemotherapy prior to receiving larotrectinib. Conventional chemotherapy also is associated with infectious risks and the need for a central venous catheter, particularly in younger patients. Sparing these young patients the known acute and late effects of conventional cytotoxic chemotherapy is a high priority. We acknowledge that the late effects of larotrectinib remain undefined, including in young children, given that the first pediatric patient treated with larotrectinib enrolled in December 2015. Additional data will be needed to define the role of larotrectinib in children with TRK fusion sarcomas, balancing antitumor activity and potential late effects. Toward this end, we currently are developing a successor clinical trial that will evaluate larotrectinib in children with newly diagnosed TRK fusion infantile fibrosarcoma or other newly diagnosed TRK fusion sarcomas.
Funding Support
The clinical trial was funded by Loxo Oncology Inc. Writing support was provided by Loxo Oncology Inc and Bayer AG to Jim Heighway of Cancer Communications. Additional support was provided by an Alex’s Lemonade Stand Foundation Center of Excellence Award (to Steven G. DuBois) and the National Institutes of Health National Center for Advancing Translational Sciences University of California at Los Angeles Clinical and Translational Science Institute grant UL1TR001881 (to Noah Federman).
Conflict Of Interest Disclosures
Steven G. DuBois has received honorarium for advisory board participation and consulting and travel expenses from Loxo Oncology Inc and travel expenses from Roche/Genentech for work performed outside of the current study. Theodore Laetsch has acted as a paid member of the advisory board for Loxo Oncology Inc and as an unpaid member of the advisory board of and received travel reimbursement from Bayer for work performed as part of the current study and has acted as a member of the advisory boards for Novartis and Lilly and received a research grant to his institution from Pfizer for work performed outside of the current study. Noah Federman has received fees for consulting and advisory board roles from Loxo Oncology Inc and fees for advisory board roles and travel expenses from Bayer AG for work performed outside of the current study. Jessica Davis has participated in a pharmaceutical advisory board for Loxo Oncology Inc/Bayer Pharmaceuticals for work performed outside of the current study. Hope E. Qamoos is an employee of and stockholder in Loxo Oncology Inc. Mark E. Reynolds has received fees for Chemistry, Manufacturing, and Controls consulting from Loxo Oncology Inc for work performed outside of the current study and has a patent US15622544 pending. Scott Cruickshank has acted as a paid consultant and received fees for study design and statistical analysis from Loxo Oncology Inc for work performed as part of the current study. Michael Cox is an employee of and stockholder in Loxo Oncology Inc and a stockholder in Bayer AG. In addition, Dr. Cox has US Patent number 62/318,041 licensed to Loxo Oncology Inc. Douglas S. Hawkins has received travel expense reimbursement from Loxo Oncology Inc, Bayer, Celgene, and Bristol‐Myers Squibb for work performed outside of the current study. Leo Mascarenhas reports that Loxo Oncology Inc is a sponsor of the Larotrectinib Phase 1 clinical trial, with all funds paid to the Children’s Hospital Los Angeles.
Author Contributions
Steven G. DuBois: Conceptualization, investigation, methodology, resources, writing–original draft, and writing–review and editing. Theodore W. Laetsch: Conceptualization, investigation, methodology, resources, and writing–review and editing. Noah Federman: Conceptualization, investigation, methodology, resources, and writing–review and editing. Brian K. Turpin: Validation and writing–review and editing. Catherine M. Albert: Investigation and resources. Ramamoorthy Nagasubramanian: Investigation and resources. Megan E. Anderson: Investigation and writing–review and editing. Jessica L. Davis: Investigation and writing–review and editing. Hope Qamoos: Investigation, methodology, and writing–review and editing. Mark E. Reynolds: Investigation and writing–review and editing. Scott Cruickshank: Investigation, methodology, and writing–review and editing. Michael C. Cox: Conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing–original draft, and writing–review and editing. Douglas S. Hawkins: Conceptualization, investigation, methodology, resources, and writing–review and editing. Leo Mascarenhas: Investigation and methodology. Alberto S. Pappo: Conceptualization, investigation, methodology, resources, and writing–review and editing.
We thank the participating patients and their families and contributing clinical staff across all sites. We also thank Alturas Analytics Inc for providing real‐time bioanalytical assessments. Medical writing services were provided by Jim Heighway, PhD, of Cancer Communications and Consultancy Ltd (Knutsford, United Kingdom).
References
- 1. Skaper SD. The neurotrophin family of neurotrophic factors: an overview. Methods Mol Biol. 2012;846:1‐12. [DOI] [PubMed] [Google Scholar]
- 2. Davis JL, Lockwood CM, Albert CM, Tsuchiya K, Hawkins DS, Rudzinski ER. Infantile NTRK‐associated mesenchymal tumors. Pediatr Dev Pathol. 2018;21:68‐78. [DOI] [PubMed] [Google Scholar]
- 3. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378:731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: a multicentre, open‐label, phase 1 study. Lancet Oncol. 2018;19:705‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eisenhauer EA, Therasse P, Bogaerts J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 6. Hermanek P, Sobin LH, eds. TNM Classification of Malignant Tumors, 4th edn Berlin: Springer‐Verlag; 1987. [Google Scholar]
- 7. Orbach D, Brennan B, De Paoli A, et al. Conservative strategy in infantile fibrosarcoma is possible: the European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer. 2016;57:1‐9. [DOI] [PubMed] [Google Scholar]
- 8. Orbach D, Rey A, Cecchetto G, et al. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol. 2010;28:318‐323. [DOI] [PubMed] [Google Scholar]
- 9. Drilon A, Nagasubramanian R, Blake JF, et al. A next‐generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion–positive solid tumors. Cancer Discov. 2017;7:963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]