

Abstract
Background:
Autoimmune diseases have increased in incidence and prevalence worldwide. While genetic predispositions play a role, environmental factors are a major contributor. Atmospheric particulate matter (PM) is a complex mixture composed of metals, nitrates, sulfates and diverse adsorbed organic compounds like polycyclic aromatic hydrocarbons (PAHs) and dioxins. Exposure to atmospheric PM aggravates autoimmune diseases such as type 1 diabetes, rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus, among others. PAHs and dioxins are known aryl hydrocarbon receptor (AHR) ligands. The AHR modulates T cell differentiation and directs the balance between effector and regulatory T cells in vitro and in experimental autoimmune encephalomyelitis (EAE), a murine model of autoimmune disease. This study aims to identify pathways that contribute to autoimmune disease and their potential use as therapeutic targets to alleviate symptoms and the need for global immunosuppression. This study tests the hypothesis that atmospheric PM enhances effector T cell differentiation and aggravates autoimmune disease.
Results:
An atmospheric ambient urban dust PM sample, standard reference material (SRM)1649b, was tested for its effects on autoimmunity. SRM1649b PM enhanced Th17 differentiation in an AHR-dependent manner in vitro, however intranasal treatment of SRM1649b PM delayed onset of EAE and reduced cumulative and peak clinical scores. Chronic and acute intranasal exposure of SRM1649b PM delayed onset of EAE. Chronic intranasal exposure did not reduce severity of EAE while acute intranasal exposure significantly reduced severity of disease. Acute intranasal treatment of low dose SRM1649b PM had no effect on clinical score or day of onset in EAE. Delayed onset of EAE by intranasal SRM1649b PM was AHR-dependent in vivo. Oral gavage of SRM1649b PM, in the absence of AHR ligands in the diet, had no effect on day of disease onset or severity of EAE. Day 10 analysis of T cells in the CNS after intranasal treatment of SRM1649b PM showed a reduction of pathologic T cell subsets in vivo. Moreover, MOG-specific splenocytes require AHR to generate or maintain IL-10 producing cells and reduce IFNγ producing cells in vitro.
Conclusions:
These results identify the AHR pathway as a potential target for driving targeted immunosuppression in the CNS in the context of atmospheric PM-mediated autoimmune disease. The effects of SRM1649b PM on EAE are dependent on route of exposure, with intranasal treatment reducing severity of EAE and delaying disease onset while oral gavage has no effect. Intranasal SRM1649b PM reduces pathologic T cells in the CNS, specifically Th1 cells and Th1Th17 double positive cells, leading to reduced severity of EAE and AHR-dependent delayed disease onset. Additionally, SRM1649b PM treatment of antigen-specific T cells leads to AHR-dependent increase in percent IL-10 positive cells in vitro. These findings may shed light on the known increase of infection after exposure to atmospheric PM and serve as the first step in identifying components of the AHR pathway responsible for Th1-mediated immunosuppression in response to atmospheric PM exposure.
Keywords: autoimmune disease, particulate matter, aryl hydrocarbon receptor, T cell differentiation, experimental autoimmune encephalomyelitis
1. Introduction:
Autoimmune diseases have dramatically increased in prevalence and incidence worldwide (Brauer et al., 2016; Health, 2005; Institute, 2018; Lerner et al., 2015). In the United States alone, approximately 24.5 million people have been diagnosed with autoimmunity, and more than 80 autoimmune diseases have been identified (Miller et al., 2012). Currently, genetic predisposition accounts for less than half of all autoimmune disease, leaving environmental factors as a major contributor (Rosenblum et al., 2015; Vojdani et al., 2014). Epidemiologic studies strongly suggest that exposure to air pollution can increase both the incidence and severity of autoimmune disease (Farhat et al., 2011; Gawda et al., 2017). Specifically, exposure to PM1 in the atmosphere aggravates type 1 diabetes, rheumatoid arthritis, and multiple sclerosis, among others (Angelici et al., 2016; Beyerlein et al., 2015; Brook et al., 2013; Chang et al., 2016; Di Ciaula, 2016; Gonzalez et al., 2013; Gregory et al., 2008; Hathout et al., 2002; Malmqvist et al., 2015; Oikonen et al., 2003; Roux et al., 2017; Vojinovic et al., 2015). Although strong correlations exist connecting PM exposure to autoimmune disease, identifying mechanisms leading to this disease process has proven particularly difficult.
Immunopathology of many autoimmune diseases is mediated by autoreactive T cells that contribute to a loss of tolerance. The AHR2 pathway directs the balance between effector and regulatory T cells (Ehrlich et al., 2018; Mohinta et al., 2015; Quintana et al., 2008; Veldhoen et al., 2008). The AHR is a ligand-activated transcription factor that responds to both endogenous and exogenous ligands, including toxicants present in PM. The organic fraction of PM contains high-affinity AHR ligands such as PAHs3 and dioxins, both of which are disease and cancer-causing agents (Cheung et al., 2011; Kelly and Fussell, 2012; Valavanidis et al., 2008; Vincent et al., 1997). In the context of autoimmune disease, some ligands of the AHR have been shown to enhance Treg4 generation and ameliorate autoimmunity, while others augment Th175 differentiation and aggravate autoimmunity (Quintana et al., 2008; Veldhoen et al., 2008). Recent data suggest the extent and duration of AHR activation by high-affinity AHR ligands are primary factors driving the fate of T cell differentiation and disease outcome (Ehrlich et al., 2018).
The current study addresses a gap in knowledge regarding identification of pathways that contribute to autoimmune disease and their potential use as therapeutic targets to prevent or mitigate symptoms and alleviate the need for global immunosuppression. The specific focus was to define the role of AHR in autoimmune responses to PM given its known effects on T cell differentiation. We hypothesized that intranasal treatment of a standardized sample of ambient urban dust particles, SRM1649b6 PM, containing PAHs and dioxins, would enhance Th17 differentiation and worsen autoimmune disease. Surprisingly, intranasal exposure to SRM1649b PM delayed onset of EAE7 in an AHR-dependent manner and reduced pathologic T cells present in the CNS8. These results implicate the AHR pathway in PM-mediated suppression of autoimmune disease using a model of murine EAE. These data offer promising potential of the AHR pathway as an early target to delay onset of disease and as a tool to investigate immunosuppressive factors that mitigate symptoms during the chronic disease phase. Ultimately, work in this area will yield new molecular pathway targets for more specific and targeted immunotherapies and treatments and lend insight into immunosuppressive pathways.
2. Materials and methods:
2.1. Mice
Wild-type (WT), C57BL/6J mice were obtained from Jackson Laboratories (stock# 000664) or bred in house in a Specific Pathogen Free facility. The 2D2 TCR9 transgenic mice, which have a T cell receptor (TCR) specific for the MOG35–5510, were also obtained from Jackson Laboratories (stock# 006192). Christopher Bradfield provided Ahrnull (Ahr−/−) (Schmidt et al., 1996) mice on a C57BL/6J background. All these genotypes have been backcrossed into the C57BL/6J background for eight generations, ensuring that the knockout genotypes reside in a genetic background that is >99.8% C57BL/6J (Nebert et al., 2000). All mice were maintained under specific, pathogen free conditions. All animal experiments were performed in accordance with protocols approved by the School of Medicine and Public Health Institutional Animal Care and Use Committee at the University of Wisconsin-Madison.
2.2. Particulate Matter (PM) Sample Preparation
The SRM1649b PM was obtained from the NIST11 (Gaithersburg, MD). Dispersed suspensions of SRM1649b PM were created by sonication in sterile PBS12 for 15 minutes in a cooling water bath. SRM1649b PM was used at 40mg/mL or 800μg PM per dose for in vivo experiments or used at 40μg/mL PM at the highest concentration in vitro.
2.3. Isolation of naïve T cells and T cell differentiation
Naïve CD4+ T cells were isolated by negative selection and purified from male or female adult WT (C57BL/6J), or Ahr−/− mice using CD4+ Isolation Kit (Miltenyi) in conjunction with QuadroMACS separator (Miltenyi). Purified naïve CD4+ T cells were plated in 96-well plates at 150,000 cells per well in 100μL and stimulated with plate-coated anti-CD3 (1μg/ml; R&D Systems) at 4°C for 24 hours and by soluble anti-CD28 (1μg/mL, BD) added at time 0. Cells were differentiated under Th17 conditions (human TGF-p (5ng/mL; R&D Systems), murine IL-6 (50ng/mL; R&D Systems)), Th1 conditions (murine IL-12 (10ng/mL; R&D systems), or Treg conditions (human TFG-β 5ng/mL; R&D Systems) for 72 hours at 37°C and 5% CO2. Treatments included positive controls FICZ13 (200nM; Enzo Life Sciences) or BNFAdjuvant (2μM; Sigma Aldrich) as well as 5 concentrations of SRM1649b PM (NIST) or PBS controls added to the culture at time 0. Media used for cultures was RPMI 1640 (Cell Gro) supplemented with Hepes buffer (Cell Gro), non-essential amino acids (Cell Gro), sodium pyruvate (Cell Gro), penicillin/streptomycin/glutamine (Cell Gro), 2-Mercaptoethanol (Life Technologies) and 5% FBS (Hyclone). All cultures included two positive controls, 6-formylindolo[3,2-b] carbazole (FICZ) (200nM; Enzo Life Sciences), which is a tryptophan photoproduct and high affinity AHR ligand and β-naphthoflavone (BNF) another AHR ligand (2μM; Sigma Aldrich). The positive controls were used to determine whether the differentiation cultures were prepared appropriately, and naïve cells responded and differentiated (Supplementary Figure 1). All treatments were done in duplicate or triplicate on each 96-well plate.
PM treatments
Cells were exposed to 5 concentrations of SRM1649b PM (NIST) or PBS vehicle control added to the culture at time 0. The treatments were in 100μL, making the final volume in each well of the 96-well plate 200μL. The concentrations were based on mass of PM per volume. The highest concentration was 40μg/mL PM and the lowest concentration was 0.78μg/mL PM. The concentrations were chosen to be 1:1000 less than the in vivo dose and in an effort to obtain a complete concentration response higher and lower concentrations were added to the experiment.
2.4. 2D2 Splenocyte Isolations and Cultures
Total splenocytes were isolated from male and female 2D2 mice. Splenocytes were treated with 1X Red Blood Cell Lysis Buffer (eBioscience) and washed with RPMI 1640 (Cell Gro) media supplemented with Hepes buffer (Cell Gro), non-essential amino acids (Cell Gro), sodium pyruvate (Cell Gro), penicillin/streptomycin/glutamine, 2-Mercaptoethanol (Life Technologies) and 5% FBS. The splenocytes were plated at 200,000 cells per well in 100μL. The splenocytes were treated with MOG35–55 peptide (20μg/ml; Tocris), 5 concentrations of SRM1649b PM (NIST) and PBS control, and 10μM AHR antagonist CH-223191. The cells were exposed to CH-223191 or nothing, and for each of those conditions, the cells were exposed to both SRM1649b PM and PBS. The cells were cultured for 5 days at 37°C and 5% CO2.
2.5. Intracellular Cytokine Staining
For intracellular cytokine staining, cultured cells were stimulated with Cell Stimulation Cocktail (eBioscience) for 5 hours. Brefeldin A 1000X (eBioscience) was added for the final 4.5 hours. Cells were then fixed and permeabilized with Intracellular Fixation & Permeabilization Buffer (eBioscience) or Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and intracellular cytokines overnight at 4°C. Cells were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit for UV Excitation (Invitrogen) or Ghost 780 (Tonbo) prior to fixation. Cells were stained with CD4 (BUV395; BD or PE, PeCy5; eBioscience) and TCRp (PeCy7; eBioscience) for extracellular markers. Cells were stained with IL-17A (FITC; eBioscience), IFNγ (eFluor450; eBioscience), and/or FOXP3 (eFluor450, eFluor660; eBioscience). Stained cells were analyzed on Fortessa (BD) or Attune NxT (Invitrogen). Data was analyzed using FlowJo software (TreeStar). Flow plots show cytokine producing cells as percent cytokine producing cells of CD4+TCRβ+ Live cells.
2.6. Experimental Autoimmune Encephalomyelitis (EAE)
PM treatment
Age-matched (approximately 9-week-old) C57BL/6J WT females were exposed intranasally to SRM1649b PM or PBS control. Female mice are used because they are more susceptible to EAE than male mice (Constantinescu et al., 2011; Rahn et al., 2014). The mice were exposed to 40.0mg/mL PM or 800μg PM of SRM1649b PM in a total of 20μL per dose. Mice were anesthetized via inhalation with isoflurane for each intranasal treatment of SRM1649b PM and PBS control. Each mouse was anesthetized for a maximum of 15 minutes. EAE scoring was done prior to anesthesia via isoflurane.
EAE Disease Induction and Course
Disease was induced on day 0 by injecting a myelin oligodendrocyte glycoprotein (MOG35–55) emulsion and pertussis toxin. First, 500μL of pertussis toxin was injected intraperitoneally (IP) at 200ng per mouse. Next, 100μL of the MOG35–55 emulsion was injected subcutaneously between the shoulder blades of each mouse. For the emulsion, 50mg MOG35–55 peptide (Tocris) was prepared and mixed in Complete Freund’s Adjuvant15 (CFA) (BD) augmented with 4mg/mL heat-killed M. tuberculosis (Difco) at a 1:1 ratio. The heat-killed M. tuberculosis was present in the emulsion at 200μg/mouse and MOG35–55 peptide was present in the emulsion at 50μg/mouse. Mice were anesthetized with isoflurane and weighed prior to injection on day 0.
Additionally, on day 2, or 44–52 hours after the initial pertussis injection, the mice were injected with another 500μL of pertussis toxin IP as a booster. Mice were scored and weighed daily starting day 7 after induction and sacrificed on day 28. The mice were scored according to a standard procedure as follows: 0, no clinical symptoms; 0.5, partially limp/flaccid tail, 1, limp/flaccid tail; 2, hind limb weakness with incomplete paralysis, loss in coordinated movement, hind impaired righting reflex; 3, partial paralysis of hind limbs; 4, complete hind limb paralysis and 5, paraplegia or moribund. Mice were monitored and managed for pain and discomfort. Mice were analyzed solely for clinical endpoints.
Clinical Endpoints
Peak clinical score was calculated as the highest clinical score each mouse achieved during the experiment. Cumulative clinical score is the sum of all the clinical scores for each mouse throughout the experiment duration. For cumulative clinical score, mice that died due to disease were given a score of 5 and that score was carried until the end of the experiment. Day of onset represents the first day after EAE induction that a score greater than 0 is noted. The scores are calculated for each mouse on each day and in the case of peak score, the highest score is used, for cumulative score, the sum of the scores from day 7 to day 28 is used, and day of onset is the first day the mouse receives an EAE score. Each mouse has its own score that is included in analysis.
In vivo Dosing Relevance
The mice were dosed with 40.0mg/mL PM of SRM1649b PM. The 40.0mg/mL PM or 800μg dose, equates to 26,666.7μg/m3, based on the amount of air a mouse would breathe in 24hrs. This is extremely high when considering the mass of PM per dose when compared to the 35.0μg/m3 24-hour primary and secondary United States Environmental Protection Agency National Ambient Air Quality Standards for PM, however the dose chosen for the current study was based on the mass of PAHs present in PM per dose.
The nanogram PAH per milliliter of exposure based on a 20μL dose of 40.0mg/mL SRM1649b PM was calculated (Table 1). The calculations show the ng PAHs per exposure (0.02mL). The 8hr and 24hr air exposures were calculated based on the amount of air a mouse would breathe in 8hrs and 24hrs (Raşid et al., 2012) (Table 1). The calculated amounts were 0.01m3 for 8hrs and 0.03m3 for 24hrs. The permissible exposure level of PAHs set for humans by Occupational Safety and Health Administration (OSHA) is 0.2mg/m3 (200,000ng/m3) which is an 8hr time- weighted average permissible exposure limit (ATSDR, 2008). This demonstrates that the in vivo exposure the mice received is within the OSHA standard for humans and can be considered a relevant exposure.
Table 1: In vivo Exposure of PAHs.
The nanogram PAH per milliliter of exposure calculations are based on a 20μL dose of 40.0mg/mL SRM1649b PM). The calculations show the ng PAHs/mL of exposure (0.02mL). The 8hr and 24hr air exposures were calculated based on the amount of air a mouse would breath in 8hrs and 24hrs. The amounts were 0.01m3 for 8 hours and 0.03m3 for 24hrs (Raşid et al., 2012). The permissible exposure level of PAHs set for humans by OSHA is 0.2mg/m3 (200,000ng/m3) which is an 8-hr time-weighted average permissible exposure limit (ATSDR, 2008). This table demonstrates that the in vivo exposure the mice receive is within the OSHA standard for humans and can be considered a relevant exposure. Abbreviations: PAH, polycyclic aromatic hydrocarbon(s); SRM, standard reference material; OSHA, Occupational Safety and Health Administration.
| ng PAH per in vivo PM exposure | |||
|---|---|---|---|
| PAHs | ng PAH/exposure | 8hrs | 24hrs |
| Phenanthrene | 3.22 | 322.40 | 107.47 |
| Anthracene | 0.41 | 41.04 | 13.68 |
| Fluoranthene | 4.99 | 499.20 | 166.40 |
| Pyrene | 3.98 | 398.40 | 132.80 |
| Benz[a]anthracene | 1.69 | 168.80 | 56.27 |
| Chrysene | 2.44 | 243.60 | 81.20 |
| Benzo[b]fluoranthene | 4.94 | 494.40 | 164.80 |
| Benzo[k]fluoranthene | 1.36 | 136.16 | 45.39 |
| Benzo[e]pyrene | 2.38 | 237.92 | 79.31 |
| Benzo[a]pyrene | 1.98 | 197.60 | 65.87 |
| Perylene | 0.49 | 49.12 | 16.37 |
| Indeno[1,2,3-cd]pyrene | 2.31 | 231.20 | 77.07 |
| Benzo[g,h,i]perylene | 3.18 | 317.60 | 105.87 |
| Dibenz[a,h]anthracene | 0.24 | 23.52 | 7.84 |
| Picene | 0.32 | 31.92 | 10.64 |
| Sum of PAHs | 33.93 ng/m3 |
3392.88 ng/m3 |
1130.96 ng/m3 |
Dosing Regimens of PM
The dosing regimens chosen in the current study aimed to account for long-term exposures, acute exposures around the time of disease induction, and every day exposures which would occur before and after disease induction. The mice received 40mg/mL PM (800μg in 20μL) of SRM1649b PM or PBS control intranasally 8 times starting at day −12 every 3 days until day 9 after induction for regular dosing. This was used to assess every day exposures and did not include additional doses at the time of disease induction. For chronic intranasal exposure, mice received 40.0mg/mL (800μg in 20μL) SRM1649b PM or PBS control intranasally 17 times starting at day −27 every 3 days until day 15 after induction. The mice were given intranasal doses on days 1 and 2 in addition to the pre-exposure. This regimen was used to assess long-term chronic exposures and two additional doses were added at the time of disease induction to assess whether chronic exposures to PM would alter severity of EAE. For acute exposure, mice received 4.0mg/mL (80μg in 20μL) or 40.0mg/mL (800μg in 20μL) SRM1649b PM or PBS control intranasally 10 times starting at day −7, every day until day 2 after induction. This regimen was used to assess a short exposure centered around the time of disease induction.
Isoflurane Anesthesia
Mice were anesthetized via inhalation with isoflurane for each intranasal treatment of SRM1649b PM and PBS. Each mouse was anesthetized for less than 15 minutes. The mice were anesthetized one time per day and all injections or intranasal treatments were conducted at one time. EAE scoring was done prior to anesthesia via isoflurane. The effects of isoflurane anesthesia on mice has been well-studied. One study demonstrated that intermittent-repeated isoflurane exposure for 45 minutes at an interval of 3–4 days for a total of 6 times, had mild effects on the mice, based on the Mouse Grimace Scale in C57BL/6Rj mice (Hohlbaum et al., 2017). In the current study, the mice are anesthetized with isoflurane for a maximum of 15 minutes and the regular dosing regimen is similar. Moreover, another study showed that that 0.5% of isoflurane for 4 hours per day, 5 days a week, for 9 weeks, caused no change in body weight, organ weight, concentration of hepatic cytochrome P450 enzymes, or the rates of hepatic microsomal metabolism and concluded that since there was no evidence of organ toxicity or enhanced or inhibited microsomal metabolism, the regimen of isoflurane was relatively non-toxic under the conditions of the study (Rice et al., 1986). This dosing regimen is similar to the acute and chronic based on the daily use of isoflurane and the number of days the mice were anesthetized, but one clear difference is how long the mice are anesthetized for. These studies suggest that the isoflurane exposure to the mice is relatively non-toxic and has mild effects on the mice. On the other hand, it has been shown that 2hrs of 1.8% isoflurane in C57BL/6J mice led to attention deficit in the mice based on a battery of behavior tests (Yonezaki et al., 2015). In the current study, any potential effects of repeated isoflurane exposure in the EAE model are controlled for by using the PBS vehicle control in which all of the mice also get anesthetized. Tolerance to isoflurane was not observed over time and no treatment effects were observed. Therefore, the effects observed after SRM1649b PM treatment are likely not due to residual isoflurane effects but the PM exposure.
Gavage PM Treatment
For AHR diet experiments, age matched female WT (C57BL/6J) mice were started on base diet (no AHR ligands) or I3C16 (enriched AHR ligands) at day −38. The base diet is a diet of essential amino acids that contains no AHR ligands and the diet enriched in AHR ligands is base diet supplemented with indole-3-carbinol, is a tryptophan derivative found in cruciferous vegetables and a precursor to high affinity AHR ligands (Hooper, 2011). Once exposed to stomach acid, I3C is converted into high affinity AHR ligands and can elicit anti-inflammatory and anti-cancer effects (Benson and Shepherd, 2011; Hooper, 2011). Mice received 4.0mg/mL in 200μL (800μg per dose or 40μg/g/day) PM or 1.0mg/mL in 200μL (200μg or 10μg/g/day) PM of SRM1649b PM or Saline control by oral gavage 8 times starting at day −12 every 3 days until day 9 after induction. The calculated dose of PM per mass of mouse was based on the average size of a mouse being 20 grams. Mice were anesthetized via inhalation with isoflurane for each gavage treatment
2.7. Spleen and lymph node isolation from EAE mice
Mice were anesthetized with isoflurane and cervical dislocation. Spleens and cervical lymph nodes were removed from EAE treated mice. Tissues were pushed through a 100μm nylon filter on top of a 50mL tube. Cells were centrifuged for 10 minutes at 4°C at 1880 RPM. Splenocyte and lymph node cells were treated with 1X Red Blood Cell Lysis Buffer (eBioscience) and washed with RPMI 1640 (Cell Gro) media supplemented with Hepes buffer (Cell Gro), non-essential amino acids (Cell Gro), sodium pyruvate (Cell Gro), penicillin/streptomycin/glutamine, 2-Mercaptoethanol (Life Technologies) and 5% FBS. The cells were washed with media and stained for flow cytometry.
2.8. Isolation of mononuclear CNS cells by percoll gradient from EAE mice
This protocol was adapted from (Pino and Cardona, 2011).
Reagent preparation:
Isotonic percoll (SIP)17 was prepared at 70% in 1X HBSS without Ca++ and Mg++. For each brain, 2mL was dispensed into a 15mL polypropylene conical tube, leaving lower phase of the gradient. The lower phase of the gradient was left at room temperature while tissue was harvested.
Tissue collection and homogenization:
Mice were anesthetized via isoflurane, and the left ventricle was perfused with ice-cold 1X HBSS. The brain and spinal cord were dissected and maintained in 15mL conical tube with 3–4mL RPMI on ice until all mice were sacrificed. The tissues were placed in 100μm nylon filters on top of a 50mL tubes. The tissue was pushed through the filter using a syringe plunger and the filter was washed periodically with RPMI to a final volume of 7mL.
Gradient set up:
3mLs of SIP were added to the 7mLs of cell suspension to make a final 30% SIP. The 10ml cell suspension was slowly layered on top of the 70% SIP. A very clear flat line was visible at the 70%−30% junction. The tube was centrifuged for 30 minutes at 500G 18°C and stopped without brakes. Next, the debris on top the tube was removed and 2.0–3.0mLs of the 70%−30% interphase was collected into a clean tube containing FACS buffer, mixed by inversion and centrifuged for 7 minutes at 500G at 18°C. The pellet was resuspended in FACS buffer and prepped for flow cytometry and intracellular cytokine staining was conducted.
2.9. Statistics
Statistics are run using GraphPad Prism. A test of normality was conducted to determine whether the statistics would be parametric or nonparametric. For all in vitro analyses, a 2-way repeated measures ANOVA was performed with p value < 0.05. For in vivo EAE experiments, Mann-Whitney T tests or unpaired T tests were conducted with a p value <0.05. Standard error of the mean (SEM) is reported for the results in each figure.
3.0. Results:
3.1. SRM1649b PM enhances Th17 differentiation in an AHR-dependent manner and has no effect on Th1 or Treg differentiation in vitro
Previously our lab showed that SRM1649b PM enhanced Th17 differentiation in an AHR- dependent manner (van Voorhis et al., 2013). Ahr is expressed in T cell subsets, including Th17, Treg, and Th1 (Julliard et al., 2017b; Julliard et al., 2016; Mascanfroni et al., 2015; Veldhoen et al., 2008). Given this, we tested whether SRM1649b PM altered T cell differentiation under different conditions in an AHR concentration-dependent manner. Naïve CD4 positive T cells were isolated from spleens of WT (C57BL/6J) mice and exposed to 5 concentrations of SRM1649b PM or PBS on day 0 and cultured under Th17, Th1, or Treg conditions for 3 days. SRM1649b PM enhanced Th17 differentiation, measured by a significant increase in the percent IL-17 positive cells, at 40 (Figure 1A (top)), 20, 12.5, and 0.78 μg/mL PM (Figure 1B (top)) but had no observable effect on Th1 (measured by the increase in the percent IFNγ positive cells) (Figure 1C (top) and 1D (top)) or Treg differentiation (measured by the percent FOXP3 positive cells) (Figure 1E and 1F) at the concentrations tested. In Ahr−/− naïve T cells, SRM1649b PM did not enhance Th17 cell differentiation (Figure 1A (bottom) and 1B (bottom)). Additionally, in Ahr’-’ naïve T cells, the percent of IFNγ positive cells trended towards a reduction at high concentrations (Figure 1C (bottom) and 1D (bottom)).
Figure 1: SRM1649b PM enhances Th17 differentiation in an AHR-dependent manner and has no effect on Th1 or Treg differentiation in vitro.
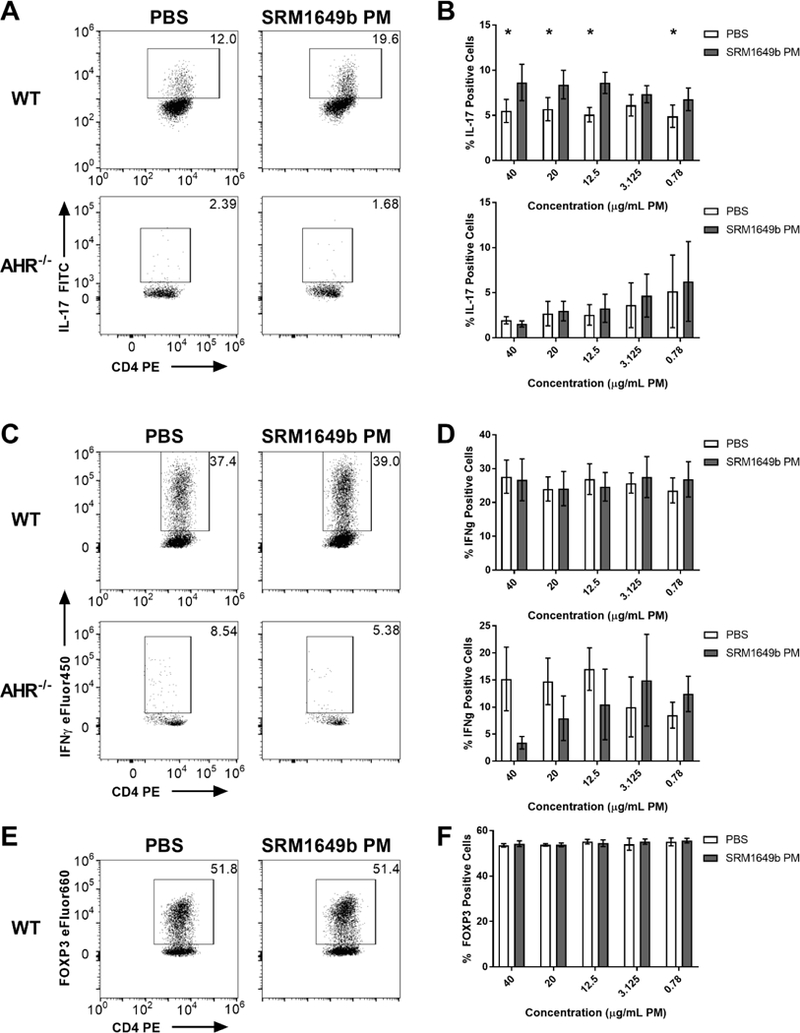
Naive CD4+ T cells were isolated from WT (C57BL/6J) or Ahr−/− mice. At time 0 cells were exposed to 5 concentrations of SRM1649b PM or PBS control and cultured for 3 days. (A) Representative flow plots of WT (top) and Ahr−/− (bottom) Th17 differentiation at 400μg/mL PM, measured by percent IL-17 positive cells. (B) SRM1649b PM enhanced WT Th17 differentiation at all concentrations except 3.125ug/mL PM compared to PBS control (top). In Ahr−/− cells, SRM1649b PM had no effect on percent IL-17 positive cells (bottom). (C) Representative flow plots of WT (top) and Ahr−/− (bottom) Th1 differentiation at 400μg/mL PM, measured by percent IFNγ positive cells. (D) SRM1649b PM had no effect on Th1 differentiation at any concentrations tested (top). In Ahr−/− cells, SRM1649b PM suppressed Th1 differentiation at 40, 20, and 12.5μg/mL PM (bottom). (E) Representative flow plots of WT Treg differentiation at 40.0μg/mL PM, measured by percent FOXP3 positive cells. (F) SRM1649b PM had no effect on Treg differentiation in WT cells. Results are mean ± SEM of (WT Th17 n=8), (Ahr−/− Th17 n=5), (WT Th1 n=5), (Ahr−/− Th1 n=2), and (WT Treg n=2). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: AHR, aryl hydrocarbon receptor; SRM, standard reference materials; PM, particulate matter; SEM, standard error of the mean.
3.2. Intranasal treatment of SRM1649b PM delays onset of EAE and reduces cumulative and peak clinical scores
Given that EAE is driven by Th17 and Th1 cells (Dardalhon et al., 2008; Korn et al., 2007; Langrish et al., 2005), and SRM1649b PM enhanced Th17 differentiation, we hypothesized that intranasal exposure to SRM1649b PM would worsen EAE. To test this hypothesis, mice received intranasal doses of 40mg/mL SRM1649b PM or PBS control starting at day −12 of EAE induction and continuing every 3 days until day 9 after induction (Figure 2A). SRM1649b PM lessened severity of EAE (Figure 2B and 2C). Exposure to SRM1649b PM significantly delayed onset of disease, the first day the mice receive a clinical score, as compared to PBS control (Figure 2C and 2D). SRM1649b PM trended towards a reduced peak score (p=0.09) representing the highest clinical score experienced during the 28-day period (Figure 2C and 2E). There was a significant reduction in cumulative score, representing the sum of the clinical scores for each individual mouse from day 7 to day 28 (Figure 2C and 2F).
Figure 2: Intranasal treatment of SRM1649b PM delays onset of EAE and ameliorates severity of EAE in vivo.
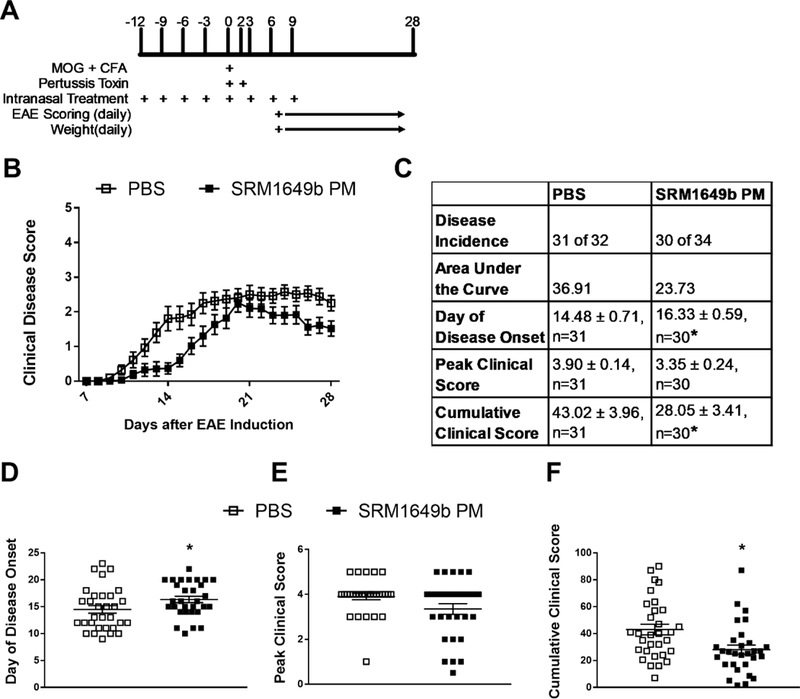
(A) Age matched female WT (C57BL/6J) received 40.0mg/mL SRM1649b PM or PBS control intranasally 8 times starting at day −12 every 3 days until day 9 after induction. Mice were weighed and scored starting on day 7 until day 28. (B, C) SRM1649b PM lessened severity of EAE. (D) SRM1649b PM delayed onset of disease represented by the first day a score is achieved. (E) SRM1649b PM significantly decreased peak clinical score over the 28-day period. (F) SRM1649b PM significantly reduced cumulative clinical score which represents the sum of scores over the 28-day period. Results are mean ± SEM of (PBS n=31) and (SRM1649b n=30). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: SRM, standard reference materials; PM, particulate matter; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
3.3. Chronic intranasal exposure of SRM1649b PM delayed onset of EAE and trended towards a reduction of disease
Intranasal exposure of SRM1649b PM lessened severity of disease unexpectedly, so we tested whether a longer, chronic exposure to intranasal SRM1649b PM would worsen severity of EAE. Mice received intranasal doses of 40mg/mL SRM1649b PM or control starting at day −27 and repeating every 3 days until day 15 after induction (Figure 3A). The mice were given intranasal doses on days 1 and 2 in addition to the pre-exposure (Figure 3A). Chronic intranasal exposure to SRM1649b PM delayed onset of EAE (Figure 3B, 3C, and 3D), as previously seen with a regular dosing regimen (Figure 2B, 2C, and 2D). Additionally, chronic intranasal exposure to SRM1649b PM trended towards reduced peak clinical score of disease (p=0.06) (Figure 3E) and had no effect on cumulative clinical score (Figure 3F).
Figure 3: Chronic and acute intranasal exposure of SRM1649b PM delayed onset of EAE and trended towards reduced severity in vivo.

(A) Age matched female WT (C57BL/6J) received 40.0mg/mL SRM1649b PM or PBS control intranasally 17 times starting at day −27 every 3 days until day 15 after induction. The mice were given intranasal doses on days 1 and 2 in addition to the pre-exposure. Mice were weighed and scored starting on day 7 until day 28. (B, C) Chronic exposure to SRM1649b PM lessened severity of EAE. (D) Chronic exposure to SRM1649b delayed onset of disease. (E) Chronic exposure to SRM1649b PM trended toward decreased peak clinical score (p=0.06) over the 28-day period. (F) Chronic exposure to SRM1649b PM had no effect on cumulative clinical score. (G) For acute exposure, mice received 40mg/mL SRM1649b PM or PBS control intranasally 10 times starting at day −7, every day until day 2 after induction. (H, I) Acute exposure to SRM1649b PM lessens severity of EAE. (J) Acute exposure to SRM1649b PM delayed onset of disease, (K) reduced peak clinical score, and (L) reduced cumulative clinical score. Results are mean ± SEM of (PBS n=9) and (SRM1649b PM n=7) for chronic exposure and (PBS n=7) and (SRM1649b PM n=9) for acute exposure. Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: SRM, standard reference materials; PM, particulate matter; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
3.4. Acute intranasal exposure of SRM1649b PM delayed onset of EAE and reduced severity of disease
Next, we tested whether acute intranasal exposure of SRM1649b PM to EAE would worsen severity of disease. We hypothesized that the acute exposure would make disease worse given that the higher exposure occurs only at disease induction. This dosing paradigm would prevent the immune system from acclimating to the presence of PM in the lung. Mice received 40mg/mL SRM1649bPM or PBS control starting at day −7, every day until day 2 after induction (Figure 3G). Acute intranasal exposure to SRM1649b PM delayed onset of EAE, as previously seen with a regular dosing regimen (Figure 3H, 3I, and 3J). Additionally, acute intranasal exposure to SRM1649b PM reduced peak clinical score of disease (Figure 3I and3K) and cumulative clinical score (Figure 3I and 3L).
3.5. Acute intranasal treatment of low dose SRM1649b PM has no effect on clinical score or day of onset in EAE
Given that acute intranasal exposure to 40mg/mL SRM1649b PM lessened severity and delayed onset of disease similar to the regular dosing regimen, we tested whether the resulting acute effect was dose-dependent. Mice received 4.0mg/mL or 40.0mg/mL SRM1649b PM or PBS control intranasally starting at day −7 every day until day 2 after induction (Figure 4A). At the 4mg/mL dose, SRM1649b PM did not significantly delay onset of disease (Figure 4C and4D) or reduce peak (Figure 4C and 4E) or cumulative disease score (Figure 4C and 4F).
Figure 4: Acute intranasal treatment of low dose SRM1649b PM has no effect on clinical score or day of onset in EAE in vivo.
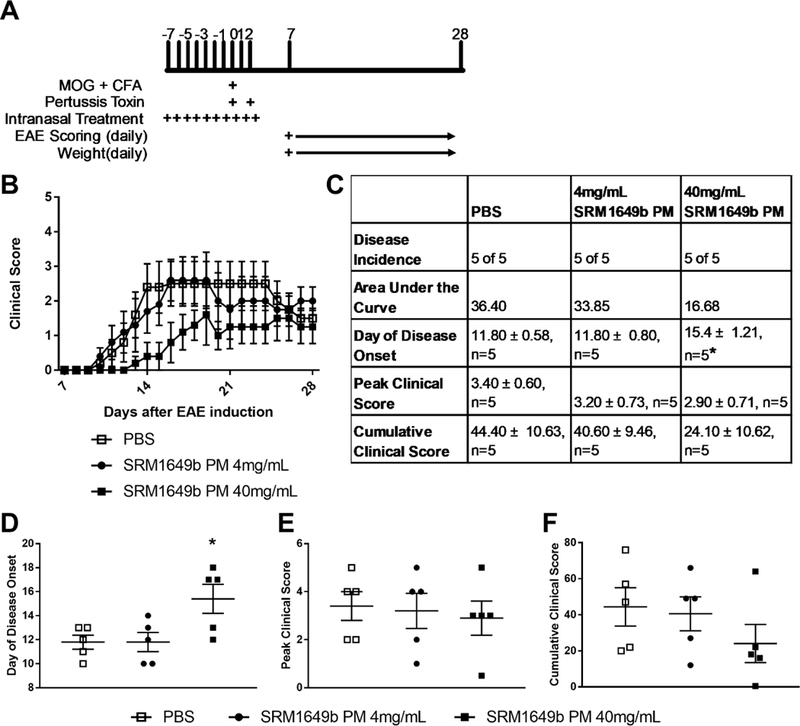
(A) Age matched female WT (C57BL/6J) mice received 4.0mg/mL or 40mg/mL SRM1649b PM diluted in PBS or PBS control intranasally 10 times starting at day −7, every day until day 2 after induction. Mice were weighed and scored starting on day 7 until day 28. (B) Low dose of SRM1649b PM had no effect on EAE and high dose of SRM1649b PM lessened severity of EAE (D) Low dose of SRM1649b PM had no effect on disease onset. High dose of SRM1649b PM significantly delayed onset of disease. (E) Low dose SRM1649b PM had no effect on peak clinical score and high dose of SRM1649b PM trended to decrease peak clinical score (p=0.82) (F) Low dose of SRM1649b PM had no effect on cumulative clinical score and high dose exposure trended towards reduced cumulative clinical score (p=0.30). Results are mean ± SEM of (PBS n=5) and (SRM 1649b PM n=5, 4mg/mL PM) and (SRM1649b PM n=5, 40mg/mL PM). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: SRM, standard reference materials; PM, particulate matter; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
However, at 40mg/mL, SRM1649b PM significantly delayed onset of disease (Figure 4C and 4D) but there was no significant difference in peak clinical score or cumulative clinical score compared to control (Figure 4C, 4E, and 4F).
3.6. Oral gavage of SRM1649b PM, in the absence of AHR ligands in the diet, has no effect on severity of EAE at high or low doses
It is known that route of exposure of AHR ligands can alter the immune response observed (Duarte et al., 2013; Veldhoen et al., 2008) and AHR ligands in the diet play a role in the balance of the immune system (Ito et al., 2007; Julliard et al., 2017a; Murray and Perdew, 2017). Based on this, we wanted to test whether altering AHR ligands in the diet would change disease status of EAE. For initial experiments, age-matched female mice were put on a diet free of AHR ligands (base diet) or on a diet enriched with AHR ligand, I3C diet starting on day −38. Mice received saline 8 times starting at day −12 every 3 days until day 9 after induction of EAE via oral gavage. Mice on base diet had more severe EAE than mice on I3C diet (Figure 5A and 5B). This finding supports previously published data demonstrating that pretreatment starting on day −1 and proceeding every other day until day 25 of I3C by oral gavage resulted in mitigated EAE symptoms compared to control (Rouse et al., 2013).
Figure 5: Oral gavage of SRM1649b PM, in the absence of AHR ligands in the diet, has no effect on severity of EAE at high or low doses in vivo.
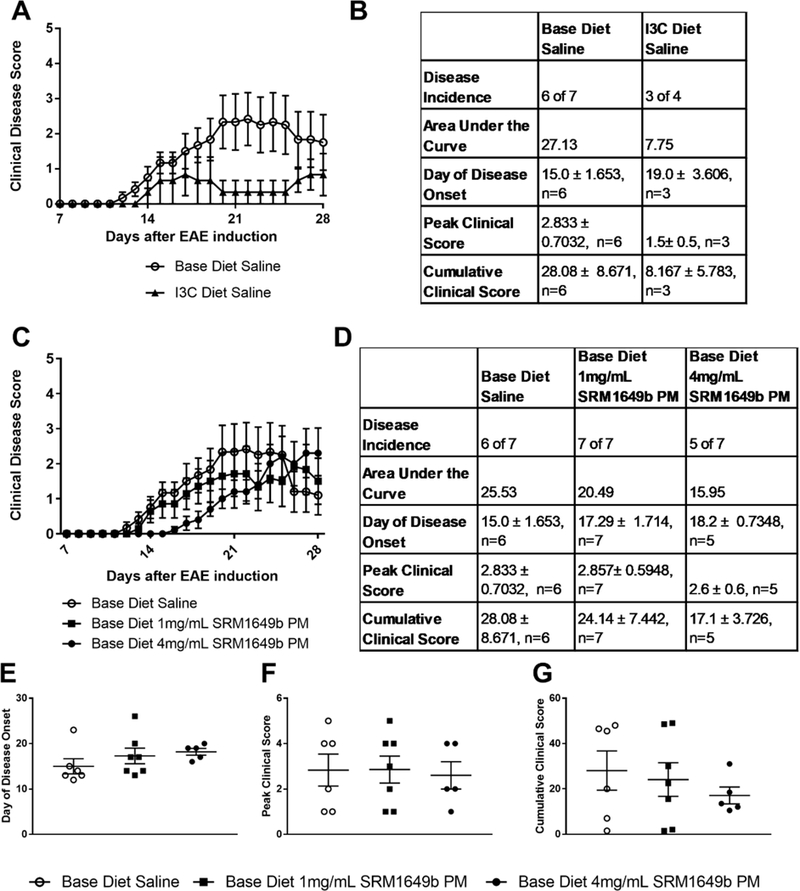
Age matched female WT (C57BL/6J) mice were started on base diet (no AHR ligands) or I3C (enriched AHR ligands) at day −38. Mice received 4.0mg/mL PM (800μg per dose) or 1.0mg/mL PM (200μg per dose) of SRM1649b PM or Saline control by oral gavage 8 times starting at day −12 every 3 days until day 9 after induction. Mice were weighed and scored starting on day 7 until day 28. (A, B) Mice were placed on base or I3C diet and administered saline by gavage. I3C diet lessened severity of EAE. (C, D) Mice on base diet were administered 4.0 or 1.0mg/mL SRM1649b PM or Saline control by oral gavage. High or low dose SRM1649b PM did not worsen severity of EAE. (E) There was no difference in day of disease onset at high or low doses of SRM1649b PM after gavage (F) There was no difference in peak clinical score at either dose of SRM1649b PM. (G) There was no difference in cumulative clinical score at either dose of SRM1649b PM. Results are mean ± SEM of (Base diet Saline n=6), (I3C diet n=3), (Base diet SRM1649b PM 4mg/mL PM, n=5), and (Base diet SRM1649b PM 1mg/mL PM, n=7). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: SRM, standard reference materials; PM, particulate matter; I3C, indole-3-carbinol; AHR, aryl hydrocarbon receptor; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
Given that a significant route of human exposure to PM and PAHs is through the GI tract (Menzie et al., 2002; Thompson, 2018) we tested whether oral gavage of SRM1649b PM to mice on a diet absent of AHR ligands would alter disease severity at high (4mg/mL PM) and low doses (1mg/mL PM). The high dose equates to 800μg per dose which is the equivalent to the high intranasal dose and the low dose is 200μg per dose. Mice were started on base diet on day −38. WT (C57BL/6J) age-matched female mice received 4mg/mL or 1mg/mL of SRM1649b PM or saline 8 times starting at day −12 every 3 days until day 9 after induction via oral gavage. Oral gavage of 4mg/mL SRM1649b PM did not delay disease onset (Figure 5D and 5E), reduce peak clinical score (Figure 5D and 5F), or cumulative clinical score (Figure 5D and5G). Oral gavage of 1mg/mL SRM1649b PM had no observable effect on peak clinical score (Figure 5D and 5F), cumulative clinical score (Figure 5D and 5G) or day of disease onset (Figure 5D and 5E). These data suggest that route of exposure does impact the severity of EAE, as intranasal treatment significantly reduced severity of EAE and delayed onset and ingestion had no significant effect on disease severity or day of disease onset.
3.7. Day 10 analysis of of T cells in the CNS after intranasal treatment SRM1649b PM show a reduction of pathologic T cell subsets in vivo
Initially, we hypothesized that intranasal treatment of SRM1649b PM would worsen EAE due to enhanced AHR-dependent Th17 and Th1 differentiation, however intranasal treatment of SRM1649b PM delays onset of disease and lessens severity of EAE. Moreover, variations in the dose, timing of dose, and route of exposure do not worsen severity of EAE. Given this, we wanted to investigate the mechanism behind the delayed onset of EAE and ameliorated disease. Mice received 40mg/mL SRM1649b PM diluted in PBS or PBS control intranasally 8 times starting at day −12 every 3 days until day 9 after induction. Mice were sacrificed on day 10 and spleen, lymph nodes, and CNS (brain and spinal cord) were harvested and processed for flow cytometry. Results show that there were no differences in the spleen or cervical lymph nodes between Th1 cells (percent IFNγ positive cells) (Figure 6A), Th17 cells (percent IL-17 positive cells) (Figure 6B), Th1Th17 double positive cells (percent IFNγ IL-17 double positive cells) (Figure 6C), or Treg cells (percent FOXP3 positive cells) (Figure 6D). There was a significant reduction in Th1 cells in mice that received intranasal SRM1649b PM compared to PBS control in the CNS (Figure 6A) and a slight reduction in Th17 cells, although not statistically different (Figure 6B). Additionally, mice exposed to SRM1649b PM had a significant reduction in the Th1Th17 double positive cells in the CNS compared to PBS control (Figure 6C). No difference in Treg cells in the CNS between mice exposed to SRM1649b PM and PBS control was seen
Figure 6: Day 10 analysis of T cells in the CNS after intranasal treatment of SRM1649b PM show a reduction of pathologic T cell subsets after EAE induction in vivo.
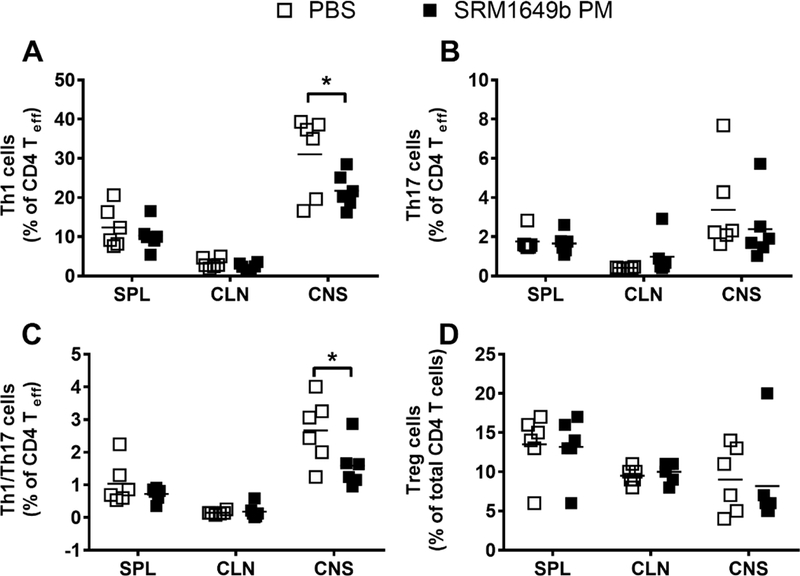
Age matched female WT (C57BL/6J) received 40.0mg/mL SRM1649b PM or PBS control intranasally 8 times starting at day −12 every 3 days until day 9 after induction. Mice were sacrificed on day 10 andthe spleen, lymph nodes, and CNS (brain and spinal cord) were harvested and processed for flow cytometry. (A) Intranasal exposure to SRM1649b PM reduced Th1 cells in the CNS, measured by percent IFNγ producing cells (B) Intranasal exposure to SRM1649b PM reduced Th17 cells in the CNS, measured by percent IL-17 positive cells, but was not statistically different. (D) SRM1649b PM intranasal exposure significantly decreased Th1/Th17 double positive cells, measured by percent of IFNγ and IL-17 producing cells, in the CNS (D) Intranasal exposure to SRM1649b PM had no impact on Treg cells in the CNS. Results are mean ± SEM of (PBS n=6) and (SRM1649b PM n=6). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: SRM, standard reference materials; PM, particulate matter; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
3.8. Delayed onset of EAE by intranasal SRM1649b PM is AHR-dependent in vivo
SRM1649b PM is a complex mixture of PM, with multiple sources collected over time, made up of numerous components including known AHR-activating PAHs and dioxins. The effects of AHR ligands on T cell differentiation and immune response can be protective as seen with TCDD18 (Quintana et al., 2008; Veldhoen et al., 2008), so we tested whether the protection seen with intranasal treatment of SRM1649b PM was AHR-dependent. Age-matched Ahr−/− or Ahr−/− female mice received 40mg/mL SRM1649b PM diluted in PBS or PBS control 8 times starting at day −12 every 3 days until day 9 after induction. In Ahr+/− mice, SRM1649b PM had no effect on peak clinical score (Figure 7A). SRM1649b PM trended toward delayed onset of disease (p=0.05) (Figure 7A and7C) and significantly reduced cumulative clinical score mice (Figure 7A). In Ahr −/− mice SRM1649b PM did not significantly delay disease onset as compared to control (Figure 7B and 7D). Additionally, there was no significant difference in peak clinical score (Figure 7B) or cumulative clinical score (Figure 7B). Of note, Ahr−/− mice are known to get less disease overall than WT C57BL/6J or Ahr+/− controls (Veldhoen et al., 2008) and this remained true in our model (Figure 7B). The disease magnitude in Ahr−/− mice is significantly reduced compared to WT C57/BL6J or Ahr+/− and the Ahr−/− mice have delayed disease onset overall regardless of treatment. making interpretation of this data challenging. Despite the reduced disease magnitude, the Ahr+/− mice trended to have delayed disease onset (p=0.05) and the Ahr−/− mice did not have delayed disease onset.
Figure 7: Delayed onset of EAE by intranasal SRM1649b PM is AHR-dependent in vivo.
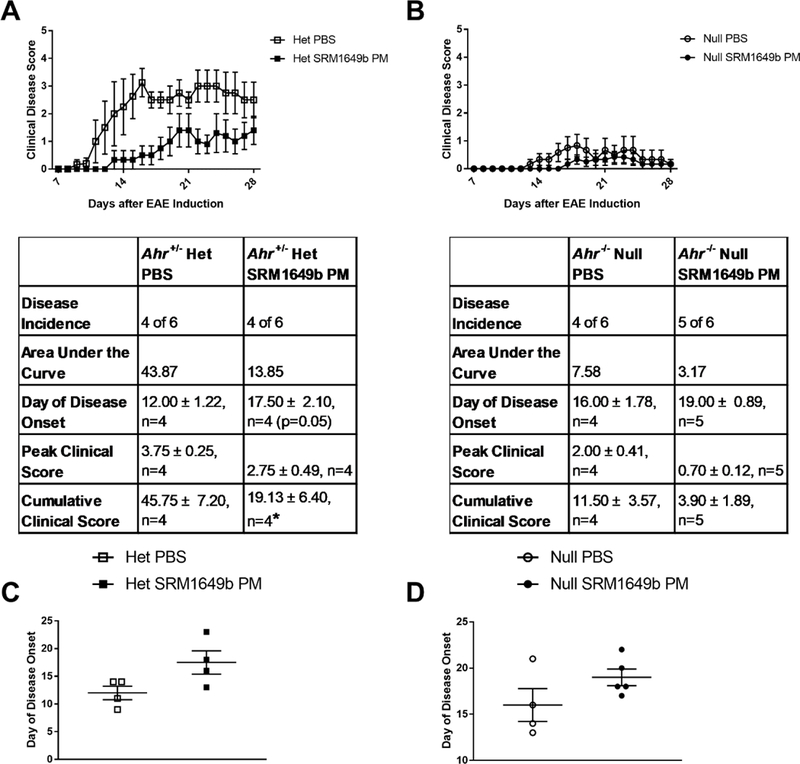
Age matched female Ahr+/− or Ahr−/− mice received 40.0mg/mL SRM1649b PM or PBS control intranasally 8 times starting at day −12 every 3 days until day 9 after induction. Mice were weighed and scored starting on day 7 until day 28. (A) In Ahr het mice, SRM1649b PM lessened severity of EAE. SRM1649b PM had no effect on peak clinical score, but significantly reduced cumulative clinical score. (B) In Ahr null mice, SRM1649b PM trended toward reduced severity of EAE, decreased area under the curve, and had no effect on cumulative clinical score or peak score. (C) In Ahr het mice, SRM1649b PM trended toward delayed onset of disease (p=0.05) and in (D) Ahr null mice SRM1649b PM had no effect on disease onset compared to PBS control. Results are mean ± SEM of (Ahr+/− PBS n=4), (Ahr+/− SRM1649b n=4), (Ahr−/− PBS n=4), and (Ahr−/− SRM1649b n=5). Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: AHR, aryl hydrocarbon receptor; SRM, standard reference materials; PM, particulate matter; PBS, phosphate buffered saline; EAE, experimental autoimmune encephalomyelitis; SEM, standard error of the mean.
3.9. MOG specific splenocytes generate or maintain IL-10 producing cells and reduce IFNγ producing cells in an AHR-dependent manner
Previous in vitro assays have focused on naïve T cells alone, which limits the physiologic relevance. We sought to identify an in vitro assay that would better assess the effects of SRM1649b PM exposures on T cells in a less artificial construct. We chose to use 2D2 mice with a transgenic T cell receptor specific for the MOG peptide. In this way we could assess influence of SRM1649b PM on the immune response that is more physiologically relevant and using an AHR antagonist CH-223191, interrogate the role of AHR. Instead of isolating naïve T cells, total splenocytes (which includes dendritic cells, macrophages, naïve, and effector T cells) were isolated and cultured for 5 days. On day 0 the cells were exposed to 5 concentrations of SRM1649b PM with or without 10μM the AHR antagonist CH-223191 and on day 5 cells were harvested for flow cytometry. There were no measurable differences of the percent IL-17 positive cells with or without AHR antagonist, CH-223191 (data not shown). In the absence of CH-223191, the percent IFNγ positive cells was significantly suppressed at the highest concentration tested 40μg/mL PM and trended to be reduced at 20 and 12.5μg/mL PM (Figure 8A). The addition of CH-223191, abrogated this suppression (Figure 8B). When examining the suppressive cytokine IL-10, SRM1649b PM treatment had no significant effect on percent IL-10 positive cells (Figure 8C). However, in the presence of CH-223191, SRM1649b PM treatment significantly reduced the percent IL-10 positive cells at 40 and 20μg/mL PM (Figure 8D) suggesting that AHR is important for the maintenance or generation of IL-10 positive cells during toxic exposures.
Figure 8: MOG specific splenocytes generate or maintain IL-10 producing cells and reduce matter IFNγ producing cells in an AHR-dependent manner in vitro.
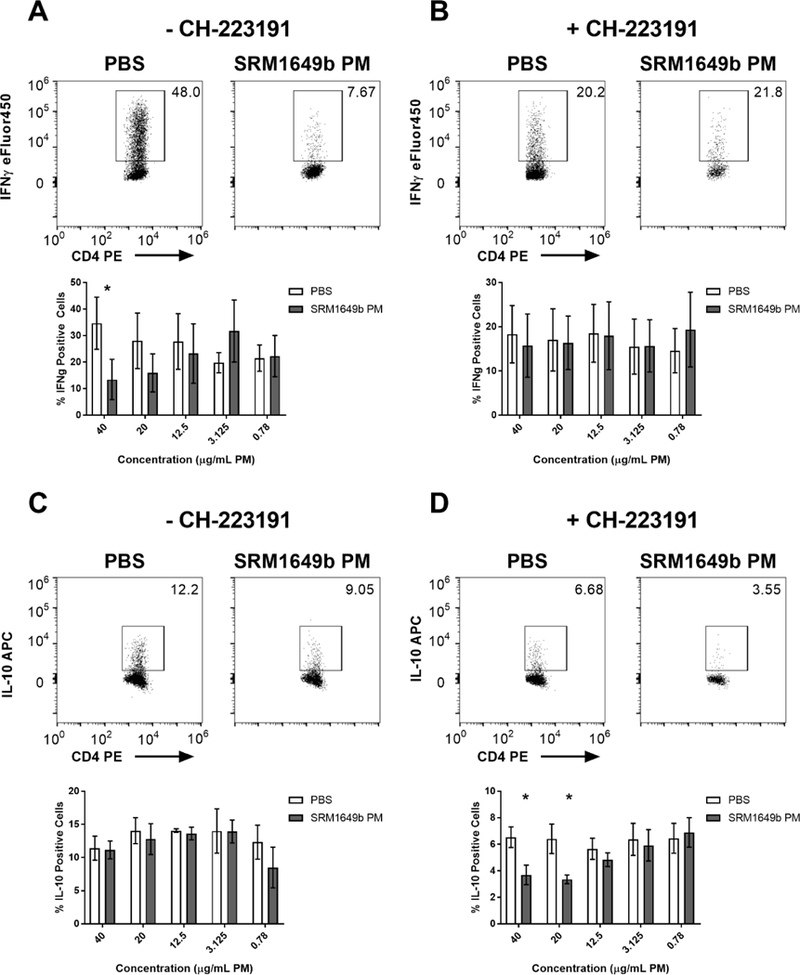
Totalsplenocytes were obtained from 2D2 mice (transgenic T cell receptor specific for the MOG peptide). At time 0, cells were exposed to 10uM CH-223191 (AHR antagonist) and 5 concentrations of SRM1649b PM or PBS (vehicle) control and cultured for 5 days. (A) SRM1649b PM reduced the percent of IFNγ positive cells in the absence of CH-223191. (B) The suppression of IFNγ positive cells was alleviated when AHR was antagonized. (C) In the absence of CH-223191, SRM1649b PM treatment had no significant increase in percent IL-10 positive cells. (D) SRM1649b PM treatment significantly reduced the percent IL-10 positive cells at 40 and 20μg/mL PM when AHR was suppressed. Results are mean ± SEM of (PBS n=4) and (SRM1649b PM n=4) for all conditions. Significant differences among groups (p<0.05) are indicated by an asterisk. Abbreviations: MOG, myelin oligodendrocyte peptide; AHR, aryl hydrocarbon receptor; SRM, standard reference materials; PM, particulate matter; SEM, standard error of the mean.
4.0. Discussion:
Autoimmune diseases are characterized by a loss of self-tolerance. Although some elements of disease etiology remain unknown, there is an obvious role for autoreactive T cells (Dardalhon et al., 2008). Suppression of regulatory responses by self-reactive, effector T cells can contribute to the pathology of autoimmune disease (Dardalhon et al., 2008). One model proposes that Th17 cells are responsible for creating an inflammatory milieu of cytokines and chemokines that recruit Th1 cells to target tissues that maintain chronic inflammation (Dardalhon et al., 2008). Normally, Tregs suppress inflammation and regulate the immune response, but in the context of autoimmune disease, functional Tregs become dysfunctional due to exacerbated inflammation (Dardalhon et al., 2008; Komatsu et al., 2014; Korn et al., 2007; Langrish et al., 2005; Rosenblum et al., 2015; Zhou et al., 2009). Due to poor mechanistic understanding, most autoimmune diseases are treated via global immunosuppression.
The lack of mechanistic understanding of autoimmune disease results in traditional therapies relying on immunosuppressive medications that globally dampen the immune response. This leaves patients vulnerable to life-threatening infections and at risk for malignancy (Chandrashekara, 2012; Rosenblum et al., 2012; Vial and Descotes, 2003). The initial response to immunosuppressive agents is 60–70% effective but is not maintained over time (Chandrashekara, 2012). Recently, immunosuppressive drugs have become more specific to reduce collateral toxicity, but even these targeted therapies have unwanted side effects, need to be continued indefinitely, and are costly (Feldmann and Steinman, 2005; Rosenblum et al., 2012). However, even these more specific and less toxic immunosuppressive drugs have not significantly reduced remission rates (Feldmann and Steinman, 2005). To create more targeted treatments, specific pathways that augment symptoms and exacerbate autoimmunity or pathways that mitigate symptoms and delay disease onset need to be identified. This study aims to identify novel pathways that alter autoimmune disease after PM exposure, to allow therapies that may be particularly applicable to patients who are exposed to high levels of PM.
The AHR pathway was identified as a likely candidate for mediating autoimmunity given its role in balancing effector and regulatory T cells (Ehrlich et al., 2018; Mohinta et al., 2015; Quintana et al., 2008; Veldhoen et al., 2008). AHR activation can have differential effects on autoimmune outcomes depending on the extent and duration of activation (Ehrlich et al., 2018). Under some conditions, the AHR pathway has been shown to aggravate autoimmunity by augmenting Th17 differentiation (Quintana et al., 2008; Veldhoen et al., 2008). Other conditions result in AHR-mediated amelioration of disease via enhanced Treg differentiation (Quintana et al., 2008; Veldhoen et al., 2008). This study further explores AHR-ligand relationships in the context of atmospheric PM to determine the role of AHR in T cell-mediated autoimmune disease and how PM exposure alters these biological responses.
Previously, we published that SRM1649b PM enhanced Th17 differentiation and increased production of IL-17 in the lung in vivo (van Voorhis et al., 2013). Based on this, we hypothesized that SRM1649b PM enhances pathologic T cell differentiation in an AHR-dependent manner leading to worsened autoimmune disease in a murine model of EAE. SRM1649b PM enhanced Th17 differentiation and generated Th1 cells in an AHR-dependent manner. Intranasal treatment of SRM1649b PM delayed onset of EAE and reduced cumulative clinical score. Chronic dosing of intranasal SRM1649b PM was not sufficient to worsen severity of EAE but did delay onset of EAE. Acute dosing of intranasal SRM1649b PM reduced severity of EAE, measured by a significant reduction in peak clinical score and cumulative clinical score, and delayed disease onset. Additionally, acute intranasal treatment of low dose SRM1649b PM had no effect on clinical score or day of onset. Overall, these results demonstrate that both chronic and acute intranasal dosing regimens of SRM1649b PM were sufficient to delay onset of EAE, but acute intranasal exposure of a lower dose was not. This suggests that the component of SRM1649b PM responsible for reduced severity of disease may not be concentrated enough in the low dose exposure to alter disease onset. SRM1649b PM enhanced Th17 differentiation in vitro, yet SRM1649b PM ameliorated EAE after intranasal treatment suggesting that differences in Th17 differentiation do not entirely explain the in vivo findings. This concept has previously been demonstrated (Duarte et al., 2013). Specifically, in vitro T cell effects of AHR do not always correlate with in vivo AHR-effects on T cells underscoring the complexity of the AHR pathway and suggesting the effects can be influenced by mode of exposure as well as metabolism (Duarte et al., 2013).
To address the previous assertion that mode of exposure can alter effects of AHR ligands, we administered SRM1649b PM via oral gavage in the absence of dietary AHR ligands. We found that this route of exposure does not significantly impact EAE progression at any dose or alter severity of EAE. To understand the mechanism in which suppression of disease was acting, EAE induced mice were sacrificed on day 10 and analysis of T cells in the CNS after intranasal treatment of SRM1649b PM showed a reduction of pathologic T cell subsets in vivo.
Published literature reports that TCDD, a canonical AHR ligand, suppresses EAE in an AHR- dependent manner (Quintana et al., 2008; Veldhoen et al., 2008) so we tested whether SRM1649b PM-mediated delayed onset of disease was AHR dependent. Delayed EAE onset following intranasal installment of SRM1649b PM was AHR-dependent in vivo. Using splenocytes from 2D2 mice that have T cells specific for the MOG35–55 peptide and exposing them to the AHR antagonist CH-223191 we discovered that MOG specific splenocytes treated with SRM1649b PM generate or maintain IL-10-producing cells and reduce IFNγ-producing cells in an AHR-dependent manner. These data support the in vivo finding of a suppression of Th1 cells and Th1Th17 double positive cells in the CNS on day 10 prior to disease symptoms. This also explains the delayed onset of disease observed in mice exposed intranasal to SRM1649b PM. The implications of this correlation are significant - in addition to explaining some portion of the mechanism behind disease reduction and delayed disease onset, these data also suggest that the in vitro 2D2 splenocyte assay, which include T cells transgenic for the MOG35–55 peptide, may be a novel way to investigate the effect of PM exposures on T cell responses and correlate those findings with outcomes of autoimmune diseases.
We recently reported the effects of intranasal treatment of SRM1649b PM on skin graft rejection in a minor mismatch model. Intranasal treatment of SRM1649b PM significantly prolonged graft survival in an AHR-dependent manner and significantly reduced IFNγ-producing cells in the graft (Julliard et al., 2017b). AHR expression was significantly upregulated in IFNγ-producing Th1 cells in the graft suggesting a role for AHR ligands in the presence and differentiation of this cell type (Julliard et al., 2017b). These findings are similar to our results in the EAE model where delayed onset of disease is AHR-mediated, and prior to symptoms there is a reduction of pathologic Th1 and Th17Th1 double positive cells in the CNS where pathology occurs. Although previous literature on the role of the AHR in autoimmunity have focused on the balance of Th17 and Treg cells, our data suggests that a primary effect may be on Th1 and Th1Th17 double positive effector cells. This will need to be further characterized for different ligands and doses, different routes of exposure, and other disease models. At the very least It would appear, that in addition to modulating Th17 and Treg cells, AHR is playing a role in Th1 cells as well.
Although the initial goal of this study was to identify a mechanism through which atmospheric PM aggravates autoimmune disease, we ended up identifying a novel pathway through which atmospheric PM can reduce immune responses in the CNS. The results of this study demonstrate that SRM1649b PM may suppress pathologic T cells responses in the CNS, based on increase in IL-10 positive MOG35–55 specific T cells, leading to reduced disease severity and delayed onset of disease. Although SRM1649b PM alleviates autoimmune disease by suppressing pathologic T cells, primarily Th1 cells, and may provide insights into new targeted therapies, it opens the door for opportunistic infection if the immunosuppression is chronic or non-reversible. At the same time, findings with SRM1649b PM and AHR may also shed light on the known increase in risk of infection and malignancy seen after chronic exposure to PM (Ghio, 2014). While previous studies have focused on DNA damage and direct toxicity behind this association (Lawal, 2017; Risom et al., 2005; Valavanidis et al., 2008), these data suggest future studies should be considered in appropriate disease models to look for a role in AHR-mediated immunosuppression, particularly on Th1 cells.
This study identifies effector Th1 cells as a primary target of AHR-mediated immunosuppression induced by PM in the context of an autoimmune response. Understanding what specific components of the AHR pathway are required for SRM1649b PM-mediated suppression of autoimmune disease via effector T cells would contribute to a more comprehensive understanding of the specific pathway targets that could be used for therapies to direct immunosuppression to specific cell types or tissues. Identifying underlying mechanisms of PM- induced AHR mediated-suppression of Th1 cells could lead to new therapies focused on reducing pathologic effector T cell responses after exposure to PM in the context of autoimmune disease as well as other inflammatory diseases. The data presented here serves as a first step in unlocking the components of the AHR pathway responsible for immunosuppression via effector Th1 cells in response to PM exposure.
Conclusions:
In summary, we present a model of autoimmune disease in which the AHR suppresses pathologic T cell responses resulting in immunosuppression and amelioration of disease. The AHR is established as playing a role in driving T cell responses to both inflammatory and regulatory phenotypes. In the case of PM exposure, the specific nature of the PM and the mixtures of constituents are likely to shift the immune balance. The finding that SRM1649b PM reduces pathologic T cell subsets through the AHR can be used to identify immunosuppressive agents that specifically target Th1 effector cells, and the described in vitro 2D2 splenocyte assay may serve as a tool to help test the potential of these immunosuppressive agents to reduce pathologic effector T cells or enhance regulatory T cells.
Supplementary Material
Highlights:
Atmospheric PM enhances Th17 differentiation in an AHR-dependent manner in vitro
Intranasal exposure of atmospheric PM lessened severity and delayed onset of EAE
Reduced severity of EAE was dependent on exposure route, dose, and timing of dose
Inhalation of atmospheric PM reduced pathologic Th1 and Th1Th17 in the CNS in vivo
MOG-specific splenocytes reduce IFNγ producing cells via the AHR in vitro
5.0. Acknowledgments
Thank you to the University of Wisconsin-Carbone Cancer Center Flow Cytometry Laboratory [University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520 and Special BD LSR Fortessa Project Grant 1S100OD018202–01]. Thank you to Brandon Shelton at the Wisconsin State Lab of Hygiene for analysis and preparation of PM samples.
Funding
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) [RO1 ES023842 to J.D.M, R21 ES025304 to J.D.M], and [T32 ES007015 (C.A.O)]. Additional support by the National Institute of Diabetes and Digestive Kidney Diseases (NIDDK) [T32 DK007665–21 (A.A)]. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NIDDK.
Footnotes
Ethics approval and consent to participate
All animal studies were performed in accordance with protocols approved by the School of Medicine and Public Health Institutional Animal Care and Use Committee at the University of Wisconsin-Madison.
Particulate matter
Aryl hydrocarbon receptor
Polycyclic aromatic hydrocarbons
Regulatory T cell
T helper 17
Standard reference material 1649b
Experimental autoimmune encephalomyelitis
Central nervous system
T cell receptor
Myelin oligodendrocyte glycoprotein 35–55 peptide
National Institute of Standards and Technology
Phosphate buffered saline
6-formylindolo[3,2-b] carbazole
Beta-napthoflavone
Complete Freund’s adjuvant
Indole-3-carbinol
Isotonic percoll
2,3,7,8-Tetrachlorodibenzo-p-dioxin
Declarations of interest: The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Chelsea A. O’Driscoll, Madison, WI 53792 caodriscoll@wisc.edu
Leah A. Owens, Madison, WI 53792 leah.owens@wisc.edu
Erica J. Hoffmann, Madison, WI 53792 ejhoffmann@wisc.edu
Madeline E. Gallo, Madison, WI 53792 megallo@wisc.edu
Amin Afrazi, Madison, WI 53792 afrazi@wisc.edu.
Mei Han, Madison, WI 53705 hanm@surgery.wisc.edu.
John H. Fechner, Madison, WI 53792 fechner@surgery.wisc.edu
James J. Schauer, Madison, WI 53706 jjschauer@wisc.edu
Christopher A. Bradfield, Madison, WI 53706 bradfield@oncology.wisc.edu
6.0 References
- Angelici L, et al. , 2016. Effects of particulate matter exposure on multiple sclerosis hospital admission in Lombardy region, Italy. Environ Res. 145, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR, Toxicity of Polycyclic Aromatic Hydrocarbons (PAHs): Standards and Regulations for PAHs Exposure | ATSDR - Environmental Medicine & Environmental Health Education - CSEM; 2008. [Google Scholar]
- Benson JM, Shepherd DM, 2011. Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol Sci. 124, 327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyerlein A, et al. , 2015. Ambient air pollution and early manifestation of type 1 diabetes. Epidemiology. 26, e31–2. [DOI] [PubMed] [Google Scholar]
- Brauer M, et al. , 2016. Ambient Air Pollution Exposure Estimation for the Global Burden of Disease 2013. Environ Sci Technol. 50, 79–88. [DOI] [PubMed] [Google Scholar]
- Brook RD, et al. , 2013. Long-term fine particulate matter exposure and mortality from diabetes in Canada. Diabetes Care. 36, 3313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekara S, 2012. The treatment strategies of autoimmune disease may need a different approach from conventional protocol: A review. Indian J Pharmacol. 44, 665–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, et al. , 2016. Air pollution exposure increases the risk of rheumatoid arthritis: A longitudinal and nationwide study. Environ Int. 94, 495–499. [DOI] [PubMed] [Google Scholar]
- Cheung K, et al. , 2011. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area. Atmospheric Environment. 45, 2651–2662. [Google Scholar]
- Constantinescu CS, et al. , 2011. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 164, 1079–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardalhon V, et al. , 2008. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 31, 252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciaula A, 2016. Type I diabetes in paediatric age in Apulia (Italy): Incidence and associations with outdoor air pollutants. Diabetes Res Clin Pract. 111, 36–43. [DOI] [PubMed] [Google Scholar]
- Duarte JH, et al. , 2013. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS One. 8, e79819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich AK, et al. , 2018. TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD4+ T Cell Differentiation. Toxicol Sci. 161, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat SC, et al. , 2011. Air pollution in autoimmune rheumatic diseases: a review. Autoimmun Rev. 11, 14–21. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Steinman L, 2005. Design of effective immunotherapy for human autoimmunity. Nature. 435, 612. [DOI] [PubMed] [Google Scholar]
- Gawda A, et al. , 2017. Air pollution, oxidative stress, and exacerbation of autoimmune diseases. Cent Eur J Immunol. 42, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghio AJ, 2014. Particle exposures and infections. Infection. 42, 459–67. [DOI] [PubMed] [Google Scholar]
- Gomez-Mejiba SE, et al. , 2009. Inhalation of Environmental Stressors & Chronic Inflammation: Autoimmunity and Neurodegeneration. Mutat Res. 674, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RN, et al. , 2013. [Association of the incidence of type 1 diabetes mellitus with environmental factors in Chile during the period 2000–2007]. Rev Med Chil. 141, 595–601. [DOI] [PubMed] [Google Scholar]
- Gregory AC, et al. , 2008. Multiple Sclerosis disease distribution and potential impact of environmental air pollutants in Georgia. Science of The Total Environment. 396, 42–51. [DOI] [PubMed] [Google Scholar]
- Hathout EH, et al. , 2002. Role of exposure to air pollutants in the development of type 1 diabetes before and after 5 yr of age. Pediatr Diabetes. 3, 184–8. [DOI] [PubMed] [Google Scholar]
- Health N. I. o., Progress in Autoimmune Disease Research. National Institute of Health, 2005. [Google Scholar]
- Hohlbaum K, et al. , 2017. Severity classification of repeated isoflurane anesthesia in C57BL/6JRj mice-Assessing the degree of distress. PLoS One. 12, e0179588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, 2011. You AhR What You Eat: Linking Diet and Immunity. Cell. 147, 489–491. [DOI] [PubMed] [Google Scholar]
- Institute HE, State of Global Air 2018. Health Effects Institute, Boston, MA, 2018. [Google Scholar]
- Ito S, et al. , 2007. Dietary phytochemicals regulate whole-body CYP1A1 expression through an arylhydrocarbon receptor nuclear translocator-dependent system in gut. J Clin Invest. 117, 1940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julliard W, et al. , 2017a. Amelioration of Clostridium difficile Infection in Mice by Dietary Supplementation With Indole-3-carbinol. Ann Surg. 265, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julliard W, et al. , Modeling the Effect of the Aryl Hydrocarbon Receptor on Transplant Immunity. Transplant Direct, 2017b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julliard W, et al. , 2016. Environmental ExposuresThe Missing Link in Immune Responses After Transplantation. American Journal of Transplantation. 16, 1358–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly FJ, Fussell JC, 2012. Size, source and chemical composition as determinants of toxicity attributable to ambient particulate matter. Atmospheric Environment. 60, 504–526. [Google Scholar]
- Komatsu N, et al. , 2014. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 20, 62–8. [DOI] [PubMed] [Google Scholar]
- Korn T, et al. , 2007. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 13, 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, et al. , 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 201, 233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawal AO, 2017. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol Lett. 270, 88–95. [DOI] [PubMed] [Google Scholar]
- Lerner A, et al. , 2015. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. International Journal of Celiac Disease. 3, 151–155. [Google Scholar]
- Malmqvist E, et al. , 2015. Maternal exposure to air pollution and type 1 diabetes--Accounting for genetic factors. Environ Res. 140, 268–74. [DOI] [PubMed] [Google Scholar]
- Mascanfroni ID, et al. , 2015. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 21, 638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzie CA, et al. , 2002. Exposure to carcinogenic PAHs in the environment. [Google Scholar]
- Miller FW, et al. , 2012. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun. 39, 259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohinta S, et al. , 2015. Differential regulation of Th17 and T regulatory cell differentiation by aryl hydrocarbon receptor dependent xenobiotic response element dependent and independent pathways. Toxicol Sci. 145, 233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Perdew GH, 2017. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr Opin Toxicol. 2, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, et al. , 2000. “Gene-swap knock-in” cassette in mice to study allelic differences in human genes. Ann N Y Acad Sci. 919, 148–70. [DOI] [PubMed] [Google Scholar]
- Oikonen M, et al. , 2003. Ambient air quality and occurrence of multiple sclerosis relapse. Neuroepidemiology. 22, 95–9. [DOI] [PubMed] [Google Scholar]
- Pino PA, Cardona AE, 2011. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, et al. , 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 453, 65–71. [DOI] [PubMed] [Google Scholar]
- Rahn EJ, et al. , 2014. Sex differences in a mouse model of multiple sclerosis: neuropathic pain behavior in females but not males and protection from neurological deficits during proestrus. Biol Sex Differ. 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasid O, et al. , 2012. Assessment of Routine Procedure Effect on Breathing Parameters in Mice by Using Whole-Body Plethysmography. J Am Assoc Lab Anim Sci. 51, 469–74. [PMC free article] [PubMed] [Google Scholar]
- Rice SA, et al. , 1986. Effects of subchronic intermittent exposure to isoflurane in Swiss Webster mice. J Environ Pathol Toxicol Oncol. 6, 285–93. [PubMed] [Google Scholar]
- Risom L, et al. , 2005. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res. 592, 119–37. [DOI] [PubMed] [Google Scholar]
- Rosenblum MD, et al. , 2012. Treating Human Autoimmunity: Current Practice and Future Prospects. Sci Transl Med. 4, 125sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum MD, et al. , 2015. Mechanisms of human autoimmunity. J Clin Invest. 125, 2228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse M, et al. , 2013. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br J Pharmacol. 169, 1305–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux J, et al. , 2017. Air pollution by particulate matter PM10 may trigger multiple sclerosis relapses. Environ Res. 156, 404–410. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, et al. , 1996. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A. 93, 6731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JE, 2018. Airborne Particulate Matter: Human Exposure & Health Effects. J Occup Environ Med. [DOI] [PubMed] [Google Scholar]
- Valavanidis A, et al. , 2008. Airborne particulate matter and human health: toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 26, 339–62. [DOI] [PubMed] [Google Scholar]
- van Voorhis M, et al. , 2013. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS One. 8, e82545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, et al. , 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 453, 106–9. [DOI] [PubMed] [Google Scholar]
- Vial T, Descotes J, 2003. Immunosuppressive drugs and cancer. Toxicology. 185, 229–40. [DOI] [PubMed] [Google Scholar]
- Vincent R, et al. , 1997. Acute pulmonary toxicity of urban particulate matter and ozone. Am J Pathol. 151, 1563–70. [PMC free article] [PubMed] [Google Scholar]
- Vojdani A, et al. , 2014. Environmental Triggers and Autoimmunity. Autoimmune Dis. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojinovic S, et al. , 2015. Disease relapses in multiple sclerosis can be influenced by air pollution and climate seasonal conditions. Vojnosanit Pregl. 72, 44–9. [DOI] [PubMed] [Google Scholar]
- Yonezaki K, et al. , 2015. Postanesthetic effects of isoflurane on behavioral phenotypes of adult male C57BL/6J mice. PLoS One. 10, e0122118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2009. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 10, 1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.