

Abstract
Mitochondria are best known for their role in ATP generation. However, studies over the past two decades have shown that mitochondria do much more than that. Mitochondria regulate both necrotic and apoptotic cell death pathways, they store and therefore coordinate cellular Ca2+ signaling, they generate and metabolize important building blocks, by-products and signaling molecules, and they also generate and are targets of free radical species that modulate many aspects of cell physiology and pathology. Most estimates suggest that although the brain makes up only 2% percent of body weight, utilizes about 20 percent of the body’s total ATP. Thus, mitochondrial dysfunction greatly impacts brain functions and is indeed associated with numerous neurodegenerative diseases. Furthermore, a number of abnormal disease-associated proteins have been shown to interact directly with mitochondria, leading to mitochondrial dysfunction and subsequent neuronal cell death. Here, we discuss the role of mitochondrial dynamics impairment in the pathological processes associated with neurodegeneration and suggest that a therapy targeting mitochondrial dysfunction holds a great promise.
Graphical Abstract
Introduction:
Mitochondria, which descended from prokaryotic bacteria through endosymbiotic evolution, mediate diverse functions within the cell (1, 2). In addition to their central bioenergetic task of ATP regeneration, the organelles are the main source both of reactive oxygen species and of the cell’s antioxidant defenses (3). Mitochondria are abundant where energy-requiring processes take place, such as the brain, and skeletal and cardiac muscles (4). Mitochondrial form/structure and function are intimately linked. Mitochondria show a highly complicated and dynamic behavior even in unicellular organisms such as yeast. An increasing number of studies indicate that the changes in mitochondrial size and form, together termed mitochondrial dynamics, which is mediated by processes of fission and fusion of the organelles, play a critical role in controlling mitochondrial function. Thus, an imbalance of mitochondrial fusion and fission impacts a diverse range of cellular biological processes.
Mitochondrial fission and fusion are mediated by the action of large and multi-domain dynamin-related GTPases (2). Loss of either fission or fusion results in mitochondrial dysfunction; fragmented mitochondrial morphology correlates with apoptotic cytochrome c release, whereas tubular (large) morphology promotes resistance to apoptotic stimuli (5–8). Mitochondrial fusion is essential, as it provides a means for exchange of intermembrane and matrix contents, including mitochondrial DNA (mtDNA), between mitochondria (9). Increase in mitochondrial size requires the fusion of both the outer and inner mitochondrial membrane and is regulated by at least three large GTPase proteins: mitofusins (Mfn1 and Mfn2) for outer mitochondrial membrane fusion, and optic atrophy 1 (OPA1), for inner mitochondrial membrane fusion (10, 11). The outer mitochondrial membrane fusion is mediated through interactions of the coiled-coil domains of Mfn1 and Mfn2 to form either homo-oligomeric or hetero- oligomeric complexes that tether membranes together (12–14). The mechanism by which OPA1 stiches together the inner mitochondrial membrane may involve specific post-translational modifications, and possibly oligomerization as well (15, 16). As will be discussed later, dynamin related protein 1 (Drp1), a large cytosolic GTPase that mediates mitochondrial fission, also undergoes protein modifications when activated, and fission is subsequently mediated by the binding of oligmerized Drp1 to the outer mitochondrial membrane. Screens in yeast have been instrumental in identifying a set of proteins required for the maintenance of mitochondrial morphology, (17) and additional proteins were identified as being involved in vertebrates [reviewed in (18–22)]. Here, we focus on the role of the mitochondrial fission machinery in neurodegenerative diseases and provide evidence for possible therapeutic strategies to regulate these pathologies through inhibition of excessive fission and the resulting mitochondrial dysfunction.
Mitochondrial fission
Dynamin related protein 1 (Drp1/Dmnl1):
Drp1 induces fission (mitochondrial division) by polymerizing and forming helical structures that wrap around mitochondria, providing the necessary mechanical fission force (23, 24). The role of Drp1/ Dmnl1 in mitochondrial fission was first identified in a screen for yeast mutants with defective mitochondrial morphology (25, 26). Drp1, a predominantly cytosolic protein, forms punctate structures on the mitochondria upon activation by signals that promote mitochondrial fission (27). However, not all the translocated Drp1 leads to functional fission events; most are aborted (26, 28). When the fission event is triggered, Drp1 rapidly oligomerizes into a ring-like structure to sever the mitochondrial membrane, mediated by self-assembly and GTP hydrolysis (29).
Drp1 is critical for mouse embryonic and brain development, and mice lacking Drp1 die by embryonic day 11.5 (30). These Drp1-null embryos fail to undergo developmentally regulated apoptosis during neural tube formation in vivo (30). In humans, two probands with distinct de novo missense mutations in Drp1 have been reported. One presented with severe neonatal encephalopathy, microcephaly, optic atrophy, and abnormal brain development in the form of demyelination and altered gyral pattern, and died at 37 days of age (31). The second proband presented at 6 months of age with global developmental delay, developed refractory epilepsy at one year of age with multiple subsequent episodes of status epilepticus, and displayed a profound developmentally delay (32). Recently, another Drp1 mutation in humans was identified in two unrelated individuals and had a remarkably similar phenotype: delayed onset yet rapid progression (33). The critical role of the GTPase activity of Drp1 was also demonstrated in an experimental model, (34) and overexpressing a dominant negative mutant Drp1K38A, or depleting Drp1 down-regulation by siRNA, led to elongated interconnected networks of mitochondria (35, 36), indicative of a critical role for Drp1 in mitochondrial fission.
Dynamin2 (Dyn2; Dnm2):
Dynamin2 is another member of the conventional dynamin family that regulates mitochondrial morphology. In a recent study, it was shown that Dyn2 further constricts membrane tubules that were first constricted by Drp1 to complete mitochondrial fission (37). Knockdown of Dyn2 results in elongated mitochondria with the occasional presence of a long, highly constricted tubule between two populations of preassembled Drp1 polymers (37). In the same study, Dyn2 was shown to only localize transiently to facilitate membrane scission, in contrast to Drp1, which is abundant on most constricted mitochondrial sites. Furthermore, Dyn2 and Drp1 are differentially segregated to daughter organelles – Drp1 remains present on both daughter mitochondria following scission, while Dyn2 appears only on one of the two (37).
Fission1 (Fis1):
Fis1, a protein anchored to the mitochondrial outer membrane through its C-terminus, exposed to the cytosol, and evenly distributed on the mitochondrial surface was first identified to be an essential regulator of mitochondrial fission (38, 39). Fis1, in yeast, is vital for the recruitment and assembly of cytosolic Drp1 on mitochondrial outer membrane by interacting with Drp1 through one of two other adaptor proteins, Mdv1 or its paralog Caf4 (40, 41). In mammals, over-expression of Fis1 induces mitochondrial fragmentation, whereas inhibition of its expression results in mitochondrial elongation (38, 39). However, whereas Fis1 is uniformly distributed throughout the mitochondrial outer membrane, Drp1 is localized to punctate structures along the mitochondrial tubules, indicating that there might be other mitochondrial adaptor proteins involved in anchoring Drp1 to enable mitochondrial fission (42). Additionally, recent studies showed that altered levels of Fis1 have no effect on the distribution or amount of Drp1 along mitochondria (42). Interestingly, Fis1 is present throughout the animal kingdom, but its functions in metazoans have been unclear. Fis1 can bind to Drp1 in vitro, promoting mitochondrial fission when overexpressed, and as such has been implicated in a number of mitochondrial fission-dependent processes, such as apoptosis and autophagy (43). However, mammalian Fis1−/− cells have mild or no fission defect under basal conditions. Although several reports suggest that Fis1 exogenous expression induces mitochondrial fragmentation and that Fis1 knockdown affects mitochondrial morphology, Fis1 appears to be dispensable for basal/physiological mitochondrial fission. Further, Fis1 and its interacting mitochondrial Rab GTPase-activating proteins (GAPs), TBC1D15 and TBC1D17, have been implicated to play an important for autophagosomal biogenesis during mitophagy thus having a satellite function independent of Drp1 (44–46). Nevertheless, while these results provide further evidence for role of other proteins in physiological fission, we and others found that Fis1 has a crucial role under pathological, stress-induced mitochondrial fission in multiple cell types (47–50). Further, studies in postmortem HD and AD patients indicate an increase in the protein levels of Fis1 concurrently with an increased Drp1 level (51–53). We found increased interaction of Fis1 with Drp1 and recruitment of Drp1 to mitochondria under hypoxic/ischemic conditions, in cell and animal models of Parkinson’s disease (PD), Huntington’s disease (HD), Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS) models, and in patients-derived cells (5, 54–57). Although the physiological/basal level of interaction between Drp1 and Fis1 is very low in healthy cells, we found an increase in Drp1 association with Fis1 under pathological stress (49, 54–56). The use of a pharmacological tool that inhibits this interaction is discussed later in the review.
Mitochondrial fission factor (Mff):
Mff was identified by high-throughput screening of a drosophila RNA interference (RNAi) library for mitochondrial morphology alterations (58). The silencing of Mff (CG30403/Tango11) induced a phenotype similar to that of Drp1 depletion with perinuclear clustering of mitochondria (58). Mff, the human ortholog, is a mitochondrial adaptor of Drp1 conserved in metazoans, but not found in yeast (28, 43). Mff is anchored to the outer mitochondrial membrane through a C-terminal transmembrane domain, while the majority of the protein is exposed to the cytosol (59). siRNA mediated depletion of Mff in mammalian cells leads to an interconnected tubular network of mitochondria, indicative of pro-fusion phenotype (43, 59). In contrast, overexpression of Mff induces extensive mitochondrial fragmentation and mitochondrial dysfunction (43, 59). Moreover, Mff and Fis1 occur in separate complexes, indicating that they may have distinct roles in regulating mitochondrial fission (20, 43, 58); Mff is localized in punctate structures on mitochondria in a manner independent of Drp1 and Fis1 and in contrast to the uniform distribution of Fis1 on the mitochondrial outer membrane (43, 59). Mff mostly co-localizes with Drp1 in dot-like structures along the mitochondrial tubules; over-expression of Mff promotes Drp1-mitochondrial association and subsequent mitochondrial fragmentation while the knock-down of Mff by siRNA reduces the recruitment of Drp1 to mitochondria, resulting in mitochondrial elongation (43, 59). Finally, Mff requires Drp1 oligomerization to physically associate with Drp1 (60). Thus, Mff plays a major role in physiological mitochondrial fission, which is independent of Fis1 (59, 60).
Mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51):
As part of a cellular localization screen of uncharacterized human proteins, the expression of SMCR7L (Mid51) in yeast was found to cause unique changes in mitochondrial cellular distribution and to result in formation of fused mitochondrial tubules (61). In the study, when MiD49/51 were expressed as carboxy‐terminal green fluorescent protein (GFP) fusions, they both localized to mitochondria (61). Similar to Mff, MiD51 and MiD49 also recruit Drp1 to the mitochondrial outer membrane (61). Knockdown of both MiD49 and MiD51 result in an irregular distribution of the network and fused mitochondria (61, 62), and MiD49 and MiD51 mediate Drp1-dependent mitochondrial fission in Fis1/Mff double-KO MEFs (61, 62). In addition, ADP-bound MiD51 assembles Drp1 into spirals and promotes Drp1 GTP hydrolysis, supporting its role in mitochondrial fission (63). Overexpression of either MiD49 or MiD51 triggered increased recruitment of Drp1 to the mitochondrial outer membrane, but blocked Drp1 activity, thus blocking mitochondrial fission (63, 64). Based on these studies, it appears that MiD51 or MiD49 alone are sufficient to act as a suppressor to sequester Drp1 and inhibits Drp1 -mediated fission. However, MiD49/ 51 expression is increased in pulmonary arterial hypertension, which is associated with accelerated Drp1-mediated mitotic fission, increased cell proliferation and decreased apoptosis (65). Silencing MiD49/ 51 (but not other Drp1 binding partners, Fis1 or MFF) promotes mitochondrial fusion and causes G1-phase cell cycle arrest, through ERK1/2 and CDK4-dependent mechanisms (65). In contrast to Mff-null cells, MiD49/MiD51-KO and Drp1-KO cells are resistant to cytochrome c release during apoptosis (62, 65). This phenotype, seen in MiD49/51-KO cells but not Drp1-KO cells, was completely abolished by treatments that disrupt mitochondrial inner cristae structure, such as OPA1 depletion, indicating that Drp1-dependent mitochondrial fission through MiD49/MiD51 regulates cristae remodeling during intrinsic apoptosis (62). Adding to this complexity, another study reported increased mitochondrial connectivity following loss of either of adaptors, and this was further enhanced following the combined loss of MiD51 and Mff (66). Moreover, loss of adaptors also conferred increased resistance of cells to intrinsic apoptotic stimuli, with MiD49 and MiD51 showing a more prominent role (66, 67). Using a proximity-based biotin labeling approach, close associations between MiD51, Mff and Drp1, but not Fis1, were observed, and MiD51 can suppress Mff-dependent enhancement of Drp1 GTPase activity (66, 67). In summary, Mid49 and Mid51 have important roles in mitochondrial fission, which may vary by stimuli and perhaps even cell types.
Other potential protein partners of mitochondrial fission:
In addition to Drp1, Fis1, Mff and MiD49/ Mid51, several other proteins have been proposed to regulate mitochondrial fission in mammals.
Fatty acyl transferase endophilin B1 (Bif-1) directly affects mitochondrial morphology. Its down-regulation or overexpression of the protein lacking N-terminal lipid-modifying domain leads to striking alterations of the mitochondrial morphology in HeLa and COS-7 cells (68). Knockdown or overexpression of endophilin B1 in cells resulted in the dissociation of the outer mitochondrial membrane compartment from that of the matrix, and led to the formation of vesicles and tubules of the outer mitochondrial membrane, indicating that endophilin B1 may be essential for the regulation of outer mitochondrial membrane dynamics (68, 69).
Overexpression of mitochondrial protein 18 kDa (MTP18), a nuclear-encoded mitochondrial membrane protein, altered mitochondrial morphology from filamentous to punctate-like structures indicating increased mitochondrial fission (70, 71). Overexpression of MTP18 blocked mitochondrial fragmentation in cells co-expressing either Mfn1 or Drp1K38A. Loss-of-function of endogenous MTP18 by RNAi resulted in highly fused mitochondria (70) and expression of MTP18 blocked excessive fission in cells overexpressing Fis1 (70).
Ganglioside-induced differentiation-associated protein 1 (GDAP1), an integral mitochondrial outer-membrane protein, and mitochondrial targeting GxxxG-motif protein (MTGM), an integral inner membrane protein, have also been implicated in the regulation of Drp1-dependent mitochondrial fission. Their over-expression causes mitochondrial fragmentation, while their downregulation results in mitochondrial elongation (72–75). A coordination of the outer and inner mitochondrial membranes is essential for mitochondrial fission. However, the inner membrane proteins and machinery that are involved in the fission process remain a mystery. It is possible that this role is mediated by MTP18 and/or MTGM, and further research will help determine if they coordinate with the key proteins involved in promoting/inhibiting outer membrane fission: Drp1, Fis1, Mff, Mid49/ 51.
Mitochondrial dysfunction in neurodegeneration
Mitochondrial Dysfunction and Defective Mitochondrial Dynamics in Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is the most common form of dementia, affecting millions globally, and is characterized by the progressive loss of neurons, ultimately leading to the onset of severe behavioral and cognitive impairments (76). The salient pathological features in AD patients include amyloid/senile plaques (SPs), neurofibrillary tangles (NFTs), granulovacuolar degeneration, and dystrophic neurites (76, 77). NFTs are intracellular aggregates composed of the hyper-phosphorylated form of the microtubule-associated protein tau, while SPs are extracellular lesions, usually composed of bundles of amyloid-β (Aβ) peptide fibrils (76–78).
Mitochondrial functional impairments were found in cultured neurons isolated from Tg mice that overexpress a mutant form of AβPP and Aβ-binding alcohol dehydrogenase (ABAD; Tg mAβPP/ABAD), and display reduced levels of brain ATP and COX activity, diminished glucose utilization, as well as electrophysiological abnormalities in hippocampal slices compared with Tg mAβPP mice (79). Similarly, mitochondrial dysfunction has been proposed as a key mechanism in the early stages of AD, since both neuronal as well as peripheral cells derived from AD patients are characterized by energy loss (80–82). The mitochondrial accumulation of Aβ in human brain tissues is correlated with altered activities in key mitochondrial enzymes, including cytochrome c oxidase (COX, complex IV), pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase complex (αKGDH), as well as with reduction in the import of nuclear-encoded proteins (82, 83). Such impairment of mitochondrial oxidative phosphorylation in the brains of AD patients is directly proportional to their clinical disability (84). In model systems, expression of a mutant APP associated with AD (APP K670M/N671L) leads to an enhanced vulnerability of PC12 cells to oxidative stress and mitochondrial dysfunction, activation of caspases, and stress-signaling pathway (85). Mitochondrial functions are also defective in APP mice (54, 86, 87), and lipid peroxidation and hydrogen peroxide levels are significantly higher in tissues of the brain in mice carrying 5 AD-associated human mutations, 5XFAD, relative to healthy mice (54, 87). Apolipoprotein (apo) E4, a major genetic risk factor for late-onset Alzheimer disease (AD) (88), assumes a pathological conformation through an intramolecular interaction mediated by Arg61 in the amino-terminal domain and Glu255 in the carboxyl-terminal domain, referred to as apoE4 domain interaction (89). In humans, PET studies detect an AD-like regional pattern of glucose hypometabolism in the brains of cognitively normal apoE4 carriers, decades before the age of onset of clinical AD (90). This result raises the possibility that apoE4 may perturb mitochondrial respiratory function in the brain, rendering subjects with apoE4 more susceptible to AD neuropathology (90). Transgenic expression of apoE4, but not apoE3, in neurons using a neuron-specific enolase (NSE) promoter, induces age-dependent learning and memory deficits and neurodegenerative changes in mice (91, 92). Further, an apoE4 fragment (amino acids 1–272) is localized to mitochondria when transiently expressed in N2A cells and can bind to subunits of mitochondrial respiratory complexes III, IV, and V and perturb the activities of complexes III and IV (93). The same study reported that expression of apoE4 in N2A cells reduced the levels of mitochondrial gene transcripts from both the nuclear genome (complex V subunit α) and mtDNA (complex IV subunit 1). Thus, regardless of the genetic cause of AD, data from cell and animal models of AD and from cells and brain tissue derived from AD patients indicate major mitochondrial metabolic dysfunctions.
These metabolic dysfunctions correlate with abnormal mitochondrial morphology. Mitochondria fragmentation is noted in many models of AD as well as in cells and brain tissue derived from AD patients. Additionally, mitochondrial fragmentation is an early feature normally preceding AD pathology in transgenic animal models, suggesting the key role of mitochondrial structural and functional abnormalities in disease progression (94, 95). The structural abnormalities are associated with accelerated mitochondrial degradation and a significant decrease in mitochondrial numbers in AD models and in cells and tissue derived from AD patients (54, 95). There is also altered expression of mitochondrial fusion and fission proteins, such as Drp1, OPA1, Mfn1/2, and Fis1 along with abnormal post-translational modifications of Drp1 in animal models and in brains of AD patients (53, 96–99). Increased levels of mitochondrial fission proteins, Drp1 and Fis1 and decreased levels of mitochondrial fusion proteins, Mfn1, Mfn2 and Opa1 were found in 12-month-old tau mice relative to age-matched WT mice, indicating that the presence of abnormal mitochondrial dynamics in tau mice as well (100, 101). These abnormalities in mitochondrial dynamics are not restricted to brain tissue; we observed increase mitochondrial fission even in cultured fibroblasts of sporadic and familial AD patients (54, 102). In further support of the role of abnormal mitochondrial dynamics in neurodegeneration, recent studies reported that the inhibition of mitochondrial fragmentation by partial Drp1 deficiency, or through inhibition of Drp1 hyperactivation, is sufficient to alleviate mitochondrial dysfunction and synaptic loss in multiple mouse models (96, 103), and we have reported similar beneficial effects in patient-derived cells and in 5XFAD mouse model of AD when using a pharmacological inhibitor of pathological fission ((54); vide infra).
Mitochondrial Dysfunction and Defective Mitochondrial Dynamics in Amyotrophic Lateral Sclerosis (ALS)
ALS, also called Lou Gehrig’s disease, is a fatal neurodegenerative disease resulting from the loss of upper motor neurons in the cerebral cortex and lower motor neurons in the brainstem and spinal cord (104). Similar to other neurodegenerative diseases, the prominent pathological hallmark of ALS is the presence of inclusion bodies of aggregated proteins in degenerating motor neurons (105–108). Abnormal mitochondrial morphology was noted in neurons and peripheral cells of sporadic or familial ALS patients, and, in the past decade, mitochondrial fragmentation has been well documented in ALS cell and animal models (109, 110). Genetic mutations in Cu/Zn superoxide dismutase 1 (SOD1) were the first mutations identified in ALS patients (111). This enzyme binds copper and zinc ions and forms a homodimer whose main known function is as a dismutase, removing dangerous superoxide radicals by metabolizing them to molecular oxygen and hydrogen peroxide, thus providing a defense against oxygen toxicity. Recently, SOD1 has been found to be critical for repressing respiration and directing energy metabolism through integrating responses to O2, glucose and superoxide levels (112). In experimental models expressing ALS- associated mutant SOD1, mitochondrial fragmentation is concurrent with the changed expression of several mitochondrial fusion and fission proteins, including Drp1, OPA1, Mfn1, and Fis1, and these all these changes are also observed in the presymptomatic phase (113, 114). TDP-43 is a nuclear protein with transcriptional repressor activity. It is highly conserved and ubiquitously expressed in a variety of tissues including the brain. Although, its physiological function in the nervous system is not currently known, a recent study suggests that that it is involved in the regulation of neuronal plasticity (115). Similarly, TDP-43 mutant neurons also show mitochondrial fragmentation and altered expression of mitochondrial fusion and fission regulators (116, 117). In neurons expressing another ALS-associated mutant, FUS/TLS (Fused in Sarcoma/Translocated in Sarcoma, FUS), an RNA/DNA binding protein, excessive mitochondrial fission is observed (117–119). As discussed in the following, we observed the same abnormal morphology in three lines of patient-derived fibroblasts (55), suggesting that abnormal mitochondrial dynamics and excessive fission is also a hallmark of ALS pathology.
Mitochondrial mobility from neuron bodies down the axon to the synapses ensures mitochondrial recycling and functioning (120). In ALS, a disease of the longest neurons in our body, the motor neurons, is associated with impairment in this mitochondrial mobility. Mitochondria accumulate in the soma and proximal axon hillock of spinal cord motor neurons of sporadic ALS patients (121). Similarly, abnormal mitochondrial clusters in proximal axons or around the peri-nuclear area were also observed in transgenic animals expressing ALS-associated SOD1 or TDP-43 mutant, strongly suggesting impaired mitochondrial transportation in ALS (116, 122–124). Furthermore, cultured neurons expressing ALS-associated SOD1 or TDP-43 mutants showed deficits in axonal trafficking of mitochondria (125–127).
What is the connection between the mutant proteins in ALS and mitochondria? Both wild type and mutant SOD1 are found in the mitochondrial outer membrane, intermembrane space, and matrix (128), and both wild type and mutant TDP-43 reside in the mitochondrial inner mitochondrial membrane facing matrix (129). Likewise, RNA-binding protein FUS/TLS is found to enter mitochondria and interact with the mitochondrial chaperonin HSP60 in the matrix (118). The presence of ALS-associated SOD1, TDP-43, and FUS in mitochondria indicates the possibility of their direct interaction with mitochondrial fusion, fission, and trafficking machineries. Blocking mitochondrial fission by overexpression of either Drp1 or Mfn2 mutants abolishes mitochondrial trafficking defects in motor neurons expressing disease SOD1 or TDP-43 mutants, indicating that mitochondrial fusion and fission dynamic abnormalities may be responsible for impaired mitochondrial movement in ALS (116, 130). Importantly, TDP-43 can be imported into mitochondria and directly interfere with OXPHOS complex assembly (129). However, a direct physical association between ALS-associated proteins and mitochondrial dynamic regulators has yet to be investigated. Due to the close interplay of mitochondrial dynamics and bioenergetics, an indirect effect of ALS-associated proteins on mitochondrial dynamics is plausible.
NEK1 was associated in 3% of the ALS cases, and it was present in both the inherited and sporadic form of the disease (131, 132). NEK1 is a member of the NIMA- (never in mitosis A) related kinase family of serine/threonine kinases and is involved in the early cellular response to genotoxic stress and plays an important role in preventing cell death induced by DNA damage (133, 134). NEK1 also plays a role in mitochondrial function regulating a pathway of mitochondrial cell death through phosphorylation of voltage-dependent anion channel 1 (VDAC1) on serine 193 (134). Thus, mutations in NEK1 may be associated with mitochondrial dysfunction in these ALS patients. Dominant mutations in CHCHD10 also cause amyotrophic lateral sclerosis (ALS)/frontotemporal dementia, and mutations in CHCHD2 have been associated with Parkinson’s disease, although the function of these proteins remains unknown (135–139). The coiled-helix coiled-helix domain containing protein 10, CHCHD10, and its paralogue CHCHD2, belong to a family of twin CX9C motif proteins, most of which localize to the intermembrane space of mitochondria (140). Multiple different functions and activities have been suggested for CHCHD2. These include a role as a transcription factor that regulates the expression of a COX subunit during stress, as an inhibitor of Bax oligomerization through its interaction with Bcl-xL, and as a protein that sequesters SMAD4 to mitochondria, suppressing the activity of the TGFβ signaling pathway (141–143). Abnormalities in any of these individual functions may provide a mechanistic insight to the role of CHCHD2 in mitochondrial dysfunction and neurodegeneration. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis (140). Respiratory chain deficiency was also observed, suggesting that CHCHD10 is critical for maintaining ATP production and oxygen consumption (140). Furthermore, repair of the mitochondrial genome after oxidative stress is impaired in CHCHD10 mutant fibroblasts (140).
Recently, TANK-binding kinase 1 (TBK1) mutations have been reported in eight independent human genetics studies linking them with ALS (144). TBK1 has a critical role in modulating autophagy, including the phosphorylation of the autophagy adaptors p62, Optineurin (OPTN), and nuclear dot protein 52 kDa (NDP52) (144). TBK1 interacts with OPTN, which binds to ubiquitin chains on mitochondria (145). This recruits TBK1 to mitochondria and promotes its kinase activation. Thus, a mutation in TBK detrimentally affects autophagy and mitophagy, potentially leading to mitochondrial dysfunction and bioenergetic failure (145). OPTN is involved in a numerous cellular processes, but its function as an autophagy receptor is possibly the most relevant to ALS pathogenesis (146). Recently, OPTN regulates PINK1-Parkin-mediated mitophagy through nucleation of the autophagosome by recruitment of LC3 (147, 148). ALS-causing mutations in OPTN disable this process, implicating inefficient mitochondrial clearance in ALS, leading to accumulation of dysfunctional mitochondria (147, 148).
We and other groups reported that the inhibition of mitochondrial dynamics abnormalities improve mitochondrial and neuronal dysfunction caused by mutant SOD1/ TDP-43/ FUS1 in neurons (3, 114), in cells derived from ALS patients, and in a SOD1 ALS mouse model (55). Together, these data indicate that mitochondrial fragmentation and dysfunction in ALS patients and experimental models is likely the downstream event or consequence of disease onset.
Mitochondrial Dysfunction and Defective Mitochondrial Dynamics in Huntington’s Disease (HD)
HD is a rare and fatal autosomal-dominant neurodegenerative disease caused by an expanded trinucleotide CAG (cytosine-adenine-guanine) repeat in the gene encoding the huntingtin protein (149, 150). The hallmark of HD includes the extensive loss or degeneration of striatal and cortical neurons, along with the presence of intracellular inclusion bodies composed of ubiquitinated or truncated Htt-containing long polyglutamine, that is progressively accompanied by a loss of voluntary and involuntary movements as well as psychiatric and cognitive disturbances (149, 150). Mitochondria isolated from the lymphoblasts of HD patients, brain tissue of mtHtt mice, and mtHtt-expressing cells all exhibit decreased mitochondrial membrane potential (ΔΨm), increased susceptibility to calcium-induced mitochondrial depolarization, and reduced mitochondrial calcium uptake capacity (51, 57, 151). Further, an increase in mitochondrial fission proteins and a corresponding decrease in mitochondrial fusion proteins was observed in frontal cortex of HD patients. Importantly, the levels of fission proteins were significantly increased in correlation with disease progression (152, 153). Additionally, increased levels of s-nitrosylation in Drp1, a detrimental post- translational modification associated with mitochondrial fission, were found in the striatum of an HD transgenic mouse as well as in the neurons from patients with HD (154, 155). Unsurprisingly, numerous studies demonstrated abnormal mitochondrial morphology in experimental models for HD. For example, mitochondrial fragmentation following mitochondrial dysfunction accompanied by an increase in ROS was observed in neuronal cells treated with 3-nitropropionic acid (3-NP), a mitochondrial complex II inhibitor (156, 157). Furthermore, iPSC-derived human GABAergic neurons from an HD patient, and neuronal cells expressing Htt protein containing expanded poly-glutamine tracts, also displayed fragmented mitochondria neurites with a decreased membrane potential, increased ROS, and enhanced apoptosis (158, 159).
Mitochondrial dynamic abnormalities in toxin-based models could be prevented by antioxidant treatment, indicating that mitochondrial dynamic changes might be the consequence of impaired mitochondrial biogenetics (156, 157). However, mutant Htt directly interacts with Drp1 on mitochondria, indicating that there is an interplay between mutant Htt and mitochondrial dynamics (51, 151). The dominant negative Drp1 (51) or pharmacological inhibition of Drp1 and Fis1 interaction (57) significantly reduce mitochondrial defects in HD neurons, suggesting the direct involvement of mutant Htt in regulating mitochondrial fusion and fission dynamics. Further, mutant Htt impairs vesicular and mitochondrial trafficking in neurons in vitro and in vivo, and this impairment corresponds with the length of the polyglutamine tract and occurs in the absence of cellular toxicity. Mutant Htt directly interacts with HAP1 to disrupt the association of motor proteins with microtubules as well as inactivates motor proteins such kinesis and dynactin (160, 161). Overall, mutant Htt affects both mitochondrial dynamics and motility, and might lead to failure of ATP synthesis, energy depletion, and, ultimately, cell death in HD.
Mitochondrial Dysfunction and Defective Mitochondrial Dynamics in Parkinson’s Disease (PD)
PD is a long-term degenerative disorder of the central nervous system that predominantly affects the motor system, causing tremors or trembling, slow movement, body rigidity and stiffness, and problems walking (162). Pathologically, PD is characterized by the progressive loss or degeneration of the dopaminergic (DA) neurons in the substantia nigra and by the presence of intracytoplasmic inclusions bodies (i.e., Lewy bodies) of which fibrillar aggregates of misfolded αSynuclein are the major components in DA neurons (163–165). MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) was found to cause DA neurodegeneration and progressive and levodopa-responsive parkinsonism resembling sporadic PD when several young intravenous drug addicts mysteriously developed a profound parkinsonian syndrome. It was later found that certain illicit street preparations of meperidine analogs were contaminated with MPTP (166, 167). Once in the brain, MPTP is metabolized to 1‐methyl‐4‐phenyl‐2,3‐dihydropyridinium (MPDP+) by the enzyme monoamine oxidase B within non-dopaminergic cells, and subsequently to 1‐methyl‐4‐ phenylpyridinium (MPP+), the active toxic compound. MPP+ (1-methyl-4-phenylpyridium ion), concentrates within DA neurons via the dopamine transporter (DAT) and specifically targets and inhibits OXPHOS complex I (50, 168). Interestingly, MPP+ tips the mitochondrial fusion and fission balance towards excessive fission, resulting in mitochondrial fragmentation concurrent with mitochondrial dysfunction, but preceding neuronal death (50). Further, impaired mitochondrial function is a classical feature of PD with decreased activity in OXPHOS complexes and/or high levels of mitochondrial DNA deletions observed in PD-affected neurons. Thus, in addition to suggesting an unexpected important role of mitochondrial fusion and fission dynamics in mediating MPP+ toxicity, these data directly link mitochondrial dysfunction to mitochondrial dynamics in PD neurotoxin models (50, 169–171).
Mitochondrial structural and functional defects have also been reported in peripheral cells from PD patients (169–171). As such, mitochondrial fragmentation in experimental models of PD has been extensively investigated. Loss of, or mutations in, PINK1, Parkin, or DJ-1 result in fragmented mitochondrial morphology in muscle and DA neurons (172). Further, mitochondrial fragmentation, associated dysfunction and energy depletion have been extensively reported in models associated with autosomal dominant PD forms (50). α-synuclein, the major structural constituent of cytoplasmic inclusion bodies (Lewy bodies) and neurites (Lewy neurites) that are characteristic of both familial and sporadic PD cases, mis-localizes to mitochondria to induce mitochondrial function and fragmentation (173, 174). The expression of disease-causing leucine-rich repeat kinase 2 (LRRK2) mutants, a common cause of PD, results in mitochondrial fragmentation in a Drp1-dependent manner (175). In the same study, co-expression of Drp1 mutant or WT Mfn2 blocked LRRK2-induced mitochondrial fragmentation, dysfunction and neuronal toxicity. Consistently, progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice were reported with changes in Drp1 phosphorylation (176). In addition, S-nitrosylation of Parkin reduced its ability as a suppressor of Drp1 expression, leading to upregulation of Drp1 in neurotoxin-based PD models, in vitro and in vivo (177). In most PD models (both genetic and toxin based), the inhibition of mitochondrial fission machinery alone is sufficient to reduce mitochondrial dysfunction (50, 175, 178), indicative of the critical role of mitochondrial dynamics in mediating neurotoxicity observed in PD.
Innate Immunity and Mitochondrial Dysfunction in Neurodegenerative diseases
Two main cell types, astrocytes and microglia, regulate the health and function of the brain through a variety of processes. Their role in neuroinflammation and the contribution of neuroinflammation to neurodegenerative disease is highlighted by many recent discoveries. In the following, we describe the contribution of mitochondrial dysfunction and imbalanced mitochondrial dynamics to neuroinflammation in these cells.
Astrocytes
Astrocytes are the main neural cell type responsible for the maintenance of brain homeostasis (179). They form extensive networks that modulate neuronal activity through the expression of various receptors for neurotransmitters, several transporters, cytokines, and growth factors. Astrocytes undergo a pronounced transformation called ‘reactive astrocytosis’ after brain injury and disease, whereby they upregulate many genes (180–182). ‘Reactive astrocytes’ release an array of molecules, including inflammatory modulators, chemokines and cytokines, as well as neurotrophic factors which are either neuroprotective or neurotoxic (180–182). Reports show the presence of reactive astrocytes at the site of Aβ deposits in postmortem human AD brains and in animal models of AD (182–185), and astrocytes can internalize Aβ from their surrounding in vitro and ex vivo (183, 186). For example, astrocytes plated on Aβ-bearing brain sections from an AD mouse model bind and degrade Aβ deposits, thereby decreasing Aβ levels (187). AD astrocytes show perturbations in energy metabolism and oxidative stress (188), which make them toxic to neurons (189, 190). Pre-treatment of astrocytes with Aβ activates them and leads to decreased neuronal viability in co-culture models while sensitizing neurons further to Aβ treatment (191). Furthermore, multiple reports provide evidence for astrocytic contribution to cellular and functional degeneration, disrupting glial–neuronal and glial–vascular signaling in AD (192–195).
As in AD, mutant huntingtin (Htt) in astrocytes, being ubiquitously expressed is damaging to both astrocytic and, subsequently, neuronal health. Mutant huntingtin-expressing astrocytes are toxic to WT neurons and show reduced brain-derived neurotrophic factor (BDNF) expression which is critical for neuronal health; Mutant Htt in cultured astrocytes decreased their protection of neurons against glutamate excitotoxicity (196, 197). Further, expression of mutant Htt in astrocytes leads to its accumulation in nuclei and subsequently decreases the expression of glutamate transporter through specificity protein 1 (Sp1) (198). Mutant huntingtin also hinders the suppression of production and secretion of the chemokine Ccl5/RANTES, which is another major trophic function of astrocytes (199). Furthermore, inflammation‐prone HD astrocytes were reported to provide less pericyte coverage by promoting angiogenesis and reducing the number of pericytes, thereby impairing vascular reactivity. This impairment possibly hinders cerebral hemodynamics and increases brain atrophy during HD progression (200).
Astrocytes are thought to be involved in both pathological stages of PD: contributing to triggering inflammation prior to neuronal loss and subsequent progressive neurodegeneration. Recently a study demonstrated that many of the genes where monogenic mutations have been identified are expressed in astrocytes at levels comparable to, or in some cases higher than, those in neurons (201). Increased expression levels of astrocytic PARK7 as compared to neurons have been observed in postmortem PD samples human (202). Further, reducing the levels of PARK7 in astrocytes results in a reduced ability to protect neurons against neurotoxicity in rotenone and 6-hydroxydopamine neurotoxin models (203, 204). In the same study, knockdown of PARK7 reduced astrocytic mitochondrial motility and further decreased mitochondrial membrane potential in response to rotenone treatment. While the expression of α-synuclein in astrocytes is low, non-fibrillized α-synuclein accumulates in the cytoplasm of protoplasmic (but not fibrous) astrocytes early in disease (205, 206). α-synuclein-containing astrocytes are found in regions lacking Lewy bodies (e.g., in striatum and dorsal thalamus, where there are likely to be dysfunctional neuron terminals) (205). A53T mutant α-synuclein, when selectively expressed in astrocytes, induced rapidly progressed paralysis in mice and produced increased levels of proinflammatory cytokines and chemokines and neuroinflammatory mediators, such as IFN-γ and TNF-α, synergistically activating microglia (206). In the same study, pre-symptomatic and symptomatic accumulation of α-synuclein aggregated in astrocytes disrupted astrocytic glutamate transporters as well as the ability of astrocytic regulation of the blood–brain barrier. Thus, a variety of astrocytic functions are impaired in PD to directly and indirectly contribute to the disease.
While ALS is a predominantly motor-neuron-specific disease, expression of human ALS- causing mutant genes specifically in motor neurons does not lead to typical ALS-like disease in mice (207). In the time since this observation, experiments revealed that non-neuronal cells expressing mutant ALS-causing SOD1 transgenes damage nearby WT motor neurons, while WT non-neuronal cells can delay degeneration of nearby neurons that express mutant SOD1 (208, 209). ALS astrocytes directly contribute to motor neuron death in vitro, and primary astrocytes from the SOD1 G93A mouse model of ALS adversely affect motor neuron viability of both WT and ALS motor neurons (210, 211). In vivo experiments in mouse models of ALS in which the mutant SOD1 transgene was deleted in a cell-specific manner revealed that reducing the levels of SOD1 expression in astrocytes alone was sufficient to delay disease onset and /or progression (212, 213). Conversely, transplanting precursors of mutant SOD1 astrocytes into the spinal cord of WT rats lead to the degeneration of motor neurons, which is thought to be mediated in part by the activation of host microglia (214). Thus, ALS astrocytes, by acting through microglia either directly and/or indirectly, can be toxic to motor neurons in vivo.
Microglia
A critical function of microglia is their ability to rapidly respond to immune-mediated insults and physical damage in the brain (215–217). Microglia modulate the stress response to a variety of pathological triggers in CNS diseases. Microglial activation in neuroinflammatory conditions is mediated by a host of interconnected signaling pathways (215–217). The role of microglia in AD has recently gained renewed interest due to identification of rare coding variants associated with AD in genes highly expressed in these cells (218). In AD brain, microglia (or peripherally-derived macrophages) cluster around neuritic plaques but appear to have a loss of phagocytic capacity and possibly a gain of toxic function as well (219). Recent evidence suggests that microglia’s neuroprotective functions are impaired in individuals with a triggering receptor expressed on myeloid cells 2 (TREM2) variants, resulting in increased AD risk (220, 221). TREM2 has been implicated in microglial phagocytosis of dead neurons, damaged myelin, and Aβ plaques (220, 221). TREM2-deficient microglia adopt a severely divergent cellular state that does not reflect homeostasis during neurodegeneration, resulting in a robust induction of autophagy, which may reflect mitochondrial defects (220, 221). Hence, abnormal microglial mitochondrial dynamics may contribute to mitochondrial dysfunction and neuronal dysfunction in the AD brain. Furthermore, many AD risk genes, in addition to TREM2, are preferentially or selectively expressed in microglia; e.g., CD33, INPP5D, MS4A6A, and PLCG2, and could impact on the same microglial activities and pathways regulated by TREM2 and have been recently reviewed (222). Additionally, given their pattern of microglial expression, several AD risk-related genes analyzed in neurons in the context of APP trafficking, Aβ production, or tau pathology should also be considered for potential roles in microglia (222).
Microglia, once activated, accumulate in all grades of HD patients’ brains, with their density correlating with the degree of neuronal loss (223). 11C-(R)-PK11195 (PK) positron emission tomography (PET) in HD patients revealed widespread microglial activation in preclinical HD which correlated with striatal neuronal dysfunction (224). Since over-activated microglia release neurotoxins, a decreased number of reactive microglia coupled with downregulation of inflammatory cytokines are thought to represent an improvement in HD (225). iNOS, IL-1β, IL-6, and TNF-α are significantly elevated after LPS treatment of primary glia cells including microglia, isolated from R6/2 transgenic mouse model (226) and higher levels of IL-1β and IL-8 are secreted by microglia in HD transgenic porcine model (227). Pro-inflammatory cytokines IL-1β, IL-6, TNF-α and IL-8 are elevated both in the striatum and cerebrospinal fluid as well as in the plasma of HD patients (228–231). However, healthy microglia can contribute towards neuroprotection in HD: adding exogenous primary microglia to mHtt-expressing neurons increases survival that is proportional to the amount of healthy microglia (232). Similarly, supplementing normal human glia to transgenic R6/2 HD mice in vivo produce neuronal protection as well as phenotypic improvement (233). Thus, healthy microglia can protect, and over- activated microglia can exacerbate, HD progression in multiple models.
Microglia-mediated neuroinflammation is a signature of PD. Similar to the findings in HD, in the brains of patients with PD, microglia have both neurotoxic and neuroprotective effects, depending on their activation state (234, 235). Recently, GWAS indicate that variants in the HLA protein (involved in immune-surveillance) are linked to sporadic PD (236). In addition, levels of pro-inflammatory mediators, including TNFα, IL-1β, IL-6, and eicosanoids are elevated in the brains and peripheral PBMC of patients with PD (237). Microglial phagocytosis occurs in response to aggregated α-synuclein, the major component of LBs in PD (205, 238, 239) and ATP, released from damaged neurons, activate microglia by binding to purinergic receptors on microglia (240, 241). Activated microglia are found the brains of both monkeys and mice/rats after systemic injection of PD-inducing agents, MPTP as well as hydroxy dopamine (242–247). Infiltration of T-lymphocytes has also been detected in the brains of MPTP-treated mice and ROS produced by activated microglia plays an important role in MPTP-induced neurotoxicity (248–250).
Together, both microglia and astrocytes are activated in a number of neurodegenerative diseases, and although known first for their neuroprotective role, in the context of the aforementioned diseases these cells appear to hyperactivated, thus contributing to neurodegeneration. However, a potential role for mitochondrial dysfunction and abnormal mitochondrial dynamics in these cells remains to be determined.
Mitochondrial Dynamics as Common Therapeutic Targets for Neurodegeneration
Numerous studies have already demonstrated the feasibility of using the inhibition of mitochondrial fragmentation as a novel approach to prevent neuronal loss and to improve behaviors in different experimental models for neurodegenerative diseases. Mdivi‐1 (mitochondrial fission inhibitor‐1), which contains a quinazolinone core substituted with a thiol moiety and an aryl (2,4‐dichloro‐5‐methoxyphenyl) side chain attached to the N3 position, is the first inhibitor of the mitochondrial fission protein Drp1 (251). Mdivi‐1 was shown to target Drp1 in mammalian cells by binding to an allosteric site and suppressing its ability to catalyze GTP hydrolysis and to self-assemble into ring‐like structures around the mitochondria (251). Mdivi-1 delays mitochondrial permeability transition pore opening, preserves mitochondrial membrane potential, increases adenosine levels, attenuates oxidative stress, and reduces endoplasmic reticulum stress (252). Mdivi‐1 also increases release of the neuroprotective agent, adenosine, through the cAMP/PKA/CREB pathway (253). However, recent studied indicated that Mdivi-1 is not a specific Drp1 inhibitor and that the ability of Mdivi-1 to reversibly inhibit complex I and modify mitochondrial ROS production may contribute to effects observed [e.g., (254)].
Inhibition of mitochondrial division with Mdivi‐1 attenuates mitochondrial functional defects observed in AD cybrid cells (255). Mdivi-1 treatment rescues both mitochondrial fragmentation and distribution deficits and improves mitochondrial function in the CRND8 neurons both in vitro and in vivo (256). Mdivi-1 treatment significantly reduces extracellular amyloid deposition and Aβ1–42/Aβ1–40 ratio, prevents the development of cognitive deficits in Y-maze test and improves synaptic parameters (256). Mdivi-1 pre- and post-treated cells treated with Aβ exhibit a reduced mitochondrial dysfunction, and maintain cell viability, mitochondrial dynamics, mitochondrial biogenesis, and synaptic activity (257). In another study, inhibition of Drp1 by Mdivi-1 restored amyloid-β (Aβ)-mediated mitochondrial dysfunctions and synaptic depression in neurons and significantly reduced lipid peroxidation, the expression of the Ab processing enzyme, BACE1, as well as Aβ deposition in the brain of AD mice (103). Treatment with Mdivi-1 in a cell culture model rescues toxicity in stem cell-derived neurons as well as functional and structural mitochondrial defects in a PINK1 mutant Parkinson’s cell culture model (258, 259). Mdivi-1 reduces α-synuclein aggregates, mitochondrial fragmentation, mitochondrial dysfunction and oxidative stress and normalized motor function in α-synuclein overexpression mouse model (260). Additionally, Mdivi-1 is protective in other models of PD by limiting mitochondrial structural damage (261, 262). In an ALS model, Mdivi-1 treatment improves skeletal muscle function by reducing mitochondrial defects (114). Treatment with Mdivi-1 also partially improves mitochondrial structure and function in primary osteocytes (263). Unfortunately, general toxicity and developmental defects are associated with Mdivi-1 treatment in animal models (264–268). Based on the reported non-Drp1 related targets, it is likely that the effects of Mdivi-1 are not only through inhibition of fission, but rather are likely dependent on its effect as a complex I inhibitor. In addition to potential effects of Mdivi-1 on molecular targets other than Drp1, the inherit limitation of these inhibitors may relate to attenuation physiological fission. Whereas excessive fission is pathological, tightly-regulated physiological fission is necessary for cell survival (2). However, the mechanism distinguishing between physiological and pathological fission are only now being uncovered (269).
Since an interaction between Drp1 and its mitochondrial adaptor/s is essential for mitochondrial fission, an inhibitor of this interaction may have a therapeutic utility. To this end, we developed a specific inhibitor of excessive mitochondrial fragmentation: P110. P110, a seven-amino acid peptide, specifically blocks the interaction of Drp1 and Fis1 while having no effect on Drp1’s interaction with Mff, MiD49 or MiD51 (269, 270). In response to treatment with MPP+, P110 reduced dopaminergic neuronal degeneration by inhibiting Drp1-mediated mitochondrial excessive fission and dysfunction (270). In another study, using the subacute MPTP model of PD in vivo, P110 treatment completely blocked MPTP-induced Drp1 mitochondrial translocation both in SN and striatum and reduced disease severity (50). P110 treatment corrects LRRK2 G2019S-induced mitochondrial dysfunction, inhibits excessive autophagy, and reduces cell death in various cell culture models, including dopaminergic neurons derived from LRRK2 G2019S PD patient-derived pluripotent stem cells (270). P110 inhibits mtHtt-induced excessive mitochondrial fragmentation, improves mitochondrial function, and increases cell viability in HD cell culture models (271). Moreover, we found that sustained treatment of R6/2 HD transgenic mice with P110 from the age of 5 weeks, for two months, reduces mitochondrial dysfunction, motor deficits, neuropathology, and mortality (5, 271). We also found that P110 treatment attenuates Aβ42-induced mitochondrial recruitment of Drp1 and prevents mitochondrial structural and functional dysfunction in cultured neurons, in cells expressing mutant amyloid precursor protein (KM670/671NL), and in five different AD patient-derived fibroblasts, including from individuals familial forms of the disease (54). Additionally, sustained P110 treatment significantly improved behavioral deficits, and reduced Aβ accumulation, energetic failure and oxidative stress in 5XFAD AD mouse model (54). We also found that P110 treatment rescues mitochondrial bioenergetic defects in fibroblasts of ALS patients carrying pathogenic mutations in SOD1 (I113T), in FUS1 (fused in sarcoma; R521G) or in TDP43 (TAR DNA‐binding protein 43; G289S) genes, by improving mitochondrial structure (55). Importantly, sustained P110 treatment that begins after the onset of paralysis in SOD1-G93A model significantly improves motor activity by improving muscle mitochondrial integrity and prolongs life span and shows no negative side effects even after sustained delivery in wildtype mice for 5 months (55).
Concluding remarks:
Mitochondria are dynamic organelles that undergo continuous cycles of fission and fusion. These dynamic processes allow mitochondria to communicate, migrate and adapt to changing energy demands and cellular conditions. It also ensures segregation of defective and damaged parts for removal (mitophagy) and maintenance of quality mitochondria. Since mitochondria provide most of the ATP required for neuronal function, it is important to consider the possible link between changes in mitochondrial dynamics and bioenergetic failure. Such a link between mitochondrial dynamics and function and neuroinflammation is also likely to be present. Overall, it is becoming increasingly clear that mitochondrial dysfunction leads to neurodegeneration and aging; AD, PD, ALS, HD, and many other neurodegenerative diseases are all characterized by mitochondrial structural and functional defects. All these diseases are expected to account for a greater socioeconomic pressure as the world population grows and individuals live longer than before, and yet, there is no cure or effective treatment for neurodegeneration. Mitochondrial excessive fission as such may provide an attractive therapeutic target to improve mitochondrial and neuronal function that will slow down or even prevent neurodegeneration.
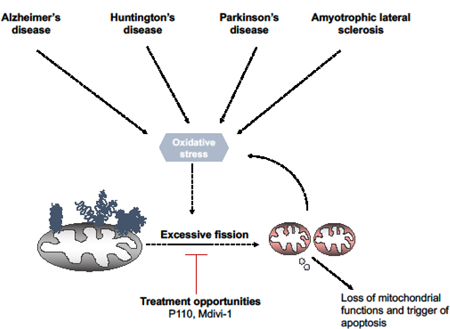
Figure 1. All roads lead to mitochondrial dysfunction.
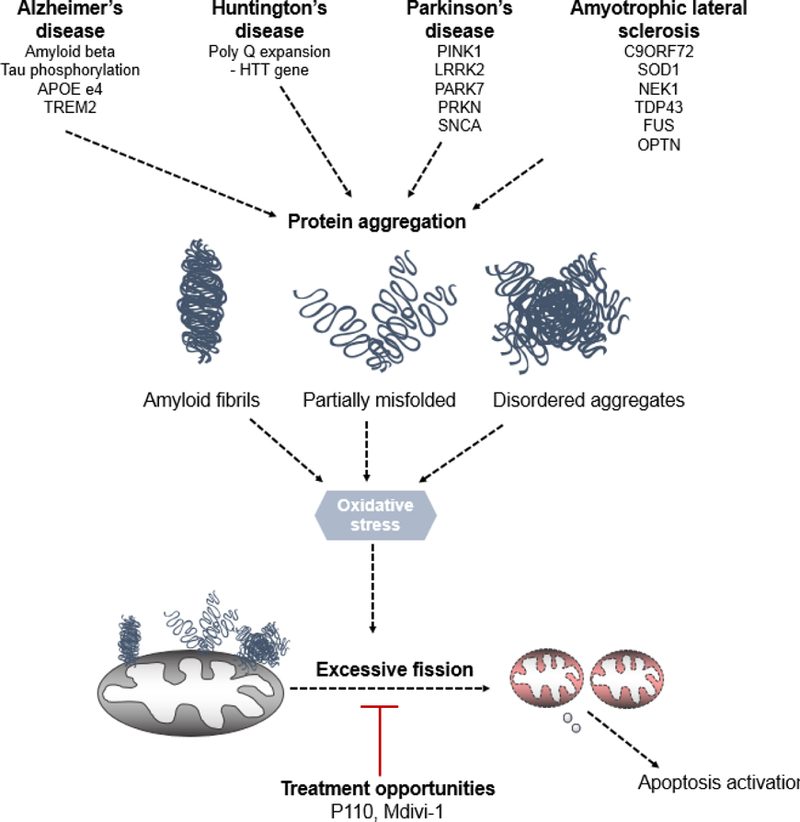
Genetic mutations in proteins leading to Alzheimer’s disease, Huntington’s disease, Parkinson’s disease as well as Amyotrophic lateral sclerosis lead to protein aggregation. Through either direct/ indirect interactions with mitochondria or mitochondrial proteins, these toxic gain of function proteins lead to oxidative stress. This stress eventually culminates into mitochondrial dysfunction, depolarization and subsequent fission. Currently, Mdivi-1 and P110 present two opportunities to rescue both mitochondrial structural and functional deficits in these diseases. However, recent studies indicate that Mdivi-1 is not a specific Drp1 inhibitor; it has multiple off target effects which may affect its utility in both pre-clinical and clinical studies.
Figure 2. Mitochondrial damage and oxidative stress occur in all CNS cells.
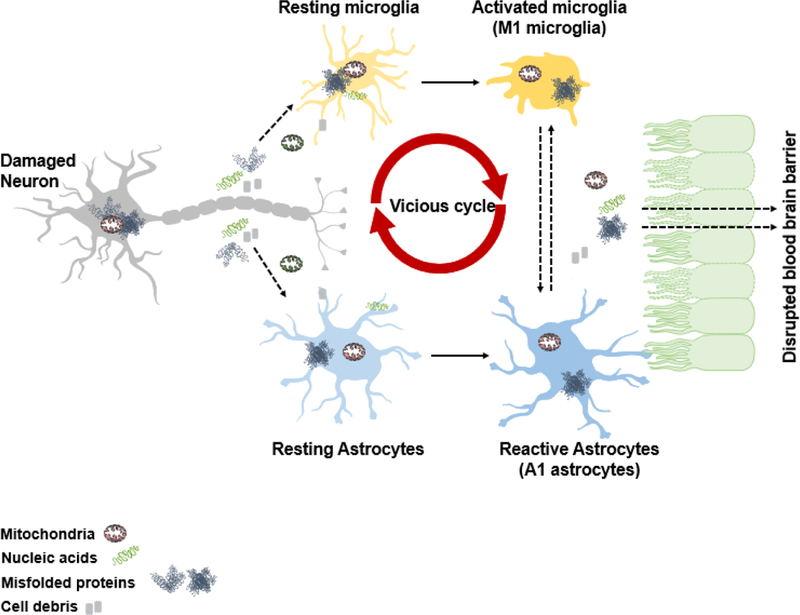
Neurons are sensitive to oxidative stress caused by misfolded proteins. These proteins then lead to mitochondrial dysfunction in neurons as well as the release of nucleic acids, cell debris as well as damaged mitochondria. This then leads to the activation of both microglia as well as astrocytes which leads to enhanced oxidative stress. A vicious cycle then occurs when additional damage occurs in these important cell type. This eventually leads to disruption of blood brain barrier allowing for the immune cells in some diseases to infiltrate eventually leading to complete bioenergetic failure.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants NIAAA11147 to Daria Mochly-Rosen. A patent on P110 and its utility in ALS has been filed by AUJ and DM-R and P110 was recently licensed to Mitoconix Bioscience, a company that DM-R founded, that develops new treatment for Huntington’s disease. None of the work was supported by Mitoconix Bioscience. The authors have no additional financial interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures None
Conflict of Interest
A patent on P110 and its utility in ALS has been filed by AUJ and DM-R and P110 was recently licensed to Mitoconix Bioscience, a company that DM-R founded, that develops new treatment for Huntington’s disease. None of the work was supported by Mitoconix Bioscience. The authors have no additional financial interest.
The authors have read and approved the final article. Dr. Mochly-Rosen is the founder of Mitoconix, a company that has licensed P110 as a treatment for Huntington’s disease. However, none of the research in her lab is supported by or is in collaboration with the company.
References:
- 1.Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol 2013;23(2):64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012;337(6098):1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS- induced ROS release. Physiol Rev 2014;94(3):909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics 2006;5(4):608– 619. [DOI] [PubMed] [Google Scholar]
- 5.Disatnik MH, Joshi AU, Saw NL, Shamloo M, Leavitt BR, Qi X, Mochly-Rosen D. Potential biomarkers to follow the progression and treatment response of Huntington’s disease. J Exp Med 2016;213(12):2655–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benedetti A, Brunelli E, Risicato R, Cilluffo T, Jezequel AM, Orlandi F. Subcellular changes and apoptosis induced by ethanol in rat liver. J Hepatol 1988;6(2):137–143. [DOI] [PubMed] [Google Scholar]
- 7.Toescu EC, Myronova N, Verkhratsky A. Age-related structural and functional changes of brain mitochondria. Cell calcium 2000;28(5–6):329–338. [DOI] [PubMed] [Google Scholar]
- 8.Wakabayashi T Structural changes of mitochondria related to apoptosis: swelling and megamitochondria formation. Acta Biochim Pol 1999;46(2):223–237. [PubMed] [Google Scholar]
- 9.Pernas L, Scorrano L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu Rev Physiol 2016;78:505–531. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology 2003;160(2):189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Annals of the New York Academy of Sciences. 2010;1201:21–25. [DOI] [PubMed] [Google Scholar]
- 12.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harbor perspectives in biology 2013;5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to Metabolism. Molecular cell 2016;61(5):683–694. [DOI] [PubMed] [Google Scholar]
- 14.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. The Journal of cell biology 2016;212(4):379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belenguer P, Pellegrini L. The dynamin GTPase OPA1: more than mitochondria? Biochimica et biophysica acta 2013;1833(1):176–183. [DOI] [PubMed] [Google Scholar]
- 16.Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, MacLaurin JG, Park DS, McBride HM, Trinkle-Mulcahy L, Harper ME, Germain M, Slack RS. OPA1- dependent cristae modulation is essential for cellular adaptation to metabolic demand. The EMBO journal 2014;33(22):2676–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Molecular biology of the cell 2002;13(3):847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Lendahl U, Nister M. Regulation of mitochondrial dynamics: convergences and divergences between yeast and vertebrates. Cell Mol Life Sci 2013;70(6):951–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem 2007;76:751–780. [DOI] [PubMed] [Google Scholar]
- 20.Westermann B Mitochondrial fusion and fission in cell life and death. Nature reviews Molecular cell biology 2010;11(12):872–884. [DOI] [PubMed] [Google Scholar]
- 21.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009;89(3):799–845. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 2005;39:503–536. [DOI] [PubMed] [Google Scholar]
- 23.Pitts KR, McNiven MA, Yoon Y. Mitochondria-specific function of the dynamin family protein DLP1 is mediated by its C-terminal domains. The Journal of biological chemistry 2004;279(48):50286–50294. [DOI] [PubMed] [Google Scholar]
- 24.Pitts KR, Yoon Y, Krueger EW, McNiven MA. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Molecular biology of the cell 1999;10(12):4403–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nature cell biology 1999;1(5):298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. The Journal of cell biology 1998;143(2):333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 2001;1(4):515–525. [DOI] [PubMed] [Google Scholar]
- 28.Kraus F, Ryan MT. The constriction and scission machineries involved in mitochondrial fission. Journal of cell science 2017;130(18):2953–2960. [DOI] [PubMed] [Google Scholar]
- 29.Michalska B, Duszynski J, Szymanski J. [Mechanism of mitochondrial fission - structure and function of Drp1 protein]. Postepy Biochem 2016;62(2):127–137. [PubMed] [Google Scholar]
- 30.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. The Journal of cell biology 2009;186(6):805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 2007;356(17):1736–1741. [DOI] [PubMed] [Google Scholar]
- 32.Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, Harper ME, Michaud J, Sell E, Chakraborty P, Tetreault M, Care4Rare C, Majewski J, Baird S, Boycott KM, Dyment DA, MacKenzie A, Lines MA. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur J Hum Genet 2016;24(7):1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A 2016;170(8):2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francy CA, Alvarez FJ, Zhou L, Ramachandran R, Mears JA. The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. The Journal of biological chemistry 2015;290(18):11692–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. The Journal of cell biology 1998;143(2):351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. Journal of cell science 2010;123(Pt 6):917– 926. [DOI] [PubMed] [Google Scholar]
- 37.Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016;540(7631):139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. The Journal of biological chemistry 2003;278(38):36373–36379. [DOI] [PubMed] [Google Scholar]
- 39.Jofuku A, Ishihara N, Mihara K. Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochemical and biophysical research communications 2005;333(2):650–659. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proceedings of the National Academy of Sciences of the United States of America 2007;104(47):18526–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. The Journal of cell biology 2005;170(2):237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagliuso A, Cossart P, Stavru F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci 2018;75(3):355–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of cell biology 2010;191(6):1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 2014;3:e01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onoue K, Jofuku A, Ban-Ishihara R, Ishihara T, Maeda M, Koshiba T, Itoh T, Fukuda M, Otera H, Oka T, Takano H, Mizushima N, Mihara K, Ishihara N. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. Journal of cell science 2013;126(Pt 1):176–185. [DOI] [PubMed] [Google Scholar]
- 46.Yamano K, Wang C, Sarraf SA, Munch C, Kikuchi R, Noda NN, Hizukuri Y, Kanemaki MT, Harper W, Tanaka K, Matsuda N, Youle RJ. Endosomal Rab cycles regulate Parkin- mediated mitophagy. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian L, Potus F, Wu D, Dasgupta A, Chen K- H, Mewburn J, Lima P, Archer SL. Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension. Frontiers in Physiology 2018;9(828). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 2011;44(4):532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, Chen KH, Hong Z, Sharp WW, Kutty S, Archer SL. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J Mol Med (Berl) 2017;95(4):381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filichia E, Hoffer B, Qi X, Luo Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Scientific reports 2016;6:32656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet 2012;21(2):406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 2011;20(7):1438–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 2011;20(13):2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joshi AU, Saw NL, Shamloo M, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018;9(5):6128–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joshi AU, Saw NL, Vogel H, Cunnigham AD, Shamloo M, Mochly-Rosen D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO molecular medicine 2018. [DOI] [PMC free article] [PubMed]
- 56.Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. Journal of cell science 2013;126(Pt 3):789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. The Journal of clinical investigation 2013;123(12):5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Molecular biology of the cell 2008;19(6):2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Otera H, Mihara K. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2011;2(3):167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Molecular biology of the cell 2015;26(24):4466–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. Embo Rep 2011;12(6):565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. The Journal of cell biology 2016;212(5):531– 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 2014;22(3):367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell 2013;24(5):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen KH, Dasgupta A, Lin J, Potus F, Bonnet S, Iremonger J, Fu J, Mewburn J, Wu D, Dunham-Snary K, Theilmann AL, Jing ZC, Hindmarch C, Ormiston ML, Lawrie A, Archer SL. Epigenetic Dysregulation of the Drp1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018. [DOI] [PMC free article] [PubMed]
- 66.Yu R, Liu T, Jin SB, Ning C, Lendahl U, Nister M, Zhao J. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Scientific reports 2017;7(1):880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. Journal of cell science 2016;129(11):2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. The Journal of cell biology 2004;166(7):1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ, Wang HG. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Molecular and cellular biology 2005;25(21):9369–9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. Journal of cell science 2005;118(Pt 14):3049–3059. [DOI] [PubMed] [Google Scholar]
- 71.Tondera D, Santel A, Schwarzer R, Dames S, Giese K, Klippel A, Kaufmann J. Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein, affects mitochondrial morphology and induces apoptosis. The Journal of biological chemistry 2004;279(30):31544–31555. [DOI] [PubMed] [Google Scholar]
- 72.Barneo-Munoz M, Juarez P, Civera-Tregon A, Yndriago L, Pla-Martin D, Zenker J, Cuevas- Martin C, Estela A, Sanchez-Arago M, Forteza-Vila J, Cuezva JM, Chrast R, Palau F. Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet 2015;11(4):e1005115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niemann A, Ruegg M, La Padula V, Schenone A, Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. The Journal of cell biology 2005;170(7):1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niemann A, Wagner KM, Ruegg M, Suter U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiology of disease 2009;36(3):509–520. [DOI] [PubMed] [Google Scholar]
- 75.Zhao J, Liu T, Jin SB, Tomilin N, Castro J, Shupliakov O, Lendahl U, Nister M. The novel conserved mitochondrial inner-membrane protein MTGM regulates mitochondrial morphology and cell proliferation. Journal of cell science 2009;122(Pt 13):2252–2262. [DOI] [PubMed] [Google Scholar]
- 76.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med 2010;77(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochimica et biophysica acta 2005;1739(2– 3):331–354. [DOI] [PubMed] [Google Scholar]
- 79.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. The Journal of cell biology 2003;161(1):41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eckert A, Schmitt K, Gotz J. Mitochondrial dysfunction - the beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-beta toxicity. Alzheimers Res Ther 2011;3(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. Journal of Alzheimer’s disease : JAD 2010;20 Suppl 2:S401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochimica et biophysica acta 2010;1802(1):2– 10. [DOI] [PubMed] [Google Scholar]
- 83.Pavlov PF, Hansson Petersen C, Glaser E, Ankarcrona M. Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med 2009;13(10):4137–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochimica et biophysica acta 2010;1802(1):122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, Eckert A. Neurotoxic mechanisms caused by the Alzheimer’s disease-linked Swedish amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. The Journal of biological chemistry 2003;278(30):28294–28302. [DOI] [PubMed] [Google Scholar]
- 86.Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, Siddiqui A, Tamura Y, Sesaki H, Wengenack TM, Dzeja PP, Poduslo JF. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PloS one 2012;7(2):e32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu LL, Shen Y, Wang X, Wei LF, Wang P, Yang H, Wang CF, Xie ZH, Bi JZ. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. Neuroreport 2017;28(4):222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013;9(2):106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mahley RW, Rall SC, Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 2000;1:507–537. [DOI] [PubMed] [Google Scholar]
- 90.Marcus C, Mena E, Subramaniam RM. Brain PET in the diagnosis of Alzheimer’s disease. Clin Nucl Med 2014;39(10):e413–422; quiz e423–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. The Journal of biological chemistry 2011;286(7):5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform- specific effects on neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience 1999;19(12):4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener 2009;4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Miller SW Jr, Davis RE, Parker WD Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Experimental neurology 2000;162(1):37–50. [DOI] [PubMed] [Google Scholar]
- 95.Silva DF, Selfridge JE, Lu J, E L, Cardoso SM, Swerdlow RH. Mitochondrial abnormalities in Alzheimer’s disease: possible targets for therapeutic intervention. Adv Pharmacol 2012;64:83–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manczak M, Kandimalla R, Fry D, Sesaki H, Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Human molecular genetics 2016;25(23):5148– 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS, Ko SH, Han HJ. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochimica et biophysica acta 2016;1863(11):2820–2834. [DOI] [PubMed] [Google Scholar]
- 98.Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu N, Li T, Su B, Chen ZY. Blockage of GSK3beta- mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiology of aging 2015;36(1):211–227. [DOI] [PubMed] [Google Scholar]
- 99.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009;324(5923):102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-beta with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. Journal of Alzheimer’s disease : JAD 2013;36(2):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Human molecular genetics 2012;21(11):2538–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez MJ, Ponce DP, Osorio-Fuentealba C, Behrens MI, Quintanilla RA. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front Neurosci 2017;11:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, Baik SH, Choi Y, Choi BY, Park JS, Bahn G, Shin JH, Jo DS, Lee JY, Jang CG, Arumugam TV, Kim J, Han JW, Koh JY, Cho DH, Jo DG. Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. The Journal of neuroscience : the official journal of the Society for Neuroscience 2017;37(20):5099–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 2015;6:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mackenzie IR, Rademakers R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 2008;21(6):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015;12(2):352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 2010;30(2):170–181. [DOI] [PubMed] [Google Scholar]
- 108.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. European journal of neurology 2008;15(8):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 2005;5(2):77–87. [DOI] [PubMed] [Google Scholar]
- 110.Chung MJ, Suh YL. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol 2002;26(1):3–7. [DOI] [PubMed] [Google Scholar]
- 111.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362(6415):59–62. [DOI] [PubMed] [Google Scholar]
- 112.Reddi AR, Culotta VC. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013;152(1–2):224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu W, Yamashita T, Tian F, Morimoto N, Ikeda Y, Deguchi K, Abe K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr Neurovasc Res 2013;10(3):222–230. [DOI] [PubMed] [Google Scholar]
- 114.Luo G, Yi J, Ma C, Xiao Y, Yi F, Yu T, Zhou J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PloS one 2013;8(12):e82112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. Journal of neurochemistry 2018. [DOI] [PMC free article] [PubMed]
- 116.Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. The ALS disease- associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Human molecular genetics 2013;22(23):4706–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Altanbyek V, Cha SJ, Kang GU, Im DS, Lee S, Kim HJ, Kim K. Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochemical and biophysical research communications 2016;481(3–4):259–264. [DOI] [PubMed] [Google Scholar]
- 118.Deng J, Yang M, Chen Y, Chen X, Liu J, Sun S, Cheng H, Li Y, Bigio EH, Mesulam M, Xu Q, Du S, Fushimi K, Zhu L, Wu JY. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet 2015;11(9):e1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stoica R, Paillusson S, Gomez-Suaga P, Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay- Barrena G, De Vos KJ, Shaw CE, Hanger DP, Noble W, Miller CC. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. Embo Rep 2016;17(9):1326–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nature reviews Neuroscience 2012;13(2):77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2007;66(1):10–16. [DOI] [PubMed] [Google Scholar]
- 122.De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CCJ, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Human molecular genetics 2007;16(22):2720– 2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Human molecular genetics 2014;23(6):1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol 2013;48(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ. Amyotrophic lateral sclerosis- associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Human molecular genetics 2017;26(23):4668–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Vos KJ, Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiology of disease 2017;105:283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. The Journal of cell biology 2014;204(7):1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tafuri F, Ronchi D, Magri F, Comi GP, Corti S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Frontiers in cellular neuroscience 2015;9:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, da Rocha EL, Sheng M, Choi H, Lerou PH, Li H, Wang X. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 2016;22(8):869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiology of disease 2013;51:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brenner D, Muller K, Wieland T, Weydt P, Bohm S, Lule D, Hubers A, Neuwirth C, Weber M, Borck G, Wahlqvist M, Danzer KM, Volk AE, Meitinger T, Strom TM, Otto M, Kassubek J, Ludolph AC, Andersen PM, Weishaupt JH. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016;139(Pt 5):e28. [DOI] [PubMed] [Google Scholar]
- 132.Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, Jones AR, Keagle P, Shatunov A, Sproviero W, Smith BN, van Es MA, Topp SD, Kenna A, Miller JW, Fallini C, Tiloca C, McLaughlin RL, Vance C, Troakes C, Colombrita C, Mora G, Calvo A, Verde F, Al-Sarraj S, King A, Calini D, de Belleroche J, Baas F, van der Kooi AJ, de Visser M, Ten Asbroek AL, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Munoz-Blanco JL, Strom TM, Meitinger T, Morrison KE, Consortium S, Lauria G, Williams KL, Leigh PN, Nicholson GA, Blair IP, Leblond CS, Dion PA, Rouleau GA, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Boylan KB, Van Blitterswijk M, Rademakers R, Esteban- Perez J, Garcia-Redondo A, Van Damme P, Robberecht W, Chio A, Gellera C, Drepper C,Sendtner M, Ratti A, Glass JD, Mora JS, Basak NA, Hardiman O, Ludolph AC, Andersen PM, Weishaupt JH, Brown RH Jr, Al-Chalabi A, Silani V, Shaw CE, van den Berg LH, Veldink JH, Landers JE. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 2016;48(9):1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stoss H, Beinder E, Abou Jamra R, Ekici AB, Schroder-Kress N, Aigner T, Kirchner T, Reis A, Brandstatter JH, Rauch A. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet 2011;88(1):106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell cycle 2009;8(2):257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Straub IR, Janer A, Weraarpachai W, Zinman L, Robertson J, Rogaeva E, Shoubridge EA. Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Human molecular genetics 2018;27(1):178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen S, He J, Tang L, Zhang N, Fan D. CHCHD10 mutations in patients with amyotrophic lateral sclerosis in Mainland China. Neurobiology of aging 2017;54:214 e217–214 e210. [DOI] [PubMed] [Google Scholar]
- 137.Zhou Q, Chen Y, Wei Q, Cao B, Wu Y, Zhao B, Ou R, Yang J, Chen X, Hadano S, Shang HF. Mutation Screening of the CHCHD10 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Mol Neurobiol 2017;54(5):3189–3194. [DOI] [PubMed] [Google Scholar]
- 138.Chaussenot A, Le Ber I, Ait-El-Mkadem S, Camuzat A, de Septenville A, Bannwarth S, Genin EC, Serre V, Auge G, French research network on FTD, Ftd ALS, Brice A, Pouget J, Paquis- Flucklinger V. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiology of aging 2014;35(12):2884 e2881–2884 e2884. [DOI] [PubMed] [Google Scholar]
- 139.White MA, Sreedharan J. Amyotrophic lateral sclerosis: recent genetic highlights. Curr Opin Neurol 2016;29(5):557–564. [DOI] [PubMed] [Google Scholar]
- 140.Genin EC, Plutino M, Bannwarth S, Villa E, Cisneros-Barroso E, Roy M, Ortega-Vila B, Fragaki K, Lespinasse F, Pinero-Martos E, Auge G, Moore D, Burte F, Lacas-Gervais S, Kageyama Y, Itoh K, Yu-Wai-Man P, Sesaki H, Ricci JE, Vives-Bauza C, Paquis-Flucklinger V. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO molecular medicine 2016;8(1):58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Woo JA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P, Maslar D, Bukhari A, Zhao X, Deonarine A, Westerheide SD, Kang DE. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nature communications 2017;8:15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Meng H, Yamashita C, Shiba-Fukushima K, Inoshita T, Funayama M, Sato S, Hatta T, Natsume T, Umitsu M, Takagi J, Imai Y, Hattori N. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nature communications 2017;8:15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu L, Gomez-Duran A, Saretzki G, Jin S, Tilgner K, Melguizo-Sanchis D, Anyfantis G, Al- Aama J, Vallier L, Chinnery P, Lako M, Armstrong L. The mitochondrial protein CHCHD2 primes the differentiation potential of human induced pluripotent stem cells to neuroectodermal lineages. The Journal of cell biology 2016;215(2):187–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 2017;10(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proceedings of the National Academy of Sciences of the United States of America 2016;113(15):4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol 2012;294:223–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wong YC, Holzbaur EL. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015;11(2):422–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America 2014;111(42):E4439– 4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Paulson HL, Albin RL. Huntington’s Disease: Clinical Features and Routes to Therapy. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington’s Disease: Applications to Drug Discovery Boca Raton (FL); 2011. [PubMed] [Google Scholar]
- 150.La Spada AR, Weydt P, Pineda VV. Huntington’s Disease Pathogenesis: Mechanisms and Pathways. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington’s Disease: Applications to Drug Discovery Boca Raton (FL); 2011. [Google Scholar]
- 151.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E. Mutant huntingtin binds the mitochondrial fission GTPase dynamin- related protein-1 and increases its enzymatic activity. Nat Med 2011;17(3):377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, Beal MF, Ferrante RJ. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Human molecular genetics 2010;19(20):3919–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guedes-Dias P, de Proenca J, Soares TR, Leitao-Rocha A, Pinho BR, Duchen MR, Oliveira JM. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochimica et biophysica acta 2015;1852(11):2484– 2493. [DOI] [PubMed] [Google Scholar]
- 154.Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, La Spada AR, Lipton SA. S- nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal 2013;19(11):1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today 2014;19(7):951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gonzalez-Polo RA, Bravo-San Pedro JM, Gomez-Sanchez R, Pizarro-Estrella E, Niso- Santano M, Fuentes JM. Autophagy, mitochondria and 3-nitropropionic acid joined in the same model. British journal of pharmacology 2013;168(1):60–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS- dependent pathway. Cell death and differentiation 2009;16(6):899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bahmad H, Hadadeh O, Chamaa F, Cheaito K, Darwish B, Makkawi AK, Abou-Kheir W. Modeling Human Neurological and Neurodegenerative Diseases: From Induced Pluripotent Stem Cells to Neuronal Differentiation and Its Applications in Neurotrauma. Front Mol Neurosci 2017;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu L, Huang JS, Han C, Zhang GX, Xu XY, Shen Y, Li J, Jiang HY, Lin ZC, Xiong N, Wang T. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol Neurobiol 2016;53(10):6698–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proceedings of the National Academy of Sciences of the United States of America 2007;104(24):10045–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Trushina E, Dyer RB, Badger JD, 2nd, Ure D Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Molecular and cellular biology 2004;24(18):8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Alexander GE. Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin Neurosci 2004;6(3):259–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2012;2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Whitworth AJ, Pallanck LJ. Genetic models of Parkinson’s disease: mechanisms and therapies. SEB Exp Biol Ser 2008;60:93–113. [PubMed] [Google Scholar]
- 165.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39(6):889–909. [DOI] [PubMed] [Google Scholar]
- 166.Hisahara S, Shimohama S. Toxin-induced and genetic animal models of Parkinson’s disease. Parkinson’s disease 2010;2011:951709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005;2(3):484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Keeney PM, Xie J, Capaldi RA, Bennett JP Jr, Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006;26(19):5256–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bhattacharjee N, Borah A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson’s disease. Neurochem Int 2016;101:48–55. [DOI] [PubMed] [Google Scholar]
- 170.Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. Journal of neurochemistry 2016;139 Suppl 1:216–231. [DOI] [PubMed] [Google Scholar]
- 171.Hu Q, Wang G. Mitochondrial dysfunction in Parkinson’s disease. Translational neurodegeneration 2016;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015;85(2):257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha- synuclein protein in alpha-synuclein overexpressing cells. Neuroscience letters 2008;439(2):125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Protter D, Lang C, Cooper AA. alphaSynuclein and Mitochondrial Dysfunction: A Pathogenic Partnership in Parkinson’s Disease? Parkinson’s disease 2012;2012:829207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Human molecular genetics 2012;21(9):1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yue M, Hinkle KM, Davies P, Trushina E, Fiesel FC, Christenson TA, Schroeder AS, Zhang L, Bowles E, Behrouz B, Lincoln SJ, Beevers JE, Milnerwood AJ, Kurti A, McLean PJ, Fryer JD, Springer W, Dickson DW, Farrer MJ, Melrose HL. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiology of disease 2015;78:172–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang Z, Liu L, Jiang X, Zhai S, Xing D. The Essential Role of Drp1 and Its Regulation by S- Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease. Antioxid Redox Signal 2016;25(11):609–622. [DOI] [PubMed] [Google Scholar]
- 178.Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Human molecular genetics 2013;22(22):4545–4561. [DOI] [PubMed] [Google Scholar]
- 179.Belanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci 2009;11(3):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pekny M, Pekna M. Reactive gliosis in the pathogenesis of CNS diseases. Biochimica et biophysica acta 2016;1862(3):483–491. [DOI] [PubMed] [Google Scholar]
- 181.Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017;46(6):957–967. [DOI] [PubMed] [Google Scholar]
- 182.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain research 2003;971(2):197–209. [DOI] [PubMed] [Google Scholar]
- 184.Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010;58(7):831–838. [DOI] [PubMed] [Google Scholar]
- 185.Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB, Function MRCC, Ageing Neuropathology Study G. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiology of aging 2010;31(4):578– 590. [DOI] [PubMed] [Google Scholar]
- 186.Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M. Multiple cellular and molecular mechanisms are involved in human Abeta clearance by transplanted adult astrocytes. Glia 2011;59(11):1643–1657. [DOI] [PubMed] [Google Scholar]
- 187.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 2003;9(4):453–457. [DOI] [PubMed] [Google Scholar]
- 188.Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, Serrano G, Beach TG, Craig DW, Valla J, Reiman EM, Liang WS. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiology of aging 2015;36(2):583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Birch AM. The contribution of astrocytes to Alzheimer’s disease. Biochem Soc Trans 2014;42(5):1316–1320. [DOI] [PubMed] [Google Scholar]
- 190.Avila-Munoz E, Arias C. When astrocytes become harmful: functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing research reviews 2014;18:29– 40. [DOI] [PubMed] [Google Scholar]
- 191.Phatnani H, Maniatis T. Astrocytes in neurodegenerative disease. Cold Spring Harbor perspectives in biology 2015;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jana A, Pahan K. Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010;30(38):12676– 12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009;323(5918):1211–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ. Amyloid- beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010;30(9):3326–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 2010;58(3):300–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. The Journal of cell biology 2005;171(6):1001– 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hong Y, Zhao T, Li XJ, Li S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. The Journal of neuroscience : the official journal of the Society for Neuroscience 2016;36(34):8790–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proceedings of the National Academy of Sciences of the United States of America 2009;106(52):22480– 22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chou SY, Weng JY, Lai HL, Liao F, Sun SH, Tu PH, Dickson DW, Chern Y. Expanded- polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience 2008;28(13):3277–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hsiao HY, Chen YC, Huang CH, Chen CC, Hsu YH, Chen HM, Chiu FL, Kuo HC, Chang C, Chern X. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol 2015;78(2):178–192. [DOI] [PubMed] [Google Scholar]
- 201.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, , Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016;89(1):37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Booth HDE, Hirst WD, Wade-Martins R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci 2017;40(6):358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mullett SJ, Hinkle DA. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiology of disease 2009;33(1):28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lev N, Barhum Y, Ben-Zur T, Melamed E, Steiner I, Offen D. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J Mol Neurosci 2013;50(3):542–550. [DOI] [PubMed] [Google Scholar]
- 205.Bruck D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiology of disease 2016;85:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson’s disease- related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 2010;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52(1):39–59. [DOI] [PubMed] [Google Scholar]
- 208.Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proceedings of the National Academy of Sciences of the United States of America 2008;105(21):7594–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tripathi P, Rodriguez-Muela N, Klim JR, de Boer AS, Agrawal S, Sandoe J, Lopes CS, Ogliari KS, Williams LA, Shear M, Rubin LL, Eggan K, Zhou Q. Reactive Astrocytes Promote ALS- like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-beta1. Stem Cell Reports 2017;9(2):667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Phatnani HP, Guarnieri P, Friedman BA, Carrasco MA, Muratet M, O’Keeffe S, Nwakeze C, Pauli-Behn F, Newberry KM, Meadows SK, Tapia JC, Myers RM, Maniatis T. Intricate interplay between astrocytes and motor neurons in ALS. Proceedings of the National Academy of Sciences of the United States of America 2013;110(8):E756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thangavelu SR, Tripathi PP, Arya U, Mishra HK, Subramaniam JR. ALS associated mutant SOD1 impairs the motor neurons and astrocytes and wild type astrocyte secreted-factors reverse the impaired motor neurons. Ann Neurosci 2011;18(2):48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nature neuroscience 2008;11(3):251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Human molecular genetics 2011;20(2):286– 293. [DOI] [PubMed] [Google Scholar]
- 214.Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nature neuroscience 2008;11(11):1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Microglia Cunningham C. and neurodegeneration: the role of systemic inflammation. Glia 2013;61(1):71–90. [DOI] [PubMed] [Google Scholar]
- 216.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014;10(4):217–224. [DOI] [PubMed] [Google Scholar]
- 217.Kofler J, Wiley CA. Microglia: key innate immune cells of the brain. Toxicol Pathol 2011;39(1):103–114. [DOI] [PubMed] [Google Scholar]
- 218.Condello C, Yuan P, Grutzendler J. Microglia-Mediated Neuroprotection, TREM2, and Alzheimer’s Disease: Evidence From Optical Imaging. Biol Psychiatry 2018;83(4):377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, Kennedy M, Kozauer N, Margolin RA, Molinuevo JL, Mueller R, Ransohoff RM, Wilcock DM, Bain L, Bales K. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimers Dement (N Y) 2016;2(2):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017;170(4):649–663 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015;160(6):1061– 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. The Journal of cell biology 2018;217(2):459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 2001;60(2):161–172. [DOI] [PubMed] [Google Scholar]
- 224.Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007;130(Pt 7):1759–1766. [DOI] [PubMed] [Google Scholar]
- 225.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. Journal of neuroinflammation 2014;11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hsiao HY, Chiu FL, Chen CM, Wu YR, Chen HM, Chen YC, Kuo HC, Chern Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Human molecular genetics 2014;23(16):4328–4344. [DOI] [PubMed] [Google Scholar]
- 227.Valekova I, Jarkovska K, Kotrcova E, Bucci J, Ellederova Z, Juhas S, Motlik J, Gadher SJ, Kovarova H. Revelation of the IFNalpha, IL-10, IL-8 and IL-1beta as promising biomarkers reflecting immuno-pathological mechanisms in porcine Huntington’s disease model. J Neuroimmunol 2016;293:71–81. [DOI] [PubMed] [Google Scholar]
- 228.Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 2008;205(8):1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Andre R, Carty L, Tabrizi SJ. Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr Opin Pharmacol 2016;26:33–38. [DOI] [PubMed] [Google Scholar]
- 230.Weiss A, Trager U, Wild EJ, Grueninger S, Farmer R, Landles C, Scahill RI, Lahiri N, Haider S, Macdonald D, Frost C, Bates GP, Bilbe G, Kuhn R, Andre R, Tabrizi SJ. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. The Journal of clinical investigation 2012;122(10):3731–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, Moller T, Stella N, Akassoglou K, Tabrizi SJ, Muchowski PJ. Mutant huntingtin impairs immune cell migration in Huntington disease. The Journal of clinical investigation 2012;122(12):4737–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kraft AD, Kaltenbach LS, Lo DC, Harry GJ. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiology of aging 2012;33(3):621 e617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, Burm HB, Toner M, Osipovitch M, Jim Xu Q, Ding F, Wang F, Kang N, Kang J, Curtin PC, Brunner D, Windrem MS, Munoz-Sanjuan I, Nedergaard M, Goldman SA. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nature communications 2016;7:11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Le W, Wu J, Tang Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front Mol Neurosci 2016;9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Subramaniam SR, Federoff HJ. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front Aging Neurosci 2017;9:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 2010;42(9):781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Qian L, Flood PM, Hong JS. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. Journal of neural transmission 2010;117(8):971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. Journal of neuroinflammation 2005;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PloS one 2010;5(1):e8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Inoue K Microglial activation by purines and pyrimidines. Glia 2002;40(2):156–163. [DOI] [PubMed] [Google Scholar]
- 241.Sperlagh B, Illes P. Purinergic modulation of microglial cell activation. Purinergic Signal 2007;3(1–2):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Barcia C, Sanchez Bahillo A, Fernandez-Villalba E, Bautista V, Poza YPM, Fernandez- Barreiro A, Hirsch EC, Herrero MT. Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia 2004;46(4):402– 409. [DOI] [PubMed] [Google Scholar]
- 243.Kanaan NM, Kordower JH, Collier TJ. Age and region-specific responses of microglia, but not astrocytes, suggest a role in selective vulnerability of dopamine neurons after 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure in monkeys. Glia 2008;56(11):1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol 2003;54(5):599–604. [DOI] [PubMed] [Google Scholar]
- 245.Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996;5(2):137–143. [DOI] [PubMed] [Google Scholar]
- 246.Huang D, Xu J, Wang J, Tong J, Bai X, Li H, Wang Z, Huang Y, Wu Y, Yu M, Huang F. Dynamic Changes in the Nigrostriatal Pathway in the MPTP Mouse Model of Parkinson’s Disease. Parkinson’s disease 2017;2017:9349487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Yagi K, Suzuki K, Fujita S. The effects of MPTP on the activation of microglia/astrocytes and cytokine/chemokine levels in different mice strains. J Neuroimmunol 2008;204(1–2):43–51. [DOI] [PubMed] [Google Scholar]
- 248.Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Experimental neurology 2006;197(2):275–283. [DOI] [PubMed] [Google Scholar]
- 249.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 2007;13(10):1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of clinical investigation 2009;119(1):182– 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008;14(2):193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Rosdah AA, J KH, Delbridge LM, Dusting GJ, Lim SY. Mitochondrial fission - a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect 2016;4(3):e00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cui M, Ding H, Chen F, Zhao Y, Yang Q, Dong Q. Mdivi-1 Protects Against Ischemic Brain Injury via Elevating Extracellular Adenosine in a cAMP/CREB-CD39-Dependent Manner. Mol Neurobiol 2016;53(1):240–253. [DOI] [PubMed] [Google Scholar]
- 254.Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 2017;40(6):583–594 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochimica et biophysica acta 2014;1842(2):220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, Torres S, Fujioka H, Xu Y, Perry G, Zhu X. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Human molecular genetics 2017;26(21):4118–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Reddy PH, Manczak M, Yin X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid- beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD 2017;58(1):147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. The Journal of biological chemistry 2010;285(15):11740–11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Twaroski DM, Yan Y, Zaja I, Clark E, Bosnjak ZJ, Bai X. Altered Mitochondrial Dynamics Contributes to Propofol-induced Cell Death in Human Stem Cell-derived Neurons. Anesthesiology 2015;123(5):1067–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bido S, Soria FN, Fan RZ, Bezard E, Tieu K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson’s disease. Scientific reports 2017;7(1):7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, Zhuang X, Bowers WJ, Tieu K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nature communications 2014;5:5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Saez-Atienzar S, Bonet-Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J, Galindo MF. The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell death & disease 2014;5:e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Wang H, Yi J, Li X, Xiao Y, Dhakal K, Zhou J. ALS-associated mutation SOD1(G93A) leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018;106:126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Akita M, Suzuki-Karasaki M, Fujiwara K, Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y, Suzuki-Karasaki Y. Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced apoptosis. International journal of oncology 2014;45(5):1901–1912. [DOI] [PubMed] [Google Scholar]
- 265.Wang J, Hansen K, Edwards R, Van Houten B, Qian W. Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochemical and biophysical research communications 2015;456(1):7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox biology 2013;1(1):542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kim B, Kim J-S, Yoon Y, Santiago MC, Brown MD, Park J-Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. American Journal of Physiology- Regulatory, Integrative and Comparative Physiology 2013;305(8):R927–R938. [DOI] [PubMed] [Google Scholar]
- 268.Ji-Yeong Y, Sung-Hun M, Hyo-Jin P, Jin-Woo K, Yong-Hee L, Soo-Yong P, Pil-Soo JEONG HP, Dong-Seok L, Sun-Uk K, Kyu-Tae CHANG D- BK. Mdivi-1, mitochondrial fission inhibitor, impairs developmental competence and mitochondrial function of embryos and cells in pigs. The Journal of reproduction and development 2015;61(2):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kornfeld OS, Hwang S, Disatnik M-H, Chen C-H, Qvit N, Mochly-Rosen D. Mitochondrial Reactive Oxygen Species at the Heart of the Matter: New Therapeutic Approaches for Cardiovascular Diseases. Circulation Research 2015;116(11):1783–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Qi X, Qvit N, Su Y-C, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. Journal of cell science 2013;126:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Guo X, Disatnik M-h, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington disease – associated neurodegeneration 2013;123. [DOI] [PMC free article] [PubMed] [Google Scholar]