

Abstract
Amyloid fibrils generally display chirality, a feature which has rarely been exploited in the development of therapeutics against amyloid diseases. Here we report, for the first time, the use of mesoscopic chiral silica nanoribbons against the in vivo amyloidogenesis of human islet amyloid polypeptide (IAPP), the peptide whose aggregation is implicated in type 2 diabetes. Our thioflavin T assay and transmission electron microscopy showed accelerated IAPP fibrillization through elimination of the nucleation phase and shortening of the elongation phase by the nanostructures. Coarse-grained simulations offered complementary molecular insights into the acceleration of amyloid aggregation through their nonspecific binding and directional seeding with the nanostructures. This accelerated IAPP fibrillization translated to reduced toxicity, especially for the right-handed silica nanoribbons, as revealed by cell viability, helium ion microscopy, as well as zebrafish embryo survival, developmental and behavioral assays. This study has implicated the potential of employing chiral nanotechnologies against the mesoscopic enantioselectivity of amyloid proteins and their associated diseases.
Keywords: human IAPP, silica nanoribbons, chirality, toxicity, amyloidogenesis
Graphical Abstract
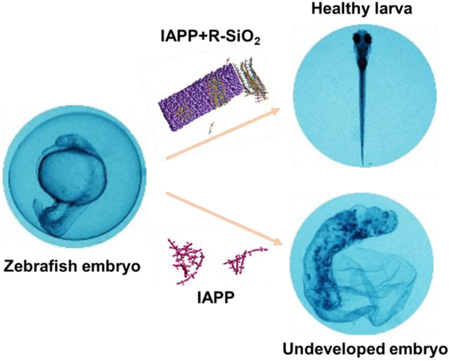
Silica nanoribbons rescued the toxicity of oppositely-handed amyloid protein IAPP in an embryonic zebrafish model, pointing to the potential of exploiting nanoscale chirality against amyloidogenesis.
INTRODUCTION
The aggregation of human islet amyloid polypeptide (IAPP) into insoluble amyloid fibrils and plaques is a hallmark of type 2 diabetes (T2D) [1, 2], a metabolic disease and a global epidemic impacting an estimated population of 360 million. IAPP is a 37-residue amphiphilic peptide secreted by pancreatic beta cell islets, and is stabilized intracellularly by the presence of insulin, low pH, physiological metal ions (such as zinc), as well as zinc-coordinated complexation of C-peptide and IAPP at a specific stoichiometric ratio [3–7]. A major strategy against IAPP amyloidogenesis has involved the use of small molecules (e.g. polyphenols such as curcumin, resveratrol and epigallocatechin gallate), chaperone proteins, or engineered nanostructures of dendritic polymers, graphene oxide nanosheets and gold nanoparticles, exploiting the capacities of these ‘ligands’ in mediating hydrogen-bonding, hydrophobic interaction or π-stacking with the amyloid protein [8]. While these approaches are designed to simultaneously inhibit protein aggregation and toxicity, and almost exclusively in vitro, we have recently shown that star polymer poly (2-hydroxyethyl acrylate) (PHEA) could accelerate the nucleation and elongation phases of IAPP fibrillization, while eliminating the production of toxic IAPP oligomers in vitro and ex vivo [9]. In addition, we have demonstrated that the amyloid fragments of beta-lactoglobulin coated on multi-walled carbon nanotubes, sequestered toxic IAPP in vivo in an embryonic zebrafish model [10]. However, strategies aiming at in vivo mitigation of amyloidogenesis remain extremely limited.
Chiral structures are prevalent in nature, ranging from phospholipids in cell membranes to D-sugars, L-amino acids and B-, A- and Z-form deoxyribonucleic acids. These chiral structures are building blocks of biological systems and play essential roles in cell recognition and uptake, metabolism, protein synthesis and genetic coding. Recently, chiral molecules such as the enantiomers of tartaric acid [11], N-isobutyryl cysteine (NIBC) [12], cysteine [13], lysine, phenylalanine and monometallic units have been used to functionalize mica, gold, carbon dots and graphene oxide nanosheets, and the resulting surfaces have elicited differential effects on the aggregation of insulin and amyloid-β (Aβ) as well as on cell response [14–16]. While much remains to be understood, salt-bridge interaction, for example, has been proposed as a major mechanism for serum albumin interacting with gold nanoparticles coated with L- and D-penicillamine [17]. The implications of this type of research range from catalysis, sensing and bioengineering to the inhibition of amyloid protein aggregation at pseudo-homochiral interfaces, such as ligand-modified surfaces and cell membranes. However, the use of chirality of either the inhibitors or the amyloid fibrils against amyloidogenesis, especially chirality on the nanoscale, remains a rarity.
In this study, we synthesized both left- and right-handed silica nanoribbons (L/R-SiO2), whose pitch sizes (~90 nm) were on the same order of magnitude as that of IAPP amyloid fibrils (20~50 nm) [8, 18]. It is known from the literature that IAPP fibrils, as well as fibrils of beta-amyloid (Aβ) and tau, generally adopt left-handedness, although right-handed IAPP fibrils have been observed occasionally [8, 18]. This biased amyloid chirality is understood as a result of the dominance of left-handed amino acid species, the building blocks of amyloid peptides and proteins. On the other hand, silica nanostructures, including silica nanoribbons, have been widely applied in electronics, drug delivery, sensing and catalysis [19, 20]. The combination of inorganic nano-objects and chirality, either by grafting chiral molecules to silica nanoparticles or forming silica nanostructures of chiral shapes, along with the well-known surface chemistry of silica and their easy functionalization by other molecules or nanoparticles, enables chiral recognition or chiral separation [21, 22], meme of nano-periodic patterns in biology [23], chiroptical nanomaterials [24, 25], nanosensors [26], or chiral catalysis [27].
Here the mesoscopic chirality of silica nanoribbons is utilized for the first time to inhibit IAPP toxicity. The effects of the nanoribbons on IAPP aggregation were first evaluated using a thioflavin T (ThT) kinetic assay and transmission electron microscopy (TEM). The efficacies of the L/R-SiO2 on IAPP toxicity inhibition were examined with βTC-6 pancreatic beta cells and a high-throughput in vivo embryonic zebrafish model. The R-SiO2 were more potent than the L-SiO2 in inhibiting IAPP aggregation and toxicity, due to fibrillization along the perpendicular direction and hence a higher density of seeding IAPP on the silica nanostructures, as corroborated by TEM and coarse-grained computer simulations. This study points to the potential of exploiting the mesoscopic enantioselectivity of amyloid proteins for the prevention and treatment of a range of human amyloid diseases.
MATERIALS AND METHODS
Chiral silica nanoribbons were fabricated as previously described [28, 29]. Briefly, the self-assembly of dicationic surfactant, 1,2-ethane-bis(dimethyldecylammonium) with chiral counterions, L or D tartrate forming nanometric helices (right-handed or left-handed respectively) were used as templates to form silica nanoribbons through sol-gel polycondensation with controlled dimensions and handedness. The silica nanoribbons were cut and individualized by sonication (Vibra-cell 75186, Sonics & Materials) to be colloidal suspensions [30]. Lyophilized white powder of human islet amyloid polypeptide (IAPP; KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY; 2–7 disulfide bridge, 3,904.5 Da, purity >95%; AnaSpec; HPLC and mass spectroscopy characterizations of IAPP refer to Figs. S1&S2, Supplementary Information) was freshly dissolved in Milli-Q water at room temperature. ThT dye (319 Da) was acquired from Sigma-Aldrich. Propidium iodide (PI, excitation/emission: 535 nm/617 nm) was purchased from ThermoFisher. Poly L-lysin (0.01%) was acquired from Sigma-Aldrich and applied for cell staining.
Zeta potential measurement
The zeta potentials of the L/R-SiO2 in Milli-Q water were determined using a Zetasizer Nano-ZS (Malvern Instruments, UK). The measurement was conducted using disposable folder capillary cells and analyzed by Zetasizer Software 7.02.
Scanning electron microscopy (SEM)
For morphology imaging, 5 μL of L/R-SiO2 (0.2 mg/mL) was pipetted and air dried on a carbon tape and the nanostructure surfaces were coated with a thin layer of sputtered gold (Bal-Tec SCD 005 Sputter) and visualized by a scanning electron microscope (FEI, Nova NanoSEM 450).
Thioflavin T (ThT) kinetic assay
A kinetic assay was performed with 50 μM ThT dye and 50 μM IAPP in the presence and absence of the L/R-SiO2 (0.2 mg/mL, for 1:1 IAPP/nanostructure mass ratio) in a 96-well plate (Costar black/clear bottom). Changes in ThT fluorescence, indicating the β-sheet content in the IAPP sample, were recorded at room temperature from the plate bottom side every 10 min over 14 h to reach the full saturation phase, using a PerkinElmer EnSight HH33400 plate reader (excitation/emission: 440 nm/485 nm). The data was stored by software Kaleido 1.2. The assay was performed in triplicate and average spectra of the measurements were presented.
Transmission electron microscopy (TEM)
For this measurement, 5 μL of the L/R-SiO2 (0.2 mg/mL) was allowed to interact with IAPP monomers (5 μL of 50 μM; freshly dissolved in water, termed as ‘0 h IAPP’ from here onwards), oligomers/protofibrils (1 h into fibrillization, termed as ‘1 h IAPP’), and fibrils (48 h into fibrillization, termed as ‘48 h IAPP’) for 24 h of incubation. The samples were then pipetted onto 15 s glow-discharged 400 mesh copper grids (Formvar film, ProSciTech) for 60 s of adsorption. Excess samples were drawn off by filter paper and the grids were washed using 10 μL of Milli-Q water, with excess drawn off. The grids were then negatively stained with 5 μL of 1% uranyl acetate (UA) for 30 s with excess stain drawn off and air-dried. Samples were characterized on a Tecnai G2 F20 transmission electron microscope (FEI, Eindhoven, The Netherlands) operated at a voltage of 200 kV. Images were recorded using an UltraScan 1000 P 2k CCD camera (Gatan, California, USA) and Gatan Digital Micrograph 3.9.5 software.
Statistical analysis of amyloid fibrils
To investigate the effects of the L/R-SiO2 on the morphology of IAPP fibrils, TEM images were analyzed with open source software FiberApp [31]. The values of IAPP fibril length and thickness were estimated for 30 samples per condition.
Coarse-grained simulations
A coarse-grained amyloid peptide model was developed in discrete molecular dynamics (DMD) simulations to study the formation of amyloid fibrils. DMD is a particular type of molecular dynamics algorithm, where the inter-atom interaction potentials are modeled by step functions [32]. Briefly, in DMD simulations any atom moves with a constant velocity until an interatomic interaction potential step is encountered, i.e., a collision event. New velocities of two colliding atoms are determined by conservation laws of energy, linear and angular momenta. DMD simulations have been widely used to study protein amyloid aggregation [33–37]. Here, each coarse-grained peptide was represented by 11 beads (Fig. S3A). Two donors (atoms N1 & N5) and two acceptors (atoms O3 & O7) were able to form inter-chain peptide hydrogen bonds. The angular and distance-dependent hydrogen bond formed between C-N and C-O, where C denoted the carbon atom (atoms C2 or C6) covalently linked to either donor or acceptor with a bond length of 1.5 Å, was modeled by a reaction-like algorithm [38]. A hydrogen bond with the lowest potential energy had a linear alignment between C-N and C-O and a donor-acceptor distance of ~ 2 Å. Two hydrophobic beads – atoms C4 and C8 attached to C2 and C6 with a bond length of 3 Å, respectively – were introduced to model side-chain interactions between different peptides. Each peptide also included three hydrophilic beads, atoms O8, O9 and O9, which were collinear with C2 and C6 with an average separation of 5 Å forming the coarse-grained peptide “backbone”. Both N1-C2-O3 and N5-C6-O7 in a peptide were collinear and perpendicular to the backbone. C2-C4 (or C6-C8) was perpendicular to both the backbone and N1-C2-O3 (or N5-C6-O7). The conformation of a peptide was, thus, determined by the dihedral angle between C4-C2-C6-C8, modeled by a multiple-well step function [39]. Motivated by a previous coarse-gained peptide model [40–42], we allowed the peptide to adopt two conformations: amyloid-competent (β) and amyloid-protected (π) states (Fig. S3B-D). In the β-state, the dihedral angle with minimum free energy was ~15o, and thus, both donors and acceptors (N1-C2-O3 and N5-C6-O7) in the peptide were approximately parallel to each other, compatible with a long fibrillar state. On the other hand, the π-state had the dihedral angle ~ 90o and was incompatible with the linear fibril. In this study, the π-state of an isolated peptide was more favorable than the β-state with a lower free energy of ~ 1.0 KBT, where KB denotes the Boltzmann constant and T corresponds to the simulation temperature. We adopted an HP-like interaction potential model [43] for non-bonded interactions – an attractive potential, ~2 KBT, was assigned among hydrophobic C4 and C6 atoms, and a hard-core only interaction potential was assigned among hydrophilic atoms (O9, O10, O11) and between hydrophobic and hydrophilic atoms. The net energy gain for a hydrogen bond was ~ 5.5 KBT.
To model the chiral silica, a ~2.1×4.3×13 nm3 crystal structure was first built along the x, y & z dimensions using a unit cell structure of SiO2 from the Materials Studio Software. We then followed the central axis in the x-y dimensions along the z-axis and rotated all atoms’ coordinates with angles proportional to their z-values, ~ +/− 2πz/L, for the L/R-SiO2, respectively (Fig. S3E,F). Here, L is the pitch length of ~ 90 nm. In the simulations, the chiral silica structures were kept static while the coarse-grained peptides were allowed to freely move. Since silica was polar and the oxide on the surface was able to form hydrogen bonds with the peptides, this effect was modeled by assigning a weak attraction between all heavy atoms in silica (Si and O atoms) and the polar beads in the coarse-grained peptides (excluding hydrophobic C4&C8), ~0.075 KBT.
We studied three sets of molecular systems, including the control of peptide self-assembly, and in the presence of either the L-SiO2 or R-SiO2. 100 peptides w/o silica nanoribbons were randomly positioned in a 20×20×20 nm3 cubic box with inter-molecular distance being kept at least 1 nm. The peptide concentration was ~20.8 mM in all cases, with the excluded volume of nanoparticles negligible. A periodic boundary condition was used. Each DMD simulation of peptide aggregation lasted 3 μs. To avoid potential bias of the initial states, the starting conformation of each peptide was randomly assigned. During the early aggregation stage, the distribution of peptides in either the π- or β-state (~80% in π-state and ~20% in β-state) was consistent with the corresponding free energy difference of isolated peptides (Fig. S4); the initially non-aggregated peptides predominantly adopted the π-state, also consistent with the experimental starting condition. Indeed, the coarse-grained peptides spontaneously formed left-handed fibrils (Fig. S5).
For the analysis of the coarse-grained aggregation simulations, the number of hydrogen bonds, the number of peptides in β-sheet conformations, and the size of β-sheet aggregates were monitored. A peptide belonged to a β-sheet only if it was in the β-conformation and was stabilized by at least two inter-peptide hydrogen bonds. The aggregates of multiple β-sheets were defined by inter-peptide contacts with an inter-atomic distance cutoff of 0.75 nm.
Cell culture and viability assay
Pancreatic βTC-6 (ATCC) beta cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM) with 15% fetal bovine serum (FBS). For the viability assay, a 96 well plate (Costar black/clear bottom) was coated with 70 μL Poly-L-lysine (Sigma, 0.01%), incubated at 37 °C for 30 min and cells at a density of ∼50,000 cells per well in 200 μL DMEM with 15% FBS were added to the wells. Cells were incubated for 48 h at 37 °C and 5% CO2 to reach 70–80% confluency. The cell culture medium was then refreshed, and 1 μM PI dye in DMEM was added to the wells and incubated for 30 min. After optimization of concentrations, samples of 20 μM IAPP and 0.08 mg/mL L/R-SiO2 (at a 1:1 mass ratio) were added into the wells. All samples were examined in triplicate and measured by Operetta (PerkinElmer, 20× PlanApo microscope objective, numerical aperture NA=0.7) in a live cell chamber (37 ºC, 5% CO2) after 14 h of treatment. The percentage of dead cells (PI-positive) relative to total cell count was determined by a built-in bright-field mapping function of Harmony High-Content Imaging and Analysis software (PerkinElmer). The measurement was conducted at 5 reads per well and performed in triplicate. Untreated cells were recorded as controls.
Helium ion microscopy (HIM)
βTC-6 cells were incubated with 20 μM IAPP monomers, oligomers and amyloid fibrils in the presence and absence of the L/R-SiO2 for 30 min, and were then stabilized by 2.5% paraformaldehyde. The samples were incubated at 4 ºC overnight. In the next step, samples were gently centrifuged and paraformaldehyde/medium was replaced with gradient concentrations of ethanol in 5 steps: 20%, 40%, 60%, 80% and 95%, with 2 h incubation at each gradient. 30 µL of treated βTC-6 cells was air-dried on a carbon tape and the morphologies of treated cells and untreated cells, as controls, were visualized by HIM (Orion NanoFab, Zeiss, USA).
Zebrafish embryo toxicity assay
IAPP of 1 nL dissolved in Milli-Q water for 0 h, 1 h and 48 h as controls and mixed with the L/R-SiO2 (10 μM IAPP and 40 µg/mL L/R-SiO2 in final concentration) were microinjected to the yolk of zebrafish embryos at the age of 2 h post fertilization (hpf). Injection was conducted using a pneumatic picopump (PV830 Pneumatic Picopump, WPI) and capillary needles. After injection each embryo was placed in 96-well plate, one embryo per well containing 200 μL Holtfreter’s medium (H buffer) [44]. To investigate the toxic effect of IAPP fibrillization w/o L/R-SiO2, the development, hatching and abnormality of the embryos were recorded (Olympus SZ61) every 24 h up to 80 hpf. The experiments were performed in triplicate, for 12 samples of each treatment condition.
Fluorescence imaging of embryonic IAPP fibrillization
Similarly to the toxicity assay, the L/R-SiO2 were brought to interact with IAPP dissolved in 20 μM ThT solution for 10 min and were then microinjected to the yolk of zebrafish embryos. The intensity related to IAPP fibrillization in the green fluorescence channel, due to the binding of the ThT dye to β-sheet rich protofibrils and fibrils, was tracked in treated embryos up to 4 days post fertilization (dpf) (Olympus MVX10, OCULAR software version 2.0). The experiments were performed in triplicate for 12 samples of each treatment condition.
Behavioral experiment
Monomeric and fibrillar IAPP of 10 µM mixed for 10 min with 40 µg/mL of L/R-SiO2 (1 nL in total volume) were microinjected to the yolk of 2 hpf zebrafish embryos and behavioral parameters were recorded on the surviving larvae 4 days after treatment. Each larva was put in one well of a 96 well plate and their real-time behavior was recorded using an automated behavior analysis system, ZebraBox (Viewpoint, France). The initial head-to-tail positions were set as the reference angles of 0° for each larva. Linear swimming distance (cm) and number of rotations per hour (<180° clock-wise or anti-clockwise; rotations beyond one full circle or >180° were rare events and were excluded in data collection to avoid miscounting by the automated system) were measured and analyzed with software VideoTrack version 3.5.
RESULTS AND DISCUSSION
Characterization of chiral silica nanoribbons
The morphology and dimensions of the L/R-SiO2 are summarized in Fig. 1, which appeared highly comparable except for the handedness. Specifically, the average width and half pitch size were 19.5 ± 2 nm and 44.7 ± 3.9 nm for L-SiO2 and 19.4 ± 1.9 nm and 44.3 ± 3.5 nm for R-SiO2, respectively. The L/R-SiO2 were negatively charged at −23 ± 0.4 mV and −22.2 ± 0.5 mV, which enabled their electrostatic interactions with the cationic IAPP peptide [8].
Figure 1.

Characterizations of the silica nanoribbons show half pitch size, length and width of the (A) L-SiO2 and (B) R-SiO2. Images acquired with scanning electron microscopy.
IAPP fibrillization inhibition and remodeling by chiral silica nanoribbons
TEM imaging revealed associations of the L/R-SiO2 with IAPP monomers, oligomers and amyloid fibrils (Fig. 2). Comparison of IAPP in the presence of the L/R-SiO2 with control IAPP fibrils clearly indicates that the peptides were attracted to the nanoribbons, causing an increase in local IAPP concentration and hence a rapid transition from nucleation to elongation. This attraction markedly affected the formation of fibrils. In the case of the silica nanoribbons with preformed IAPP protofibrils/fibrils (48 h IAPP), the R-SiO2 showed more interaction with the helical IAPP fibrils than the L-SiO2 (Fig. 2H-I).
Figure 2.

Transmission electron microscopy images of (A, B) the L/R-SiO2 and (C) IAPP control. (D-G) Attractions between the peptide and the nanoribbons caused shortening and inhibition of the fibrils compared with the control. (H-I) The R-SiO2 was effective in remodeling the IAPP fibrils. All samples were incubated for 24 h. (J) ThT kinetic assay of IAPP fibrillization over 14 h show a shortened lag phase and inhibition of fibrillization in the presence of the L/R-SiO2. Arrows in panels D-F, H and I indicate discernible L/R-SiO2 in contact with IAPP protofibrils/fibrils. Scale bars: 100 nm. The experiments was carried out in triplicate and error bars show the standard deviations of the averaged data sets. IAPP concentration: 50 µM for the ThT assay and 20 µM for TEM, at room temperature and pH 7.
Statistics analysis of the TEM images using FiberApp [31] offered additional insights into IAPP aggregation and fibril remodeling by the nanostructures. Both the L-SiO2 and R-SiO2 were effective in preventing the formation of full IAPP fibrils of micrometers in length [18] and, instead, yielded IAPP protofibrils and short fibrils of 100–150 nm in length. Regular IAPP fibrils, 13.6 ± 2.9 nm in width, were split into much thinner fibrils of 6.7 ± 1.8 nm in the presence of the R-SiO2, indicating strong remodeling by the mismatched pitches (90 nm for R-SiO2 vs. 20–50 nm for IAPP fibrils [18]) and opposite handedness of the two interactants, while L-SiO2 did not show notable remodeling of the established IAPP fibrils. The greater capacity of the R-SiO2 in remodeling IAPP amyloid fibrils, in comparison with the L-SiO2, can be attributed to their morphological mismatches than with the latter, and hence breakage of H-bonding and hydrophobic interactions within the IAPP fibrils to render protofibrils. More discussion on this aspect can be found in the following simulation section concerning fibril remodeling by the L/R-SiO2.
In the absence of the chiral nanoribbons, the ThT kinetics displayed an initial lag phase due to IAPP nucleation, followed by a rapid aggregation of the peptide before reaching the saturation phase in ~8 h (Fig. 2J). This result is consistent with IAPP fibrillization kinetics reported in literature [45, 46]. In the presence of the L/R-SiO2, the ThT intensity significantly dropped compared to the IAPP control, indicating the lower β-sheet contents due to the inhibition of fibril formation. In addition, in the presence of the nanoribbons, the lag time of IAPP fibrillization was significantly shortened. This phenomenon is related to charge attraction between the N-terminus of the peptide and the anionic silica nanoribbons, in addition to hydrogen bonding and hydrophobic interaction between the two species, which elevated local peptide concentration to accelerate their nucleation through seeding [34, 46] on the silica surfaces, similarly to the catalytic role of lipid membranes for amyloid protein aggregation [2, 8]. Such interactions converted IAPP from disordered monomers to α-helix and then β-sheet rich oligomers and protofibrils and, eventually, cross-beta amyloid fibrils of compromised lengths (Fig. S6). TEM imaging corroborated the observation, revealing less densely populated IAPP fibrils. The rigidity of the IAPP protofibrils and fibrils in the presence of the nanoribbons was difficult to determine by statistical analysis, however, due to the much shorter lengths of the peptide structures (compared to the micrometer lengths of full IAPP fibrils). Interestingly, IAPP fibrillization in the presence of the L-SiO2 exhibited higher ThT intensities than with the R-SiO2. In the saturation phase, specifically, the ThT fluorescence intensity was ~36% lower with the R-SiO2 than with the L-SiO2, indicating a higher efficiency of IAPP aggregation inhibition with the oppositely-handed silica nanoribbons. A plausible reason for such discrepancy is provided in later sections.
Coarse-grained simulation of fibrillization and fibril remodeling by L/R-SiO2 nanoribbons
To understand the different effects of the L/R-SiO2 on IAPP oligomerization and fibrillization at the molecular level, we developed a coarse-grained 11-bead peptide model capable of capturing general features of amyloid aggregation, including peptide conformational changes upon aggregation, the mesoscopic morphology of amyloid fibrils, and aggregation kinetics, which cannot be fully captured by existing coarse-grained models (details of the peptide model, DMD simulation, and data analysis in Methods). DMD simulations with 100 coarse-grained model peptides were performed for peptides alone and in the presence of either an L- or R-SiO2. A peptide in our model can adopt either the aggregation-incompetent π-state representing random coil or helical conformations (Fig. S3B), or the aggregation-prone β-state representing β-sheets (Fig. S3C). We assigned the π-state with a lower free energy than the β-state (Fig. S3D). Isolated peptides before aggregation in our simulations mainly adopted the aggregation-incompetent π-state (Fig. S4). The time evolution of the total number of hydrogen bonds (Fig. 3A) and the number of peptides in β-sheet conformations (Fig. 3B) indicated that the nanoribbons accelerated peptide aggregation by significantly reducing the initial lag phase of the peptide alone (0–0.3 μs). The peptide in the presence of L-SiO2 reached its saturation state faster than those with R-SiO2. Hence, the aggregation kinetics results from the simulations were consistent with the ThT assay (Fig. 2J). The time evolution of the average size of fibril aggregates was computed, by estimating the β-state aggregates weighted by their aggregate sizes (i.e., the number of peptide in an aggregate). Peptides in the presence of the R-SiO2 tended to form smaller aggregates compared to both the control and the L-SiO2 (Fig. 3C). The final aggregates of peptide alone or in the presence of the L-SiO2 could reach the size of ~80, corresponding to the majority of the simulated peptides, while remained at ~40 in the presence of the R-SiO2. Since the final aggregates were about the same for different systems (Fig. 3B), these results suggest that the R-SiO2 promoted the formation of multiple smaller aggregates than the control or the L-SiO2.
Figure 3.

Coarse-grained modelling of fibrillization and fibril remodeling in the presence of chiral silica nanoribbons. (A-C) Time evolution of total number of hydrogen bonds, average β-sheet sizes, and average fibrillar aggregate sizes. (D-F) Fibrillization in the absence and presence of the L/R-SiO2 during 3 µs of simulation. (G-H) Remodeling of pre-formed fibril in the presence of the L/R-SiO2.
Snapshots taken along the simulation trajectories (Fig. 3D, Video S1) indicated that peptides in solution first formed smaller aggregates, which then merged into larger fibrillar oligomers either along the elongation direction or on the nanostructure surface, forming left-twisted multi-layer aggregates. In the presence of the L-SiO2 (Fig. 3E, Video S2), peptides first bound to the nanostructure surface in the forms of both fibrillar and non-fibrillar oligomers, which further rearranged and merged into a single fibrillar aggregate on the nanostructure surface and along its axis due to their matching left-handed morphologies. Interestingly, the fibrils could form branches upon binding and conformational re-arrangement (e.g., 2.4–3 μs in Fig. 3E). Due to the morphological mismatch between fibrils and the R-SiO2, small aggregates formed on the nanoparticles surface preferred to align perpendicularly to the nanostructure axis and did not self-assemble into larger aggregates as in the control or with the L-SiO2 (Fig. 3F, Video S3). These small aggregates on the nanoparticle surfaces could function independently as seeds for further elongation. With the same total number of peptides but more independently formed fibrils, the average size/length of fibrils in the presence of the R-SiO2 were smaller than with L-SiO2, consistent with the contour length analysis of the IAPP fibrils (Fig. 2). Comparison of fibrils originated from nanostructure surfaces in TEM imaging also suggests that fibrils near the R-SiO2 surface tended to be perpendicular to the nanostructure axes (Fig. 2E,G) while fibrils near the L-SiO2 surface were more aligned along the nanostructure axes (Fig. 2D,F), as predicted by the coarse-grained DMD simulations (Fig. 3).
In addition, we performed simulations of a pre-formed fibril interacting with the L/R-SiO2 in order to understand their differential capacities of remodeling fibrils (Fig. 3G,H). A pre-formed fibril was initially positioned away from the nanoribbons with randomly generated orientation. The pre-formed fibril could first adsorb onto the surfaces of both the L- and R-SiO2. Due to the morphological match between the fibril and the L-SiO2, the left-handed fibril could re-arrange itself and align with the nanoribbon to increase the fibril-nanoparticle contacts (Fig. 3G). On the other hand, the fibril on the surface of the R-SiO2 could not fully align with the nanoparticle due to the mismatch of morphologies, and instead broke into shorter fibrils bound to the nanoparticle surface perpendicularly (Fig. 3H). The latter phenomenon took place because the energy gain of surface contact between shorter fibrils and the R-SiO2 was more favorable than the energetic cost of breaking the fibril (e.g., local interactions between stacked peptides in the fibril). In cases of excessive fibrils with respect to silica nanoribbons, as in the experiments, the L-SiO2 surfaces were fully covered by fibrils aligned along the nanoribbon axes due to matching morphologies. Fibrils, on the other hand, tended to bind the R-SiO2 without alignments as shown in simulations (Fig. 3). Hence, the R-SiO2 had more available surface areas for interacting with fibrils than the L-SiO2. Fibrils in solution undergo constant dynamics [18], including the bundling/un-bundling equilibrium between thin protofibrils and thick fibrils. Compared to the L-SiO2, more available surface areas on the R-SiO2 for fibril binding shifted the bundling/un-bundling equilibrium toward thin protofibrils. Hence, the coarse-grained simulations offered molecular insights to the differential capabilities of the L- and R-SiO2 in remodeling amyloid fibrils, as observed in TEM imaging (Fig. 2H,J).
In vitro IAPP toxicity with silica nanoribbons
The toxicity of IAPP is a main cause for the degeneration of pancreatic beta cell islets [47, 48] and, as with amyloid proteins such as Aβ and alpha synuclein, the oligomeric forms of IAPP are believed to be the most toxic species [1]. Therefore, a viability assay was performed using pancreatic βTC-6 cells exposed to the L/R-SiO2 in interaction with various IAPP structures. The results of 14 h cell treatment are shown in Fig. 4A for IAPP monomers, oligomers and amyloid fibrils. In the case of IAPP monomers, silica nanoribbons were potent in alleviating IAPP toxicity, by ~75% for both the L/R-SiO2. As monomeric IAPP possessed no handedness, the effect of nanostructure chirality was understandably negligible. With IAPP protofibrils, in contrast, their interactions with chiral nanoparticles appeared handedness dependent. Specifically, the R-SiO2 were over twice more effective than the L-SiO2 in reducing IAPP cytotoxicity, indicating a stronger binding affinity between the R-SiO2 and toxic IAPP species. In the case of IAPP amyloid fibrils, the R-SiO2 were 1.7 times more efficient than the L-SiO2 in suppressing IAPP toxicity, suggesting cooperative binding between the R-SiO2 and the oppositely handed IAPP fibrils over length scales of tens to hundreds of nanometers (Fig. 2I,H). Consistently, a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay revealed that preformed IAPP fibrils were less toxic than monomeric and oligomeric IAPP. In the presence of the silica nanostructures, the R-SiO2 was effective in decreasing the toxicity of oligomeric and preformed IAPP, while both the L/R-SiO2 were effective in reducing the toxicity of monomeric IAPP (Fig. S7).
Figure 4.

(A) Determination of in vitro toxicities of 0 h, 1 h and 48 h aged IAPP on βTC-6 cells in the presence and absence of L/R-SiO2 (ns: P > 0.05, ***: P ≤ 0.001 and ****: P ≤ 0.0001). Visualization of βTC-6 cell (B) damage induced by 0 h IAPP (C), 1 h IAPP (F) and 48 h IAPP (I) and their mitigation by the L/R-SiO2 with helium ion microscopy (D, E; G, H; J, K). Scale bars: 2 µm. Here the 0 h, 1 h and 48 h aged IAPP refer to IAPP freshly dissolved in water (0 h IAPP), oligomers/protofibrils (1 h into fibrillization), and fibrils (48 h into fibrillization), respectively. The incubation time of the IAPP species in the cell toxicity assay was 14 h. The experiments was carried out in triplicate and error bars show the standard deviations of the averaged data sets. IAPP concentration: 20 µM, at room temperature and pH 7.
As expected, helium ion microscopy revealed significant damage (including pore formation) and deformation of βTC-6 cell membranes exposed to IAPP monomers and oligomers (Fig. 4C,F). No remarkable cell damage was found in exposure to IAPP amyloid fibrils, and coating of cell surfaces by the protein was notable due to attraction between the anionic lipid membranes and cationic IAPP. The damage was unnoticeable when the cells were exposed to IAPP in the presence of the L/R-SiO2, especially for the R-SiO2 (Fig. 4I-K). Some cells displayed rugged morphologies in exposure to IAPP oligomers/amyloid fibrils together with the L-SiO2 (Fig. 4D,G). However, no cell deformation was observed in exposure to IAPP oligomers/fibrils together with the R-SiO2 (Fig. 4E-K), which is consistent with the viability results (Fig. 4A).
In vivo IAPP toxicity with silica nanoribbons
The use of zebrafish embryos as an in vivo model has led to remarkable progress in toxicology and genetic studies [49], and has recently been applied to the study of amyloidogenesis taking advantage of its high fecundity, well characterized developmental stages, transparency of embryos, and multi-cellular and multi-organ compositions [10, 50, 51]. Here an in vivo toxicity assay was performed by microinjecting the L/R-SiO2 and the three IAPP species into the yolk of 2 hpf zebrafish embryos. The results on the survival and phenotypic abnormalities are shown in Fig. 5A-C. In the case of the 0 h IAPP, silica nanoribbons increased the survival of the embryos, from 25 ± 6% for the control to 43.7 ± 5.2% and 51.2 ± 5.5% in the presence of the L/R-SiO2, respectively. In the case of the 1 h IAPP, where unstructured monomers were converting into oligomers and protofibrils [8], the survival rate increased from 19 ± 6% for the control to 40 ± 9% and 66 ± 9% in the presence of the L-SiO2 and R-SiO2, respectively. In the case of the 48 h IAPP fibrils, the R-SiO2 were 100 ± 8.5% more efficient in suppressing IAPP cytotoxicity than the control, while the L-SiO2 decreased the survival of the embryos induced by IAPP fibrils by 40% in comparison with the control. Consistent with the survival results, the R-SiO2 in interaction with all IAPP structures showed lower abnormalities compared to the control and with the L-SiO2.
Figure 5.

Determination of in vivo toxicity assay (A) survival, (B) abnormality on 2 hpf zebrafish embryos 80 h after treatments (related to fully hatched, untreated embryos) by microinjection of 1 nL of 10 µM IAPP of 0 h, 1 h and 48 h in the presence and absence of the L/R-SiO2 (40 µg/mL) to the yolk of 2 hpf zebrafish embryos. (C) Representative images of healthy, dead and abnormal embryos. The experiments were carried out in triplicate and error bars show the standard deviations of the averaged data sets (ns: P > 0.05, *: P ≤ 0.05 and **: P ≤ 0.01).
Fig. 6 compares non-treated embryos with the treated embryos based on the green fluorescence of ThT from 4 hpf until 4 dpf (A-E). The strongest signal (corresponding to the highest IAPP aggregation) occurred in the absence of the silica helices, which was concomitant with the appearance of phenotypic abnormalities (panel B). Consistent with the ThT assay, the IAPP fluorescence intensity was decreased upon interaction with the L-SiO2, and in the presence of the R-SiO2 the ThT intensity remained comparable to that of the control. In general, the strongest ThT intensity was observed at 4 dpf indicating saturation of IAPP fibrillization.
Figure 6.

Fluorescence imaging of (A) untreated and (B-D) treated embryos during 4 days after treatment (saturation phase of IAPP fibrillization in embryos). IAPP (10 µM) dissolved in ThT dye (20 µM) pre-incubated w/o L/R-SiO2 (40 µg/mL) was microinjected to the yolks of 2 hpf embryos. The intensities of ThT fluorescence were indicative of IAPP fibrillization in embryonic development from 10 hpf to 4 dpf. (E) The mean intensities (a.u.) of ThT fluorescence were measured for IAPP pre-incubated w/o L/R-SiO2. (F) Linear swimming distance and number of rotations per hour for untreated and treated embryos 4 days after microinjection. The area of circle is proportional to the frequency of rotation per hour.
Many studies have shown the physiological mechanisms of zebrafish larvae behaviors in terms of their vision, swimming activity and responses to touching resulting from early toxicant exposure [52–56]. In this study, the effects of IAPP in the presence and absence of the L/R-SiO2 on the swimming behavior of larvae, in the observable forms of swimming distance and rotation, were recorded (Fig. 6F). The 96 hpf larvae were able to freely swim in linear paths and change their swimming directions spontaneously. The zebrafish larvae (4 dpf) treated with 0 h IAPP and 0 h IAPP/L-SiO2 showed more rotational movement (<180°, video S4) compared with the control (video S5) and with the R-SiO2. One plausible cause for the elevated rotational movement is IAPP-induced stress. The larvae displaying more pronounced rotations were visually deformed due to the impact of the peptide toxicity on their development. Such behavioral abnormality may be closely related to the phenotype but not necessary damage to the neurons. Interestingly, the larvae injected with 48 h IAPP/L-SiO2 exhibited the lowest activities both in rotation and swimming distance (video S6), indicating the highest toxicity upon interactions between peptide fibrils and nanoribbons of the same handedness (refer to Fig. 4A). However, the larvae treated with 48 h IAPP and the R-SiO2 showed significant improvements on activities, signifying their recovery from the amyloid toxicity.
CONCLUSION
The mesoscopic-scale chirality of silica nanoribbons strongly influenced IAPP fibrillization and toxicity in vitro and in vivo, especially in the case of the R-SiO2. The nanoribbons attracted IAPP monomers through H-bonding and electrostatic and hydrophobic interactions and acted as nucleation sites for the conversion of IAPP monomers to oligomers and protofibrils, thereby reducing the concentrations of these species in solution to mitigate IAPP toxicity. Due to steric constraints, the R-SiO2 nanoribbons allowed their surface-associated IAPP to elongate into left-handed amyloid fibrils in the directions away from or perpendicular to the silica backbone, while IAPP fibrils formed along the L-SiO2 surfaces, as evidenced by TEM and DMD simulations. As a result of such directional IAPP aggregation on the nanoribbon surfaces, the R-SiO2 possessed more binding and nucleation sites per surface area than the L-SiO2 in sequestering toxic IAPP species from the solution (Fig. 7). In addition, the R-SiO2 was significantly more effective than their left-handed counterpart in remodeling mature IAPP fibrils, through complementary handedness of the interactants, mismatch of their pitch sizes and directional aggregation of the peptide on the nanostructures. On-pathway protein aggregation, from disordered monomers to toxic oligomers and protofibrils and eventually to mostly left-handed β-sheet rich fibrils, may be steered off-pathway by interference from chiral molecules [11–15, 17, 57], as shown in the literature, or from chiral nanostructures, as first demonstrated by this study through biophysical, toxicological and behavioral characterizations. In light of the nonspecific nature of nanoparticle-amyloid protein binding, it is conceivable that the current findings may be applicable to other classes of chiral nanostructures (e.g. plasmonic chiral gold nanoparticles [58–60]) and other types of amyloid proteins (e.g. Aβ associated with Alzheimer’s disease and alpha synuclein associated with Parkinson’s disease). Hence, exploiting mesoscopic-scale chirality may prove an exciting new avenue for the synthesis and development of potent nanostructures against the aggregation and toxicity of a range of amyloid diseases.
Figure 7.

Adsorption of IAPP monomers and oligomers accelerated their nucleation and aggregation on silica nanoribbon surfaces. Such interactions decreased the net concentrations of free IAPP monomers and oligomers in solution to mitigate their associated toxicity. Due to the steric constraints imposed by the chiral silica nanoribbons, left-handed IAPP amyloid fibrils rendered by surface-adsorbed monomers and oligomers elongated away from or perpendicular to the R-SiO2 backbone. In contrast, left-handed IAPP fibrils extended nearly parallel to the L-SiO2 surfaces, thereby shielding the nanostructures from being further accessed by the peptide. Accordingly, the R-SiO2 possessed much more binding sites per surface area than the L-SiO2 for IAPP adsorption and aggregation, as also evidenced by TEM and DMD simulations, and hence were far more effective in preventing IAPP induced toxicity.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by ARC Project No. CE140100036 (Davis), the Recruitment Program of “1000plan Youth” and Startup Funds from Tongji University (Lin), NSF CBET-1553945 (Ding) and NIH R35GM119691 (Ding). TEM imaging was performed at Bio21 Advanced Microscopy Facility, University of Melbourne and SEM imaging at Ramaciotti Centre for Cryo-Electron Microscopy, Monash University. HIM imaging was performed at the MCFP platform, University of Melbourne by Dr. Babak Nasr. We thank Dr. Kevin M. McPeak for discussions.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
SUPPLEMENTARY INFORMATION
Methods and data of CD and MTT assays, as well as HPLC and mass spectroscopy (Figs. S1&S2, S6&S7). Coarse-grained simulation setup (Figs. S3-S5). Videos of fibrillization simulations (videos 1–3: Fibrilization-control.mpg, Fibrillization-L-SiO2.mpg, and Fibrillization-R-SiO2.mpg) and zebrafish behavior experiment (videos 4–6: control.avi, 0h IAPP-L-SiO2.avi, and 48h IAPP-L-SiO2.avi).
REFERENCES
- 1.Haataja L; Gurlo T; Huang CJ; Butler PC, Endocr. Rev 2008. 29, 303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao P; Abedini A; Wang H; Tu LH; Zhang X; Schmidt AM; Raleigh DP, Proc. Natl. Acad. Sci. USA 2013. 110, 19279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brender JR; Hartman K; Nanga RPR; Popovych N; de la Salud Bea R; Vivekanandan S; Marsh ENG; Ramamoorthy A, J. Am. Chem. Soc 2010. 132, 8973–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sciacca MF; Milardi D; Messina GM; Marletta G; Brender JR; Ramamoorthy A; La Rosa C, Biophys. J 2013. 104, 173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson JL; Miranker AD, J. Mol. Biol 2004. 335, 221–31. [DOI] [PubMed] [Google Scholar]
- 6.Ge X; Kakinen A; Gurzov EN; Yang W; Pang L; Pilkington EH; Govindan-Nedumpully P; Chen P; Separovic F; Davis TP; Ke PC; Ding F, Chem. Commun 2017. 53, 9394–9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nanga RP; Brender JR; Vivekanandan S; Ramamoorthy A, Biochim. Biophys. Acta 2011. 1808, 2337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ke PC; Sani MA; Ding F; Kakinen A; Javed I; Separovic F; Davis TP; Mezzenga R, Chem. Soc. Rev 2017. 46, 6492–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilkington EH; Lai M; Ge X; Stanley WJ; Wang B; Wang M; Kakinen A; Sani M-A; Whittaker MR; Gurzov EN; Ding F; Quinn JF; Davis TP; Ke PC, Biomacromolecules 2017, 18, 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Javed I; Yu T; Peng G; Sánchez-Ferrer A; Faridi A; Kakinen A; Zhao M; Mezzenga R; Davis TP; Lin S; Ke PC, Nano Letters 2018. DOI 10.1021/acs.nanolett.8b02446. [DOI] [PubMed] [Google Scholar]
- 11.Wei W; Xu C; Gao N; Ren J; Qu X, Chem. Sci 2014. 5, 4367–4374. [Google Scholar]
- 12.Gao G; Zhang M; Lu P; Guo G; Wang D; Sun T, Angew. Chem. Int. Ed 2015. 54, 2245–50. [DOI] [PubMed] [Google Scholar]
- 13.Qing G; Zhao S; Xiong Y; Lv Z; Jiang F; Liu Y; Chen H; Zhang M; Sun T, J. Am. Chem. Soc. 2014. 136, 10736–10742. [DOI] [PubMed] [Google Scholar]
- 14.Du Z; Guan Y; Ding C; Gao N; Ren J; Qu X, Nano Research 2018, DOI: 10.1007/s12274-018-1995-y. [Google Scholar]
- 15.Li M; Howson SE; Dong K; Gao N; Ren J; Scott P; Qu X, J. Am. Chem. Soc 2014. 136, 11655–11663. [DOI] [PubMed] [Google Scholar]
- 16.Malishev R; Arad E; Bhunia SK; Shaham-Niv S; Kolusheva S; Gazit E; Jelinek R, ChemComm. 2018, 54, 7762–7765. [DOI] [PubMed] [Google Scholar]
- 17.Wang X; Wang X; Wang M; Zhang D; Yang Q; Liu T; Lei R; Zhu S; Zhao Y; Chen C, Small 2018, 14, e1703982. [DOI] [PubMed] [Google Scholar]
- 18.Kakinen A; Adamcik J; Wang B; Ge X; Mezzenga R; Davis TP; Ding F; Ke PC, Nano Research 2017, 11, 3636–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin D; Banerjee D, Int. J. Heat and Mass Transfer 2011. 54, 1064–1070. [Google Scholar]
- 20.Slowing II; Trewyn BG; Giri S; Lin VSY, Adv. Func. Mat 2007. 17, 1225–1236. [Google Scholar]
- 21.Ichinose W; Miyagawa M; An Z; Yamaguchi M, Org. Lett 2012, 14, 3123–3125. [DOI] [PubMed] [Google Scholar]
- 22.Song L; Wang S; Kotov NA; Xia Y, Anal. Chem 2012, 84, 7330–7335. [DOI] [PubMed] [Google Scholar]
- 23.Das RK; Zouani OF; Labrugère C; Oda R; Durrieu M-C, ACS Nano 2013, 7, 3351–3361. [DOI] [PubMed] [Google Scholar]
- 24.Gansel JK; Thiel M; Rill MS; Decker M; Bade K; Saile V; von Freymann G; Linden S; Wegener M Science 2009, 325, 1513–5. [DOI] [PubMed] [Google Scholar]
- 25.Cheng J; Le Saux G; Gao J; Buffeteau T; Battie Y; Barois P; Ponsinet V; Delville M-H; Ersen O; Pouget E; Oda R, ACS Nano 2017, 11, 3806–3818. [DOI] [PubMed] [Google Scholar]
- 26.Delgado-Perez T; Bouchet LM; de la Guardia M; Galian RE; Perez-Prieto J, Chem. 2013, 19, 11068–76. [DOI] [PubMed] [Google Scholar]
- 27.Shah E; Soni HP, RSC advances 2013, 3 , 17453–17461. [Google Scholar]
- 28.Sugiyasu K; Tamaru S-i.; Takeuchi M; Berthier D; Huc I; Oda R; Shinkai S, Chem. Comm 2002, 1212–1213. [DOI] [PubMed] [Google Scholar]
- 29.Delclos T; Aimé C; Pouget E; Brizard A; Huc I; Delville M-H; Oda R, Nano Lett. 2008, 8, 1929–1935. [DOI] [PubMed] [Google Scholar]
- 30.Okazaki Y; Cheng J; Dedovets D; Kemper G; Delville M-H; Durrieu M-C; Ihara H; Takafuji M; Pouget E; Oda R, ACS Nano 2014, 8, 6863–6872. [DOI] [PubMed] [Google Scholar]
- 31.Usov I; Mezzenga R, Macromolecules, 2015. 48, 1269–1280. [Google Scholar]
- 32.Rapaport DC, The Art of Molecular Dynamics Simulation. 2 ed. 2004, Cambridge: Cambridge University Press. [Google Scholar]
- 33.Auer S; Trovato A; Vendruscolo M, PLoS. Comput. Biol 2009. 5, e1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radic S; Davis TP; Ke PC; Ding F, RSC Adv. 2015. 5, 105498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen HD; Hall CK, Proc. Natl. Acad. Sci. U. S. A 2004. 101, 16180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urbanc B; Cruz L; Yun S; Buldyrev SV; Bitan G; Teplow DB; Stanley HE, Proc. Natl. Acad. Sci. U.S.A 2004. 101, 17345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding F; Furukawa Y; Nukina N; Dokholyan NV, J. Mol. Biol 2012. 421, 548–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding F; Borreguero JM; Buldyrey SV; Stanley HE; Dokholyan NV, Proteins 2003. 53, 220–8. [DOI] [PubMed] [Google Scholar]
- 39.Ding F; Tsao D; Nie H; Dokholyan NV, Structure 2008. 16, 1010–1018.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pellarin R; Schuetz P; Guarnera E; Caflisch A, J. Am. Chem.l Soc 2010. 132, 14960–14970.. [DOI] [PubMed] [Google Scholar]
- 41.Pellarin R; Caflisch A, J. Mol. Biol 2006. 360, 882–92. [DOI] [PubMed] [Google Scholar]
- 42.Pellarin R; Guarnera E; Caflisch A, J. Mol. Biol, 2007. 374, 917–24 [DOI] [PubMed] [Google Scholar]
- 43.Dill KA, Biochemistry 1985. 24, 1501–1509. [DOI] [PubMed] [Google Scholar]
- 44.Lin S; Zhao Y; Xia T; Meng H; Ji Z; Liu R; George S; Xiong S; Wang X; Zhang H; Pokhrel S; Mädler L; Damoiseaux R; Lin S; Nel AE, ACS Nano 2011. 5, 7284–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sudhakar S; Kalipillai P; Santhosh PB; Mani E, J. Phys. Chem. C 2017. 121, 6339–6348. [Google Scholar]
- 46.Knowles TP; Vendruscolo M; Dobson CM, Nat. Rev. Mol. Cell Biol 2014. 15, 384–96. [DOI] [PubMed] [Google Scholar]
- 47.Wiltzius JJ; Sievers SA; Sawaya MR; Eisenberg D, Protein Sci. 2009. 18, 1521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X; St. Clair JR; London E; Raleigh DP, Biochemistry 2017. 56, 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin S; Zhao Y; Nel AE; Lin S, Small 2013. 9, 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalueff AV; Stewart AM; Gerlai R, Trends Pharmacol Sci. 2014. 35, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Best JD; Alderton WK, Neuropsychiatr Dis Treat. 2008. 4, 567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brustein E; Saint-Amant L; Buss RR; Chong M; McDearmid JR; Drapeau P, Physiol J. Paris 2003. 97, 77–86. [DOI] [PubMed] [Google Scholar]
- 54.Granato M; van Eeden FJ; Schach U; Trowe T; Brand M; Furutani-Seiki M; Haffter P; Hammerschmidt M; Heisenberg CP; Jiang YJ; Kane DA; Kelsh R [DOI] [PubMed] [Google Scholar]
- 55.Levin ED; Swain HA; Donerly S; Linney E, Neurotoxicol Teratol 2004. 26, 719–23.. [DOI] [PubMed] [Google Scholar]
- 57.Geng J; Li M; Wu L; Chen C; Qu X, Adv. Healthc. Mater 2012. 1, 332–6. [DOI] [PubMed] [Google Scholar]
- 58.Ma W; Xu L; de Moura AF; Wu X; Kuang H; Xu C; Kotov NA, Chem. Rev 2017. 117, 8041–8093. [DOI] [PubMed] [Google Scholar]
- 59.McPeak KM; van Engers CD; Blome M; Park JH; Burger S; Gosálvez MA; Faridi A; Ries YR; Sahu A; Norris DJ, Nano Lett. 2014. 14, 2934–2940. [DOI] [PubMed] [Google Scholar]
- 60.Lee HE; Ahn HY; Mun J; Lee YY; Kim M; Cho NH; Chang K; Kim WS; Rho J; Nam KT, Nature 2018. 556, 360–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.