

Abstract
Claudins are a family of transmembrane proteins integral to the structure and function of tight junctions (TJ). Disruption of TJ and alterations in claudin expression are important features of invasive and metastatic cancer cells. Expression of CLDN18.1, the lung-specific isoform of CLDN18, is markedly decreased in lung adenocarcinoma (LuAd). Furthermore, we recently observed that aged Cldn18−/− mice have increased propensity to develop LuAd. We now demonstrate that CLDN18.1 expression correlates inversely with promoter methylation and with LuAd patient mortality. In addition, when restored in LuAd cells that have lost expression, CLDN18.1 markedly attenuates malignant properties including xenograft tumor growth in vivo as well as cell proliferation, migration, invasion and anchorage-independent colony formation in vitro. Based on high throughput analyses of Cldn18−/− murine lung alveolar epithelial type II cells, as well as CLDN18.1-repleted human LuAd cells, we hypothesized and subsequently confirmed by Western analysis that CLDN18.1 inhibits insulin-like growth factor-1 receptor (IGF-1R) and AKT phosphorylation. Consistent with recent data in Cldn18−/− knockout mice, expression of CLDN18.1 in human LuAd cells also decreased expression of transcriptional co-activator with PDZ-binding motif (TAZ) and Yes-associated protein (YAP) and their target genes, contributing to its tumor suppressor activity. Moreover, analysis of LuAd cells in which YAP and/or TAZ are silenced with siRNA suggests that inhibition of TAZ, and possibly YAP, is also involved in CLDN18.1-mediated AKT inactivation. Taken together, these data indicate a tumor suppressor role for CLDN18.1 in LuAd mediated by a regulatory network that encompasses YAP/TAZ, IGF-1R and AKT signaling.
Keywords: lung alveolar epithelial type II cells, xenograft, AKT, IGF-1 receptor, YAP/TAZ
Introduction
Lung cancer is the leading cause of cancer death worldwide, with ~1.8 million new cases diagnosed each year, and prognosis remains poor (1). Despite advances in treatment and incorporation of new drugs and targeted therapies, 5-year survival of LuAd patients is only ~15% (1). Therefore, improved understanding of disease biology and development of novel diagnostic, prognostic and therapeutic tools would enhance management of LuAd.
Claudins (CLDNs) are transmembrane protein components of both epithelial and endothelial tight junctions (TJs) (2). They are critical for the assembly and function of TJs and play essential roles in regulating paracellular permeability and maintaining polarity. Claudins are altered in various cancers and their role in carcinogenesis is context-dependent (3). CLDN3 and CLDN4 are frequently upregulated in ovarian, breast, prostate and pancreatic tumors (4–6), while CLDN7 is downregulated in breast cancer but elevated in stomach cancer (7,8). There is as yet no unifying hypothesis as to the role of claudins in carcinogenesis.
The lung-specific CLDN18.1 is one of the most highly expressed claudin family members in alveolar epithelial cells (9). Another splicing isoform, CLDN18.2, is normally expressed in stomach (10,11) and ectopically in pancreatic, esophageal, ovarian and lung cancers (12–14). Whereas numerous studies have focused on ectopic expression of CLDN18.2, investigation of CLDN18.1 in cancer has been limited (15,16). Using Cldn18−/− mice, in which both Cldn18.1 and Cldn18.2 isoforms are globally deleted, we recently assigned a role for CLDN18 in regulating not only epithelial permeability and ion transport, but also proliferation of lung alveolar epithelial type II (AT2) cells, organ size and tumorigenicity (17,18). In the present study, we address in vivo and in vitro the contributions of CLDN18.1 to the malignant phenotype of human LuAd.
Molecular mechanisms underlying lung carcinogenesis involve interplay of multiple signaling pathways. The insulin-like growth factor (IGF) pathway has been implicated in induction and maintenance of different malignancies including lung cancer (19,20). A major downstream effector of IGF signaling is the phosphoinositide 3-kinase (PI3K)/AKT pathway, increased activity of which is frequently observed in NSCLC (21,22). Aberrant activation of the PI3K/AKT/mTOR pathway has been reported in more than 40% of LuAd cases from The Cancer Genome Atlas (TCGA) network (cBioportal.org) (23). Further underscoring the importance of the PI3K/AKT pathway in lung cancer, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) was among 18 genes recurrently mutated in a cohort of 412 LuAd patients (24).
Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are well-known critical regulators of organ size and tumorigenesis (25,26). Both YAP and TAZ are highly expressed in LuAd (25), and knockdown of either is sufficient to inhibit cell proliferation, invasion and clonogenicity of LuAd cell lines (27,28). As a key downstream effector of the Hippo pathway, nuclear YAP/TAZ signaling is involved in context-dependent combinatorial regulation of a rich transcriptional program that mediates diverse biological processes (29,30). YAP/TAZ activity is regulated in both Hippo kinase-dependent and -independent manners (25,31). We have recently reported that CLDN18 interacts with YAP in murine AT2 cells, resulting in nuclear exclusion and downregulation of YAP target genes (18).
In the present study, we investigated the role for CLDN18 in lung carcinogenesis using complementary in vivo and in vitro experimental models, as well as publicly available high-throughput data from LuAd patients. Our data indicate a tumor suppressor role for CLDN18.1 in human LuAd cells which involves interplay among IGF-1R, YAP/TAZ and AKT.
Materials and methods
Analysis of data from the LuAd patient cohort of TCGA
DNA methylation data and matching gene-level RNA-sequencing (RNA-seq) data were retrieved from the data portal of TCGA (https://tcga-data.nci.nih.gov/tcga/). DNA methylation data were generated using the Illumina Infinium HumanMethylation450 platform and Level 3 data are represented by β-values which define the ratio of the intensity of the methylated bead type to the combined locus intensity. The TCGA Level 3 RNA-seq dataset quantifies transcript levels by normalized counts using the RNA-seq by Expectation-Maximization (RSEM) method. Kaplan Meier curves were generated using Partek Genomics Suite 6.6 (Partek, St. Louis, MO) for LuAd patients based on clinical data and isoform-specific RNA-seq data retrieved from the National Cancer Institute Genomic Data Commons data portal (https://portal.gdc.cancer.gov). Normalized counts of CLDN18.1 (uc003erp.1) were standardized by mean-centering and scaling to dichotomize tumors into groups expressing either high or low levels of this isoform.
Animal procedures
Isolated lung AT2 cells and whole lung extracts were derived from mice with global deletion of both Cldn18 isoforms as previously described (17). In vivo tumorigenicity assays were carried out in female athymic nude mice (Jackson Laboratories, Bar Harbor, ME). Briefly, LuAd cells were grafted subcutaneously in the flanks of eight-week-old mice (1x106 cells per flank). Mice were fed a Dox-containing diet (625 mg/kg) (Teklad Diets, Madison, WI; #TD.01306) for doxycycline (Dox)-induced gene expression in the xenografts. Tumor length (l) and width (w) were measured weekly, and tumor volume (V) was calculated as V = lw2/2 (32). Mice were euthanized after 6 weeks and tumors were excised and weighed. All animal studies were performed in compliance with the University of Southern California Institutional Animal Care and Use Committee guidelines.
Preparation of plasmids and lentiviral particles
For promoter methylation studies, the CLDN18.1 sequence between positions −300 and −1 relative to the transcription start site was PCR-amplified using human genomic DNA as template and the primer pair 5′-AGTCTGGTTTAAGACAGAGCAC-3′ and 5′-GCCGAAGGTGTGAAGCTAA-3′. The amplicon was ligated into the TA cloning vector (Invitrogen, Carlsbad, CA; #K4500-01) and the insert was excised using Acc65I and BamHI and directionally cloned into Acc65I/BamHI-digested CpG-free pCpGL-basic luciferase vector (gift from Dr. Peter Jones, Van Andel Research Institute, Grand Rapids, MI) to yield pCpGL-300. For Dox-inducible CLDN18.1 expression, the Myc-Flag-tagged coding sequence of CLDN18.1 was cloned into the SpeI/XbaI-digested lentiviral entry vector pEN_TmiRc3 (ATCC, Manassas, VA; MBA-248), and the resulting plasmid was recombined using the Gateway system (Invitrogen; #11791-020) with the pSLIK-Hygro destination vector (ATCC; MBA-237) to yield pSLIK-Tet-rtTA-CLDN18.1-Hygro. All constructs were verified by sequencing. For preparation of lentiviral particles, HEK293T cells were co-transfected with pSLIK-Tet-rtTA-CLDN18.1-Hygro, the pCMV8.91 packaging plasmid and the pMDG envelope plasmid, using the calcium chloride method (33). Culture medium containing virus particles was harvested after 48 hours and virus titer (typically 108 lentiviral particles/ml) was determined using a p24 Elisa Assay Kit (Cell Biolabs, San Diego, CA; #VPK-108-H).
Cell culture procedures and generation of stable cell lines
Primary human AT2 cells were obtained from remnant transplant lungs in compliance with Institutional Review Board-approved protocols for the use of human source material in research (HS-07-00660). Human H23 (#CRL-5800) and H358 (#CRL-5807) LuAd cell lines from the American Type Culture Collection (ATCC, Manassas, VA) were cultured in RPMI-1640 medium (Invitrogen; #11875-119) supplemented with 4 mM L-glutamine, 10% fetal bovine serum (FBS) (Hyclone, Logan, UT; #SH30071.02), 100 U/ml penicillin and 100 μg/ml streptomycin. Both cell lines were authenticated at the Genetic Core of the University of Arizona based on polymorphic short tandem repeat (STR) loci. To generate stable H23 and H358 subclones with Dox-inducible CLDN18.1, cells were transduced with the pSLIK-Tet-rtTA-CLDN18.1-Hygro lentivirus in the presence of 10 μg/ml polybrene at multiplicity of infection (MOI)=2. Transduced cells were selected with 100–150 μg/ml of hygromycin (Life Technologies, Carlsbad, CA; #10687-010) for 10 days, resulting in H23/C18 and H358/C18 Dox-inducible CLDN18.1-expressing sublines. All studies were performed with cells pretreated with Dox for 2 days to induce CLDN18.1 expression. Mouse lung epithelial (MLE-15) cells (gift from Dr. Jeffrey Whitsett, University of Cincinnati) were cultured in HITES medium (RPMI 1640, 10 nM hydrocortisone, 5 μg/ml insulin, 5 μg/ml human transferrin, 10 nM β-estradiol, 5 μg/ml selenium, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin and 100 μg/ml streptomycin) supplemented with 2% FBS (34). Human embryonic kidney HEK293T cells (ATCC; #CRL-1573) were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, St. Louis, MO; #5796) supplemented with 4 mM L-glutamine, 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Human AT2 cells were isolated as previously described (35).
Methylation, transient transfections and luciferase assays
The plasmids pCpGL and pCpGL-300 (20 μg) were each incubated for 48 hours at 37°C with 50 units of methyltransferase SssI (New England Biolabs, Ipswich, MA; #M0226L) and 160 μM S-adenosylmethionine (SAM) (New England Biolabs; #B9003S). Control, unmethylated DNA was prepared by incubating the plasmids as above, except the methyltransferase was omitted. Completeness of methylation was verified using the methylation-sensitive restriction enzyme HhaI. MLE-15 cells were seeded in 24-well plates (6x104 cells/well) and transfected after 24 hours with 0.75 μg/well of either methylated or unmethylated pCpGL or pCpGL-300 firefly luciferase plasmids, along with 50 ng Renilla luciferase control vector using Superfect reagent (Qiagen, Valencia, CA; #301307). Reporter activity was determined 48 hr later with the Dual-Luciferase Reporter System (Promega, Madison, WI; #E1960). siRNA transfections of H23/C18 cells were performed with Lipofectamine RNAi-MAX (Thermo Fisher, Waltham, MA; #13778030) according to the manufacturer’s instructions. Non-targeting (control) and YAP and TAZ siRNAs were purchased from Dharmacon (Chicago, IL). Sequences of siRNAs used are provided in Supplemental Table 1.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from cultured H23/C18 cells and from human AT2 cells using RNeasy Plus Mini Kit (Qiagen, Germantown, MD; #74134). cDNA synthesis was carried out with Superscript (Invitrogen; #18080-051). qRT-PCR was performed with a thermal cycler (Applied Biosystems, Foster City, CA; 7900HT). Primer sequences are provided in Supplemental Table 2.
Western analysis
Protein extracts (10–60 μg) were resolved by SDS-PAGE and transferred to PVDF membranes (Bio-Rad; #162-0177) as previously described (36). Sources of antibodies and their dilution factors were as follows: anti-CLDN18 (Life Technologies, Grand Island, NY; #700178; 1:500), anti-lamin A/C (Santa Cruz Biotechnology, Santa Cruz, CA; #sc20681; 1:2000), anti-α-tubulin (Sigma-Aldrich, #6-11B-1; 1:5000), anti-β-actin (Abcam, Cambridge, MA; #ab8227; 1:5000) and anti-GAPDH (Ambion, Foster City, CA; #AM4300; 1:2000). Anti-phospho-IGF-1R (#3027; 1:200), anti-IGF-1R (#3018; 1:200), anti-phospho-S127-AKT (#4911; 1:200), anti-AKT (#3477; 1:500), anti-Erk1/2 (#4695; 1:200), anti-phospho-Erk1/2 (#4370; 1:200), anti-YAP (#4912; 1:200), anti-p-YAP (#4911; 1:200) and anti-TAZ ((#4883; 1:200) were from Cell Signaling Technology (Beverly, MA).
Immunofluorescence and confocal microscopy
For immunostaining, 6x104 H23/C18 or H23 cells were seeded on 12 mm Transwell filters (Corning, Corning, NY; #3401) and cultivated for two days before fixation with 4% paraformaldehyde (PFA). After permeabilization with 0.1% Triton-X, filters were incubated with primary Abs overnight at 4°C followed by 1hr incubation with biotinylated anti-rabbit or -mouse IgG (Vector Laboratories, Burlingame, CA; #BA-1000 or #BA-2000, 1:300) and 10 min incubation with Cy3- or Alexa 488-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA; #016-160-084 or Invitrogen, #S11223; 1:300). Primary Abs for CLDN18, YAP and TAZ were the same as those used for Western analysis; anti-E-cadherin was from BD Biosciences (Franklin Lake, NJ; #610181; 1:1000). For CLDN18.1/ZO-1 double staining, filters were incubated overnight with mouse anti-cMyc, targeting Myc-tag of CLDN18.1 (Sigma; #M4439; 1:100) and rabbit anti-ZO-1 (Invitrogen; #40-2200; 1:100), followed by sequential incubations with biotinylated goat anti-rabbit IgG, Cy3-conjugated streptavidin and Alexa 488-labeled goat anti-mouse Ab (Invitrogen; #A-10680; 1:300). Images were captured using a Nikon Eclipse 80i microscope (QImaging, Surrey, BC, Canada) and confocal images were captured using a ZEISS LSM 510 confocal system (Carl Zeiss, Jena, Germany).
Cell counting and proliferation assay
For cell growth assays, H23/C18 and H23 cells pretreated with Dox or vehicle were seeded in 24 well plates at a density of 6x104 cells/well. Cells were then counted daily using a Z1 coulter particle counter (Beckman Coultier, Brea, CA) up to day 5. To evaluate proliferation of H23 and H23/C18 cells, 5-ethynyl-2′-deoxyuridine (EdU; 10 μM) was added to the culture medium 1 hr before fixation with 4% PFA. EdU incorporation was detected with the EdU Imaging Kit (Life Technologies; #10337).
Soft agar anchorage-independent growth assay
H23 and H23/C18 cells were suspended in growth medium containing 0.3% agar (Fisher Scientific, Hampton, NH; #BP1423-500) and 1.5 ml (4x104 cells) was added per well (22 mm) in 12-well plates. Prior to seeding, each well was prepared with a 1.5 ml basal layer of 0.6% agar in growth medium. Cells were incubated at 37°C with a change of media supplemented with Dox or vehicle every other day. Colonies were fixed in 4% PFA after 14 days and stained with crystal violet.
Migration and invasion assays
For migration assays, 6x104 H23/C18 cells were seeded in the upper chamber of a non-coated FluoroBlok 12 mm insert (Corning; #62406-504). Inserts coated with Matrigel (Corning; #354234) were used for invasion assays. After incubation at 37°C for 48 hrs in the presence or absence of Dox, cells were stained with 4 μg/ml Calcein-AM (VWR, Radnor, PA; #89044-502) for 1 h and fluorescence was measured from the bottom of the plates using the CytoFluor4000 fluorescence plate-reader (Applied Biosystems) at wavelengths of 490/520 (Ex/Em).
Microarray analysis
Total RNA was extracted from AT2 cells from wild type and Cldn18−/− mice using an RNeasy kit (Qiagen) and global expression profiling was performed in quadruplicate using Mouse Ref8 v2.0 BeadChips (IIlumina, San Diego, CA) by the Southern California Genotyping Consortium at the University of California, Los Angeles. Raw data processing was done using GenomeStudio (Illumina) and extended analyses were carried out using R (version 2.11.1) as previously described (35). The complete microarray dataset has been deposited to Gene Expression Omnibus (GEO) with the accession number GSE106233.
Reverse phase protein array (RPPA) analysis
H23/C18 cells with and without Dox and wild type and Cldn18−/− knockout mouse AT2 cells were subjected to RPPA analyses at the Functional Proteomics RPPA Core Facility, MD Anderson Cancer Center. Detailed descriptions for sample processing and data analysis as well as antibody information are provided on their website: https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html.
Statistical analyses
Unless otherwise specified, data are presented as mean ± standard error of the mean (SEM). Significance (p < 0.05) was determined by two-sided t-test for comparison of two group means and Z-test for comparisons between normalized data. Significance (p < 0.05) for ≥ 3 group means with one and two factors was determined by one-way and two-way analysis of variance (ANOVA), respectively. Post-hoc analyses were performed with Fisher’s least significant difference (LSD) test. Significance (p < 0.05) for Kaplan-Meier curves was determined using the log-rank test. Ingenuity Pathway Analysis (IPA) package (http://www.ingenuity.com) was used for analysis of differentially expressed RNAs (microarray) and proteins (RPPA). Fisher’s exact test as implemented in IPA software was used to calculate p-values of significance level 0.05.
Results
Methylation and expression patterns validate a tumor suppressor role for CLDN18.1 in human LuAd
Consistent with a tumor suppressor role in human lung alveolar epithelial cells, methylation of a CpG island near the CLDN18.1 transcription start site was increased in tumor versus normal tissue in the TCGA LuAd patient cohort (Figure 1A), while in vitro methylation of the CLDN18.1 promoter resulted in strong transcriptional inhibition of a linked luciferase reporter (Figure 1B). Methylation of the CpG island near the CLDN18.1 transcription start site was inversely correlated with CLDN18 mRNA expression in the TCGA LuAd patient cohort (Figure 1C). Furthermore, comparison of the survival rates of patients expressing CLDN18.1 mRNA at levels above versus below the mean normalized counts (mean = 911 ± 1396) in the TCGA LuAd patient cohort indicates that higher CLDN18.1 expression is associated with significantly better survival (Figure 1D). Similar to pancreatic and other cancers, a small fraction of tumors in the TCGA LuAd patient cohort exhibited ectopic expression of the stomach-specific CLDN18.2 isoform, which was associated with decreased methylation (Supplemental Figure 1A–C). In contrast to CLDN18.1, there was no significant inverse association between CLDN18.2 expression (mean = 1132 ± 3998) and patient survival in the TCGA LuAd cohort (Supplemental Figure 1D). These results validate a tumor suppressor role for CLDN18.1 in human LuAd consistent with the development of such tumors in the Cldn18−/− mouse model (18).
Figure 1. CLDN18.1 expression in LuAd is inversely correlated with patient mortality and promoter methylation.

A. Methylation of the CLDN18.1 promoter in the TCGA LuAd patient cohort. Data are from 286 LuAd samples and 19 adjacent non-tumor tissues, with boxes representing the 25% to 75% quartiles and lines within the boxes depicting median values. Data are for probe cg10602180 (depicted in the schematic) (*, p < 0.0001, unpaired two-tailed t-test). Schematic of CLDN18 gene located in chromosome 3 indicating its two isoforms (lung-specific CLDN18.1 and stomach-specific CLDN18.2). A CpG island is located in the CLDN18.1 promoter region (modified from NCBI Reference Sequence (RefSeq)). B. In vitro methylation inhibits activity of the CLDN18.1 promoter. CpGL-300 contains the CpG-rich 300-bp CLDN18.1 promoter fragment in the CpG-less-luciferase vector CpGL. The indicated plasmids were methylated or mock-methylated and transfected into MLE-15 cells. * indicates p < 0.05 compared to unmethylated CpGL-300, n = 3, two-way ANOVA. C. Analysis of the correlation between CLDN18.1 mRNA expression and methylation of its promoter CpG island (r = 0.5476, p < 0.0001). D. Kaplan-Meier curves for 502 patients in the TCGA LuAd cohort (p < 0.003, log-rank test).
Expression of CLDN18.1 in human LuAd cells suppresses xenograft tumor growth
To address the role of CLDN18 in suppressing human lung carcinogenesis in vivo, we conditionally expressed CLDN18.1 in the H23 human LuAd cell line and tested tumorigenicity in a mouse xenograft model. Cells were transduced with a doxycycline (Dox)-inducible CLDN18.1 lentiviral vector, yielding cells termed H23/C18. Dox-induced CLDN18.1 expression was confirmed by qRT-PCR and Western analysis, and confocal microscopy verified its co-localization with zonula occludens (ZO-1) at tight junctions (Figure 2A–C). Expression of CLDN18.1 affected neither intracellular distribution of junctional proteins ZO-1 and E-cadherin nor any gross cell morphology features in both H23/C18 and H358/C18 human LuAd cell lines (Supplemental Figure 2A–C). CLDN18.1 was induced two days prior to implantation by treating cells with 0.5 μg/ml Dox, a concentration that increased CLDN18.1 mRNA and protein expression to levels comparable to primary human AT2 cells (Figure 2A–B). Dox-treated and control cells were inoculated subcutaneously into the flanks of nude mice and CLDN18.1 induction continued in vivo by administration of Dox in the diet (Supplemental Figure 3). Tumors were detectable in the control group after 2 weeks and their mean volume reached 314 ± 19.90 mm3 by 6 weeks. In contrast, H23/C18 cells in which CLDN18.1 was induced by Dox did not form tumors until 3 weeks, and their mean volume was only 98 ± 19.36 mm3 at 6 weeks (Figure 2D–E). Tumor weight at 6 weeks was 136 ± 20.35 mg in the control group versus 49 ± 9.22 mg in the CLDN18.1-expressing group (Figure 2F). As expected, Dox affected neither the size nor the weight of xenograft tumors formed by the parental (non-transduced) H23 cells, attributing the inhibition of xenograft tumor growth to CLDN18.1 induction (Figure 2D–F). The ~70% decrease in tumor volume and weight (p<0.05) in the Dox-treated group (Figure 2D–F) directly indicates that CLDN18.1 plays a tumor suppressor role in human LuAd cells in vivo.
Figure 2. Induction of CLDN18.1 suppresses xenograft tumor growth.
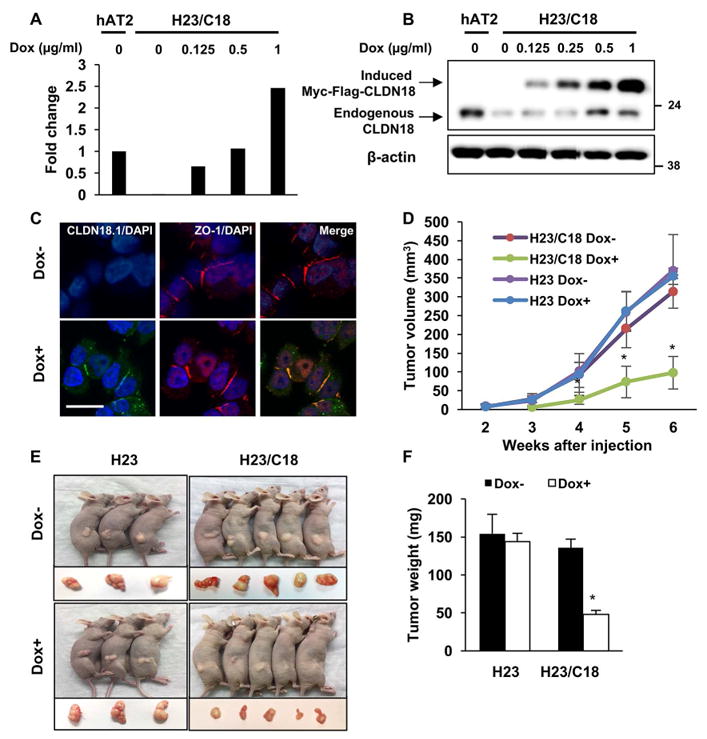
A–B. H23/C18 cells were treated with Dox at the indicated concentrations and CLDN18.1 expression was assessed by qRT-PCR (A) and Western analysis (B). Human lung alveolar type II (hAT2) cells were used as a reference. C. Co-staining of CLDN18.1 (green) and ZO-1 (red) on Dox-treated versus control H23/C18 cells. Bar = 2 μm. Mouse anti-cMyc antibody was used to detect Myc-tagged CLDN18.1. D–F. Nude mice were injected with H23/C18 or parental H23 cells and CLDN18.1 expression was induced by Dox. Tumor volumes (D) were measured every week and compared among Dox-treated H23/C18 cells and other conditions at the same time point. (* indicates p < 0.05, n ≥ 3, two-way ANOVA). Mice were euthanized 6 weeks after injection and nodules were photographed (E), excised and weighed (F). * indicates p < 0.05 compared to H23 cells and untreated H23/C18 cells, n ≥ 3, two-way ANOVA).
CLDN18.1 inhibits malignant properties of LuAd cells in vitro
In pursuit of cellular mechanisms underlying the tumor suppressor role of CLDN18 in human LuAd cells, we investigated effects of Dox-induced CLDN18.1 expression on malignant properties of cultured LuAd cells. Dox significantly reduced cell growth rate in both the H23/C18 (Figure 3A) and the H358/C18 model (Supplemental Figure 4). The decreased growth rate was not attributable increased apoptosis (Supplemental Figure 5). Rather, assessment of cell proliferation based on EdU incorporation demonstrated Dox-mediated decrease in EdU+ cells (Figure 3B–C). Dox treatment did not affect the number of Edu+ cells in cultures of parental H23 cells (Supplemental Figure 6A).
Figure 3. CLDN18.1 suppresses malignancy of LuAd cells.

H23/C18 cells were treated with Dox to induce CLDN18.1. A. Cell numbers were counted on days 1–5. * indicates p < 0.05 compared to untreated cells at the same time, n = 3, two-way ANOVA. B–C. Cell proliferation was assessed based on Edu incorporation. Shown are representative fluorescence images with EdU staining in green and CLDN18.1 staining of the same cells in red (B; bar = 20 μm) and quantitative analysis of the percentage of EdU+ cells (C). D–G. The effects of Dox on migration (D–E) and invasion (F–G) were assessed in Transwell assays by staining of cells that crossed the membrane with Calcein AM dye. Shown are representative images of cells that crossed the membrane (D, F; bar = 100 μm) and quantitative analyses of the data (E, G). H–J. Dox decreases anchorage-independent growth. Colonies formed in soft agar in the absence and presence of Dox are shown at low (top, whole well), medium (middle, bar = 200 μm) and high (bottom, bar = 50 μm) magnification, and the data are quantitated for colony number (I) and size (J). In C, E, G, I and J, * indicates p < 0.05, n = 3, unpaired two-tailed t-test.
We next investigated the effects of CLDN18.1 on cell migration and invasion using Boyden chamber assays. Dox-induced CLDN18.1 expression decreased the number of cells that crossed the membrane in the migration assay by ~2-fold (Figure 3D–E). Remarkably, Dox decreased the number of cells that crossed the Matrigel-coated membrane in the invasion assay by ~16-fold (Figure 3F–G). We next tested the effect of CLDN18.1 on anchorage-independent growth in the H23/C18 culture model. As shown in Figure 3H–J, CLDN18.1 markedly inhibited both the number and size of colonies formed by H23/C18 cells in soft agar. As control, Dox treatment did not affect anchorage-independent growth of parental H23 cells (Supplemental Figure 6B–D). These data suggest that CLDN18.1 suppresses LuAd tumor growth by inhibiting cell proliferation, both anchorage-dependent and -independent, and by decreasing cell migration and invasion.
CLDN18.1 suppresses IGF-1R/AKT signaling
We employed two experimental systems, primary AT2 cells from Cldn18−/− mice and H23/C18 cells with Dox-inducible CLDN18.1, to pursue pathways involved in the tumor suppressor function of CLDN18 in lung alveolar epithelial cells in an unbiased fashion. First, we investigated global gene expression in AT2 cells freshly isolated from Cldn18−/− versus wild-type control mice. Ingenuity Pathway Analysis (IPA) of 135 differentially expressed genes (Supplemental Table 3) suggests that IGF-1 signaling is the most altered pathway in AT2 cells isolated from Cldn18−/− mice (Figure 4A). Indeed, Western blot analysis with anti-phospho-IGF-1R and pan-IGF-1R Abs indicates specific upregulation of the phosphorylated active receptor in AT2 cells from Cldn18−/− as compared to wild type mice (Figure 4B). Quantitative analysis indicates ~1.7-fold increase in IGF-1R phosphorylation/activation upon loss of Cldn18 (Figure 4B). We also used the same Western blot to assess the effect of Cldn18 loss on phosphorylation (activation) of AKT, a major downstream effector of IGF-1R. As expected, activation of IGF-1R was associated with a 2-fold activation of AKT (Figure 4B).
Figure 4. CLDN18.1 regulates IGF-1R signaling.

A. IPA analysis of 135 genes differentially expressed in AT2 cells freshly isolated from Cldn18−/− and WT mice (FC > 2, p < 0.05). Line graph represents the ratio between the number of genes in a given pathway that are present in the list of differentially regulated genes divided by the total number of genes that make up that pathway in the reference gene set. The significance values for the canonical pathways represented by bars were calculated using right-tailed Fisher’s exact test. B. Western analysis and quantitation of IGF-1R and AKT phosphorylation in wild type mouse and Cldn18−/− E18 lung. C. The top 77 proteins affected by Dox in H23/C18 cells were interrogated using IPA as in Panel A. D. Representative Western blots and quantitative analysis of IGF-1R and AKT phosphorylation in Dox-treated versus control H23/C18 cells. In B and D, * indicates p < 0.05, n = 3, Z-test. Western blot data shown in figures 4D, 5B and S7A of the manuscript are from the same experiment.
To further identify pathways potentially mediating the tumor suppressor function of CLDN18.1 in LuAd, we subjected Dox-treated and control H23/C18 human LuAd cells to functional proteomics analysis using RPPA of over 300 antibodies. Supplemental Table 4 lists 77 proteins that were altered by Dox treatment (fold change ≥ 1.2), and IPA of these proteins again suggested that IGF-1 and AKT signaling were among the pathways most strongly affected by CLDN18.1 (Figure 4C). Indeed, Western analysis demonstrated that Dox-mediated CLDN18.1 induction in H23/C18 cells resulted in a ~2 fold decrease in the phosphorylation of both IGF-1R and AKT (Figure 4D). In contrast, CLDN18.1 induction in the H23/C18 cells did not alter ERK1/2 phosphorylation (Supplemental Figure 7).
To further understand how CLDN18.1 regulates the IGF-1R/AKT axis in human LuAd cells, we tested whether IGF-1 would overcome CLDN18.1-mediated inhibition of IGF-1R/AKT signaling. Treatment of H23/C18 cells with recombinant IGF-1 (5 ng/ml) resulted in the expected increase in both IGF-1R and AKT phosphorylation (Figure 5A, lane 3 versus 1), along with an activation-induced decrease in total Akt (37). Consistent with Figure 4D, Dox-mediated CLDN18.1 expression in the absence of IGF-1 resulted in decreased phosphorylation of both IGF-1R and AKT (Figure 5A, lane 2 versus 1). Interestingly, however, Dox-mediated CLDN18.1 expression in the presence of IGF-1 resulted in downregulation of AKT phosphorylation without any significant change in IGF-1R phosphorylation (Figure 5A, lane 4 versus 3), suggesting that CLDN18.1-mediated inactivation of AKT may employ an alternative pathway in addition to the well-established IGF-1R/AKT axis.
Figure 5. Role for YAP/TAZ in CLDN18.1-mediated AKT inactivation in H23/C18 cells.

A. H23/C18 cells were treated with Dox to induce CLDN18.1 in the presence or absence of exogenous IGF-1, followed by Western analysis with anti-phospho-IGF-1R, anti-phospho-AKT and the respective pan-Abs. B. Representative western blot and quantitative analysis of TAZ and YAP in Dox-treated versus control H23/C18 cells. * indicates p < 0.05, n = 3, Z-test. Western blot data shown in figures 4D, 5B and S7A of the manuscript are from the same experiment. C–D. Representative immunostaining for TAZ and YAP in H23/C18 cells grown in the presence or absence of Dox. Bar = 5 μm, n = 3. E. qRT-PCR analysis of YAP/TAZ target gene expression in Dox-treated versus untreated H23/C18 cells. * indicates p < 0.05, n = 3, Z-test. F. H23/C18 cells were treated with Dox to induce CLDN18.1 while YAP and/or TAZ were silenced with siRNA, followed by Western analysis with the indicated antibodies. G. Quantitative analysis of AKT activation based on optical density of the p-AKT and total AKT bands. *, p < 0.05 compared to control with no Dox treatment; ┼, p < 0.05 compared to siTAZ/YAP with no Dox treatment, n = 4, two-way ANOVA. H. Working model for the tumor suppressor activity of CLDN18.1 in LuAd. CLDN18.1 attenuates a number of tumorigenic properties in lung alveolar cells. Inhibition of AKT is mediated via IGF-1R and YAP/TAZ. Additional potential interactions between the CLDN18.1—YAP/TAZ—AKT and the CLDN18.1—IGF1-R—AKT axes, depicted by the dotted line, remain to be elucidated.
Contribution of YAP/TAZ to CLDN18.1-mediated regulation of AKT
Because CLDN18 downregulates YAP in murine AT2 cells (18) and TAZ in the human H23/C18 cells (RPPA data; Supplemental Table 4), and given the known cross-talk between AKT and YAP/TAZ (38–42), we wondered whether YAP and/or TAZ could also play a role in CLDN18.1-mediated AKT regulation in human LuAd cells. We first tested the effect of CLDN18.1 expression in LuAd cells on the levels of YAP and TAZ. Western analysis (Figure 5B) demonstrates downregulation of total levels of TAZ and YAP in CLDN18.1-expressing cells. Furthermore, confocal images show the proportion of nuclear YAP and TAZ decreases in Dox-treated cells (Figure 5C–D) and accordingly, qRT-PCR analysis indicates 3–5 fold inhibition of YAP/TAZ target gene expression by CLDN18.1 (Figure 5E). These results are consistent with our previous in vivo data indicating negative regulation of YAP by CLDN18 (18). We then tested by siRNA silencing the potential roles of YAP and TAZ in regulating AKT. As shown in Figure 5F–G, TAZ depletion resulted in AKT inactivation (lane 3) to levels comparable to those observed after Dox-induced CLDN18.1 expression (lane 5), indicating that TAZ stimulates AKT and that its inhibition by CLDN18.1 (Figure 5B) may thus play a role in CLDN18.1-mediated AKT inactivation (Figure 4D and 5F). Compared to TAZ silencing, the effect of YAP silencing on AKT phosphorylation was difficult to assess because, consistent with previous studies (43), YAP silencing strongly induced TAZ (Figure 5F, lane 2 and 6), potentially resulting in two opposing effects: direct AKT inactivation as a result of decreased YAP and indirect (TAZ-dependent) AKT activation. Nevertheless, stimulation of AKT by YAP is suggested by lack of AKT activation in siYAP-transfected cells (Figure 5F, lane 2 versus 1) despite a robust (likely compensatory) increase in TAZ expression upon YAP silencing. In addition to establishment of a CLDN18.1—YAP/TAZ—AKT axis, the YAP/TAZ silencing study also demonstrates that CLDN18.1 further inhibits AKT activation in cells depleted of both YAP and TAZ (Figure 5F, lane 8 versus 4), indicating that inhibition of YAP/TAZ is insufficient for execution of the full negative regulation of AKT by CLDN18.1 and consistent with residual activation via a CLDN18.1—IGF1-R—AKT axis. These results suggest that CLDN18.1—YAP/TAZ—AKT (Figure 5) and CLDN18.1—IGF1-R—AKT (Figure 4) axes regulate AKT activity via two parallel pathways (Figure 5H), although we cannot rule out additional interactions between the two pathways.
Discussion
CLDN18.1 is emerging as a tumor suppressor in lung alveolar epithelial cells (15,18). Compared to normal tissue, LuAd displays increased CLDN18.1 promoter methylation (Figure 1) and decreased mRNA expression (Figure 1 and Ref. 29). Furthermore, low levels of CLDN18.1 expression are associated with poor survival in patients with LuAd (Figure 1). These features of CLDN18.1 contrast with those of CLDN18.2, which is ectopically expressed in a small proportion of tumors in the TCGA LuAd cohort and shows no association with patient survival (Supplemental Figure 1). Supporting the tumor suppressor role of CLDN18.1 in in human LuAd, restoration of its expression in human LuAd cell lines to levels approximating those observed in normal AT2 cells resulted in strong inhibition of tumor growth in a mouse xenograft model (Figure 2). Contributing to this tumor suppressor activity, repletion of CLDN18.1 in LuAd cells resulted in decreased proliferation, migration, invasion and anchorage-independent colony formation in vitro (Figure 3). While our recent work demonstrates direct interaction between CLDN18.1 and YAP (18), the present results and those of Shimobaba et.al. (15) suggest that additional signaling pathways are involved in the tumor suppressor activity of CLDN18.1, including TAZ, IGF-1R and AKT.
A role for AKT downstream of CLDN18.1 was indicated by functional proteomics analysis of >300 proteins in H23/C18 human LuAd cells, in which CLDN18.1 was induced by Dox. PI3K/AKT was the pathway most enriched in the set of 77 proteins altered in response to CLDN18.1 (Figure 4C). A 2-fold decrease in AKT phosphorylation, with no change in ERK phosphorylation, was confirmed by Western analysis (Figure 4 and Supplemental Figure 7). Interestingly, studies in Cldn18−/− mice showed that AKT phosphorylation is also regulated by CLDN18 in non-malignant cells from both embryonic lungs (Figure 4B) and adult AT2 cells (Supplemental Table 5), suggesting that the CLDN18.1/AKT axis is operative in normal cells and may therefore be involved in tumor initiation associated with loss of CLDN18. These results are consistent with inhibition of AKT recently observed in A549 LuAd cells upon CLDN18.1 expression (15).
Several upstream regulators could account for attenuation of AKT activity by CLDN18. EGFR, which is recurrently mutated in LuAd (24), does not appear to play such a role in this context ((15) and Supplemental Table 4). The tumor suppressor role of CLDN18.1 in LuAd cells likely involves interactions at tight junctions, although cytoplasmic and nuclear localization of over-expressed CLDN18.1 (Figure 2C and (15)) does not exclude additional mechanisms. Adherens junctions, tight junctions and associated polarity proteins are well-known regulators of the Hippo pathway (31,44), and our recent work demonstrates that CLDN18 directly inhibits YAP activity in murine AT2 cells (18). While inhibition of YAP by CLDN18.1 in human H23/C18 LuAd cells is modest, we observe stronger inhibition of its paralog TAZ as well as decreased nuclear localization of both YAP and TAZ and reduced expression of YAP/TAZ target genes (Figure 5). We suggest that CLDN18.1-mediated downregulation of TAZ plays a role in AKT inactivation because silencing of TAZ mimicked the CLDN18.1-mediated AKT inactivation (Figure 5). Specific effects on YAP versus TAZ may be context-dependent and vary among species and experimental systems. Nevertheless, CLDN18.1 further inhibited AKT in cells depleted of both YAP and TAZ (Figure 5), suggesting that additional mechanisms besides YAP/TAZ are involved in AKT suppression by CLDN18.1.
A novel finding in the present work is the inhibitory effect of CLDN18.1 on IGF-1R, a major regulator of AKT. A decrease in IGF-1R phosphorylation was observed in H23/C18 LuAd cells in vitro in response to Dox-induced CLDN18.1 expression (Figure 4D), and IGF-1R is more highly phosphorylated in lungs isolated from Cldn18−/− compared to control mice (Figure 4B). In contrast to IGF-1R and AKT, ERK phosphorylation was not decreased upon CLDN18.1-mediated IGF-1R inactivation in H23 cells (Supplemental Figure 7), and recent work with A549 LuAd cells indicates no significant effects on phosphorylation of either EGFR or downstream kinases such as Raf, MEK1, ERK1/2, JNK and p38 (15). Inactivation of receptor tyrosine kinases (RTKs) by junctional proteins is not without precedent. For example, E-cadherin inactivates IGF-1R, as well as EGFR and c-Met in canine kidney epithelial MCDK and human HEK293 cells (45), and overexpression of the tight junction protein ZO-1 in multiple myeloma cells inhibits EGFR phosphorylation (46). Delineation of precise mechanisms whereby CLDN18.1 regulates IGF1-R activity will require further investigation.
Molecular mechanisms by which CLDN18.1 integrates regulation of YAP/TAZ on the one hand, and IGF-1R on the other hand, to inactivate AKT (Figure 5H) remain largely unknown. There is evidence that CLDN18 physically interacts with YAP (18) and with PDK1, which acts upstream of AKT (15), suggesting direct regulatory mechanisms. Additionally, cross-talk between YAP/TAZ and IGF-1R/AKT signaling in LuAd may result in regulation of one pathway by the other. Similar crosstalk has been observed in other contexts. For example, it was shown that activation of RTKs such as EGFR and IGF-1R results in dissociation of complexes containing PDK1 and Hippo pathway components, leading to YAP/TAZ activation (40,41,44). Reciprocally, there is evidence that YAP/TAZ target genes indirectly regulate the IGF-1R/AKT pathway by, for example, increased expression of IGF-1 and its binding proteins (38,39) as well as the RTK/PI3K adaptor protein GAB2 (47) and the PTEN-targeting miR29 (38,48). Finally, it is also plausible that PI3K/AKT stabilizes TAZ through inhibitory phosphorylation of glycogen synthase kinase-3 beta and thus attenuation of the phosphorylation of the N-terminal phosphodegron in TAZ (42). In addition to delineating the relationships among the effects of CLDN18.1 on IGF-1R, YAP/TAZ and AKT, future studies will also have to address their relative contributions to the various tumor suppressor properties of CLDN18.1 in LuAd.
In summary, our work provides a multitude of evidence for the tumor suppressor activity of CLDN18.1 in LuAd, including inhibition of xenograft tumor growth in vivo, attenuation of various malignant phenotypes in vitro and its regulation of multiple oncogenic pathways. We demonstrate that CLDN18.1 inhibits the IGF-1R/AKT and the YAP/TAZ/AKT axes, and that high CLDN18.1 expression is associated with better LuAd patient survival. Our findings suggest that the methylation and expression of CLDN18.1 may be of prognostic value in LuAd management and that effectors downstream of CLDN18.1 may constitute viable therapeutic targets for the treatment of LuAd.
Supplementary Material
Novelty and Impact.
CLDN18.1, a key component of epithelial tight junctions, functions as a tumor suppressor in human lung adenocarcinoma (LuAd). It suppresses human LuAd cell proliferation, migration and invasion and importantly, xenograft tumor growth while suppressing YAP/TAZ, IGF-1R and AKT signaling. Methylation and expression of CLDN18.1 may be of prognostic value in LuAd management. CLDN18.1 and downstream effectors may constitute viable targets for the treatment of LuAd.
Acknowledgments
This work was supported by the Hastings Foundation, Whittier Foundation and research grants R35HL113574 (ZB), HL095349 (ZB), HL114959 (ZB), HL114094 (ZB and IAO) and DK071122 (BF) from the National Institutes of Health. Critical reading of the manuscript and assistance with statistical analysis by Kwang-Jin Kim, Ph.D., is appreciated. Histology and microscopy services were provided by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases (P30 DK048522 and S10 RR022508) and Norris Comprehensive Cancer Center Core (P30 CA014089). Microarray analysis was performed at the Southern California Genotyping Consortium at the University of California, Los Angeles (UCLA) http://scgc.genetics.ucla.edu/ under the direction of Joseph DeYoung.
Functional Proteomics RPPA was performed at the MD Anderson Cancer Center (Support Grant #5 P30 CA016672-40). E.D. Crandall is Hastings Professor and Kenneth T. Norris Jr. Chair of Medicine. Z. Borok is Hastings Professor and Edgington Chair in Medicine.
Abbreviations
- TJ
tight junctions
- LuAd
lung adenocarcinoma
- IGF-1R
insulin-like growth factor-1 receptor
- YAP
Yes-associated protein
- TAZ
transcriptional co-activator with PDZ-binding motif
- NSCLC
non-small cell lung cancer
- IGF
insulin-like growth factor
- TCGA
The Cancer Genome Atlas
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- Dox
doxycycline
Footnotes
Author contributions
JL conceived and performed experiments, analyzed and interpreted experimental data and wrote the manuscript, NOC conceived and performed experiments, analyzed and interpreted experimental and bioinformatics data and wrote the manuscript, BZ conceived experiments, analyzed and interpreted experimental data and edited the manuscript, PF conceived experiments and edited the manuscript, AC conceived experiments and edited the manuscript, AF conceived experiments and edited the manuscript, YL performed western blotting, HW performed molecular cloning, CY and CNM performed DNA methylation microarray analysis, EDC edited the manuscript, IAO conceived experiments, provided cell lines and plasmids and edited the manuscript, BF and ZB conceived the study, conceived and directed experiments, interpreted experimental data and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Torre LA, Siegel RL, Jemal A. Lung Cancer Statistics. Adv Exp Med Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- 2.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–50. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh AB, Sharma A, Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol. 2010;2010:541957. doi: 10.1155/2010/541957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005;65:7378–85. doi: 10.1158/0008-5472.CAN-05-1036. [DOI] [PubMed] [Google Scholar]
- 5.Long H, Crean CD, Lee WH, Cummings OW, Gabig TG. Expression of Clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res. 2001;61:7878–81. [PubMed] [Google Scholar]
- 6.Michl P, Buchholz M, Rolke M, Kunsch S, Löhr M, McClane B, et al. Claudin-4: a new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology. 2001;121:678–84. doi: 10.1053/gast.2001.27124. [DOI] [PubMed] [Google Scholar]
- 7.Johnson AH, Frierson HF, Zaika A, Powell SM, Roche J, Crowe S, et al. Expression of tight-junction protein claudin-7 is an early event in gastric tumorigenesis. Am J Pathol. 2005;167:577–84. doi: 10.1016/S0002-9440(10)62999-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, et al. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene. 2003;22:2021–33. doi: 10.1038/sj.onc.1206199. [DOI] [PubMed] [Google Scholar]
- 9.Frank JA. Claudins and alveolar epithelial barrier function in the lung. Ann N Y Acad Sci. 2012;1257:175–83. doi: 10.1111/j.1749-6632.2012.06533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Türeci O, Koslowski M, Helftenbein G, Castle J, Rohde C, Dhaene K, et al. Claudin-18 gene structure, regulation, and expression is evolutionary conserved in mammals. Gene. 2011;481:83–92. doi: 10.1016/j.gene.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Niimi T, Nagashima K, Ward JM, Minoo P, Zimonjic DB, Popescu NC, et al. claudin-18, a novel downstream target gene for the T/EBP/NKX2. 1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol Cell Biol. 2001;21:7380–90. doi: 10.1128/MCB.21.21.7380-7390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sahin U, Koslowski M, Dhaene K, Usener D, Brandenburg G, Seitz G, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res. 2008;14:7624–34. doi: 10.1158/1078-0432.CCR-08-1547. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka M, Shibahara J, Fukushima N, Shinozaki A, Umeda M, Ishikawa S, et al. Claudin-18 is an early-stage marker of pancreatic carcinogenesis. J Histochem Cytochem. 2011;59:942–52. doi: 10.1369/0022155411420569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Micke P, Mattsson JS, Edlund K, Lohr M, Jirström K, Berglund A, et al. Aberrantly activated claudin 6 and 18. 2 as potential therapy targets in non-small-cell lung cancer. Int J Cancer. 2014;135:2206–14. doi: 10.1002/ijc.28857. [DOI] [PubMed] [Google Scholar]
- 15.Shimobaba S, Taga S, Akizuki R, Hichino A, Endo S, Matsunaga T, et al. Claudin-18 inhibits cell proliferation and motility mediated by inhibition of phosphorylation of PDK1 and Akt in human lung adenocarcinoma A549 cells. Biochim Biophys Acta. 2016;1863:1170–8. doi: 10.1016/j.bbamcr.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Akizuki R, Shimobaba S, Matsunaga T, Endo S, Ikari A. Claudin-5, -7, and -18 suppress proliferation mediated by inhibition of phosphorylation of Akt in human lung squamous cell carcinoma. Biochim Biophys Acta. 2017;1864:293–302. doi: 10.1016/j.bbamcr.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Li G, Flodby P, Luo J, Kage H, Sipos A, Gao D, et al. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am J Respir Cell Mol Biol. 2014;51:210–22. doi: 10.1165/rcmb.2013-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou B, Flodby P, Luo J, Castillo DR, Liu Y, Yu FX, et al. Claudin-18-mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J Clin Invest. 2018;128:970–84. doi: 10.1172/JCI90429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Batth IS, Qu X, Xu L, Song N, Wang R, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer. 2017;16:6. doi: 10.1186/s12943-016-0576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karamouzis MV, Papavassiliou AG. The IGF-1 network in lung carcinoma therapeutics. Trends Mol Med. 2006;12:595–602. doi: 10.1016/j.molmed.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 21.David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, et al. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res. 2004;10:6865–71. doi: 10.1158/1078-0432.CCR-04-0174. [DOI] [PubMed] [Google Scholar]
- 22.Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197–207. doi: 10.1016/j.bcp.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73–9. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–83. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Gao Y, Li F, Tong X, Ren Y, Han X, et al. YAP promotes malignant progression of Lkb1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. 2015;75:4450–7. doi: 10.1158/0008-5472.CAN-14-3396. [DOI] [PubMed] [Google Scholar]
- 28.Noguchi S, Saito A, Horie M, Mikami Y, Suzuki HI, Morishita Y, et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin Cancer Res. 2014;20:4660–72. doi: 10.1158/1078-0432.CCR-13-3328. [DOI] [PubMed] [Google Scholar]
- 29.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–27. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M, Kim T, Johnson RL, Lim DS. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015;11:270–82. doi: 10.1016/j.celrep.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 32.Kinkade R, Dasgupta P, Carie A, Pernazza D, Carless M, Pillai S, et al. A small molecule disruptor of Rb/Raf-1 interaction inhibits cell proliferation, angiogenesis, and growth of human tumor xenografts in nude mice. Cancer Res. 2008;68:3810–8. doi: 10.1158/0008-5472.CAN-07-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borok Z, Harboe-Schmidt JE, Brody SL, You Y, Zhou B, Li X, et al. Vesicular stomatitis virus G-pseudotyped lentivirus vectors mediate efficient apical transduction of polarized quiescent primary alveolar epithelial cells. J Virol. 2001;75:11747–54. doi: 10.1128/JVI.75.23.11747-11754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, et al. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11029–33. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marconett CN, Zhou B, Rieger ME, Selamat SA, Dubourd M, Fang X, et al. Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet. 2013;9:e1003513. doi: 10.1371/journal.pgen.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou B, Flodby P, Luo J, Castillo DR, Liu Y, Yu F-X, et al. Claudin-18 regulates lung stem/progenitor cell homeostasis, YAP activity and tumorigenesis. JCI. 2017 doi: 10.1172/JCI90429. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu YT, Ouyang W, Lazorchak AS, Liu D, Shen HM, Su B. mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J Biol Chem. 2011;286:14190–8. doi: 10.1074/jbc.M111.219923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6:17206–20. doi: 10.18632/oncotarget.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110:2569–74. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Straßburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev Biol. 2012;367:187–96. doi: 10.1016/j.ydbio.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, et al. The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem. 2012;287:26245–53. doi: 10.1074/jbc.M112.382036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finch-Edmondson ML, Strauss RP, Passman AM, Sudol M, Yeoh GC, Callus BA. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J Biol Chem. 2015;290:27928–38. doi: 10.1074/jbc.M115.692285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. 2014;127:709–17. doi: 10.1242/jcs.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23:1739–48. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang XD, Baladandayuthapani V, Lin H, Mulligan G, Li B, Esseltine DW, et al. Tight Junction Protein 1 Modulates Proteasome Capacity and Proteasome Inhibitor Sensitivity in Multiple Myeloma via EGFR/JAK1/STAT3 Signaling. Cancer Cell. 2016;29:639–52. doi: 10.1016/j.ccell.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Gu C, Jeong KJ, Zhang D, Guo W, Lu Y, et al. YAP/TAZ-Mediated Upregulation of GAB2 Leads to Increased Sensitivity to Growth Factor-Induced Activation of the PI3K Pathway. Cancer Res. 2017;77:1637–48. doi: 10.1158/0008-5472.CAN-15-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, et al. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. 2012;14:1322–9. doi: 10.1038/ncb2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.