

Abstract
Mitochondrial Ca2+ regulation is crucial for bioenergetics and cellular signaling. The mechanisms controlling mitochondrial calcium homeostasis have been recently unraveled with the discovery of mitochondrial inner membrane proteins that regulate mitochondrial Ca2+ uptake and extrusion. Mitochondrial Ca2+ uptake depends on a large complex of proteins centered around the Ca2+ channel protein, mitochondrial Ca2+ uniporter (MCU) in close interactions with several regulatory subunits (MCUb, EMRE, MICU1, MICU2). Mitochondrial Ca2+ extrusion is mainly mediated by the mitochondrial Na+/Ca2+/Li+ exchanger (NCLX). Here, we review the major players of mitochondrial Ca2+ homeostasis and their physiological functions.
Keywords: Ca2+ signaling, MCU, NCLX, Mitochondria associated membranes, Reactive oxygen species, Cell metabolism, physiology of mitochondrial Ca2+
Introduction
Mitochondria are vital cellular organelles which are required for adenosine triphosphate (ATP) production and are active participants in cellular Ca2+ signaling. Long before the establishment of the endoplasmic reticulum (ER) as the major reservoir of intracellular Ca2+ storage within eukaryotic cells, mitochondria were the first intracellular organelles associated with Ca2+ handling and sequestering (for review: (Rizzuto, De Stéfani, Raffaello, & Mammucari, 2012)). The high Ca2+ capacity of mitochondria is achieved mainly through Ca2+ chelation by phosphates in mitochondrial matrix, forming amorphous Ca3(PO4)2 precipitates (Lehninger, Carafoli, & Rossi, 1967). The formation of these precipitates is pH dependent and they are reversible into Ca2+ and phosphate on collapse of mitochondrial potential by protonophores, which liberate Ca2+, leading to its efflux from mitochondria into the cytosol (Chalmers & Nicholls, 2003; Zoccarato & Nicholls, 1982). Pioneering studies from the 1960s and 1970s showed that mitochondrial Ca2+ uptake was dependent on the steep mitochondrial membrane potential (Δψ ~−180mV) and was inhibited by the drug Ruthenium Red or its analog Ruthenium 360 (Carafoli & Lehninger, 1971; DeLuca & Engstrom, 1961; Ying, Emerson, Clarke, & Sanadi, 1991). The mitochondrial electron transport chain (ETC) transfers protons across the inner mitochondrial membrane (IMM) into the intermembrane space generating a huge electrochemical proton gradient which drives the synthesis of ATP. This mitochondrial membrane potential also creates a large driving force for cytosolic Ca2+ uptake by mitochondria (Rizzuto et al, 2012; Stock, Leslie, & Walker, 1999). Changes in mitochondrial Ca2+ concentrations, which can apparently range between 0.1μM to the sub-millimolar (mM) range, are important for coupling receptor activity to bioenergetics and for regulating downstream functions such as exocytosis by controlling the amount of Ca2+ available near secretory vesicles (Montera et al., 2000). Indeed, at least three dehydrogenases of the tricarboxylic acid (TCA) cycle are regulated by mitochondrial matrix Ca2+ (Hansford, 1994; McCormack & Denton, 1979; McCormack, Halestrap, & Denton, 1990). The cytosolic Ca2+ signal typically initiated by activation of plasma membrane receptors to specific agonists propagates into mitochondria, enhancing its respiratory rate, H+ extrusion and ATP synthesis, thus effectively coupling agonist stimulation to mitochondrial metabolism and ATP production. Hence, the strength and pattern of receptor-generated Ca2+ signals are decoded by mitochondria to match the energetic demands of the cell.
Despite the huge driving force for Ca2+ across the IMM, mitochondria have very low affinity for Ca2+ uptake. As such, Ca2+ transfer into mitochondria occurs at highly specialized regions of close contacts between mitochondria and ER called mitochondria associated membranes (MAMs). MAMs are the site where phosphatidyl serine (PS) synthase Eli are enriched, and where the newly synthesized PS in the ER gains the mitochondria (Mesmin, 2016). Enzymes required for cholesterol and ceramide synthesis also reside in MAMs. In addition to lipid transfer, MAMs are required for reactive oxygen species (ROS) transfer between ER and mitochondria (Booth, Enyedi, Geiszt, Vârnai, & Hajnoczky, 2016). Metabolic reprograming of CD8+ T cells was shown to be dependent on mTORC2-AKT-GSK3β signaling complex at the MAMs which promotes hexokinase-I binding to voltage-dependent anion channel (VDAC) and facilitate metabolite flux into mitochondria required for rapid production of cytokines (Bantug et al, 2018). Recent studies indicate that MAMs can regulate mitophagy in response to cellular and environmental signals (Gelmetti et al, 2017; Gomez-Suaga et al, 2017; Liu et al, 2012; W. Wu et al., 2016). MAMs are also critical for the unfolded protein response (UPR) and autophagy signaling, insulin signaling, and function as sites for glucose sensing (for review (Rieusset,2018). Miscommunication at MAMs or disruption of MAMs could result in various metabolic diseases such as obesity (Theurey et al., 2016; Tubbs et al., 2018), type 2 diabetes (Thivolet, Vial, Cassel, Rieusset, & Madec, 2017) and leptin resistance (Schneeberger et al., 2013).
Within the MAMs, Ca2+ release into the cytosol through the ER Ca2+ release channel inositol-1,4,5-trisphosphate receptor (IP3R) creates a cytosolic Ca2+ nanodomain that is sufficiently concentrated, within the μM range, to be sensed by adjacent mitochondria (Mannella, Buttle, Rath, & Marko, 1998; Rizzuto et al., 1998; Szabadkai et al., 2006) (Fig. 1). In addition to decoding Ca2+ signals to match the energetic demands of the cell, the ability of mitochondria to either shuttle Ca2+ between different areas of the cell or to buffer cytosolic Ca2+ through coordinated Ca2+ uptake and extrusion allows it to shape cytosolic Ca2+ signaling (Fig. 1). By virtue of their mobile nature, mitochondria can buffer Ca2+ at the mouth of plasma membrane voltage-gated Ca2+channels in excitable cells, thus reducing local cytosolic Ca2+ concentrations and exocytosis (Montera et al., 2000). Mitochondria can also buffer Ca2+ at the vicinity of store-operated Ca2+ entry (SOCE) channels, which are encoded by ORAI proteins and regulated by the Ca2+ sensors, stromal interaction molecule (STIM) (for review (Trebak & Putney, 2017)) (Fig. 1). In this case, the consequence is relief of Ca2+-dependent inactivation of SOCE and increased Ca2+ entry (Hoth, Button, & Lewis, 2000). Recent evidence showed that knockdown of the newly discovered mitochondrial Ca2+ uniporter protein (MCU; discussed in detail below) in HeLa cells caused a decrease in STIM1 oligomerization and inhibited SOCE in response to Phospholipase C-coupled agonists, suggesting that mitochondrial Ca2+ uptake prevents inactivation of SOCE (Deak et al, 2014). Through their specific cytosolic organization, mitochondria can form a buffering barrier between the apical and basal membranes of polarized pancreatic acinar cells. This subcellular organization effectively divides the cell into two distinct cytosolic compartments with distinct Ca2+ signatures (Park, Ashby, Erdemli, Petersen, & Tepikin, 2001) Similarly at the immunological synapse (IS) between T helper cells and antigen presenting cells, mitochondrial Ca2+ uptake prevents SOCE inactivation and maintains a sustained Ca2+ influx, supporting nuclear factor of activated T-cells (NFAT) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-қ B) mediated T cell activation (A. Quintana et al, 2007; Ariel Quintana et al, 2011). In some instances, subplasmalemmal mitochondria can take up Ca2+ entering through SOCE and extrude this Ca2+ near the Sarco/Endoplasmic Reticulum Ca2+ ATPase (SERCA) pumps at ER sites distant from the plasma membrane, thus contributing to refilling of ER stores after their depletion by agonist stimulation (Arnaudeau, Kelley, Walsh, & Demaurex, 2001).
Figure 1: Mitochondrial Ca2+ homeostasis: Ca2+ cycling between the cytosol, ER, and mitochondria.
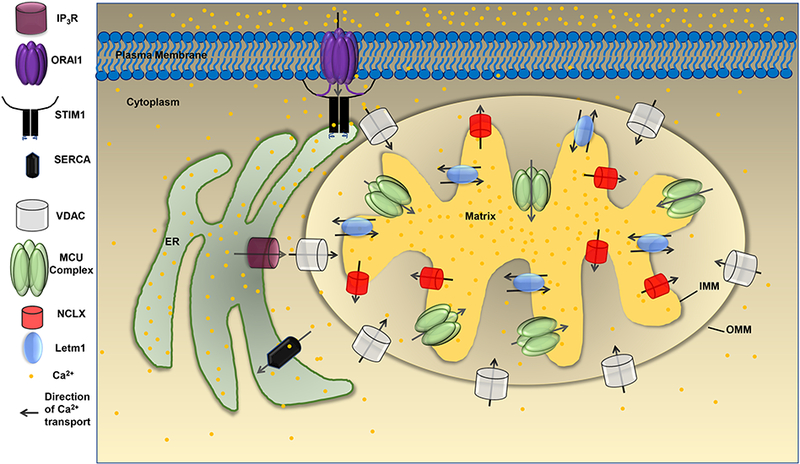
Activation of phospholipase C (PLC)-coupled agonists produce the soluble cytosolic second messenger inositol-1,4,5 trisphosphate (ΓΡ3) which binds to its receptor, the IP3R, and cause Ca2+ release from the ER to the cytosol. Through mitochondria associated membranes (MAMs), Ca2+ released from IP3R crosses the OMM through VDAC and enters the intermembrane space (IMS) before reaching the mitochondrial matrix through MCU. Depletion of ER Ca2+ concentration also triggers the activation of store-operated Ca2+ entry (SOCE) into the cytosol across the plasma membrane through ORAI1 channels. Ca2+ entry through SOCE is buffered back into the ER by the action of sarcoplasmic/endoplasmic reticulum ATPase (SERCA) pumps or can be taken up by mitochondria through MCU.VDAC resides in the outer mitochondrial membrane (OMM) and brings Ca2+ in the intermembrane space (IMS) of mitochondria. Once Ca2+ concentration in the IMS is high, it is sensed by the gatekeeper proteins MICU1/MICU2 which disinhibit MCU allowing Ca2+ uptake into the mitochondrial matrix. Matrix Ca2+ is pumped back to the IMS through the Na+/Ca2+ exchange (NCLX) and Letml.
The mechanisms of Ca2+ homeostasis in mitochondria have been studied extensively in the past few years, and several molecular players involved in mitochondrial Ca2+ uptake and extrusion have been identified. To reach the mitochondrial matrix, Ca2+ must cross both outer and inner mitochondrial membranes. The outer mitochondrial membrane (OMM) is permeable to molecules smaller than 5kDa (Madesh & Hajnoczky, 2001; Rapizzi et al, 2002), thus allowing Ca2+ to easily diffuse across the OMM. The OMM permeability is mostly the result of abundant expression of VDAC. However, the IMM ion permeability is more stringent, and Ca2+ uptake into the mitochondrial matrix is mediated by a highly Ca2+ selective channel called the mitochondrial Ca uniporter (MCU). Mitochondrial Ca extrusion is mediated by a Na /Ca exchanger-like protein termed Na+/Ca2+/Li+ exchanger (NCLX); its name is owed to the fact that unlike members of the plasma membrane Na+/Ca2+ exchangers (NCX), NCLX can function when Na+ is substituted with Li+. Interestingly, a single point mutation of threonine 103 to valine in plasma membrane NCX1 resulted in significant activation of Li+/Ca2+ exchange (Doering et al., 1998). While threonine 103 is conserved in NCX, the Na+/Ca2+/K+ exchanger (NCKX), and NCLX, only NCLX possesses a valine preceding threonine 103, which could underlie the Li+/Ca2+ exchange activity of NCLX. Nevertheless, it is not yet clear which residues are responsible for the Li+/Ca2+ exchange activity of NCLX. In some instances, the rate of mitochondrial Ca2+ extrusion and uptake is maintained by a 2H+/Ca2+ exchanger likely encoded by Letm1(Jiang, Zhao, & Clapham, 2009; Mingn Feng Tsai, Jiang, Zhao, Clapham, & Miller, 2014).
The Mitochondrial Ca2+ Uniporter (MCU)
The mitochondrial Ca2+ uniporter (MCU) is present in almost all mammalian tissues, and its activity has been known over 50 years before the discovery of its molecular identity (for review (De Stefani et al., 2016)). In the early 1990, Rizzuto et al. targeted recombinant aequorin to mitochondria and demonstrated that agonist-activated increase in cytosolic Ca2+ is rapidly coupled to increases in mitochondrial Ca2+ (Rizzuto, Simpson, Brini, & Pozzan, 1992). These authors subsequently showed efficient exposure of mitochondria to Ca2+ signals emanating from IP3Rs owing to the existence of functional sites of close ER-mitochondria contacts (Rizzuto et al., 1998). MCU currents were first recorded in 2004 using patch clamp electrophysiology on isolated mitoplasts which revealed the existence of an inwardly rectifying, highly Ca2+ selective channel in the IMM (Kirichok, Krapivinsky, & Clapham, 2004). The first protein of the mitochondrial Ca2+ uniporter complex was identified in 2010 and named MICU1 (mitochondrial calcium uptake 1) (Perocchi et al., 2010). Shortly after, two studies from two independent laboratories identified MCU as a 40 kDa protein (known as CCDC109A) that is present in the IMM and is essential for Ca2+ uptake into mitochondria (Baughman et al., 2011; De Stefani, Raffaello, Teardo, Szabô, & Rizzuto, 2011). MCU has two transmembrane domains, which are connected by a short loop with both the N- and C-termini located in the mitochondrial matrix (Fig. 2). MCU is present in nearly all vertebrate cells and is critical for mitochondrial Ca2+ homeostasis, ATP synthesis and mitochondrial metabolism (Rizzuto et al., 2012). Downregulation of MCU resulted in reduced mitochondrial Ca2+ and increased cytosolic Ca2+, while overexpression of MCU caused mitochondrial Ca2+ accumulation with a reduction in cytosolic Ca2+. Reconstitution of MCU proteins in lipid bilayers resulted in a Ca2+ current which was blocked by Ruthenium red (Rizzuto et al., 2012). These findings were subsequently confirmed with whole-mitoplast voltage-clamp recordings showing that knockdown of MCU decreased MCU Ca2+ currents whereas MCU overexpression led to increase in current (Chaudhuri, Sancak, Mootha, & Clapham, 2013). In the same study Chaudhuri et al, showed MCU current inhibition by ruthenium red and loss of ruthenium red-mediated inhibition in a form of MCU where serine 259 was mutated to alanine (S259A), consistent with the idea that MCU is the pore forming subunit of the uniporter complex (Chaudhuri et al., 2013). A more recent study in Caenorhabditis elegans showed that an analog of ruthenium red, Ruthenium 360 blocks MCU by direct interaction with the Asp240-X-X-Glu243 (DXXE) motif (D261 and E264 in human MCU). They proposed that Ruthenium 360 blocks MCU by binding to Asp240 but not to Glu243 (Cao, Wang, Cui, Su, & Chou, 2017). The same group proposed a pore domain structure of MCU from Caenorhabditis elegans based on nuclear magnetic resonance and electron microscopy. These authors suggested that MCU forms a pentameric assembly in vitro and that the DXXE motif forms the pore gate and selectivity filter (Oxenoid et al., 2016). Both Ca2+ selectivity and conductance of MCU were shown to depend on this conserved DXXE sequence. Mutation of D or E within the conserved DXXE sequence of human MCU resulted in complete loss of channel activity (Baughman et al., 2011). While these findings support the idea that MCU is the pore forming unit of the MCU complex, the idea that functional MCU channels are organized into pentameric structure remain a matter of debate. MCU was proposed to serve as a mitochondrial matrix sensor of reactive oxygen species (ROS). Recently, Dong et al. identified a conserved cysteine (Cys-97) in the N-terminus of MCU located within the mitochondrial matrix. Oxidation of this cysteine promoted oligomerization of MCU and enhanced MCU channel activity leading to enhanced mitochondrial ROS, mitochondrial Ca2+ overload, and cell death (Dong et al., 2017). The mechanisms of transcriptional and posttranslational regulation of the MCU protein complex remain scarce. One report suggested that the expression of MCU in chicken DT40 B lymphocytes is dependent on Ca2+ signals generated through the IP3/SOCE signaling nexus and controlled by the Ca2+-dependent transcription factor, cyclic adenosine monophosphate response element-binding protein (CREB); CREB was shown to interact directly with the MCU promoter to stimulate gene expression. Mitochondrial Ca2+ uptake and MCU expression were reduced in DT40 cells lacking either IP3 receptors, STIM1, or ORAI1 (Shanmughapriya et al., 2015), suggesting that SOCE is a potential regulator of MCU
Figure 2: Components of the MCU complex and its regulation by Ca2+.
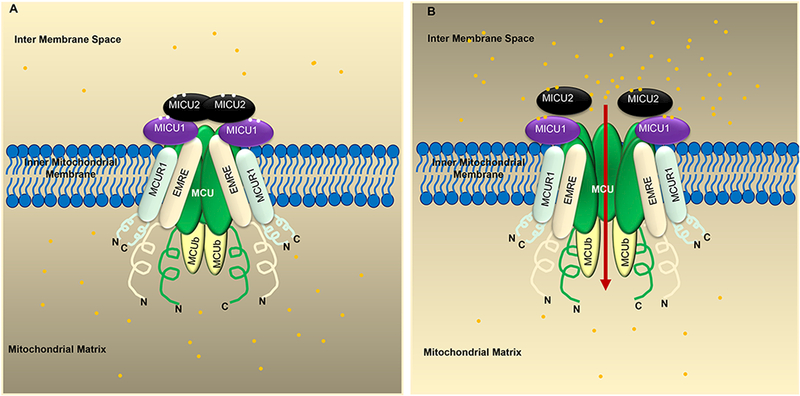
A) MCU, MICU1/2/3, EMRE, MCUR1, and MCUb form the Uniporter complex. When the Ca2+ concentration in mitochondrial intermembrane space (IMS) is low, MICU2 inhibits MCU activity. B) When the IMS Ca2+ concentration is high, Ca2+ binds to MICU1 and activates MCU to cause Ca2+ uptake into the mitochondrial matrix. EMRE is an essential subunit that is required for MCU function and helps tether MICU1/MICU2 to the MCU complex. The yellow dots represent Ca2+.
Regulators of MCU.
Since the discovery of MCU as the pore-forming unit, it is now appreciated that MCU functions as a large multi-molecular complex consisting of MCU proteins, mitochondrial calcium uptake proteins (MICU1, MICU2, and MICU3), and a 10 KDa protein called essential MCU regulator (EMRE); additional MCU partners include MCUR1, SLC25A23, and MCUb (N. E. Hoffman et al., 2014; Mallilankaraman, Cârdenas, et al., 2012; Mallilankaraman, Doonan, et al., 2012; Plovanich et al., 2013; Sancak et al., 2013).
Mitochondrial Ca2+ uptake proteins (MICU1, MICU2, and MICU3)
Mitochondrial Ca2+ uptake (MICU1) is a critical regulator of MCU (Perocchi et al., 2010), which appears to have evolved alongside MCU. The discovery of MICU1, which preceded that of MCU, emerged from an RNAi screen on mitochondria-located proteins (Perocchi et al., 2010). The compendium of approximately 1,100 mitochondrial proteins was established few years earlier and termed mitocarta (Pagliarini et al., 2008). Perocchi et al utilized RNAi strategy and identified that MICU1 is required for mitochondrial Ca uptake. Downregulation of MICU1 resulted in reduced mitochondrial Ca2+ entry without significantly affecting mitochondrial respiration or membrane potential (Perocchi et al., 2010). They proposed that MICU1 resides in the IMM and has two EF hands which are essential for its function (Perocchi et al., 2010). While most investigators agree on the location of MICU1 in the intermembrane space, one report of time lapse protein flux assay with selective permeabilization of either the OMM or both the OMM and IMM of the mitochondria showed that MICU1 is released only when both membranes are permeabilized, suggesting that MICU1 is located in the mitochondrial matrix (Nicholas E Hoffman et al., 2013). Later, two homologs of MICU1 were identified in humans, MICU2 and MICU3 (Plovanich et al., 2013). Both MICU2 and MICU3 have N-terminal mitochondrial targeting sequence. A proteomic study and microarray analysis showed that MICU1, MICU2 and MICU3 are differentially expressed in different tissues. MICU2 is strongly expressed in visceral organs while MICU3 is expressed in skeletal muscle and neuronal tissues. Similar to MICU1, both MICU2 and MICU3 have EF hands and since both MICU2 and MICU3 are paralogs of MICU1 (Plovanich et al., 2013), they likely have redundant roles depending on the tissue types where they are expressed.
MICU1 can form a functional heterodimer with MICU2 or MICU3, while MICU2 and MICU3 do not seem to heterodimerize (Patron, Granatiero, Espino, Rizzuto, & De Stefani, 2018).The original role of MICU1 as a positive regulator of Ca2+ uptake into mitochondria by MCU stems from the fact that when MICU1 is deleted MCU expression is also reduced. The prevailing model at this juncture is that MICU proteins are gate keepers for mitochondrial Ca2+ uptake. The EF hands allow MICU proteins to sense Ca2+ at the MCU channel gate and regulate its opening depending on the Ca2+ concentration in the intermembrane space (Fig. 2). In the presence of low cytosolic Ca2+ concentrations, MICU keeps MCU in a closed state preventing Ca2+ flow into the mitochondrial matrix. Increases in Ca2+ concentrations are detected by the EF hands of MICUs, which results in the opening of MCU (Csordâs et al., 2013; Nicholas E. Hoffman et al., 2013; Mallilankaraman, Doonan, et al., 2012; Troilo et al., 2014; Ming Feng Tsai et al., 2016). A recent study showed that skeletal muscle possesses a splice variant of MICU1 termed MICU1.1, which binds to Ca2+ more efficiently than MICU1. MICU1.1/MICU2 heterodimers activate MCU at lower Ca2+ concentrations than MICU1/MICU2 heterodimers (Vecellio Reane et al., 2016). Petrungaro et al. showed an important role for the mitochondrial intermembrane space (IMS) resident oxidoreductase Mia40 in MCU function. Mia40 links MICU1 and MICU2 as a heterodimer through a disulfide covalent bond, which is critical for the gate keeping function of MICU1/MICU2 heterodimer. The divalent bond allows binding of MICU1/MICU2 to MCU at low levels of Ca2+ and heterodimer dissociation from MCU at high Ca2+ concentrations. Without this MICU1/MICU2 covalent bond, MICUs gatekeeping capability is jeopardized and agonist-activated mitochondrial Ca2+ uptake is enhanced (Petrungaro et al., 2015). These studies show that the MCU channel complex is a highly regulated mitochondrial Ca2+ uptake pathway, and that its mechanisms of regulation vary in a tissue-dependent manner. The different Ca2+ binding capacity of different isoforms of MICUs can serve as a threshold-setting for mitochondrial Ca2+ and bioenergetics in different tissues and cell types, expressing different isoforms and different levels of MICU isoforms.
Studies showed that protein expression of MICU1 and MICU2, are dependent upon each other (Patron et al., 2014). Interestingly, protein knockdown of either MICU1 or MICU2 or both also resulted in reduced MCU protein levels (Plovanich et al., 2013). Double knockdown of MICU1 and MICU2 has an additive effect on impaired mitochondrial Ca2+ uptake. Differential expression and relative abundance of MICU1, MICU2 and MICU3 in different cell types likely serve to tailor mitochondrial Ca2+ uptake and kinetics to the demands of a specific cell type. Two studies proposed that MICU1 can act as an MCU gatekeeper (Csordâs et al., 2013; Mallilankaraman, Doonan, et al., 2012). These studies showed that deletion of MICU1 causes mitochondrial Ca2+ accumulation at low basal levels of cytosolic Ca2+, suggesting that MICU1 has a dual role in both activating Ca2+ uptake at high cytosolic Ca2+ concentrations and in inhibiting MCU at low resting cytosolic Ca2+ concentrations. A follow up study by Patron et al. proposed that MICU2 is the genuine gatekeeper. They showed that MICU1 and MICU2 within a functional heterodimer have opposing effects on MCU allowing for fine tuning of MCU current activity both in a lipid bilayer reconstitution system and in intact cells. They proposed that MICU1 stimulates MCU activity at higher Ca2+ concentrations, whereas at lower Ca2+ concentrations MICU2 inhibits MCU activity (Patron et al., 2014). This suggested that at low Ca2+ concentrations the inhibitory effect of MICU2 on MCU is dominant, but when cytosolic Ca2+ concentrations increase, MICU1 stimulatory effect permits rapid mitochondrial uptake of cytosolic Ca2+ by MCU. Patron et al. showed that when MICU1 expression is eliminated, MICU2 expression is also abrogated and mitochondrial Ca2+ gatekeeping ability is impaired, thus in agreement with previous studies (Patron et al., 2014). Kamer et al reported that both MICU1 and MICU2 have high affinities for Ca2+, with Kd of recombinant proteins to Ca2+ around 300 nM for MICU1 and 620 nM for MICU2. MICU1 and MICU2 form heterodimers in vitro in both Ca2+-free and Ca2+-bound conditions. Ca2+ binding to the EF hands of MICU1/MICU2 heterodimers occurs in a cooperative fashion with a Hill coefficient of ~2.1. Under Ca2+-free conditions, MICU1/MICU2 heterodimers inhibit MCU while cooperative binding of Ca2+ to MICU1/MICU2 heterodimers, at around 620 nM, stabilizes the interaction and induces a conformational change, which disinhibits MCU and allows Ca2+ uptake by mitochondria (Kamer, Grabarek, & Mootha, 2017). According to Kamer et al, MICU1 alone (in the absence of MICU2) can function as a gatekeeper to inhibit MCU at low Ca2+ concentrations (below ~350 nM). The absence of both MICU1 and MICU2 results in a lower threshold of MCU inhibition (~200 nM). Whether MICU3 is the functional equivalent of MICU2 in different cell types such as neurons and can function as a heterodimer with MICU1 to regulate the Ca2+ threshold of MCU activity remains to be determined. A recent study showed that MICU3 can form a heterodimer with MICU1 through a disulfide bond and works as a positive regulator of MCU-mediated mitochondrial Ca2+ uptake in primary cortical neurons (Patron et al., 2018).
Essential MCU regulator (EMRE)
EMRE was discovered through a quantitative mass spectroscopy analysis as a component of the MCU complex. EMRE is a small 10kDa protein with a single transmembrane domain which is present in the inner mitochondrial membrane (Sancak et al., 2013). EMRE is present in metazoans and absent in plants and protozoa. Downregulation or deletion of EMRE resulted in impaired mitochondrial Ca2+ uptake despite normal expression of the MCU protein and its oligomerization. Further overexpression of MCU in EMRE knockout cells failed to rescue mitochondrial Ca2+ uptake. Interactions between MCU and MICU1/MICU2 require EMRE leading to the proposal that EMRE is essential for MCU current activity through its ability to connect MCU channels to the calcium-sensing proteins MICU1 and MICU2 (Sancak et al., 2013). Interestingly, abrogation of MCU also resulted in significant reduction in EMRE protein levels with no discernible change in mRNA levels, suggesting that without MCU, EMRE proteins become destabilized and are likely degraded (Sancak et al., 2013). This is similar to the dependence of MCU expression on the presence of MICU1 or that of MICU2 on the presence of MICU1(Patron et al., 2014).
Using protease protection assays, Vais et al. proposed that EMRE C-terminus is located in the mitochondrial matrix and serves as a sensor of matrix Ca2+, which contributes to MCUgatekeeping activity (Vais et al., 2016). The authors proposed that regulation of MCU by EMRE requires both cytosolic Ca2+ and the presence of MICU1 and MICU2 attached to the MCU complex from the intermembrane side, suggesting that MCU activity is regulated by both cytosolic Ca2+ through MICU1/MICU2 and by matrix Ca2+ through EMRE. Deletion of the C- terminus of EMRE or neutralization of seven acidic residues within this C-terminus abrogated MCU current regulation by matrix Ca2+ without altering the expression, localization or interaction between different proteins of the MCU complex (MCU, MICU1, MICU2, EMRE) (Vais et al., 2016). However, shortly after the publication of this report, two independent studies challenged the findings of Vais et al. (Ming Feng Tsai et al., 2016; Yamamoto et al., 2016). Yamamoto et al. used a yeast expression system of Flag-tagged EMRE either at the N- or C- terminus and concluded that the topology of EMRE consists of the N-terminus (and not the C- terminus as reported by Vais et al) being located in the mitochondrial matrix side (Yamamoto et al., 2016). Tsai et al. used two independent strategies consisting of cysteine modification and mass-tagging and the expression of MCU-EMRE fusion proteins and reached the same conclusion as Yamamoto et al, that EMRE N-terminus is located in the mitochondrial matrix while its C-terminus is in the intermembrane space (Ming Feng Tsai et al., 2016). In the study by Tsai et al. the authors showed that the N-terminus of EMRE physically interacts with MCU while the C terminus interacts with MICU1. This led the authors to propose a model where EMRE has a dual role: 1) EMRE binds to MCU and acts as an essential component of the MCU pore; and 2) EMRE serves as a tether for the Ca2+-sensing MICU proteins, which serve as gatekeepers to the MCU channel. According to this model, EMRE transduces MICU1/MICU2 gatekeeping function to ensure that MCU is active only when cytosolic Ca2+ levels are high, preventing mitochondrial Ca2+ overloaded and cell death (Ming Feng Tsai et al., 2016). However, it is not clear that how interactions between EMRE and MCU or MICU1/MICU2 alter the gating of MCU. Two separate studies showed that the mitochondrial m-AAA protease, an IMM resident protein whose mutations are associated with neurodegenerative and neuromuscular disease, degrades non-assembled EMRE proteins and thus regulates MCU activity. Loss of function of m-AAA protease disrupts the stoichiometry of the MCU complex, leading to increased number of overactive MCU-EMRE channels lacking gatekeeping MICU subunits. The loss of m-AAA protease resulted in mitochondrial Ca2+ overload and neurnal death (König et al., 2016; C.-W. Tsai et al., 2017).
MCU Regulator 1 (MCUR1)
Mitochondrial Ca2+ regulator 1 (MCUR1 or CCDC90A) was reported as a member of the MCU complex and was proposed to regulate Ca2+ entry through MCU (Mallilankaraman, Cârdenas, et al., 2012). Similar to MCU, MCUR1 is ubiquitously expressed across mammalian tissues. MCUR1 has two transmembrane domains with both N- and C-termini facing the inter-membrane space (Fig. 2). The precise role of MCUR1 in mitochondrial Ca2+ uptake and whether MCUR1 directly interacts with the MCU complex are still under debate. Downregulation of MCUR1 in HEK293T, HeLa or human primary fibroblasts reduced mitochondrial Ca2+ uptake (Mallilankaraman, Cârdenas, et al., 2012). However, a study from Paupe et al. suggested that MCUR1 does not directly regulate MCU and that reduced MCU activity upon MCUR1 knockdown or deletion is rather due to decreased mitochondrial membrane potential. Instead, they proposed that MCUR1 is important for cytochrome c oxidase (COX) assembly (Paupe, Prudent, Dassa, Rendon, & Shoubridge, 2015). In response to Paupe at al., Vais et al. Provided direct MCU current measurements on isolated mitoplasts. Under these conditions where membrane potential across the IMM is controlled through the recording electrode, Vais et al. showed that knockdown of MCUR1 reduced MCU inward currents (Vais et al., 2015). Tomar et al. used pulldowns of overexpressed proteins in COS7 cells and bimolecular fluorescence complementation assays and suggested that MCUR1 functions as a scaffold protein for the MCU complex through binding to both MCU and EMRE via conserved coiled-coil domains identified in both MCU and MCUR1 (Tomar et al., 2016). Chaudhuri et al. proposed that MCUR1 confers mitochondrial permeability transition induced by Ca2+ while MCUR1 abrogation increased the Ca2+ threshold for initiation of permeability transition, thus increasing resistance to Ca2+ overload and enhancing cell survival (Chaudhuri, Artiga, Abiria, & Clapham, 2016). In a more recent study, Xing et al reported that MCUR1 expression is commonly increased in human hepatocellular carcinomas. Enhanced MCUR1 expression increases MCU-mediated mitochondrial Ca2+ uptake, enhances cell proliferation, and inhibits apoptosis (Xing et al., 2017). Enhanced expression of MCUR1 might confer a proliferative and survival advantage through enhanced mitochondrial Ca2+ uptake and bioenergetics. However, it is also conceivable that excessive increases in MCUR1 levels might drive mitochondrial permeability transition and cell death.
SLC25A23
Solute carrier family 25 member 23 (SLC25A23) is part of a family of mitochondrial proteins containing an N-terminal regulatory domain, four EF-hands, a linker domain, and C-terminal with transporter domain. SLC25A23 works as ATP-Mg/Pi exchanger, it imports adenine nucleotides into the mitochondrial matrix and effluxes inorganic phosphate. Interestingly this function of SLC24A23 is Ca2+ dependent, as the Ca2+ concentration increases in inner mitochondrial space, it binds with EF hand of SLC25A23 and activates transport of adenine nucleotides (Fiermonte et al., 2004; Harborne, King, Crichton, & Kunji, 2017).. In neurons, SLC25A23 prevents N-Methyl-D-aspartic acid (NMDA) dependent neuronal excitotoxicity (Rueda et al., 2015). SLC25A23 deletion caused a reduction in oxidative phosphorylation and mutation in SLC25A23 resulted in early age bone dysplasia of the skull and fingers (Amigo et al., 2013; Aprille, 1988; Tewari, Dash, Beard, & Bazil, 2012; Writzl et al., 2017). SLC25A23 has also been proposed to regulate mitochondrial Ca2+ uptake through the MCU complex. Downregulation of SLC25A23 resulted in reduced mitochondrial Ca2+ uptake without affecting either Ca2+ efflux or mitochondrial membrane potential. Hoffman et al. proposed that SLC25A23 interacts with MICU1 and MCU and that expression of EF-hand mutants of SLC25A23 has a dominant-negative effect on mitochondrial Ca2+ uptake. In the same report, SLC25A23 knockdown also decreased basal mitochondrial ROS and reduced cell death (N. E. Hoffman et al., 2014). Further studies are required to uncover the exact mechanisms by which SLC25A23 regulates mitochondrial Ca2+ uptake.
MCUb
MCU regulatory subunit b (MCUb), also known as CCDC109B, is a paralogue of MCU (~330aa long protein with 50% sequence identity with MCU) which was proposed to function as a part of the MCU complex (Plovanich et al., 2013; Raffaello et al., 2013). Similar to MCU, MCUb has two predicted transmembrane domains. Unlike MCU, when MCUb is reconstituted in lip bilayers it does not generate functional Ca2+ channels (Raffaello et al., 2013). Thus, it has been proposed that MCUb works as a negative regulator of MCU (Plovanich et al., 2013; Raffaello et al., 2013). MCUb physically interacts with other proteins of the MCU complex and seems to negatively regulate MCU channel activity (Fig. 2). Insertion of MCUb in the MCU complex has a dominant-negative effect manifested by inhibition of mitochondrial Ca2+ uptake in response to receptor stimulation. Further, reconstitution of MCUb with MCU in lipid bilayers largely reduced the probability of observing channel activity (Raffaello et al, 2013). The mechanisms by which MCUb regulates the MCU complex and MCU channel activity remain unknown.
Physiological role of MCU
Mitochondrial dehydrogenases such as pyruvate dehydrogenase, a-ketoglutarate dehydrogenase, and isocitrate dehydrogenase are regulated by Ca2+. Ca2+ activates a-ketoglutarate dehydrogenase and isocitrate dehydrogenase through direct binding. However, Ca2+-mediated activation of pyruvate dehydrogenase is indirect and mediated through a Ca2+-dependent phosphatase which dephosphorylates its catalytic subunit (Hansford, 1994; McCormack & Denton, 1979; McCormack et al, 1990). Activated mitochondrial dehydrogenases increase NADH level leading to increased electron supply to the ETC which increases ATP generation. Thus, mitochondrial Ca2+ uptake directly correlates with ATP production and a reduction in mitochondrial Ca2+ would inhibit ATP production. Since the MCU complex is a major regulator of mitochondrial Ca2+ uptake, MCU is expected to play a major role in controlling mitochondrial function. It has been shown in a myriad of organisms including plants, that the MCU complex plays a critical role in mitochondrial Ca2+ homeostasis by regulating Ca2+ uptake into the mitochondria (Teardo et al, 2017). MCU complex also regulates pollen tube germination and growth in Arabidopsis (Selles, Michaud, Xiong, Leblanc, & Ingouff, 2018). In trypanosomes, MCU governs bioenergetics, autophagy, and cell death (Huang, Vercesi, & Docampo, 2013). MCU is also required in the function of mushroom body neurons of Drosophila in olfactory memory formation (Drago & Davis, 2016). Accordingly, deletion of MCU in cultured cells causes reduced oxidative phosphorylation, reduced ATP generation and increased autophagy (Rizzuto et al, 2012). Remarkably, MCU global knockout mice generated using gene trap are viable, although mitochondria isolated from their skeletal muscle and heart lack the ability to rapidly uptake Ca2+. Although the number, morphology and basal metabolism of purified mitochondria from MCU knockout mice are unaffected, these mice showed reduced ability to perform energy-intensive work (Pan et al, 2013). Skeletal muscle derived from MCU knockout mice showed reduced activity of pyruvate dehydrogenase, and this coincided with muscle hypertrophy of these mice (Mammucari et al, 2015; Pan et al, 2013). A recent study showed that muscle exercise at old age results in improved muscle function and structure which correlated with increased MCU expression (Zampieri et al, 2016). MCU has a critical role in many other tissues and different cells types such as pancreatic-β cells, macrophages, and lung. MCU is critical for glucose sensing by pancreatic β cells (Tarasov et ak, 2012). One study proposed a role for mitochondrial Ca2+ in macrophage polarization (Gu, Larson-Casey, & Carter, 2017). MCU knockdown in a cystic fibrosis cell line model resulted in reduced Pseudomonas aeruginosa-induced inflammatory response due to reduced inflammasome activation (Rimessi et al 2015). Further, mitochondrial Ca2+ uptake through MCU was proposed to have a role in embryonic development. Specifically, MCU was proposed to regulate notochord axis formationby controlling blastomere movement during gastrulation, likely through regulation of microfilament dynamics (Prudent et al., 2013).
Additional roles were ascribed to MCU in several pathophysiologies including ischemia, myocardial infarction, and neurohumoral injury. Joiner et al reported co-immunoprecipitation of MCU and CaMKII from heart mitochondria, suggesting that MCU and CaMKII interact with each other. They also showed that increased Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity enhances MCU currents while CaMKII inhibition reduced currents. CaMKII inhibition in heart mitochondria protected against ischemia-reperfusion injury. The authors concluded that increased MCU current through catalytically active CaMKII in mitochondria resulted in increased myocardial cell death and heart failure (Joiner et al., 2012). Interestingly, MCU knockout mice did not show any defect in basal cardiac function under either normal or stress conditions (Holmström et al., 2015) and hearts from MCU knockout mice were not protected against ischaemia-reperfusion injury (Pan et al., 2013). Inhibition of MCU function by Ru360 in isolated sinoatrial nodal cells resulted in reduced action potential frequency in response to isoproterenol. Similarly, transgenic mice expressing a dominant negative form of MCU in the myocardium have reduced ATP generation and inhibited fight or flight-induced enhancement of heart rate, which can be rescued by dialysis of ATP (Y. Wu et al., 2015). These data suggest that altered MCU function in the heart results in mitochondrial dysfunction and reduced ATP production. It is known that Ru360 and its derivatives are nonspecific MCU inhibitors and results obtained with these inhibitors in physiological experiments should be interpreted with extreme caution. Recently, a yeast-based orthogonal interspecies chemical screen identified Mitoxantrone (an anti-cancer drug) as a specific MCU inhibitor. Mitoxantrone proved promising in blocking MCU Ca2+ currents in permeabilized cells, intact cells, and in isolated mitochondria, suggesting that it could represent a potential drug for MCU-related diseases (Arduino et al., 2017).
It is puzzling that MCU knockout and MCU dominant negative mice were viable with no overt phenotype. To address potential adaptation in these two global MCU loss of function models, Luongo et al. generated a tamoxifen-inducible conditional MCU knockout mice (Luongo et al.,2015). Interestingly, germline deletion of MCU in this model resulted in embryonic lethality, suggesting that MCU is critical for embryonic development, whereas MCU knockout after birth or in adults did not result in any significant phenotype. These findings suggest that adaptation likely plays an important role in MCU knockout and MCU dominant negative mice. Another possibility is that the absence of MCU is compensated for by Letm1 or another transporter which have partial permeability to Ca2+ and can support Ca2+ uptake into the mitochondrial matrix in the absence of MCU. Specific deletion of MCU in adult cardiomyocytes resulted in complete loss of mitochondrial Ca2+ uptake without affecting mitochondrial membrane potential. While these mice did not show any inhibition in basal cardiac function or morphology, they displayed reduced heart rate during fight or flight responses. Interestingly, ablation of MCU in adult hearts showed strong protection from ischaemia-reperfusion mediated injury (Kwong et al., 2015; Luongo et al., 2015), which is at odds with results from Pan et al using global MCU knockout mice (Pan et al., 2013). A recent study showed that diabetic mice have impaired Ca2+ handling in heart, due to altered expression of proteins of the MCU complex. MCU and EMRE levels were decreased while MCUb level was increased in mice hearts with age. Cardiac specificoverexpression of MCU improved cardiomyocyte contractility and significantly rescued ischemia reperfusion injury in diabetic mice (Suarez et al., 2018).
Mitochondrial Ca2+ overload is associated with apoptosis and necrosis. The current models show that apoptotic signals cause mitochondrial Ca2+ overload, resulting in mitochondrial fragmentation and release of pro-apoptotic proteins such as cytochrome C (Giorgi et ah, 2012). Mitochondria play a major role in cancer progression as altered mitochondrial function is one of the main drivers of tumor growth (Hanahan & Weinberg, 2011). Marchi et al showed that a cancer-related microRNA, miR-25 downregulates MCU expression when overexpressed in HeLa cells causing reduced mitochondrial Ca2+ uptake. Several human colon cancer cell lines (HCT116, RKO, SW80 and WiDr) have higher levels of miR-25 and reduced MCU levels compared to normal mucosal cells. MCU levels were also reduced in adenocarcinoma samples which have higher miR-25 expression as compared to normal mucosa. These results suggest that cancer cells which have a higher level of miR-25 downregulate MCU as means to protect against apoptosis and promote growth (S. Marchi et al., 2013). However, Zeng et al have challenged these results by showing that receptor-interacting protein kinase 1 (RIPK1) and MCU are upregulated in colorectal cancer samples from patients and that RIPK1 interacts with MCU to increase MCU activity and promote colorectal cancer through enhanced cell proliferation and energy metabolism (Zeng et al., 2018). In agreement with Zeng et al, knockdown of MCU in triple-negative breast cancer cell lines resulted in reduced cell motility, invasion and tumor growth (Tosatto et ah, 2016). Downregulation of MCU resulted in decreased ROS production, decreased expression of hypoxia-inducible factor-la (HIF-la) and reduced breast cancer cell growth and metastasis. This indicates that mitochondrial Ca2+ influx through the MCU complex regulates mitochondrial ROS, expression of HIF-la and breast cancer growth (Tosatto et ah, 2015). Another study found a correlation between MCU mRNA expression and metastatic breast cancer. Knockdown of MCU expression with siRNA or pharmacological inhibition with ruthenium red inhibited migration of MDA-MB-231 breast cancer cells in response to serum (Tang et ah, 2015). Overall, these results suggest that MCU promotes metabolism, survival and proliferation in different cancers.
Mitochondrial Na+/Ca2+/Li+ exchanger (NCLX)
The Mitochondrial Na+/Ca2+/Li+ exchanger is also known as solute carrier family 8-member B1 (SLC8B1) or solute carrier family 24 member 6 (SLC24A6). NCLX extrudes one Ca2+ from the mitochondria for every three Na+ imported and the estimated turnover rate of NCLX is 1000 cycles per second. NCLX is a ~60kDa protein present in almost all types of tissues and cells. Putative topology modeling using TopPred (von Heijne, 1992) suggested that similar to NCX, NCLX has two domains al and a2 attached by a long loop, each containing six transmembrane segments. This long loop is believed to play a regulatory role in NCLX function although its exact orientation either in the matrix side or the intermembrane space remains unknown (Kostic et al., 2015). NCLX was first cloned in 2004 and thought to be localized in the ER or plasma membrane (Palty et al., 2004). In 2010, the Sekler group showed that NCLX is enriched in mitochondria and localized in the IMM. Using Na2+ and Ca2+ fluorescent dyes, they showed that overexpression of NCLX resulted in increased mitochondrial Ca2+efflux (Fig. 1) while knockdown of NCLX resulted in reduced mitochondrial Ca2+ efflux. Inhibition of NCLX function by a pharmacological benzothiazepine compound called CGP-37157 resulted in reduced mitochondrial-Ca2+ efflux (R. Palty et al., 2010). Palty et al. expressed NCLX in SH-SY5Y cells and showed that agonist-induced mitochondrial Ca2+ efflux was higher in NCLX-overexpressing cells as compared to wild-type cells (R. Palty et ah, 2010). They also confirmed the Na+ dependence of NCLX, by showing strong Ca2+ efflux in the presence of 20mM Na+ as compared to the absence of Na+. Loading mitochondria with the Na+ dye CoroNa Red confirmed that the Ca2+ efflux is dependent on mitochondrial Na+ uptake (R. Palty et al., 2010).
Regulation of NCLX
Members of the NCX superfamily of exchangers have an allosteric Ca2+-binding site within their regulatory region, which mediates NCX regulation by cytosolic Ca2+ (Doering et al., 1998). However, NCLX does not possess such Ca2+ regulatory site (Sekler, 2015). Sequence analysis of NCLX revealed many phosphorylation sites potentially involved in regulating NCLX function. A previous study showed that PTEN-induced putative kinase 1 (PINK1 ) is one of the regulators of mitochondrial Ca2+ efflux. Using primary cortical neurons from ΡΓΝΚ1 knockout mice and human dopaminergic neuroblastoma cell line (SH-SY5Y) with PINK1 knockdown, Gandhi et al. showed that PINK1 depletion resulted in reduced mitochondrial Ca2+ efflux rate (Gandhi et al, 2009). In PINK1 knockdown neurons, neuronal induction by UV caused irreversible increase in mitochondrial Ca2+ and this enhanced mitochondrial Ca2+ was associated with reduced mitochondrial Na+ uptake (Gandhi et al, 2009). A subsequent study by Kostic et al. also confirmed that downregulation of PINK1 in SH-SY5Y cells resulted in significant reduction of mitochondrial Ca2+ efflux rate (Kostic et al, 2015). Previous studies have shown that reduced mitochondrial membrane potential was rescued by activation of protein kinase A (PKA) (Dagda et al., 2011), suggesting that PINK1 could regulate NCLX through PKA-mediated NCLX phosphorylation. Kostic et al found that mitochondrial Ca2+ efflux rate was significantly increased upon PKA activation with forskolin in PINK1 knockdown SH-SY5Y cells (Kostic et al, 2015). NCLX has a putative PKA phosphorylation site at serine 258 (S258) located in the loop connecting aland a2 domains. A phosphorylation-deficient mutant of NCLX where S258 was mutated to alanine (S258A) failed to rescue mitochondrial Ca2+ efflux in NCLX knockdown SH-SY5Y cells while a phosphomimetic mutation (S258D) rescued mitochondrial Ca2+ efflux. Interestingly, neurons isolated from PINK1 knockout mice showed reduced Ca2+ efflux rate compared to wild- type neurons and overexpression of NCLX S258D, but not NCLX S258A, in neurons from PINK1 knockout mice rescued mitochondrial membrane potential and restored mitochondrial Ca2+ efflux. PINK1 depleted neurons are very sensitive to dopamine induced cell death and this was prevented by overexpression of NCLX S258D but not NCLX S258A (Kostic et al, 2015). While NCLX transports Na+ into mitochondria, the Na+/H+ exchanger (NHE), which is responsible for Na+ efflux from mitochondria into the cytosol, is major regulator of mitochondrial Na+ in several cell types (Bemardinelli, Azarias, & Chatton, 2006; Bers, Barry, & Despa, 2003; Murphy & Eisner, 2009). NHE is therefore critical for maintaining Na+ gradients across the mitochondrial membrane and its function is critical for NCLX activity and for the maintenance of Ca2+ homeostasis in mitochondria (Nita, Hershfinkel, Lewis, & Sekler, 2015). A genome wide Drosophila RNAi screen identified CG4589, which is a mammalian orthologue of Letml, as the mitochondrial Ca2+/H+ antiporter. Furthermore, downregulation of Letml reduced Ca2+/H+ antiporter activity, while overexpression of Letml resulted in increased Ca2+/H+ antiporter activity. Reconstitution of purified Letml in lysosomes also resulted in enhanced Ca2+transport (Jiang et al., 2009). These results show that in addition to NCLX, Letml can also extrude Ca2+ from the mitochondrial matrix into cytosol (Fig. 1). Downregulation of Letml resulted in impaired basal mitochondrial oxygen consumption, increased ROS production, AMP- activated protein kinase (AMPK) activation, autophagy and cell cycle arrest (Doonan et al., 2014), suggesting that Letml plays a crucial role in regulating cellular metabolism. Nevertheless, the identity of Letml as a Ca2+ exchanger is uncertain. Originally, Letml has been proposed to be a K+/H+ exchanger that regulates mitochondrial osmolarity (Dimmer et al., 2007; Hasegawa & van der Bliek, 2007; Hashimi, McDonald, Stribrna, & Lukes, 2013; Mitchell, 2011; Nowikovsky et al., 2004). Kinetic analysis showed that the turnover rate of Letml for Ca2+ is two cycles per second which is in the non-physiological range (Mingn Feng Tsai et ak, 2014). Further, the overexpression of Letml, which is insensitive to the NCLX blocker CGP-37157, in HeLa cells did not increase the Ca2+ efflux rate of mitochondria and did not enhance mitochondrial Ca2+ efflux in response to physiological stimulation (U. De Marchi et ak, 2014). It is possible that Letml does not work alone to regulate mitochondrial Ca2+, and rather works in conjunction with MCU and NCLX to maintain Ca2+ homeostasis in the mitochondrial matrix. A detailed study is required to decipher Letml stoichiometry, mechanisms of Ca2+ transport, and communication with MCU and NCLX during homeostatic and regulated mitochondrial Ca2+ transport.
Physiological role of NCLX
Nita et al showed that downregulation of NCLX in MIN6 cells or primary pancreatic β-cells inhibited mitochondrial Ca2+ efflux induced by either high glucose or by cell depolarization. This coincided with reduced rate and amplitude of the cytosolic Ca2+ signal and delayed insulin secretion in response to high glucose or membrane depolarization (Nita et ak, 2012), suggesting that NCLX plays a critical role in the communication between the plasma membrane and mitochondria and in regulating cytosolic Ca2+ signals and insulin secretion by pancreatic β-cells. A whole-genome genotyping of single nucleotide polymorphisms and exome sequencing discovered a patient with congenital hyperinsulinemia with a mutation in the NCLX gene. This NCLX mutation is caused by a change of tyrosine at position 564 to histidine (Proverbio et ak, 2013); this residue is located in the highly conserved putative transmembrane domain 12 (Takeuchi, Kim, & Matsuoka, 2015). However, it remained to be established whether this mutation results in altered NCLX function and loss of mitochondrial Ca2+ extrusion, thus leading to altered insulin secretion of pancreatic β cells. Kim et al showed that NCLX knockdown from A20 and DT40 B lymphocytes results in increased random migration and significantly reduced chemotaxis in response to the chemokine C-X-C motif chemokine 12-CXCL12 (Kim, Takeuchi, Hikida, & Matsuoka, 2016). Similarly, B lymphocytes derived from mouse spleen also showed markedly reduced chemotaxis after pharmacological inhibition of NCLX by CGP-37157. In NCLX knockdown cells, F-actin and Racl localization was altered leading to enhanced random cell migration (Kim et al, 2016).
There is growing evidence that astrocytes release neurotransmitters in response to increased cytosolic Ca2+ (Navarrete & Araque, 2008; Serrano, 2006). It was suggested that NCLX is enriched in mitochondria of astrocytes and plays an important role in glutamate release, cell proliferation and wound healing (Parnis et al, 2013). Downregulation or pharmacological inhibition of NCLX in primary astrocytes resulted in significantly higher mitochondrial Ca2+ levels and decrease in store-operated Ca2+ entry (SOCE) when cells were challenged externally by the purinergic agonist ATP. Pamis et al showed that basal glutamate release, proliferation and migration were reduced in astrocytes with NCLX knockdown (Parnis et al, 2013). A study in Caenorhabditis elegcms showed that the NCLX like protein NCX-9 (which has similar Ca2+ handling properties as NCLX) plays a role in neural circuit development, specifically in axonal guidance (Sharma, Roy, Sekler, & O’Halloran, 2017). In fibroblasts derived from familial Parkinson’s disease patients where leucine-rich repeat kinase 2 (LRRK2) is mutated, MCU and MICU1 genes (which are located on the same chromosome in humans) were transcriptionally upregulated. In a mouse model, overexpression of LRRK2 also resulted in upregulation of MCU and MICU1. Interestingly, no change in MICU2 or NCLX levels was observed. Knockdown or pharmacological inhibition of MCU as well as overexpression of a constitutively active NCLX allele appeared to be neuroprotective, showing that tight mitochondrial Ca2+ homeostasis is required for neuronal activity and function (Verma et al, 2017). Taken together, these studies suggest that NCLX plays a role in neural development, transmission and plasticity. More detailed studies are required to fully uncover the mechanisms of NCLX regulation of neuronal signaling and plasticity.
Takeuchi et al showed that NCLX modulates action potential and regulates automaticity in HL-1 mouse atrial cardiomyocytes. Downregulation of NCLX resulted in reduced frequency of spontaneous Ca2+ oscillations and action potentials due to significantly slow and prolonged cycle length of each action potential. The authors showed that Ca2+ content and reuptake in the sarcoplasmic reticulum (SR) was reduced in NCLX knockdown HL-1 cells, likely explaining the prolonged cycle length of spontaneous action potentials (Takeuchi, Kim, & Matsuoka, 2013). Mathematical modeling of HL-1 cardiomyocyte showed that blocking NCLX caused a reduction in SR Ca2+ content leading to prolongation of the cycle length of the spontaneous action potential. These findings suggest that functional Ca2+ extrusion from mitochondria by NCLX contributes to SR Ca2+ refilling during action potentials. While inhibition of NCLX by CGP37157 resulted in slower rabbit sinoatrial (SA) node automaticity (Yaniv et al, 2012), these results are based on an NCLX inhibitor with questionable specificity. Indeed, CGP37157 is also a blocker of L-type Ca2+ channels, which are required for automaticity of sinoatrial (SA) cells (Takeuchi et al., 2015).
A recent study showed that tamoxifen-induced deletion of NCLX in adult mouse heart resulted in myocardial dysfunction leading to sudden cardiac arrest and death (Luongo et al, 2017). Adult cardiac myocytes lacking NCLX showed loss of mitochondrial Ca2+ efflux and significant reduction of mitochondrial Ca2+ uptake. These NCLX knockout cardiomyocytes showed reduced protein levels of EMRE while the expression levels of MCU, MCUb, MICU1, LETM1 or electron transport chain proteins were normal in these cells. No significant changes in cytosolic Ca2+ were observed in these NCLX knockout cells. Cardiomyocytes with NCLX knockout showed increased mitochondrial Ca2+, mitochondrial swelling (due to opening of mitochondrial permeability transition pore; MPTP), increased mitochondrial superoxide generation and enhanced necrotic cell death within two days of tamoxifen treatment. Luongo et al. also generated a mouse in which NCLX overexpression was specifically induced in cardiomyocytes with doxycycline. Overexpression of NCLX resulted in increased mitochondrial Ca2+ efflux by −38%; this enhanced Ca2+ efflux activity was sufficient to inhibit MPTP activity. Overexpression of NCLX did not affect basal respiration, ATP-linked respiration and proton leak, but it inhibited myocardial infraction, reduced post infraction fibrosis, inflammation, superoxide generation andcell death caused by mitochondrial Ca2+ overload (Luongo et al, 2017). Another study showed that cardiomyocyte-specific knockout mouse for the main transcription factor for mitochondrial DNA (transcription factor A, mitochondrial, Tfam) develop dilated cardiomyopathy associated with altered oxidative phosphorylation and this correlated with enhanced mitochondrial Ca2+ due to increased MCU and reduced NCLX expression and activity (Sommakia et al, 2017). There is no obvious reason that could explain the phenotype of cardiomyocyte-specific NCLX KO mice, which is more drastic than that of cardiomyocyte-specific MCU KO mice. One possible explanation is that another Ca2+ conducting channel can compensate for the lack of MCU. However, NCLX being a slower transporter and the rate-limiting step in Ca2+ efflux from mitochondria, cannot be easily compensated for by another transporter.
Pharmacological inhibition of mitochondrial Ca2+ extrusion by the NCLX inhibitor CGP-37157 or the use of mitochondria-depolarizing compounds partially inhibited SOCE, but when both strategies were used together SOCE was completely inhibited (Naghdi et al, 2010). When the authors physically immobilized mitochondria at the plasma membrane using an mAKAP-RFP- CAAX linker, SOCE activity was not altered, questioning the prominence of the proximity- dependent mechanism involving Ca2+ buffering by mitochondria in regulating SOCE and suggesting the existence of additional mechanisms independent of buffering (Naghdi et al, 2010). A more recent study by Ben-Kassus Nissim et al showed that molecular downregulation of NCLX proteins in HEK293 cells resulted in partial inhibition of SOCE and its biophysical correlate, the Ca2+ release activated Ca2+ (CRAC) current (Ben- Kasus Nissim et al, 2017), in agreement with the findings of Naghdi et al, Ben-Kassus Nissim et al used whole cell patch clamp electrophysiology to rule out a role for mitochondrial Ca2+ buffering in this mode of SOCE regulation by mitochondria and demonstrated that instead NCLX knockdown causes mitochondrial Ca2+ overload which in turn leads to upregulation of mitochondrial reactive oxygen species (ROS) production. ROS act directly on the redox-sensitive Cysteine 195 within the SOCE channel ORAI1 to inhibit SOCE and CRAC currents (Ben- Kasus Nissim et al, thus providing evidence for a proximity-independent role of mitochondria in regulationg plasma membrane channels through ROS-mediated ORAI1 channel inhibition.
Future Directions
The discovery of MCU and NCLX has shed light on how mitochondria contribute to shaping cellular Ca2+ signaling to control metabolism and growth. This provided a clearer picture on how distinct Ca2+ stores within the cell communicate with one another to shuttle Ca2+ ions, ensuring precise temporal and spatial Ca2+ signaling required for a myriad of cellular processes. In the past decade, much work has been focused on understanding the mechanisms of Ca2+ uptake into mitochondria through MCU. Although the discovery of components of the MCU uniporter complex provided valuable insights into the regulatory mechanisms of mitochondrial Ca2+ uptake, several questions remain unanswered. For instance, the stoichiometry of the MCU complex and whether this stoichiometry is dynamically regulated within the same cell or whether its components vary between different cell types remain unknown. The heterogeneity of the MCU complex during cell signaling or within different tissue types might be due to the existence of multiple isoforms of MICU regulators. For example, the role of MICU3, which was proposed to be invovled in the central nervous system, remain unclear (Plovanich et al, 2013). Similarly,other regulators of MCU such as SLAC25A23 and MCUb await detailed and thorough investigations. Additional studies are required to unravel the stoichiometry and signaling pathways regulating NCLX activity. The exact orientation of NCLX within IMM remains unknown and whether S258 is located in the matrix or the intermembrane space is also unknown. The mechanisms of PKA phosphorylation of NCLX are also not clear. Regardless of where S258 is located, the presence of PKA in either the mitochondrial matrix or the intermembrane space is a highly contentious idea. NCLX has been recently proposed to have an important role in heart function and neuronal plasticity (Luongo et al, 2017; Parnis et al, 2013). However, the exact mechanisms behind this regulation remain elusive. Future high resolution structures of MCU bound to its regulators in the presence and absence of Ca2+ will provide crucial insights on MCU channel gating and regulation. Similarly, structure of NCLX and Letml have not been reported. As we uncover the permeation and regulatory mechanisms of mitochondrial Ca2+ transport and identify potential cell-specific heterogeneities in these mechanisms, we will be one step closer to harness this knowledge for the purpose of disease therapy.
Acknowledgements
Research in the authors’ laboratories is supported by grants R01HL123364, R01HL097111, R21AG050072 from the National Institutes of Health, and grant NPRP8–110-3–021 from the Qatar National Research Fund (QNRF) to MT.
Abbreviations
- AMPK
AMP-activated protein kinase
- ATP
adenosine triphosphate
- CaMKII
Ca2+/calmodulin- dependent protein kinase II
- CREB
cyclic adenosine monophosphate response element-binding protein
- CXCL12
C-X-C motif chemokine 12
- EMRE
essential MCU regulator
- ER
endoplasmic reticulum
- ETC
electron transport chain
- HIF-lα
hypoxia-inducible factor-lα
- IMM
inner mitochondrial membrane
- IP3R
inositol-1,4,5-trisphosphate receptor
- IS
immunological synapse
- MAMs
mitochondria associated membranes
- MCU
mitochondrial Ca2+ uniporter
- MCUb
MCU regulatory subunit b
- MCUR1
mitochondrial Ca2+ regulator 1
- MICU1
mitochondrial calcium uptake 1
- MPTP
mitochondrial permeability transition pore
- NCLX
Na+/Ca2+/Li+ exchanger
- NCX
Na+/Ca2+ exchangers
- NCKX
Na+/Ca2+/K+ exchanger
- NFAT
nuclear factor of activated T-cells
- NF-қB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NHE
Na+/H+ exchanger
- NMDA
N-Methyl-D-aspartic acid
- OMM
outer mitochondrial membrane
- PINK1
PTEN-induced putative kinase 1
- PKA
protein kinase A
- PS
phosphatidylserine
- RIPK1
receptor-interacting protein kinase 1
- ROS
reactive oxygen species
- SERCA
sarco/endoplasmic reticulum Ca2+ ATPase
- SLC8B1
solute carrier family 8-member Bl
- SLC24A6
solute carrier family 24 member 6
- SLC25A23
solute carrier family 25 member 23
- SOCE
store-operated Ca2+ entry
- SR
sarcoplasmic reticulum
- STIM
stromal interaction molecule
- VDAC
voltage-dependent anion channel
- UPR
unfolded protein response
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amigo I, Traba J, González-Barroso MM, Rueda CB, Fernández M, Rial E, ... Del Arco A (2013). Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+ activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. Journal of Biological Chemistry, 288(11), 7791–7802. https://doi.org/10.1074/jbc.M112.409144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprille JR (1988). Regulation of the mitochondrial adenine nucleotide pool size in liver: mechanism and metabolic role. The FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 2(10), 2547–2556. [DOI] [PubMed] [Google Scholar]
- Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, Leimpek A, ... Perocchi F (2017). Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Molecular Cell, 67(4), 711–723. e7 https://doi.org/10.1016/J.MOLCEL.2017.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudeau S, Kelley WL, Walsh JV, & Demaurex N (2001). Mitochondria Recycle Ca2+ to the Endoplasmic Reticulum and Prevent the Depletion of Neighboring Endoplasmic Reticulum Regions. Journal of Biological Chemistry, 276(31), 29430–29439. https://doi.org/10.1074/jbc.M103274200 [DOI] [PubMed] [Google Scholar]
- Bantug GR, Fischer M, Grählert J, Balmer ML, Unterstab G, Develioglu L, ... Hess C (2018). Mitochondria-Endoplasmic Reticulum Contact Sites Function as Immunometabolic Hubs that Orchestrate the Rapid Recall Response of Memory CD8+T Cells. Immunity, 48(3), 542–555.e6. https://doi.org/10.1016/j.immuni.2018.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, ... Mootha VK (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature, 476(7360), 341–345. https://doi.org/10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben- Kasus Nissim T, Zhang X, Elazar A, Roy S, Stolwijk JA, Zhou Y, ... Sekler I (2017). Mitochondria control store- operated Ca 2+ entry through Na + and redox signals. [DOI] [PMC free article] [PubMed]
- The EMBO Journal, 36(6), 797–815. https://doi.org/10.15252/embj.201592481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardinelli Y, Azarias G, & Chatton JY (2006). In situ fluorescence imaging of glutamate-evoked mitochondrial Na + responses in astrocytes. Glia, 54(5), 460–470. https://doi.org/10.1002/glia.20387 [DOI] [PubMed] [Google Scholar]
- Bers DM, Barry WH, & Despa S (2003). Intracellular Na+ regulation in cardiac myocytes. Cardiovascular Research, 57(4), 897–912. https://doi.org/10.1016/S0008-6363(02)00656-9 [DOI] [PubMed] [Google Scholar]
- Booth DM, Enyedi B, Geiszt M, Várnai P, & Hajnóczky G (2016). Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Molecular Cell, 63(2), 240–248. https://doi.org/10.1016/j.molcel.2016.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, & Lytton J (2004). Molecular Cloning of a Sixth Member of the K+-dependent Na +/Ca2+ Exchanger Gene Family, NCKX6. Journal of Biological Chemistry, 279(7), 5867–5876. https://doi.org/10.1074/jbc.M310908200 [DOI] [PubMed] [Google Scholar]
- Cao C, Wang S, Cui T, Su X-C, & Chou JJ (2017). Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proceedings of the National Academy of Sciences, 114(14), E2846–E2851. https://doi.org/10.1073/pnas.1620316114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, & Lehninger AL (1971). A survey of the interaction of calcium ions with mitochondria from different tissues and species. The Biochemical Journal, 122(5), 681–90. https://doi.org/10.1042/bj1220681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers S, & Nicholls DG (2003). The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. Journal of Biological Chemistry, 278(21), 19062–19070. https://doi.org/10.1074/jbc.M212661200 [DOI] [PubMed] [Google Scholar]
- Chaudhuri D, Artiga DJ, Abiria SA, & Clapham DE (2016). Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proceedings of the National Academy of Sciences, 113(13), E1872–E1880. https://doi.org/10.1073/pnas.1602264113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, & Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. ELife, 2013(2), e00704 https://doi.org/10.7554/eLife.00704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, ... Hajnóczky G (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+uniporter. Cell Metabolism, 17(6), 976–987. https://doi.org/10.1016/j.cmet.2013.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, ... Chu CT (2011). Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death and Differentiation, 18(12), 1914–1923. https://doi.org/10.1038/cdd.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, & Demaurex N (2014). NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. Journal of Biological Chemistry, 289(29), 20377–20385. https://doi.org/10.1074/jbc.M113.540898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabó I, & Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature, 476(7360), 336–340. https://doi.org/10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R, & Pozzan T (2016). Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annual Review of Biochemistry, 85(1), 161–192. https://doi.org/10.1146/annurev-biochem-060614-034216 [DOI] [PubMed] [Google Scholar]
- Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, ... Malli R (2014). IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. Journal of Cell Science, 127(13), 2944–2955. https://doi.org/10.1242/jcs.149807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca HF, & Engstrom GW (1961). Calcium Uptake By Rat Kidney Mitochondria. Proceedings of the National Academy of Sciences, 47(11), 1744–1750. https://doi.org/10.1073/pnas.47.11.1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, ... Scorrano L (2007). LETM1, deleted in Wolf Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Human Molecular Genetics, 17(2), 201–214. https://doi.org/10.1093/hmg/ddm297 [DOI] [PubMed] [Google Scholar]
- Doering AE, Nicoll DA, Lu Y, Lu L, Weiss JN, & Philipson KD (1998). Topology of a functionally important region of the cardiac Na+/Ca2+ exchanger. Journal of Biological Chemistry, 273(2), 778–783. https://doi.org/10.1074/jbc.273.2.778 [DOI] [PubMed] [Google Scholar]
- Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, ... Madesh M (2017). Mitochondrial Ca2+Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Molecular Cell, 65(6), 1014–1028.e7. https://doi.org/10.1016/j.molcel.2017.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cárdenas C, Shanmughapriya S, ... Madesh M (2014). LETM1-dependent mitochondrial Ca2+flux modulates cellular bioenergetics and proliferation. FASEB Journal, 28(11), 4936–4949. https://doi.org/10.1096/fj.14-256453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, & Davis RL (2016). Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Reports, 16(10), 2763–2776. https://doi.org/10.1016/j.celrep.2016.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, & Palmieri F (2004). Identification of the Mitochondrial ATP-Mg/P i Transporter. Journal of Biological Chemistry, 279(29), 30722–30730. https://doi.org/10.1074/jbc.M400445200 [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, ... Abramov AY (2009). PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Molecular Cell, 33(5), 627–638. https://doi.org/10.1016/j.molcel.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, ... Valente EM (2017). PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy, 13(4), 654–669. https://doi.org/10.1080/15548627.2016.1277309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, ... Pinton P (2012). Mitochondrial Ca2+and apoptosis. Cell Calcium, 52(1), 36–43. https://doi.org/10.1016/j.ceca.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, & Miller CCJ (2017). The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Current Biology, 27(3), 371–385. https://doi.org/10.1016/j.cub.2016.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Larson-Casey JL, & Carter AB (2017). Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB Journal, 31(7), 3072–3083. https://doi.org/10.1096/fj.201601371R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, & Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. https://doi.org/10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hansford RG (1994). Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr, 26(5), 495–508. [DOI] [PubMed] [Google Scholar]
- Harborne SPD, King MS, Crichton PG, & Kunji ERS (2017). Calcium regulation of the human mitochondrial ATP-Mg/Pi carrier SLC25A24 uses a locking pin mechanism. Scientific Reports, 7, 45383 https://doi.org/10.1038/srep45383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa A, & van der Bliek AM (2007). Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Human Molecular Genetics, 16(17), 2061–2071. https://doi.org/10.1093/hmg/ddm154 [DOI] [PubMed] [Google Scholar]
- Hashimi H, McDonald L, Stříbrná E, & Lukeš J (2013). Trypanosome Letm1 Protein Is Essential for Mitochondrial Potassium Homeostasis. Journal of Biological Chemistry, 288(37), 26914–26925. https://doi.org/10.1074/jbc.M113.495119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, ... Madesh M (2013). MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Reports, 5(6), 1576–1588. https://doi.org/10.1016/j.celrep.2013.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, ... Madesh M (2014). SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Molecular Biology of the Cell, 25(6), 936–947. https://doi.org/10.1091/mbc.E13-08-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, ... Finkel T (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. Journal of Molecular and Cellular Cardiology, 85, 178–182. https://doi.org/10.1016/j.yjmcc.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Button DC, & Lewis RS (2000). Mitochondrial control of calcium-channel gating: A mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proceedings of the National Academy of Sciences, 97(19), 10607–10612. https://doi.org/10.1073/pnas.180143997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Vercesi AE, & Docampo R (2013). Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nature Communications, 4(1), 2865 https://doi.org/10.1038/ncomms3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, & Clapham DE (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca 2+/H+ antiporter. Science, 326(5949), 144–147. https://doi.org/10.1126/science.1175145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner MLA, Koval OM, Li J, Julie He B, Allamargot C, Gao Z, ... Anderson ME (2012). CaMKII determines mitochondrial stress responses in heart. Nature, 491(7423), 269–273. https://doi.org/10.1038/nature11444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Grabarek Z, & Mootha VK (2017). High- affinity cooperative Ca 2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Reports, 18(8), e201643748 https://doi.org/10.15252/embr.201643748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Takeuchi A, Hikida M, & Matsuoka S (2016). Roles of the mitochondrial Na+- Ca2+exchanger, NCLX, in B lymphocyte chemotaxis. Scientific Reports, 6(1), 28378 https://doi.org/10.1038/srep28378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, & Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature, 427(6972), 360–364. https://doi.org/10.1038/nature02246 [DOI] [PubMed] [Google Scholar]
- König T, Tröder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, ... Langer T (2016). The m -AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Molecular Cell, 64(1), 148–162. https://doi.org/10.1016/j.molcel.2016.08.020 [DOI] [PubMed] [Google Scholar]
- Kostic M, Ludtmann MHR, Bading H, Hershfinkel M, Steer E, Chu CT, ... Sekler I (2015). PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Reports, 13(2), 376–386. https://doi.org/10.1016/j.celrep.2015.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, ... Molkentin JD (2015). The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Reports, 12(1), 15–22. https://doi.org/10.1016/j.celrep.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehninger AL, Carafoli E, & Rossi CS (1967). Energy-linked ion movements in mitochondrial systems. Adv Enzymol Relat Areas Mol Biol, 29, 259–320. [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, ... Chen Q (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature Cell Biology, 14(2), 177–185. https://doi.org/10.1038/ncb2422 [DOI] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, ... Elrod JW (2017). The mitochondrial Na+/Ca2+exchanger is essential for Ca2+homeostasis and viability. Nature, 545(7652), 93–97. https://doi.org/10.1038/nature22082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, ... Elrod JW (2015). The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Reports, 12(1), 23–34. https://doi.org/10.1016/j.celrep.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madesh M, & Hajnóczky G (2001). VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. Journal of Cell Biology, 155(6), 1003–1015. https://doi.org/10.1083/jcb.200105057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, ... Madesh M (2012). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nature Cell Biology, 14(12), 1336–1343. https://doi.org/10.1038/ncb2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Müller M, Miller R, ... Madesh M (2012). MICU1 is an essential gatekeeper for mcu-mediated mitochondrial Ca 2+ uptake that regulates cell survival. Cell, 151(3), 630–644. https://doi.org/10.1016/j.cell.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, ... Rizzuto R (2015). The Mitochondrial Calcium Uniporter Controls Skeletal Muscle Trophism InVivo. Cell Reports, 10(8), 1269–1279. https://doi.org/10.1016/j.celrep.2015.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella C. a, Buttle K, Rath BK, & Marko M (1998). Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. BioFactors (Oxford, England), 8(3–4), 225–228. https://doi.org/10.1002/biof.5520080309 [DOI] [PubMed] [Google Scholar]
- Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, ... Pinton P (2013). Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Current Biology, 23(1), 58–63. https://doi.org/10.1016/j.cub.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, & Denton RM (1979). The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochemical Journal, 180(3), 533–544. https://doi.org/10.1042/bj1800533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, & Denton RM (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiological Reviews, 70(2), 391–425. https://doi.org/10.1152/physrev.1990.70.2.391 [DOI] [PubMed] [Google Scholar]
- Mesmin B (2016). Mitochondrial lipid transport and biosynthesis: A complex balance. Journal of Cell Biology, 214(1), 9–11. https://doi.org/10.1083/jcb.201606069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (2011). Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1807(12), 1507–1538. https://doi.org/10.1016/j.bbabio.2011.09.018 [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibá̃ez T, Albillos A, García AG, ... Alvarez J (2000). Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+transients that modulate secretion. Nature Cell Biology, 2(2), 57–61. https://doi.org/10.1038/35000001 [DOI] [PubMed] [Google Scholar]
- Murphy E, & Eisner DA (2009). Regulation of intracellular and mitochondiral sodium in health and disease. Circ Res, 104(3), 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghdi S, Waldeck-Weiermair M, Fertschai I, Poteser M, Graier WF, & Malli R (2010). Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1- dependent store-operated Ca2+ entry. Journal of Cell Science, 123(15), 2553–2564. https://doi.org/10.1242/jcs.070151 [DOI] [PubMed] [Google Scholar]
- Navarrete M, & Araque A (2008). Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron, 57(6), 883–893. https://doi.org/10.1016/j.neuron.2008.01.029 [DOI] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, ... Sekler I (2012). The Mitochondrial Na +/Ca 2+ Exchanger Upregulates Glucose Dependent Ca 2+ Signalling Linked to Insulin Secretion. PLoS ONE, 7(10), e46649 https://doi.org/10.1371/journal.pone.0046649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Lewis EC, & Sekler I (2015). A crosstalk between Na+channels, Na+/K+pump and mitochondrial Na+transporters controls glucose-dependent cytosolic and mitochondrial Na+signals. Cell Calcium, 57(2), 69–75. https://doi.org/10.1016/j.ceca.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, ... Schweyen RJ (2004). The LETM1 / YOL027 Gene Family Encodes a Factor of the Mitochondrial K + Homeostasis with a Potential Role in the Wolf-Hirschhorn Syndrome. Journal of Biological Chemistry, 279(29), 30307–30315. https://doi.org/10.1074/jbc.M403607200 [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, ... Chou JJ (2016). Architecture of the mitochondrial calcium uniporter. Nature, 533, 269–273. https://doi.org/10.1038/nature17656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, ... Mootha VK (2008). A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell, 134(1), 112–123. https://doi.org/10.1016/j.cell.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, ... Sekler I (2004). Lithium-calcium exchange is mediated by a distinct potassium-independent sodium- calcium exchanger. Journal of Biological Chemistry, 279(24), 25234–25240. https://doi.org/10.1074/jbc.M401229200 [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, ... Sekler I (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proceedings of the National Academy of Sciences, 107(1), 436–441. https://doi.org/10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, ... Finkel T (2013). The Physiological Role of Mitochondrial Calcium Revealed by Mice Lacking the Mitochondrial Calcium Uniporter. Nature Cell Biology, 15(12), 1464–1472. https://doi.org/10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Ashby MC, Erdemli G, Petersen OH, & Tepikin AV (2001). Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO Journal, 20(8), 1863–1874. https://doi.org/10.1093/emboj/20.8.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, ... Nolte C (2013). Mitochondrial Exchanger NCLX Plays a Major Role in the Intracellular Ca2+ Signaling, Gliotransmission, and Proliferation of Astrocytes. Journal of Neuroscience, 33(17), 7206–7219. https://doi.org/10.1523/JNEUROSCI.5721-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, VecellioReane D, Mantoan M, ... Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Molecular Cell, 53(5), 726–737. https://doi.org/10.1016/j.molcel.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Granatiero V, Espino J, Rizzuto R, & De Stefani D (2018). MICU3 is a tissue- specific enhancer of mitochondrial calcium uptake. Cell Death & Differentiation, 1 https://doi.org/10.1038/s41418-018-0113-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupe V, Prudent J, Dassa EP, Rendon OZ, & Shoubridge EA (2015). CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metabolism, 21(1), 109–116. https://doi.org/10.1016/j.cmet.2014.12.004 [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, & Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+uptake. Nature, 467(7313), 291–296. https://doi.org/10.1038/nature09358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrungaro C, Zimmermann KM, Küttner V, Fischer M, Dengjel J, Bogeski I, & Riemer J (2015). The Ca2+-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca2+ uptake. Cell Metabolism, 22(4), 721–733. https://doi.org/10.1016/j.cmet.2015.08.019 [DOI] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, ... Mootha VK (2013). MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS ONE, 8(2), e55785 https://doi.org/10.1371/journal.pone.0055785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proverbio MC, Mangano E, Gessi A, Bordoni R, Spinelli R, Asselta R, ... Battaglia C (2013). Whole Genome SNP Genotyping and Exome Sequencing Reveal Novel Genetic Variants and Putative Causative Genes in Congenital Hyperinsulinism. PLoS ONE, 8(7), e68740 https://doi.org/10.1371/journal.pone.0068740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, Popgeorgiev N, Bonneau B, Thibaut J, Gadet R, Lopez J, ... Gillet G (2013). Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nature Communications, 4 https://doi.org/10.1038/ncomms3330 [DOI] [PubMed] [Google Scholar]
- Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, ... Hoth M (2011). Calcium microdomains at the immunological synapse: How ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO Journal, 30(19), 3895–3912. https://doi.org/10.1038/emboj.2011.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, & Hoth M (2007). T cell activation requires mitochondrial translocation to the immunological synapse. Proceedings of the National Academy of Sciences, 104(36), 14418–14423. https://doi.org/10.1073/pnas.0703126104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stéfani D, Sabbadin D, Teardo E, Merli G, Picard A, ... Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominantnegative pore-forming subunit. EMBO Journal, 32(17), 2362–2376. https://doi.org/10.1038/emboj.2013.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, ... Rizzuto R (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca 2+ microdomains to mitochondria. The Journal of Cell Biology, 159(4), 613–624. https://doi.org/10.1083/jcb.200205091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieusset J (2018). The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death and Disease, 9(3), 388 https://doi.org/10.1038/s41419-018-0416-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, & Pinton P (2015). Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nature Communications, 6 https://doi.org/10.1038/ncomms7201 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, & Mammucari C (2012). Mitochondria as sensors and regulators of calcium signalling. Nature Reviews Molecular Cell Biology, 13(9), 566–578. https://doi.org/10.1038/nrm3412 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, ... Pozzan T (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+responses. Science, 280(5370), 1763–1766. https://doi.org/10.1126/science.280.5370.1763 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AWM, Brini M, & Pozzan T (1992). Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature, 358(6384), 325–327. https://doi.org/10.1038/358325a0 [DOI] [PubMed] [Google Scholar]
- Rueda CB, Traba J, Amigo I, Llorente-Folch I, Gonzalez-Sanchez P, Pardo B, ... Satrustegui J (2015). Mitochondrial ATP-Mg/Pi Carrier SCaMC-3/Slc25a23 Counteracts PARP-1-Dependent Fall in Mitochondrial ATP Caused by Excitotoxic Insults in Neurons. Journal of Neuroscience, 35(8), 3566–3581. https://doi.org/10.1523/JNEUROSCI.2702-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, ... Mootha VK (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science, 342(6164), 1379–1382. https://doi.org/10.1126/science.1242993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, ... Claret M (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell, 155(1), 172–187. https://doi.org/10.1016/j.cell.2013.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekler I (2015). Standing of giants shoulders the story of the mitochondrial Na+Ca2+ exchanger. Biochemical and Biophysical Research Communications. https://doi.org/10.1016/j.bbrc.2015.02.170 [DOI] [PubMed] [Google Scholar]
- Selles B, Michaud C, Xiong T-C, Leblanc O, & Ingouff M (2018). Arabidopsis pollen tube germination and growth depend on the mitochondrial calcium uniporter complex. The New Phytologist, 219(1), 58–65. https://doi.org/10.1111/nph.15189 [DOI] [PubMed] [Google Scholar]
- Serrano A (2006). GABAergic Network Activation of Glial Cells Underlies Hippocampal Heterosynaptic Depression. Journal of Neuroscience, 26(20), 5370–5382. https://doi.org/10.1523/JNEUROSCI.5255-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, ... Madesh M (2015). Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Science Signaling, 8(366), ra23 https://doi.org/10.1126/scisignal.2005673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Roy S, Sekler I, & O’Halloran DM (2017). The NCLX-type Na+/Ca2+exchanger NCX-9 is required for patterning of neural circuits in Caenorhabditis elegans. Journal of Biological Chemistry, 292(13), 5364–5377. https://doi.org/10.1074/jbc.M116.758953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, ... Chaudhuri D (2017). Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. Journal of Molecular and Cellular Cardiology, 113, 22–32. https://doi.org/10.1016/j.yjmcc.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock D, Leslie AGW, & Walker JE (1999). Molecular architecture of the rotary motor in ATP synthase. Science, 286(5445), 1700–1705. https://doi.org/10.1126/science.286.5445.1700 [DOI] [PubMed] [Google Scholar]
- Suarez J, Cividini F, Scott BT, Lehmann K, Diaz-Juarez J, Diemer T, ... Dillmann WH (2018). Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. Journal of Biological Chemistry, (1), jbc.RA118.002066. https://doi.org/10.1074/jbc.RA118.002066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, ... Rizzuto R (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. Journal of Cell Biology, 175(6), 901–911. https://doi.org/10.1083/jcb.200608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Kim B, & Matsuoka S (2013). The mitochondrial Na+-Ca 2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Scientific Reports, 3(1), 2766 https://doi.org/10.1038/srep02766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Kim B, & Matsuoka S (2015). The destiny of Ca2+ released by mitochondria. The Journal of Physiological Sciences, 65(1), 11–24. https://doi.org/10.1007/s12576-014-0326-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, ... Zou F (2015). Mitochondrial Ca2+uniporter is critical for store-operated Ca2+entry-dependent breast cancer cell migration. Biochemical and Biophysical Research Communications (Vol. 458). https://doi.org/10.1016/j.bbrc.2015.01.092 [DOI] [PubMed] [Google Scholar]
- Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, ... Rutter GA (2012). The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced atp increases in pancreatic β-cells. PLoS ONE, 7(7). https://doi.org/10.1371/journal.pone.0039722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teardo E, Carraretto L, Wagner S, Formentin E, Behera S, De Bortoli S, ... Szabó I (2017). Physiological Characterization of a Plant Mitochondrial Calcium Uniporter in Vitro and in Vivo. Plant Physiology, 173(2), 1355–1370. https://doi.org/10.1104/pp.16.01359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari SG, Dash RK, Beard DA, & Bazil JN (2012). A biophysical model of the mitochondrial ATP-Mg/Pi carrier. Biophysical Journal, 103(7), 1616–1625. https://doi.org/10.1016/j.bpj.2012.08.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurey P, Tubbs E, Vial G, Jacquemetton J, Bendridi N, Chauvin MA, ... Rieusset J (2016). Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. Journal of Molecular Cell Biology, 8(2), 129–143. https://doi.org/10.1093/jmcb/mjw004 [DOI] [PubMed] [Google Scholar]
- Thivolet C, Vial G, Cassel R, Rieusset J, & Madec A-M (2017). Reduction of endoplasmic reticulum- mitochondria interactions in beta cells from patients with type 2 diabetes. PloS One, 12(7), e0182027 https://doi.org/10.1371/journal.pone.0182027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, ... Madesh M (2016). MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Reports, 15(8), 1673–1685. https://doi.org/10.1016/j.celrep.2016.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, ... Mammucari C (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1a. EMBO Mol Med, 8(5), 569–585. https://doi.org/10.15252/emmm [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, & Putney JW (2017). ORAI Calcium Channels. Physiology, 32(4), 332–342. https://doi.org/10.1152/physiol.00011.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troilo A, Alexander I, Muehl S, Jaramillo D, Knobeloch K, & Krek W (2014). HIF1a deubiquitination by USP 8 is essential for ciliogenesis in normoxia. EMBO Reports, 15(1), 77–85. https://doi.org/10.1002/embr [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C-W, Wu Y, Pao P-C, Phillips CB, Williams C, Miller C, ... Tsai M-F (2017). Proteolytic control of the mitochondrial calcium uniporter complex. Proceedings of the National Academy of Sciences of the United States of America, 114(17), 4388–4393. https://doi.org/10.1073/pnas.1702938114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Jiang D, Zhao L, Clapham D, & Miller C (2014). Functional reconstitution of the mitochondrial Ca 2+ /H + antiporter Letm1. The Journal of General Physiology, 143(1), 67–73. https://doi.org/10.1085/jgp.201311096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Williams C, & Miller C (2016). Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. ELife, 5(APRIL2016), 1–17. https://doi.org/10.7554/eLife.15545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs E, Chanon S, Robert M, Bendridi N, Bidaux G, Chauvin M-A, ... Rieusset J (2018). Disruption of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes, 67(4), db170316 https://doi.org/10.2337/db17-0316 [DOI] [PubMed] [Google Scholar]
- Vais H, Mallilankaraman K, Mak DOD, Hoff H, Payne R, Tanis JE, & Foskett JK (2016). EMRE Is a Matrix Ca2+Sensor that Governs Gatekeeping of the Mitochondrial Ca2+Uniporter. Cell Reports, 14(3), 403–410. https://doi.org/10.1016/j.celrep.2015.12.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Tanis JE, Müller M, Payne R, Mallilankaraman K, & Foskett JK (2015). MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metabolism. https://doi.org/10.1016/j.cmet.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, Raffaello A (2016). A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Molecular Cell, 64(4), 760–773. https://doi.org/10.1016/j.molcel.2016.10.001 [DOI] [PubMed] [Google Scholar]
- Verma M, Callio J, Otero PA, Sekler I, Wills ZP, & Chu CT (2017). Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD- Associated LRRK2 Mutants. The Journal of Neuroscience, 37(46), 11151–11165. https://doi.org/10.1523/JNEUROSCI.3791-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G (1992). Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. Journal of Molecular Biology, 225(2), 487–494. https://doi.org/10.1016/0022-2836(92)90934-C [DOI] [PubMed] [Google Scholar]
- Writzl K, Maver A, Kovačič L, Martinez-Valero P, Contreras L, Satrustegui J, ... Hennekam RC (2017). De Novo Mutations in SLC25A24 Cause a Disorder Characterized by Early Aging, Bone Dysplasia, Characteristic Face, and Early Demise. American Journal of Human Genetics, 101(5), 844–855. https://doi.org/10.1016/j.ajhg.2017.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, ... Feng D (2016). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. The EMBO Journal, 35(13), 1368–1384. https://doi.org/10.15252/embj.201593102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval ΟM, Joiner MLA, Hall DD, Chen B, ... Anderson ME (2015). The mitochondrial uniporter controls fight or flight heart rate increases. Nature Communications, 6, 6081 https://doi.org/10.1038/ncomms7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ren T, Wang J, Zhang H, Yuan P, Zhu J, ... Yang H (2017). MCUR1-mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via ROS-dependent P53 degradation. Antioxidants & Redox Signaling, ars.20176990 https://doi.org/10.1089/ars.2017.6990 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yamagoshi R, Harada K, Kawano M, Minami N, Ido Y, ... Shinohara Y (2016). Analysis of the structure and function of EMRE in a yeast expression system. Biochimica et Biophysica Acta - Bioenergetics, 1857(6), 831–839. https://doi.org/10.1016/j.bbabio.2016.03.019 [DOI] [PubMed] [Google Scholar]
- Yaniv Y, Spurgeon HA, Lyashkov AE, Yang D, Ziman BD, Maltsev VA, & Lakatta EG (2012). Crosstalk between mitochondrial and sarcoplasmic reticulum Ca2+ cycling modulates cardiac pacemaker cell automaticity. PLoS ONE, 7(5), e37582 https://doi.org/10.1371/journal.pone.0037582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying WL, Emerson J, Clarke MJ, & Sanadi DR (1991). Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry, 30(20), 4949–52. https://doi.org/10.1021/bi00234a016 [DOI] [PubMed] [Google Scholar]
- Zampieri S, Mammucari C, Romanello V, Barberi L, Pietrangelo L, Fusella A, ... Rizzuto R (2016). Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiological Reports, 4(24). https://doi.org/10.14814/phy2.13005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F, Chen X, Cui W, Wen W, Lu F, Sun X, ... Zhang X (2018). RIPK1 binds MCU to mediate induction of mitochondrial Ca2+ uptake and promote colorectal oncogenesis. Cancer Research, canres.30822017 https://doi.org/10.1158/0008-5472.CAN-17-3082 [DOI] [PubMed] [Google Scholar]
- Zoccarato F, & Nicholls D (1982). The Role of Phosphate in the Regulation of the Independent Calcium- Efflux Pathway of Liver Mitochondria. European Journal of Biochemistry, 127(2), 333–338. https://doi.org/10.1111/j.1432-1033.1982.tb06875.x [DOI] [PubMed] [Google Scholar]