

Abstract
ABHD12 metabolizes bioactive lysophospholipids, including lysophosphatidylserine (lyso-PS). Deleterious mutations in human ABHD12 cause the neurological disease PHARC, and ABHD12(−/−) mice display PHARC-like phenotypes, including hearing loss, along with elevated brain lyso-PS and features of stimulated innate immune cell function. Here, we develop a selective and in vivo-active inhibitor of ABHD12 termed DO264 and show that this compound elevates lyso-PS in mouse brain and primary human macrophages. Unlike ABHD12(−/−) mice, adult mice treated with DO264 exhibited minimal perturbations in auditory function. On the other hand, both DO264-treated and ABHD12(−/−) mice displayed heightened immunological responses to lymphocytic choriomeningitis virus (LCMV) clone 13 infection that manifested as severe lung pathology with elevated proinflammatory chemokines. These results reveal similarities and differences in the phenotypic impact of pharmacological versus genetic blockade of ABHD12 and point to a key role for this enzyme in regulating immunostimulatory lipid pathways in vivo.
Lysophospholipids are a diverse class of chemical transmitters that includes sphingosine 1-phosphate (S1P)1, lysophoshatidic acid (LPA)2, and more emerging bioactive lipids such as lysophosphatidylserine (lyso-PS) and lysophosphatidylinositol (lyso-PI)3. Lysophospholipids are generally thought to act through discrete sets of G-protein coupled receptors (GPCRs)4, although other categories of receptor have also been described5. Diverse areas of biology are impacted by lysophospholipid signaling, leading to therapeutics, such as S1P receptor modulators, to treat multiple sclerosis and potentially other autoimmune disorders6.
Lyso-PS represents a more recently characterized class of signaling lipids that has been shown to activate both immune-restricted GPCRs7-9 and Toll-like receptors (TLRs)5. Consistent with an immunomodulatory function for lyso-PS, these lipids have been found to promote degranulation of mast cells10, suppress cytokine production11 and proliferation12 of cultured T cells, and constrain regulatory T cell function in vivo9. Lyso-PS lipids can be generated from PS lipases, including ABHD16A13 and PS-PLA114, and are degraded by the integral membrane serine hydrolase ABHD1215, which is highly expressed in the central nervous system (CNS) and in innate immune cells (e.g., macrophages, microglia). Human genetic studies have found that autosomal recessive mutations in the ABHD12 gene lead to the polymodal sensorimotor disease PHARC (Polyneuropathy, Hearing loss, Ataxia, Retinitis pigmentosa and Cataract; MIM 612674)16,17. We have found that ABHD12(−/−) mice display a subset of PHARC-related neurological abnormalities, and these phenotypes correlate with substantial elevations in lyso-PS and 20:4 PS throughout the CNS15. ABHD12(−/−) mice also have heightened brain microgliosis15, supporting a potential immunological contribution to PHARC-related pathology.
Our understanding of the functional contributions of the ABHD12-(lyso)-PS pathway to (neuro)immunology has been limited by a lack of selective and in vivo-active ABHD12 inhibitors. Indeed, PHARC-related pathologies emerge slowly throughout life in both humans16,17 and mice15 with ABHD12-inactivating mutations, raising important questions about whether i) this pathology reflects perturbations in developmental versus adult biology, ii) the immunological and neurological phenotypes caused by ABHD12 disruption are mechanistically related, and iii) acute versus chronic disruption of ABHD12 may differ in physiological effect.
Here, we describe the discovery, optimization, and biological characterization of a potent, selective, and in vivo-active ABHD12 inhibitor termed DO264. Mice treated with DO264 display dose-dependent increases in brain lyso-PS and 20:4 PS content that are qualitatively similar to the changes observed in ABHD12(−/−) mice. DO264-treated mice, however, show minimal impairment in auditory function following four weeks of drug exposure. We also found that DO264 elevated the lyso-PS and 20:4 PS content of primary human macrophages. Finally, both ABHD12(−/−) and DO264-treated mice display exacerbated immunopathology following infection with the lymphocytic choriomeningitis virus (LCMV) clone 13 that resulted in severe inflammatory lung damage, heightened chemokine production, and, in some cases, death. These phenotypes support an immunostimulatory function for the ABHD12-(lyso)-PS pathway that can be pharmacologically controlled with selective ABHD12 inhibitors.
Results
An HTS assay for discovery of ABHD12 inhibitors
Only a handful of inhibitors of ABHD12 have been described, and these compounds are either low potency (e.g., low-μM triterpenoids18) or lack selectivity (e.g., the general lipase inhibitor tetrahydrolipstatin (THL)19), and none have been shown to inhibit ABHD12 in vivo. We have also found that ABHD12 displays generally limited reactivity with prominent electrophilic scaffolds used to create irreversible inhibitors of other serine hydrolases, such as activated carbamates20 and ureas21. We therefore sought to discover new types of ABHD12 inhibitors by high-throughput screening (HTS). While an HTS-compatible assay for ABHD12 has been described that uses an arachidonoyl glycerol substrate22, we favored development of an assay based on a lysophospholipid substrate for the enzyme. Accordingly, we developed an enzyme-coupled fluorescence assay that measures ABHD12-mediated hydrolysis of lysophosphatidic acid (lyso-PA)23 (Fig. 1a, b and Supplementary Fig. 1), which we implemented in a 384-well format to screen the 16,000 compound Maybridge HitFinder™ library tested at 10 μM. The HTS assay showed excellent statistics with average Z’ and S/B values of 0.78 and 5.5, respectively, and identified ~ 200 compounds that displayed > 50% inhibition (1.2 % hit rate) (Fig. 1c and Supplementary Fig. 2). Two secondary screens were then used to remove false-positive compounds: i) an LC/MS-based lyso-PS hydrolysis assay15, and ii) a competitive ABPP assay using the serine hydrolase-directed fluorophosphonate-rhodamine (FP-Rh) probe24. Only one compound (#175) showed activity in both secondary assays (Supplementary Fig. 3). We found through resynthesis that the structure of this compound had been mis-assigned by the supplier as a semithiocarbazide (Supplementary Fig. 4) and instead represented a thiourea that we refer to hereafter as DO130 (1) (Fig. 1d and Supplementary Fig. 4). DO130 inhibited the lyso-PS lipase activity of ABHD12 with an IC50 value of 1.3 μM (Fig. 1e and Supplementary Fig. 5). In contrast, we prepared an authentic sample of the semithiocarbazide (termed DO127 (2)) and found that it showed no activity against ABHD12 (Supplementary Fig. 4). The HTS study is summarized in Supplementary Table 1.
Figure 1.
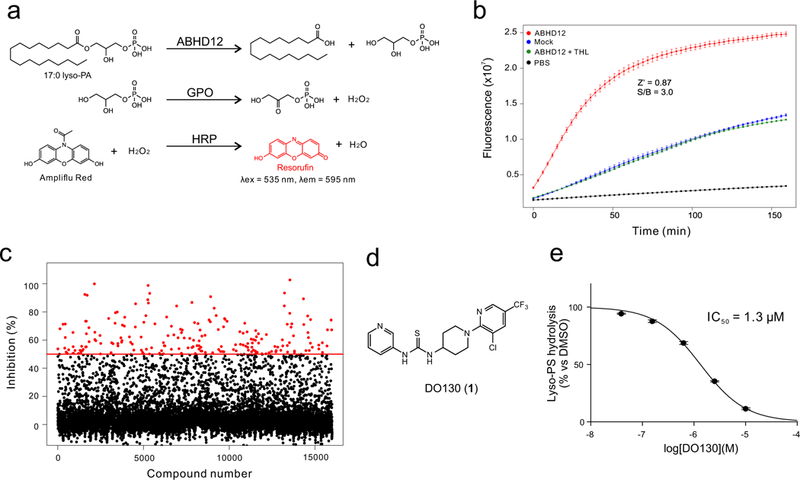
Development of a high-throughput assay (HTS) screen for ABHD12 inhibitors. (a) An enzyme-coupled assay that begins with ABHD12-mediated hydrolysis of 17:0 lyso-PA, followed by glycerol-3-phosphate oxidase (GPO)-mediated generation of H2O2, and culminating in the horseradish peroxidase (HRP)-mediated production of the fluorescent compound resorufin. (b) Kinetic performance of the enzyme-coupled HTS assay with membrane lysates from ABHD12- or mock-transfected HEK293T cells. The Z’ and S/B values at 45 min were 0.87 and 3.0, respectively. THL (10 μM) was used as a control ABHD12 inhibitor. (c) Screening data for the Maybridge HitFinder™ collection of 16,0000 compounds. Compounds showing> 50% inhibition are marked in red (198 total). The screen was performed once. (d) Structure of the hit compound DO130. (e) IC50 value for inhibition of lyso-PS hydrolysis activity of ABHD12 by DO130 measured using ABHD12-transfected cell lysates with 17:1 lyso-PS substrate (100 μM, 20 min, 37 °C). Data represent average values ± SD (n = 3 independent experiments).
Optimization of thiourea ABHD12 inhibitors
For the purposes of optimizing thiourea inhibitors of ABHD12, we took advantage of a tailored, clickable activity-based probe targeting this enzyme termed JJH350 (3) (Supplementary Fig. 6), which originated from an N-hydroxyhydantoin (NHH) carbamate library of covalent serine hydrolase inhibitors25. Even though we were unsuccessful in our efforts to create in vivo-active inhibitors of ABHD12 based on the NHH carbamate scaffold, JJH350 offered a more selective probe compared to FP-Rh for assessing ABHD12 activity in diverse biological systems (see below). Using JJH350 for competitive gel-based ABPP of ABHD12 in mouse brain membrane proteome, we measured an IC50 value of 1.1 μM for DO130 (Supplementary Fig. 7 and Supplementary Table 2), which is similar to the value measured with the lyso-PS lipase assay (Fig. 1e). A series of modifications to DO130, including conversion of the 3-chloro-5-trifluoromethyl group to a 4-trifluoromethoxyphenyloxy group (DO230 (4)) and addition of Cl groups at the 3-position of the pyridine ring (DO253 (5)) and 2-position of the phenyl ring, improved the potency of ABHD12 inhibition and culminated in a compound DO264 (6) that displayed an IC50 value of 11 nM (9.6 nM-13 nM 95% confidence interval (CI)) (Fig. 2a, Supplementary Fig. 7, and Supplementary Table 2). Modifications to the thiourea group, on the other hand, decreased potency, with N-methylthiourea- (DO276 (7)) and urea- (DO277 (8)) analogues of DO264 displaying IC50 values of 1.1 μM and 170 nM, respectively (Supplementary Fig. 7, and Supplementary Table 2).
Figure 2.
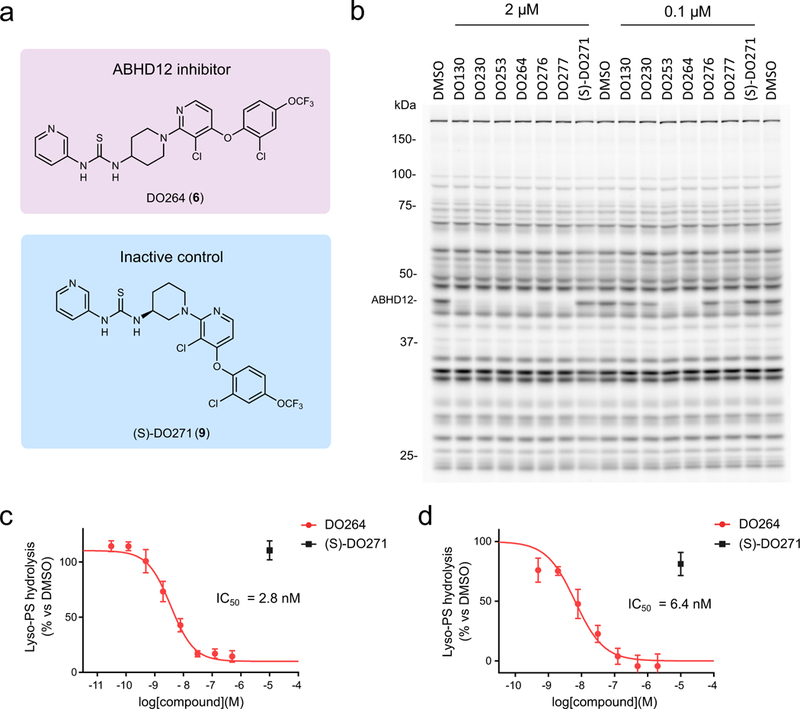
Discovery and optimization of (thio)urea inhibitors of ABHD12. (a) Chemical structures of an optimized ABHD12 inhibitor DO264 and an inactive control compound (S)-DO271. (b) In vitro potency and selectivity of (thio)urea ABHD12 inhibitors and the control compound (S)-DO271 (0.1 or 2 μM, 30 min pre-incubation 37 oC) measured in mouse brain membrane proteome (1 mg/mL) by gel-based competitive ABPP using the FP-Rh probe (1 μM, 45 min, 37 °C). The result is a representative of two independent experiments. (c, d) Concentration-dependent inhibition of lyso-PS hydrolysis activity of mouse brain (c) or THP-1 (d) membrane proteome (0.4 mg/mL) by DO264 as determined using 17:1 lyso-PS as a substrate. Data were normalized to ABHD12(−/−) brain and JJH350 (10 μM)-treated THP1 membrane proteomes, respectively, to designate the ABHD12-dependent component of lyso-PS hydrolysis in these proteomes (see Supplementary Fig. 9) and represent average values ± SD. n = 4 and 3 independent experiments for c and d, respectively.
Competitive gel- and mass spectrometry (MS)-based ABPP26 ABPP with FP-Rh or a biotinylated FP probe, respectively, revealed that DO264 and other (thio)urea analogues displayed excellent selectivity for ABHD12 over other mouse brain serine hydrolases (Fig. 2b and Supplementary Fig. 8). Our SAR analysis of ABHD12 inhibitors also identified an inactive analogue of DO264 with an (S)-3-aminopiperidine arrangement ((S)-DO271 (9), Fig. 2a, b and Supplementary Fig. 7, and Supplementary Table 2), which we surmised could serve as a useful control compound for biological studies.
We next confirmed that DO264, but not (S)-DO271, blocked lyso-PS hydrolysis activities of recombinant mouse and human ABHD12 in transfected HEK293T cell lysates (DO264 IC50 values of ~30 and 90 nM against mouse and human ABHD12, respectively) (Supplementary Fig. 9) and the ABHD12-dependent lyso-PS lipase activity of membrane lysates from mouse brain (IC50 = 2.8 nM, 2.4 nM-3.3 nM 95% CI; Fig. 2c and Supplementary Fig. 9) and human THP-1 cells (IC50 = 8.6 nM, 6.3 nM-12 nM 95% CI; Fig. 2d and Supplementary Fig. 9). We are unsure why DO264 exhibits improved potency for endogenous versus recombinant forms of ABHD12, but the difference is unlikely to relate to greater quantity of recombinant enzyme in the substrate assays, as we observed a similar IC50 value for DO264 when tested against different amounts of ABHD12-transfected cell lysates (Supplementary Fig. 9).
Further studies revealed that DO264 did not show time-dependent inhibition of ABHD12, which contrasted with the profile observed for the covalent NHH inhibitor JJH350 (Supplementary Fig. 10). We also found that dilution of DO264-treated ABHD12-transfected HEK293T lysates resulted in the recovery of ABHD12 activity (Supplementary Fig. 10). Finally, an alkyne analogue of DO264 (DO274 (10)) was generated and found to retain potent ABHD12 inhibitory activity, but did not show evidence of covalent modification of ABHD12 (Supplementary Fig. 11). These results, taken together, indicate that DO264 acts as a reversible inhibitor of ABHD12. We also attempted to assess whether DO264 acts as a competitive inhibitor. We first found that, among a panel of tested detergents, only Triton X-100 solubilized ABHD12 (Supplementary Fig. 12). But, the Triton-solubilized enzyme lost virtually all lyso-PS lipase activity (Supplementary Fig. 12). We therefore evaluated the potency of DO264 inhibition of unsolubilized mouse ABHD12 in the presence of increasing concentrations of lyso-PS substrate (12.5-200 μM) and found that the IC50 value of inhibition shifted substantially rightward (~5 to 70 nM) with greater substrate concentration (Supplementary Fig. 12). These data, along with the blockade of FP-Rh (and JJH350) labeling of ABHD12 by DO264, are consistent with this compound acting as a competitive inhibitor of ABHD12.
DO264 alters (lyso)-PS content of human macrophages
We next tested whether DO264 inhibits ABHD12 in cells. As ABHD12 is highly expressed in macrophages and microglia13,27-29, we first evaluated DO264 activity in the human monocytic THP-1 cell line. THP-1 cells were treated with DO264 for 4 h, lysed, and then analyzed by competitive gel-based ABPP with the JJH350 probe, which revealed concentration-dependent inhibition of ABHD12 with complete blockade observed at 1 μM of DO264 (Fig. 3a). In contrast, ABHD12 activity was unaffected by (S)-DO271 (1 μM) in THP-1 cells (Fig. 3a). Membrane lysates from DO264-treated cells also showed concentration-dependent reductions in lyso-PS hydrolysis activity, from which an in situ IC50 value of 26 nM (20 nM-34 nM 95% CI) was determined (Fig. 3b and Supplementary Fig. 13) and found to match the in situ IC50 value estimated by competitive ABPP (Supplementary Fig. 13).
Figure 3.
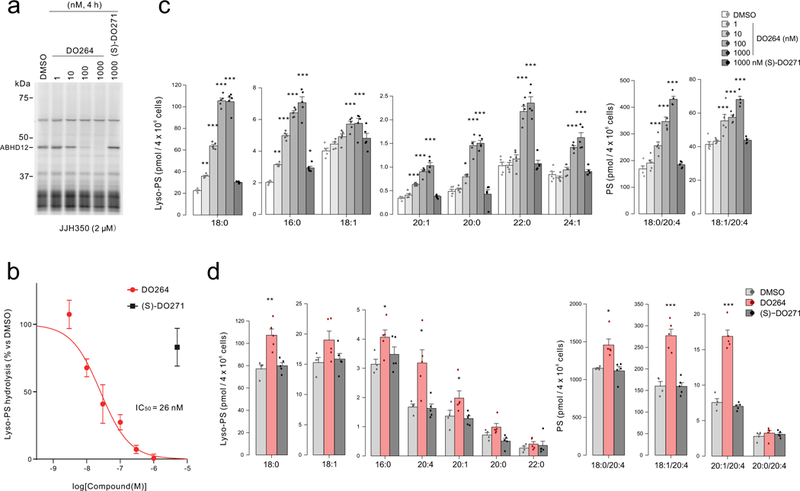
DO264 inhibits ABHD12 and increases lyso-PS content in human monocytic cells. (a, b) Concentration-dependent inhibition of ABHD12 in THP-1 cells treated with the indicated concentrations of DO264 (4 h, in situ), as determined by gel-based competitive ABPP using the JJH350 probe (a) or measurement of lyso-PS hydrolysis activity (b) with the membrane proteome of DO264-treated THP-1 cells. For a, the result is a representative of two independent experiments. For b, data represent mean ± SD (n = 3 independent experiments). (c) Treatment of THP-1 cells with DO264 (0.001-1 μM, 4 h) leads to the concentration-dependent increase in lyso-PS (left) and 20:4 PS (right) content. (S)-DO271 (1 μM, 4 h) did not alter (lyso)-PS content of THP-1 cells. (d) Primary human macrophages treated with DO264, but not (S)-DO271 (1 μM compound, 24 h) show increases in lyso-PS (left) and 20:4 PS (right). For c and d, data represent mean ± SEM. n = 5 independent samples per group except for the DMSO-treated group in d (n =4 independent samples per group). * p < 0.05; ** p < 0.01; *** p < 0.001 (Two-sided Student’s t-test performed relative to DMSO control for c and d). The p-values and other lipid species measured for c and d are provided in Supplementary Table 4.
We next confirmed that DO264 inhibits ABHD12 with excellent selectivity in THP-1 cells. In brief, THP-1 cells labeled with isotopically heavy or light amino acids were treated with DMSO and compound (DO264 or (S)-DO271, 1 μM), respectively, for 4 h and then lysed, reacted with FP-biotin, and processed for quantitative MS-based ABPP as previously described21. These experiments revealed that DO264 blocked > 95% of ABHD12 activity without affecting the activity of other serine hydrolases in THP-1 cells, while (S)-DO271 did not affect the activity of any serine hydrolases, including ABHD12 (Supplementary Fig. 14 and Supplementary Table 3).
DO264, but not (S)-DO271, produced substantial elevations in the lyso-PS content of THP-1 cells (Fig. 3c). Other measured (lyso)phospholipids were mostly unchanged in DO264-treated THP-1 cells (Supplementary Table 4), with the exception of a concentration-dependent increase in several 20:4 PS lipids and corresponding decrease in 18:1 PS lipids (Fig. 3c and Supplementary Fig. 14). These data are consistent with previous findings in ABHD12(−/−) mice, which display elevations in both lyso-PS and 20:4 PS (and reductions in 18:1 PS) content in the CNS15. Similar lyso-PS and PS changes were observed in PMA-differentiated THP-1 cells (Supplementary Fig. 15), which adopt a more macrophage-like cell state compared to undifferentiated THP-1 cells. We also found that DO264 inhibits ABHD12 activity (Supplementary Fig. 16) and alters lyso-PS/PS content in primary human macrophages, which showed elevations in both lyso-PS and 20:4 PS following 4 or 24 h treatment with DO264 (1 μM), but not (S)-DO271 (1 μM) (Fig. 3d, Supplementary Fig. 17 and 18).
These data, taken together reveal that ABHD12 controls a dynamic pool of lyso-PS in human macrophages that undergoes substantial change following acute inactivation of the enzyme. That alterations in 20:4 PS occur on a similar time scale following ABHD12 inhibition points to a rapid reacylation pathway for lyso-PS in macrophages, possibly mediated by a 20:4-preferring lysophospholipid acyltransferase30.
DO264 alters brain (lyso)-PS content in vivo
C57BL/6 mice treated with DO264 by intraperitoneal (i.p., 4 h or 24 h) or oral (p.o., 4 h) administration showed dose-dependent reductions in brain ABHD12 activity as measured by ex vivo analysis with the JJH350 probe, with complete inhibition being observed at the 30 and 60 mg/kg doses (Fig. 4a, b and Supplementary Figs. 19 and 20). In contrast, (S)-DO271 (60 mg/kg, 4 h) did not block brain ABHD12 activity in mice (Fig. 4a, b). The inhibition of ABHD12 activity was observed for 4-24 h post-treatment with DO264 (30 mg/kg, i.p.), with partial recovery observed at 48 h post-dosing (Fig. 4c, d). No changes in ABHD12 protein abundance, as measured by western blotting, were observed in DO264-treated animals (Supplementary Fig. 19). ABPP studies confirmed that DO264 displayed excellent selectivity for ABHD12 in vivo (Fig. 4b-e and Supplementary Figs. 19 and 20 and Supplementary Table 3). Among the more than 65+ other serine hydrolases quantified by MS-based ABPP, only ABHD2 and PLA2G6 showed partial (~40-50%) reductions in activity in DO264-treated mice (Fig. 4e and Supplementary Table 3), but these changes may reflect an indirect outcome of ABHD12 inhibition, as DO264 did not inhibit mouse ABHD2 in vitro (Supplementary Fig. 8) or human ABHD2 in situ (Fig. 3c), nor did the compound inhibit recombinantly expressed PLA2G6 (Supplementary Fig. 21). The MS-based ABPP results (Fig. 4e and Supplementary Table 3), along with gel-based ABPP assays with recombinant enzymes (Supplementary Fig. 21), also confirmed that the candidate lyso-PS biosynthetic enzymes ABHD16A and PS-PLA1 were not inhibited by DO264. We also determined using general and tailored ABPP probes (FP-Rh and JJH350, respectively) that DO264, but not (S)-DO271, (i.p., 30 mg/kg, 4 h), selectively inhibited ABHD12 in peripheral tissues of mice (Supplementary Fig. 22). We further found that JJH350 can function as an in vivo target engagement probe for ABHD12, as the sequential administration of DO264 (30 mg/kg, p.o., 4 h) followed by JJH350 (30 mg/kg, i.p., 4 h) confirmed inhibition of ABHD12 by DO264 in mouse brain and peripheral tissues (Supplementary Fig. 23). Finally, we measured blood and brain concentrations of DO264 in mice, which revealed good peripheral (~4-9 μM in blood) and central (~3-6 μM in brain) exposure of the compound at 4 h following i.p. or p.o. administration (30 mg/kg). (S)-DO271 showed lower exposure, but, at 60 mg/kg (i.p.), approximated exposures observed with DO264 (Supplementary Fig. 23).
Figure 4.
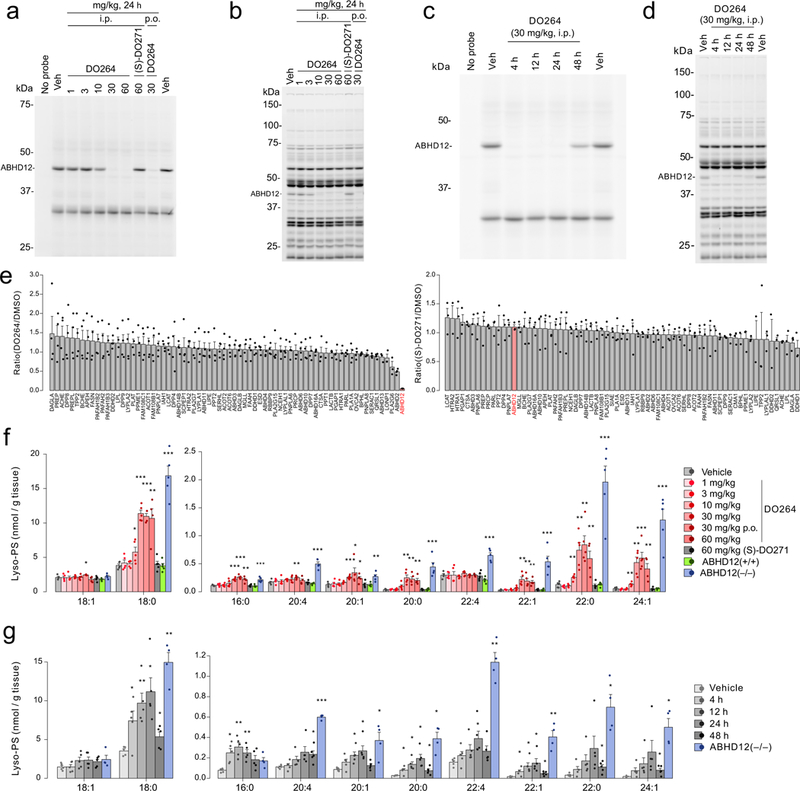
DO264 inhibits ABHD12 and increases brain lyso-PS content in vivo. (a, b) Dose-dependent inhibition of ABHD12 in mice treated with the indicated doses of DO264 (24 h, i.p.), as determined in brain membrane proteome by gel-based competitive ABPP using (a) the JJH350 probe (2 μM, 45 min, 37 °C) or (b) the FP-rhodamine probe (1 μM, 45 min, 37 °C). (c, d) Time-course analysis of inhibition of ABHD12 in DO264-treated mice (30 mg/kg, i.p., indicated time points) as determined in brain membrane proteome by gel-based competitive ABPP using (c) the JJH350 probe (2 μM, 45 min, 37 °C) or (d) the FP-rhodamine probe (1 μM, 45 min, 37 °C) (d). For a-d, the results are representative of two independent experiments. (e) Quantitative MS-ABPP of brain membrane serine hydrolase activities from mice treated with vehicle versus DO264 (left plot) or (S)-DO271 (right plot) (30 mg/kg compound, i.p., 4 h treatment). Data represent the mean of median ratios ± SEM for peptides quantified for each protein. n = 4 and 3 independent samples for DO264 and (S)-DO271 data, respectively. (f) Brain lyso-PS content from mice treated with vehicle or the indicated doses of DO264 or (S)-DO271 (i.p., 24 h, except where indicated p.o. for oral dosing groups). The lyso-PS content of brain tissue from ABHD12(+/+) and ABHD12(−/−) mice were included for comparison. (g) Time-course analysis of brain lyso-PS content from mice treated with vehicle or DO264 (30 mg/kg, i.p.). The lyso-PS content of brain tissue from ABHD12(−/−) mice was included for comparison. For f and g, data represent mean ± SEM. n = 5 mice per group except the ABHD12(−/−) mice group in g (n = 4 mice). * p < 0.05; ** p < 0.01; *** p < 0.001 (Two-sided Student’s t-test performed relative to vehicle-treated mice for f and g). The p-values and other lipid species measured for f and g are provided in Supplementary Table 4.
DO264 (1-60 mg/kg, i.p., 24 h), but not (S)-DO271 (60 mg/kg, i.p., 24 h), dose-dependently increased most brain lyso-PS species, as well as select 20:4 PS species (Fig. 4f and Supplementary Fig. 24). Maximal lyso-PS and 20:4 PS elevations were observed at 30 mg/kg of DO264, which matched the dose at which full ABHD12 inhibition was observed (Fig. 4a and Supplementary Fig. 19). Time-course studies revealed that the lyso-PS changes caused by DO264 were detectable by 4 h and maximal at 24 h post-dosing (Fig. 4g), while PS changes accumulated more slowly (Supplementary Fig. 24). ABHD12(−/−) mice also show elevations in 20:4 lyso-PI15, and an increase in this lipid was also observed in DO264, but not (S)-DO271-treated mice (Supplementary Fig. 24). ABHD12 has been shown to hydrolyze the endocannabinoid 2-arachidonoylglycerol (2-AG)31,32 and, in certain cellular setting (e.g., microglial cultures29), can regulate 2-AG content. However, brain tissue from DO264-treated or ABHD12(−/−) mice did not show changes in 2-AG (Supplementary Table 4).
The elevations in brain lyso-PS and 20:4 PS in DO264-treated mice did not generally approach the magnitude of changes observed in ABHD12(−/−) mice (Fig. 4f, g and Supplementary Fig. 24), which suggested that more chronic inhibition of ABHD12 might lead to further augmentation of brain (lyso)-PS content. To address this question, we first treated adult mice with DO264 or (S)-DO271 (30 mg/kg daily, p.o.) for two weeks and confirmed that neither compound produced overt signs of toxicity, such as weight loss (Supplementary Fig. 25) or behavioral distress. We then proceeded to treat 8-month-old mice with DO264 or (S)-DO271 (30 mg/kg daily, p.o.) or vehicle for four weeks. Prior to sacrifice and lipid analysis, we tested the mice for auditory function and found that, unlike ABHD12(−/−) mice, which showed robust auditory defects as reported previously15 (Fig. 5a), the DO264-treated mice showed only a mild trend toward impairment at lower decibels that failed to reach statistical significance in comparison to either vehicle or (S)-DO271-treated mice (Fig. 5b). Substantial elevations in both lyso-PS and 20:4 PS occurred in brain tissue from the DO264-treated mice (Fig. 5c and Supplementary Fig. 26), and these changes included species such as 18:0/20:4 and 20:0/20:4 PS that were unaltered in mice treated acutely with DO264 (Supplementary Fig. 24). Nonetheless, even after four weeks of treatment with DO264, the elevations in many brain (lyso)-PS species remained less substantial than the changes observed in ABHD12(−/−) mice (Fig. 5c and Supplementary Fig. 26).
Figure 5.
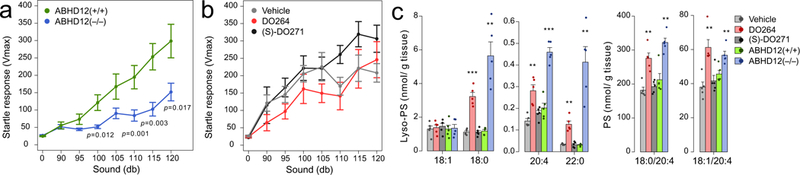
Effects of ABHD12 inhibition on mouse auditory capacity and brain lyso-PS content in vivo. (a, b) Auditory startle test results for (a) ABHD12(+/+) versus (ABHD12(−/−) mice or (b) mice treated daily for four weeks with vehicle, DO264, or (S)-DO271 (30 mg/kg, p.o.). (c) Brain lyso-PS and PS content from mice treated daily for four weeks with vehicle, DO264, or (S)-DO271 (30 mg/kg, p.o.). The lyso-PS content of brain tissue from ABHD12(+/+) and ABHD12(−/−) mice were included for comparison. Data represent average values ± SEM. For a, b, n = 9, 10, 8, 11, and 15 mice for vehicle, DO264, (S)-DO271, ABHD12(+/+) and ABHD12(−/−) group respectively. For c, n = 5mice per group. * p < 0.05; ** p < 0.01; *** p < 0.001 (Two-sided Student’s t-test performed relative to vehicle-treated mice except for the ABHD12(−/−) data, which were compared to the ABHD12(+/+) data). The p-values and other lipid species measured for c are provided in Supplementary Table 4.
These data, taken together, indicate that both acute and chronic treatment with DO264 raises brain (lyso)-PS in vivo, but these changes are less dramatic than the (lyso)-PS changes observed in ABHD12(−/−) mice and do not result in substantial auditory dysfunction.
Heightened immune responses in ABHD12-disrupted mice
Both ABHD12 and major lyso-PS receptors are strongly expressed in immune cells, and previous studies from our lab and others have pointed to an immunomodulatory function for the ABHD12-lyso-PS pathway that may involve both innate13,15 and adaptive9,11,12 immunological systems. Nonetheless, how ABHD12 specifically contributes to dynamic immune responses in vivo remains unknown. We set out to investigate this question with the LCMV clone 13 (Cl13) model of immune challenge, which produces a pan-immune suppressive state that promotes T cell exhaustion and virus persistence33. Relief of immune suppression by blocking negative regulatory molecules like PD-1 or IL-10 results in hastened control of Cl13 infection, as well as concomitant immune pathology that can produce severe morbidity and lethality34,35. We found that infection with a persistent dose of Cl13 caused a striking pathological response in ABHD12(−/−) mice indicated by hunched posture, ruffled fur, labored breathing, and death in ~50% of the animals (Fig. 6a) that correlated with gross lung damage (Supplementary Fig. 27), whereas Cl13-infected ABHD12(+/+) mice displayed much healthier cage behavior with only ~10% succumbing to infection (Fig. 6a). A similar heightened pathology was observed in mice treated with DO264 in comparison to (S)-DO271 (30 mg/kg of DO264 or (S)-DO271, daily, i.p). (Fig. 6b). Consistent with ABHD12 disruption leading to a reversal of the immune suppressive environment during Cl13 infection36,37, we found that bronchoalveolar lavage fluid (BALF) from ABHD12(−/−) or DO264-treated mice showed increases in total protein (Supplementary Fig. 28) and pro-inflammatory chemokines (Fig. 6c, d) compared to their respective control groups. To provide further evidence that DO264 produces immunostimulatory effects through inhibiting ABHD12, we tested the compound, alongside (S)-DO271 (30 mg/kg, daily, i.p.), in ABHD12(−/−) mice and evaluated immunological parameters at day 10 post-Cl13 infection. A similar fraction (~30-40%) of DO264- and (S)-DO271-treated ABHD12(−/−) mice died prior to sacrifice at day 10, consistent with the response of ABHD12(−/−) mice to Cl13 infection in previous experiments (Fig. 6a), and both groups of compound-treated animals also showed similar amounts of chemokines in BALF (Supplementary Fig. 28). The DO264-treated ABHD12(−/−) mice paradoxically showed a reduction in BALF protein compared to (S)-DO271-treated ABHD12(−/−) mice (Supplementary Fig. 28). We do not understand the basis for this difference, as it is opposite in directionality from the elevations in BALF protein observed for DO264 versus (S)-DO271 in ABHD12(+/+) mice (Supplementary Fig. 28). But, regardless, our data indicate that DO264 treatment does not further exacerbate the heightened immunological responses observed in ABHD12(−/−) mice infected with Cl13, supporting that this compound’s immunostimulatory effects occur mainly through ABHD12 inhibition.
Figure 6.
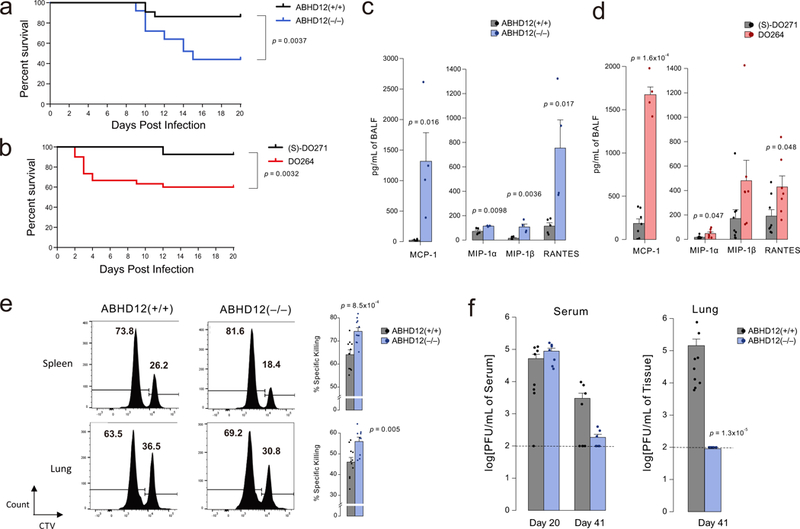
Heightened immunopathological responses to LCMV-Clone 13 (Cl13) infection of mice with genetic or pharmacological inactivation of ABHD12. (a, b) Mortality plots for (a) AHBD12(+/+) and ABHD12(−/−) mice or (b) DO264 and (S)-DO271-treated mice (30 mg/kg, i.p., daily, dosing started one day prior to infection) monitored daily post-infection with Cl13. Data are pooled from at least three experiments with n=5 mice per group. For a, data are pooled from 3 independent experiments for a total of 22 ABHD12(+/+) and 25 ABHD12(−/−) mice. For b, the plot represents four experiments for a total of 28 (S)-DO271-treated mice and 30 DO264-treated mice. Two-side log-rank test was performed to compare two survival distributions. (c, d) Overall chemokine concentrations in BALF from (c) AHBD12(+/+) and ABHD12(−/−) mice or (d) DO264 and (S)-DO271-treated mice (30 mg/kg, i.p., daily, dosing started one day prior to infection) evaluated 10 days post-infection with Cl13. Data in c are data representative of two independent experiments of n=5 mice per group, and d are plots representative of mean values of three experiments of n=5 mice per group ± SEM. Two-sided Student’s t-test performed relative to ABHD12(+/+) or (S)-DO271-treated mice). (e) ABHD12(−/−) mice kill adoptively transferred splenocytes displaying LCMV-specific antigen (GP33-41) more efficiently than ABHD12(+/+) mice. In vivo killing of immunogenic LCMV-specific GP33-41 peptide or unrelated NP118-126-peptide-pulsed target cells labeled with high or low CellTrace Violet (CTV), respectively, was measured in spleen and lung tissue from ABHD12(+/+) and ABHD12(−/−) mice 7 days post-infection with Cl13. See Supplementary Fig. 29 for diagram of FACS gating strategy. Data represent of mean values of two independent experiments of n=5 mice per group ± SEM. Two-sided Student’s t-test performed relative to ABHD12(+/+) mice). (f) Cl13 viral titers were measured by plaque assay at the indicated days post-infection in either serum (left) or lung (right). Dotted lines indicate the limit of detection of the plaque assay. Two-sided Mann-Whitney U test was performed relative to ABHD12(+/+) data.
The lung pathology caused by restoration of T cell function by, for instance, anti-PD-1 antibodies during persistent Cl13 infection depends on CD8 T cell-mediated killing36,37. We found evidence for a similar mechanism of action in ABHD12(−/−) mice, where anti-viral CD8 T cells from these animals displayed increased killing capacity compared to cells from ABHD12(+/+) mice in both spleen and lung (Fig. 6e and Supplementary Fig. 29). Finally, ABHD12(−/−) mice that survived the initial exaggerated immune response (~days 8-15) displayed a gradual reduction in viral titers in the serum and lung over the ensuing weeks (Fig. 6f). Taken together, these data indicate that the inactivation of ABHD12 promotes increased lung pathology and leakage following Cl13 viral infection. The observed lung pathology also correlates with enhanced local chemokine production, T-cell mediated killing capacity, and, over time, superior control of viremia.
Discussion
Recent years have witnessed a massive gain in our understanding of the genetic basis of human neurological disorders due principally to advances in DNA sequencing technologies38. Such studies illuminate gene products that play fundamental roles in the developing and adult nervous systems and have, more recently, pointed to a key contribution of the immune system to a range of neurological diseases39,40. Nonetheless, the biochemical basis for many genetically defined neurological diseases remains unknown, and the causative mutations often occur in proteins of poorly characterized function. The development of selective chemical probes that can perturb CNS disorder-related proteins acutely and reversibly in adult animals offers a compelling path to illuminate the functions of these proteins in physiology and disease.
We have described herein a versatile chemical tool set for the lysophospholipid hydrolase ABHD12, mutations in which cause the rare neurological disease PHARC16,17, that includes a potent, selective, and in vivo-active inhibitor DO264, along with activity-based (JJH350) and inactive control ((S)-DO271) probes for biological studies. Using these chemical probes and ABHD12(−/−) mice15, we discovered concordant and discordant effects of genetic versus pharmacological inactivation of ABHD12 in vivo. Similar alterations in brain lyso-PS and PS content were, for instance, found in DO264-treated and ABHD12(−/−) mice, but PHARC-like auditory defects were primarily observed in the latter animals. We do not yet know whether this difference reflects the greater magnitude of change in brain (lyso)-PS in ABHD12(−/−) mice, which could, in principle, be due to submaximal inhibition of ABHD12 by DO264 in vivo, or that these animals have a neurodevelopmental defect underlying their hearing dysfunction.
On the other hand, DO264-treated and ABHD12(−/−) mice exhibited similar phenotypic responses in the LCMV Cl13 model of immune challenge, pointing to an immunosuppressive function for ABHD12. While the heightened immunological responses to LCMV Cl13 displayed by ABHD12-disrupted mice may reflect the direct action of lyso-PS lipids that accumulate in these animals, we note that our current mechanistic understanding of these immunological phenotypes is limited, and past studies have provided conflicting views on whether lyso-PS pathways promote11,12 or suppress9 immune system function. Of course, the extent to which there is a functional connection between ABHD12 inactivation and individual lyso-PS lipids or specific lyso-PS receptors remains unknown, and we acknowledge the potential for several lyso-PS and 20:4 PS lipids, as well as other endogenous lysophospholipid substrates of ABHD12, such as lyso-PI lipids, which act on distinct receptors41, to contribute to phenotypes observed in DO264-treated and ABHD12(−/−) mice. We also call attention to potential signaling pathways for (lyso)-PS beyond GPCRs. Lyso-PS lipids have been found, for instance, to activate the TLR2 receptor5, which acts as a pattern recognition receptor in the innate immune system and is abundantly expressed in macrophages and microglia42. TLR2 agonists can induce microglia-mediated neurodegeneration both in primary neuronal cultures43,44 and in vivo45. Whether prolonged treatment with DO264, especially in older animals, leads to microgliosis, a phenotype observed in ABHD12(−/−) mice of 12-18 months15, is an important subject matter for future investigation. We also note that future pharmacological studies of ABHD12 in animal models would benefit from the development of additional inactive control compounds beyond (S)-DO271 that display more equivalent pharmacokinetics properties to DO264.
Finally, to what extent might our findings point to an immunological basis for PHARC? We note that hearing loss and motor dysfunction can be manifestations of autoinflammatory and autoimmune diseases46,47, and some of these disorders are responsive to immunosuppressive therapy6. Our data suggest that evaluating such therapeutic agents in ABHD12(−/−) mice is warranted as a prelude to their potential assessment in PHARC subjects. Our findings also designate ABHD12 inhibitors as a novel strategy to promote immune function in vivo, which could have translational relevance for infectious disease and immuno-oncology.
Materials and methods
Materials
All chemicals were obtained from Sigma Aldrich unless indicated otherwise. All lipids were purchased from Avanti Polar lipids except for 15:0 FFA (Sigma Aldrich), 17:1 FFA (Sigma Aldrich), and 20:4-d5 MAG (Cayman chemicals). Pharmacological and immunological studies were conducted in C57BL/6 mice. Mice were maintained in pathogen-free conditions and handled in accordance to the requirements of the National Institutes of Health and The Scripps Research Institute Animal Research Committee.
Ethical compliance
All studies using samples from human volunteers follow protocols approved by The Scripps Research Institute institutional review board (protocol no. IRB-15-6682). All mouse studies were performed following protocols that received approval from The Scripps Research Institute-Institutional Animal Care and Use Committee office.
Recombinant expression of enzymes by transient transfection of HEK293T cells
HEK293T cells were transiently transfected with the full-length cDNA of human or mouse ABHD12 (Dharmacon), mouse PLA2G6 or human PLA1A in pCMV-SPORT6 vector using the previously reported method13. Since PLA1A is a secreted enzyme, conditioned media was collected from HEK293T cells transiently overexpressing hPLA1A as described previously 48.
Lyso-PS hydrolysis assay
The lyso-PS lipase activity of ABHD12 was determined as previously described13 with some minor modifications. The proteome concentrations were adjusted to 0.25 mg/mL (membrane lysate from ABHD12-transfected HEK293T cells) or 0.5 mg/mL (mouse brain membrane proteome) in Dulbecco’s phosphate-buffered saline (DPBS) (80 μL/reaction). 20 μL of 500 μM 17:1 lyso-PS in DPBS was added to each reaction (100 μM final concentration) and incubated at 37 °C. After 20 min, the reaction was quenched with 400 μL of 2:1 CHCl3/MeOH (v/v) with 1 nmol 15:0 FFA as an internal standard. The mixture was vortexed and centrifuged at 1,400 × g to separate the aqueous and organic phase. The organic phase was analyzed following the previously reported protocol13.
Gel-based competitive ABPP for in vitro inhibitor treatment of proteomes
Gel-based ABPP assays were performed as previously reported49. Cell or tissue proteomes (50 μL, 1 mg/mL) were treated with either FP-Rh (1 μM final concentration) or JJH350 (2 μM final concentration) for 45 min at 37 °C. For FP-Rh labeled samples, the reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer. The 30 μL of the quenched samples were loaded on gel for analysis. For JJH350-labeled samples, copper-catalyzed azide-alkyne cycloaddition (CuAAC) was used for visualizing the labeled proteins following the reported protocol with slight modifications50. Briefly, TAMRA- PEG3-N3 (1 μL/reaction, 1.25 mM in DMSO), CuSO4 (1 μL/reaction, 50 mM in H2O), TBTA (3 μL/reaction, 1.7 mM in DMSO/t-BuOH [1:4, v/v]) and tris(2-carboxyethyl)phosphine (TCEP) (1 μL/reaction, 50 mM in H2O, freshly prepared) were premixed. 6 μL of this click reagents mixture was immediately added to each JJH350-labeled samples (50 μL, 1 mg/mL) and incubated for 1 h at room temperature. The reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer. The 40 μL of the quenched samples were loaded on a gel for analysis. After separation by SDS-PAGE (10% acrylamide), samples were visualized by in-gel fluorescence scanning using the ChemiDoc MP system (Bio-Rad). Band intensities were quantified using the Image Lab (5.2.1) software (Bio-Rad).
Development of mABHD12 fluorescence assay, screening against the Maybridge HitFinder™ library and hit validation
Membrane lysates from mABHD12-overexpressing HEK293F cells were used for screening and hit validation. To perform transient transfection, HEK293F cells were grown to 2 million cells/mL in 1 L of FreeStyle™ 293 Expression Medium (LifeTechnologies). 1.5 mg of mABHD12-pCMV-SPORT6 plasmid was mixed with 45 mL of FreeStyle™ 293 Expression Medium, then 5 mL of 1 mg/mL of PEI ‘MAX’ (MW 40,000; Polysciences Inc.) solution, pH 7.0, was added as a transfection reagent and incubated at room temperature for 30 min. The plasmid-PEI solution was added to the HEK293F culture, and the cells were incubated at 37 °C and 5 % CO2 with shaking for 48 h. After the 48 h incubation, the cells were pelleted by centrifugation at 6,000 × g at 4 °C for 15 min. The cell pellet was washed twice with cold DPBS, resuspended in 30 mL hypotonic buffer (20 mM HEPES pH 7.5, 10 mM NaCl, 1 mM EDTA), and was incubated at 4 °C for 30 min on a rotator. The cells were lysed by sonication using a probe sonicator (Branson Sonifier model 250) with 15 pulses (30 % duty cycle, output setting = 3). The lysate was centrifuged at 100,000 × g at 4 °C for 1 h to collect the membrane pellet. The membrane pellet was resuspended in 30 mL of ice-cold salt wash buffer (50 mM HEPES pH 7.5, 600 mM NaCl, 2 mM EDTA) and homogenized using a douncer. The homogenate was centrifuged at 100,000 × g at 4 °C for 1 h. The pellet was washed twice with DPBS and resuspended in DPBS and stored at −80 °C until further use. The assay was performed as described in Supplementary Table 1.
Of the 16,000 compounds tested in the screening, 198 of them showed inhibition >50 %. These hits were further validated by lyso-PS lipase assay and gel-ABPP using FP-Rh as described above (tested at 10 μM).
Preparation of tissue proteomes
Mouse tissues proteomes were prepared as reported previously15.
IC50 calculation
For gel-based ABPP, remaining % ABHD12 activities were determined by measuring the integrated optical intensity of the fluorescent ABHD12 bands using Image Lab 5.2.1. The relative intensity was compared to the ABHD12 band intensity from a control-treated sample, which was set to 100%. For the lyso-PS hydrolysis assay, remaining % ABHD12 activities were determined as a ratio of the area under the peak of 17:1 FFA (product, m/z = 267.20) over the area under the peak of 15:0 FFA (internal standard, m/z = 241.20). Data were then normalized by subtracting the lyso-PS hydrolase activity of ABHD12(−/−) brain (for brain membrane samples) or JJH350 (10 μM)-treated THP-1 membrane (for THP-1 membrane samples) or mock-transfected HEK293 membrane (for ABHD12-overexpressing HEK293 membrane samples) proteomes. IC50 values were determined by plotting a log(inhibitor) vs. normalized response, and the dose-response curves were generated using the Prism software (GraphPad).
Detergent solubilization of ABHD12
Membrane lysates from mABHD12-transfected HEK293F cells were diluted to 1 mg/mL in DPBS. The following detergents were added at indicated concentrations: Triton X-100 (0.1 and 1%, v/v), lauryldimethylamine oxide (LDAO, 1%, w/v), CHAPS (1%, w/v), n-dodecyl β-D-maltoside (DM, 1%, w/v), and n-octyl-β-D-glucoside (OG, 1%, w/v). The lysates were incubated on a rotator at 4 °C for 1 h and centrifuged at 100,000 × g for 1 h at 4 °C. After centrifugation, the supernatant was collected, and ABHD12 activity was tested using the FP-Rh probe (1 μM, 30 min, 37 °C). Lyso-PS hydrolysis assay was performed on Triton-solubilized mABHD12 lysate as described above.
Metabolomic analysis for cell and brain samples
For cell samples, cells were washed twice with cold DPBS, and the total cell metabolome was extracted in 4 mL 2:1:1 CHCl3/MeOH/DPBS (v/v/v) solution containing the internal standard mix (500 pmol 17:1 FFA, 100 pmol 17:1 lyso-PS, 300 pmol 17:0 lyso-PC, 300 pmol 17:1 lyso-PE, 200 pmol PS(17:0/20:4), 1 nmol PC(12:0/12:0) and 1 nmol PE(12:0/12:0)). For brain metabolomic analysis, a half brain was dounce-homogenized in a 15 mL glass douncer in 8 mL 2:1:1 CHCl3/MeOH/DPBS (v/v/v) solution containing the internal standard mix (1 nmol 17:1 FFA, 1 nmol 20:4-d5 MAG, 500 pmol 17:1 lyso-PS, 5 nmol 17:0 lyso-PC, 5 nmol 17:1 lyso-PE, 500 pmol 17:1 lyso-PI, 400 pmol PS(17:0/20:4), 10 nmol PC(12:0/12:0), 10 nmol PE(12:0/12:0), and 1 nmol PI(16:0-d31/18:1)). In both cases, the mixture was vortexed vigorously and centrifuged at 2,000 × g for 5 min at 4 °C. The bottom organic phase was collected, and the remaining aqueous phase was acidified with 20 μL (cell samples) or 40 μL (brain samples) formic acid and re-extracted by the addition of 2 mL (cell samples) or 4 mL (brain samples) CHCl3. Both of the organic phases were pooled, dried down under N2 gas, and reconstituted in 150 μL (cell samples) or 800 μL (brain samples) 2:1 CHCl3/MeOH (v/v) for LC/MS analysis. For analysis of MS data see Supplementary Table 4.
In situ treatment of THP-1 cells and macrophages with DO264
THP-1 cells were obtained from ATCC, grown and maintained in RPMI 1640 media (ThermoFisher Scientific) containing 10 % (v/v) FBS (Omega Scientific), 2 mM L-glutamine, 100 U/mL penicillin-streptomycin (GE Life Sciences), and 0.05 mM 2-mercaptoethanol at 37 °C with 5 % CO2. For in situ treatment of THP-1 cells with DO264, 6 million cells were cultured in 10 mL media, and DMSO, DO264 (1, 10, 100, and 1000 nM) and (S)-DO271 (1000 nM) were added to the desired concentrations, and the cells were incubated at 37 °C for 4 h with 5% CO2. After the incubation, the cells were washed twice with ice-cold DPBS and stored at −80 °C for gel-ABPP and metabolomic analyses. Gel-ABPPP analysis of ABHD12 inhibition and metabolomic analysis of lipid content were performed as described above. To perform metabolomic analysis on THP-1 macrophages, 6 million THP-1 cells were differentiated into macrophages by adding phorbol 12-myristate 13-acetate (PMA) to a final concentration of 150 ng/mL in a 10 cm plate. 24 h after incubation at 37 °C, the cells were washed twice with DPBS, and 10 mL of new media was added to the plate, and the cells were incubated for additional 24 h. Compound treatment and metabolomic analysis were performed in the same way as THP-1 cells. See Supplementary Table 4 for complete metabolomic data.
ABPP-SILAC sample preparation of THP-1 macrophages treated with DO264 or (S)-DO271 in situ
The SILAC experiments were performed using a THP-1 cell line generated by 10 passages in either light (100 μg/mL each of L-arginine and L-lysine) or heavy (100 μg/mL each of [13C615N4]L-arginine and [13C615N2]L-lysine) SILAC RPMI medium (ThermoFisher Scientific) supplemented with 10% dialyzed FBS (Omega Scientific). 30 million heavy or light THP-1 cells were transferred to 15 cm plates, and the cells were differentiated into macrophages by adding PMA to a final concentration of 150 ng/mL. 24 h after incubation at 37 °C, the cells were washed twice with DBPS, and 25 mL of corresponding new SILAC media was added to the plates, and the cells were incubated for additional 24 h. Light and heavy THP-1 macrophages were treated with the test compounds (1 μM DO264 or 1 μM DO271) or DMSO in situ, respectively, for 4 h at 37 °C. The cells were then washed twice with DPBS, harvested and lysed by sonication to obtain whole cell proteomes. 0.5 mL of 2 mg/mL light proteome and 0.5 mL of 2 mg/mL heavy proteome were treated with 1 μM FP-biotin separately for 30 min at room temperature, and the two solutions were combined for further MS sample preparation. The MS sample preparation was performed as described previously13. Peptides obtained from this procedure were acidified using formic acid (5% final concentration, v/v) and stored at -80 °C until MS analysis. MS run for proteomic samples was performed using a Orbitrap Velos mass spectrometer following previously described protocols51,52. MS data analysis was performed as described prevously13. See Supplementary Table 3 complete proteomic data and extracted serine hydrolase data.
Isolation and generation of primary human macrophages.
All studies with samples from human volunteers followed the protocols approved by The Scripps Research Institute Institutional Review Board. Blood from healthy donors (females aged 26-41) were obtained after informed consent. Peripheral blood mononuclear cells (PBMCs) were purified over Lymphoprep gradients (StemCell Technologies, 07851) by layering 25 mL of blood onto 12.5 mL of Lymphoprep. Samples were fractionated by centrifugation at 750 × g for 20 min at room temperature with no brake. PBMCs were harvested from the interface and washed twice with DPBS. Contaminating red blood cells (RBCs) were lysed with 1X RBC Lysis Buffer (eBioscience, 00-4300-54) for 5 min at room temperature and washed twice with DPBS. PBMCs were plated in DPBS at a density of 2.26 × 105 cells/cm2 and adherent monocytes attached after 1-2 h. Non-adherent cells were subsequently washed off the plates, and the culture media was added, which consisted of RPMI-1640 supplemented with 10% FBS, 10 mM HEPES, 1X GlutaMax (Life Technologies), 1mM sodium pyruvate (Life Technologies), and 100 U/mL penicillin-streptomycin. Monocytes are differentiated with 50 ng/mL recombinant human M-CSF (Peprotech, 300-25) for 6 days with a media change on day 3. On day 6, the macrophages turn into differentiated resting macrophages.
Compound treatment for in vivo studies
For intraperitoneal (i.p.) injection, DO264 and (S)-DO271 were dissolved in 1:1 (v/v) solution of EtOH/PEG40 (by bath sonication), and the solution was diluted with 9 volumes of DPBS followed by vortexing to obtain 1:1:18 (v/v/v) compound solution of EtOH/PEG40/DPBS. 10 μL/g mouse body weight of the freshly prepared compound solution was injected into the mouse peritoneal. For oral gavage (p.o.), DO264 and (S)-DO271 were resuspended in 20% (2-hydroxypropyl)-β-cyclodextrin in H2O (v/v) by probe and bath sonication. 10 μL/g mouse body weight of the freshly prepared compound solution was administered by oral gavage. Compound-treated mice were anesthetized with isoflurane and euthanized by cervical dislocation to harvest tissues.
Gel-based competitive ABPP for in vivo inhibitor treatment.
Tissues from mice treated with inhibitors were dounce-homogenized in DPBS (1 mL for a half brain, spleen and liver, and 1.5 mL for lung) followed by low-speed spin (1,400 × g, 3 min, 4 °C) to remove debris. For FP-Rh labeling, 2 μL of 50 μM FP-Rh was added to 50 μL of the whole tissue lysate obtained above (2 μM final) and incubated for 20 min at room temperature. For JJH350 labeling, 1 μL of 500 μM JJH350 was added to 50 μL of the whole tissue lysate obtained above (10 μM final) and incubated for 30 min at room temperature. 1.5 mL of cold DPBS was added to each probe-labeled samples and spun at 16,000 × g for 45 min at 4 °C. The resulting membrane pellet was resuspended in cold DPBS, and protein concentrations were adjusted to 1 mg/mL using the Bio-Rad DC protein assay kit. For FP-Rh labeled samples, the reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer, and the 30 μL of the quenched samples were loaded on a gel for analysis. For JJH350-labeled samples, CuAAC reactions were performed as described above and the reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer. The 40 μL of the quenched samples were loaded on gel for analysis.
Measurement of drug concentration in brain and blood
Measurement of tissue drug concentration was performed at Pharmaron (Bejing, China). DO264 and (S)-DO271 were administered as described above and incubated for 4 h. The brain tissues were homogenized with 4x volume of water (mL) by brain weight (g). Blood was collected via cardiac puncture and immediately quenched with acetonitrile at 1:4 blood:acetonitrile ratio. The brain homogenate and blood samples from drug-treated mice were diluted by 1000 to 2000-fold with 50% ACN in H2O. 5 μL of the diluted sample solutions were added to 50 μL of the blank C57BL/6J mice brain homogenate and blood respectively to make dose samples. For making calibration standards, the stock solutions of DO264 and (S)-DO271 (1 mg/mL in DMSO) were diluted with 50% ACN in H2O to generate serial concentrations of working solution. 5 μL of the working solutions (5, 10, 20, 50, 100, 500, 1000, 5000, 10000 ng/mL) were added to 50 μL of the blank C57BL/6J mice blood to achieve calibration standards of 0.5-1000 ng/mL (0.5, 1, 2, 5, 10, 50, 100, 500, 1000 ng/mL). 55 μL of calibration standards and 55 μL dose samples were added to 200 μL of ACN for precipitating protein respectively. Then the samples were vortexed for 30 s. After centrifugation at 4 °C, 4000 rpm for 15 min, the supernatant was diluted 5 times with water. 5 μL of the diluted supernatant was injected into the LC/MS/MS system for quantitative analysis (LCMS-8060, SHIMADZU).
Quantitative MS-ABPP sample preparation for mouse brain proteomes treated with DO264 in vitro and brain proteomes from mice treated with DO264 or (S)-DO271 in vivo
1 mL of brain proteomes (2 mg/mL in DPBS) were labeled with 6 μM FP-biotin for 1 h at room temperature, and the proteomes were denatured and precipitated using 4:1 MeOH/CHCl3 (v/v). The precipitates were subject to reduction, alkylation, avidin-based enrichment, and on-bead trypsin digestion as described in ABPP-SILAC sample preparation. Following the on-bead trypsin digest, reductive dimethylation was performed as previously described26,53. See Supplementary Table 3 for complete proteomic data and extracted serine hydrolase data.
Production of ABHD12 antiserum
Rabbits were immunized with a peptide fragment encoding mouse Cys 376ABHD12(376-398) coupled to keyhole limpet hemocyanin (KLH) via maleimide per manufacturer’s instructions (ThermoFisher, Waltham MA). The ABHD12 antiserum obtained from the rabbit was affinity-purified by passing over a column containing mouse Cys 376ABHD12(376-398)-agarose resin, specific antibodies eluted with acidic buffer, and then dialyzed into phosphate buffered saline. Covalent attachment of the peptide to resin (Sulfolink coupling resin; Thermo Fisher, Waltham, MA) was per manufacturer’s instructions.
Measurement of ABHD12 protein level by western blotting.
20 μg proteins of brain membrane lysates from inhibitor- or vehicle-treated mice were loaded to NovexTM 12% Tris-Glycine mini gel (Invitrogen, Catalog number XP04205BOX) and then transferred to a PVDF membrane in Towbin buffer. The membrane was blocked in 5% nonfat dry milk (w/v) in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature, incubated with anti-ABHD12 polyclonal antibody (1:2000 dilution in 5% nonfat dry milk in TBST) overnight at 4 °C. Blots were washed (3 × 5 min) with TBST, incubated with HRP-conjugated anti-rabbit secondary antibody (1:5000 in 5% nonfat dry milk in TBST) for 1 h at room temperature and washed (3 × 5 min) with TBST. Protein-antibody complexes were detected by enhanced chemiluminescence (ECL, Thermo Fisher Sci, catalog number 32106) on the ChemiDoc Imaging System (Bio-Rad).
Auditory test.
Auditory startle response testing was performed using SRLab startle chambers (San Diego Instruments) as reported previously15. Data were analyzed using SR-LAB version 6500-0091-E.
Preparation and infection of mice with LCMV-Clone 13
LCMV-Clone 13 was grown, stored, and quantified according to published methods54. For all experiments, each mouse was injected intravenously (i.v.) with 2 × 106 plaque forming units. To quantify viremia, blood was drawn retro-orbitally while mice were under isoflurane anesthesia. Blood was centrifuged to remove red and white blood cells, then the isolated serum was used to perform 10-fold serial dilutions for plaque assays on VeroE6 cells55. Lungs were harvested from euthanized mice, weighed, homogenized in media, and clarified by centrifugation, and the supernatant was used in plaque assays54. Following infection, mice were monitored daily for signs of morbidity.
Preparation and administration of compounds for immunological studies
DO264 and (S)-DO271 solutions were prepared as described above. Mice were then treated at 30 mg/kg i.p. beginning 1 day prior to infection and continuing daily for the course of each experiment.
Quantification of BALF protein levels
Overall BALF protein levels were quantified using the BCA protein assay kit (ThermoFisher Scientific) in accordance with the manufacturer’s instructions.
In vivo CTL specific killing assay
Splenocytes from naïve C57BL6/J mice were labeled with either 5 μm (CTVhi) or 667 nm (CTVlo) CellTrace Violet (ThermoFisher Scientific) for 20 minutes at 37 °C. Cells were washed, then CTVhi cells were loaded with immunodominant LCMV peptide GP33-41, and CTVlo cells were loaded with non-recognized irrelevant peptide NP118-1126 for 1 h at 37 °C. Cells were washed, then mixed at a 1:1 ratio. The cell mixture was then diluted such that each infected mouse (d7 post-infection) would be injected i.v. with at least 10 million total cells. After 30 min, animals were euthanized, and peptide-specific depletion of injected target splenocytes was analyzed via flow cytometry after the addition of 7-aminoactinomycin D (7-AAD) to discriminate live from dead cells. Percent in vivo lysis was calculated as a percent change in the ratio of live CTVhi to CTVlo cells prior to injection and post-recovery.
Statistics
Statistical analyses were performed using the R statistical programming language or GraphPad Prism (GraphPad Software, Inc.). A p-value of < 0.05 was considered statistically significant for this study.
Supplementary Material
Acknowledgements.
This work was supported by the NIH (DA033760, NS092980, AI123210) and the Skaggs Institute for Chemical Biology. J.B. is supported by a fellowship from Celgene. We thank J. Olucha for recombinant expression of mABHD12 in HEK293F cells; J. Chen (Automated Synthesis Facility at Scripps Research) for measuring enantiopurity of (S)-DO271; C. E. Moore and M. Gembicky (UCSD) for X-ray crystallographic analysis of DO253; and J. Wang and C. Chen (Pharmaron) for the measurement of tissue drug concentration.
Footnotes
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.
Competing Financial Interests.
O.U. and A.R. are employees of and B.F.C. is a founder and advisor to Abide Therapeutics, a biotechnology company interested in developing serine hydrolase inhibitors and therapeutics.
References
- 1.Rosen H, Germana Sanna M, Gonzalez-Cabrera PJ & Roberts E The organization of the sphingosine 1-phosphate signaling system. Curr Top Microbiol Immunol 378, 1–21, doi:10.1007/978-3-319-05879-5_1 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Yung YC, Stoddard NC & Chun J LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res 55, 1192–1214, doi:10.1194/jlr.R046458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makide K et al. Novel lysophosphoplipid receptors: their structure and function. J Lipid Res 55, 1986–1995, doi:10.1194/jlr.R046920 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kihara Y, Mizuno H & Chun J Lysophospholipid receptors in drug discovery. Exp Cell Res 333, 171–177, doi:10.1016/j.yexcr.2014.11.020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Kleij D et al. A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune polarization. J Biol Chem 277, 48122–48129, doi:10.1074/jbc.M206941200 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez-Cabrera PJ, Brown S, Studer SM & Rosen H S1P signaling: new therapies and opportunities. F1000Prime Rep 6, 109, doi:10.12703/P6-109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue A & Aoki J [TGFalpha shedding assay is useful for identifying ligands for orphan GPCRs]. Seikagaku 85, 1029–1033 (2013). [PubMed] [Google Scholar]
- 8.Kitamura H et al. GPR34 is a receptor for lysophosphatidylserine with a fatty acid at the sn-2 position. J Biochem 151, 511–518, doi:10.1093/jb/mvs011 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Barnes MJ et al. The lysophosphatidylserine receptor GPR174 constrains regulatory T cell development and function. J Exp Med 212, 1011–1020, doi:10.1084/jem.20141827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugo T et al. Identification of a lysophosphatidylserine receptor on mast cells. Biochem Biophys Res Commun 341, 1078–1087, doi:10.1016/j.bbrc.2006.01.069 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Shinjo Y et al. Lysophosphatidylserine suppresses IL-2 production in CD4 T cells through LPS3/GPR174. Biochem Biophys Res Commun 494, 332–338, doi:10.1016/j.bbrc.2017.10.028 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Barnes MJ & Cyster JG Lysophosphatidylserine suppression of T cell activation via GPR174 requires Galphas proteins. Immunol Cell Biol, doi:10.1111/imcb.12025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamat SS et al. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat Chem Biol 11, 164–171, doi:10.1038/nchembio.1721 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato T et al. Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family. J Biol Chem 272, 2192–2198 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Blankman JL, Long JZ, Trauger SA, Siuzdak G & Cravatt BF ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc Natl Acad Sci U S A 110, 1500–1505, doi:10.1073/pnas.1217121110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiskerstrand T et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. Am J Hum Genet 87, 410–417, doi:10.1016/j.ajhg.2010.08.002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiskerstrand T et al. A novel Refsum-like disorder that maps to chromosome 20. Neurology 72, 20–27, doi:10.1212/01.wnl.0000333664.90605.23 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Parkkari T et al. Discovery of triterpenoids as reversible inhibitors of alpha/beta-hydrolase domain containing 12 (ABHD12). PLoS One 9, e98286, doi:10.1371/journal.pone.0098286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoover HS, Blankman JL, Niessen S & Cravatt BF Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg Med Chem Lett 18, 5838–5841, doi:10.1016/j.bmcl.2008.06.091 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795, doi:10.1038/nature09472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adibekian A et al. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat Chem Biol 7, 469–478, doi:10.1038/nchembio.579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savinainen JR, Navia-Paldanius D & Laitinen JT A Sensitive and Versatile Fluorescent Activity Assay for ABHD12. Methods Mol Biol 1412, 179–189, doi:10.1007/978-1-4939-3539-0_19 (2016). [DOI] [PubMed] [Google Scholar]
- 23.van der Wel T et al. A natural substrate-based fluorescence assay for inhibitor screening on diacylglycerol lipase alpha. J Lipid Res 56, 927–935, doi:10.1194/jlr.D056390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung D, Hardouin C, Boger DL & Cravatt BF Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol 21, 687–691, doi:10.1038/nbt826 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Cognetta AB 3rd et al. Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem Biol 22, 928–937, doi:10.1016/j.chembiol.2015.05.018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inloes JM et al. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A 111, 14924–14929, doi:10.1073/pnas.1413706111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu C et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10, R130, doi:10.1186/gb-2009-10-11-r130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947, doi:10.1523/JNEUROSCI.1860-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viader A et al. A chemical proteomic atlas of brain serine hydrolases identifies cell type-specific pathways regulating neuroinflammation. Elife 5, e12345, doi:10.7554/eLife.12345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gijon MA, Riekhof WR, Zarini S, Murphy RC & Voelker DR Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem 283, 30235–30245, doi:10.1074/jbc.M806194200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blankman JL, Simon GM & Cravatt BF A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14, 1347–1356, doi:10.1016/j.chembiol.2007.11.006 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navia-Paldanius D, Savinainen JR & Laitinen JT Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). J Lipid Res 53, 2413–2424, doi:10.1194/jlr.M030411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oldstone MB Lessons learned and concepts formed from study of the pathogenesis of the two negative-strand viruses lymphocytic choriomeningitis and influenza. Proc Natl Acad Sci U S A 110, 4180–4183, doi:10.1073/pnas.1222025110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687, doi:10.1038/nature04444 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Brooks DG et al. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12, 1301–1309, doi:10.1038/nm1492 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baccala R et al. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci U S A 111, 8925–8930, doi:10.1073/pnas.1408148111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frebel H et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med 209, 2485–2499, doi:10.1084/jem.20121015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H & Inoue I Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 57, 621–632, doi:10.1038/jhg.2012.91 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Sims R et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384, doi:10.1038/ng.3916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183, doi:10.1038/nature16549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alhouayek M, Masquelier J & Muccioli GG Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol Sci 39, 586–604, doi:10.1016/j.tips.2018.02.011 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Olson JK & Miller SD Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173, 3916–3924 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Neher JJ et al. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol 186, 4973–4983, doi:10.4049/jimmunol.1003600 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Lehnardt S et al. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol 177, 583–592 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann O et al. TLR2 mediates neuroinflammation and neuronal damage. J Immunol 178, 6476–6481 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Vambutas A & Pathak S AAO: Autoimmune and Autoinflammatory (Disease) in Otology: What is New in Immune-Mediated Hearing Loss. Laryngoscope Investig Otolaryngol 1, 110–115, doi:10.1002/lio2.28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wootla B, Eriguchi M & Rodriguez M Is multiple sclerosis an autoimmune disease? Autoimmune Dis 2012, 969657, doi:10.1155/2012/969657 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference for Methods section
- 48.Suciu RM, Cognetta AB 3rd, Potter ZE & Cravatt BF Selective Irreversible Inhibitors of the Wnt-Deacylating Enzyme NOTUM Developed by Activity-Based Protein Profiling. ACS Med Chem Lett 9, 563–568, doi:10.1021/acsmedchemlett.8b00191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bachovchin DA et al. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A 107, 20941–20946, doi:10.1073/pnas.1011663107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexander JP & Cravatt BF Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol 12, 1179–1187, doi:10.1016/j.chembiol.2005.08.011 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin BR, Wang C, Adibekian A, Tully SE & Cravatt BF Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–89, doi:10.1038/nmeth.1769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE & Cravatt BF Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods 10, 259–264, doi:10.1038/nmeth.2368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogasawara D et al. Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc Natl Acad Sci U S A 113, 26–33, doi:10.1073/pnas.1522364112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borrow P, Evans CF & Oldstone MB Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol 69, 1059–1070 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmed R, Salmi A, Butler LD, Chiller JM & Oldstone MB Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160, 521–540 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.