

SUMMARY
Ca2+-permeable AMPA-type glutamate receptors (CP-AMPARs) containing GluA1 but lacking GluA2 subunits contribute to multiple forms of synaptic plasticity, including long-term potentiation (LTP), but mechanisms regulating CP-AMPARs are poorly understood. A-kinase anchoring protein (AKAP) 150 scaffolds kinases and phosphatases to regulate GluA1 phosphorylation and trafficking, and trafficking of AKAP150 itself is modulated by palmitoylation on two Cys residues. Here, we developed a palmitoylation-deficient knockin mouse to show that AKAP150 palmitoylation regulates CP-AMPAR incorporation at hippocampal synapses. Using biochemical, super-resolution imaging, and electrophysiological approaches, we found that palmitoylation promotes AKAP150 localization to recycling endosomes and the postsynaptic density (PSD) to limit CP-AMPAR basal synaptic incorporation. In addition, we found that AKAP150 palmitoylation is required for LTP induced by weaker stimulation that recruits CP-AMPARs to synapses but not stronger stimulation that recruits GluA2-containing AMPARs. Thus, AKAP150 palmitoylation controls its subcellular localization to maintain proper basal and activity-dependent regulation of synaptic AMPAR subunit composition.
In Brief
Purkey et al. uncover a requirement for palmitoylation of the postsynaptic scaffold protein AKAP150 in regulating Ca2+-permeable AMPA receptors to control synaptic plasticity.
Graphical Abstract
INTRODUCTION
AMPARs are the primary mediators of fast excitatory neurotransmission in the CNS, and regulation of the number and activity of postsynaptic AMPARs is crucial for forms of synaptic plasticity that support learning and memory, including NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) and longterm depression (LTD) (Huganir and Nicoll, 2013). AMPARs are tetramers assembled from GluA1–4 subunits, with incorporation of GluA2 subunits decreasing channel conductance and inhibiting Ca2+ influx. After the early postnatal period, the majority of AMPARs at hippocampal CA1 synapses under basal conditions are Ca2+-impermeable GluA1/2 or GluA2/3 heterotetramers (Lu et al., 2009; Rozov et al., 2012; Stubblefield and Benke, 2010). However, Ca2+-permeable GluA1 homomeric receptors (CP-AMPARs) can be recruited to hippocampal synapses from extrasynaptic and/or intracellular stores to regulate synaptic strength during LTP, LTD, and homeostatic plasticity (Aoto et al., 2008; Soares et al., 2013; Lu et al., 2007; Plant et al., 2006; Sanderson et al., 2016, 2012, 2018; Sutton et al., 2006; Thiagarajan et al., 2005; Yang et al., 2008; but see Adesnik and Nicoll, 2007; Gray et al., 2007). These recruited CP-AMPARs, because of both greater single-channel conductance and Ca2+ permeability, can in turn not only influence the level of plasticity expression but also alter the capacity of synapses to undergo subsequent plasticity, so-called metaplasticity. Importantly, CP-AMPAR-mediated metaplasticity in the nucleus accumbens and amygdala is, respectively, linked to reward learning relevant for drug addiction and fear memory extinction relevant for post-traumatic stress disorder (Clem and Huganir, 2010; Wolf, 2016). However, the roles of CP-AMPARs in regulating LTP and LTD and metaplasticity at hippocampal synapses relevant for spatial and contextual learning and memory are less clear and remain controversial.
We know that phosphorylation and dephosphorylation of S845 in the GluA1 C-terminal domain by the cAMP-dependent protein kinase PKA and the Ca2+-calmodulin-dependent protein phosphatase 2B/calcineurin (CaN) regulate CP-AMPAR synaptic insertion and removal, respectively (Esteban et al., 2003; He et al., 2009; Hu et al., 2007; Man et al., 2007; Oh et al., 2006; Qian et al., 2012; Sun et al., 2005; Yang et al., 2008). However, we still do not understand how postsynaptic PKA and CaN signaling are coordinated to control CP-AMPAR trafficking between intracellular compartments, such as recycling endosomes (REs), the extrasynaptic membrane, and the postsynaptic density (PSD). An increasing body of evidence indicates that the scaffold protein AKAP79/150 (human79/rodent150; Akap5 gene) targets both PKA and CaN to AMPARs to regulate GluA1 phosphorylation and trafficking to control LTP and LTD balance and homeostatic potentiation (Diering et al., 2014; Jurado et al., 2010; Lu et al., 2007; Sanderson et al., 2012, 2016, 2018; Tunquist et al., 2008; Zhang et al., 2013). Thus, a key question is how is the post-synaptic localization of AKAP79/150 itself regulated.
AKAP79/150 is targeted to the postsynaptic plasma membrane primarily by an N-terminal poly-basic domain that binds to PIP2, cortical F-actin, and cadherin adhesion molecules and secondarily by an internal domain that binds PSD-95, a major structural scaffold of the PSD (Colledge et al., 2000; Dell’Acqua et al., 1998; Gomez et al., 2002; Gorski et al., 2005; Robertson et al., 2009). More recently we discovered that AKAP79/150 is S-palmitoylated on two conserved Cys residues (C36 and C129 human/123 mouse) within the N-terminal targeting domain by the RE-localized palmitoyl acyltransferase DHHC2 (Keith et al., 2012; Woolfrey et al., 2015). AKAP palmitoylation is not required for its general targeting to the plasma membrane or its binding to F-actin (Gomez et al., 2002) but is required for its specific localization to dendritic REs and association with cholesterol-rich, detergent-resistant membrane lipid rafts (Delint-Ramirez et al., 2011; Keith et al., 2012). Of note, the PSD is biochemically defined by its detergent insolubility, and accordingly, many PSD proteins are palmitoylated and lipid-raft associated, including PSD-95 (El-Husseini et al., 2002; Fukata and Fukata, 2010; Fukata et al., 2013; Globa and Bamji, 2017; Keith et al., 2012; Noritake et al., 2009; Sezgin et al., 2017). However, it is not known if AKAP79/150 palmitoylation also controls its association with the PSD.
In contrast to other protein lipidations like myristoylation and prenylation, palmitoylation is reversible, with palmitate removal being catalyzed by protein palmitoyl thioesterases (Yokoi et al., 2016). Importantly, palmitoylation of PSD-95, AKAP150, and other PSD scaffolds is affected by seizures and anticonvulsants in vivo and has been implicated in regulating AMPAR trafficking and synaptic strength in cultured neurons in vitro (Brigidi et al., 2014; El-Husseini et al., 2002; Kang et al., 2008; Kay et al., 2015; Keith et al., 2012; Thomas et al., 2012; Woolfrey et al., 2015; Zhang et al., 2014). In particular, AKAP79/150 palmitoylation and dendritic spine targeting are bi-directionally regulated by neuronal activity in cultured neurons to coordinately control a number of cellular correlates of LTP and LTD, including RE exocytosis, spine morphology, GluA1 surface expression, and AMPAR synaptic activity (Keith et al., 2012; Woolfrey et al., 2015, 2018). However, we do not know whether palmitoylation controls AKAP79/150 postsynaptic localization or AMPAR regulation during synaptic plasticity in the intact circuitry of the hippocampus in vivo. In addition, despite the prominence of palmitoylation modifying PSD proteins, no in vivo models have been developed to specifically disrupt palmitoylation of a specific postsynaptic protein and then determine the impacts on synaptic function. Importantly, here we developed a palmitoylation-deficient AKAP150 C36,123S (AKAPCS) knockin mutant mouse line to characterize the role of AKAP palmitoylation in regulating its targeting to the PSD and in controlling CP-AMPAR incorporation both basally and during LTP at CA1 synapses.
RESULTS
AKAPCS Palmitoylation-Deficient Knockin Mice Exhibit Reduced AKAP150 Levels in PSD-Enriched Fractions
To study the impacts of loss of AKAP150 palmitoylation, we generated a palmitoylation-deficient AKAP150 mouse (AKAPCS) (Figure 1A) using a piggyBac transposon-based embryonic stem cell (ESC)-targeting vector strategy to introduce mutations into the mouse Akap5 gene locus (Figure S1A). The resulting Akap5CS mutant allele replaces Cys at positions 36 and 123 with Ser (Figures 1A and S1A), while simultaneously introducing a HindIII site to facilitate genotyping (Figure S1B). AKAPCS mice are viable and are visibly indistinguishable from their wild-type (WT) littermates, with no apparent physical deficits or changes in overall brain anatomy (not shown). In addition, we observed no changes in dendritic spine numbers or morphology in CA1 stratum radiatum of ex vivo brain slices (Figures S1C–S1E) or in cultured hippocampal neurons prepared from CS compared with WT mice (Figures S1F–S1H). To confirm loss of AKAP150 palmitoylation in CS mice, we used an APEGS (Acyl-PEGyl exchange gel-shift) assay of palmitoylation that exchanges palmitates for polyethylene glycol polymers to produce upward molecular weight shifts (Woolfrey et al., 2018; Yokoi et al., 2016). Using this APEGS assay we detected mono- and di-palmitoylated AKAP in whole-brain extracts of WT but not CS mice, in which only unpalmitoylated 150 kDa AKAP was detected (Figure 1B).
Figure 1. AKAP150 and PKA-RII Levels Are Reduced in PSD-Enriched Fractions from AKAPCS Palmitoylation-Deficient Mice.

(A) Schematic of AKAP150 highlighting binding partners and functional domains. AKAP150 is palmitoylated at Cys 36 and 123, and these residues are mutated to Ser to create the AKAPCS palmitoylation-deficient mutant mouse.
(B) APEGS assay showing that AKAP150 WT, but not CS, is palmitoylated in lysates from mouse brain.
(C) Subcellular fractionation and western blotting from WT and CS P21 mouse hippocampus for AKAP150, PSD-95, and PKA-RIIβ. P2, crude synaptosomes; S2, cytosol and light membranes; TxP, triton-insoluble sub-fraction of P2 = PSD-enriched fraction; TxS, triton-soluble sub-fraction of P2; WE, whole extract.
(D–F) Quantification of subcellular fractionation from (C) normalized to WT WE levels showing (D) decreased AKAP150 protein levels in P2 and TxP fractions from CS mice (P2: WT 1.36 ± 0.14, CS 0.44 ± 0.16, unpaired t test **p = 0.0033; TxP: WT 1.00 ± 0.10, CS 0.17 ± 0.05, unpaired t test ***p = 0.00028; WT n = 5, CS n = 4), (E) decreased PKA-RIIβ protein levels in TxP fractions from CS mice (WT 0.44 ± 0.063, CS = 0.22 ± 0.029, unpaired t test *p = 0.036; n = 3), but (F) no change in fractionation of PSD-95 in CS versus WT mice.
(G) AKAP150 APEGS assay of subcellular fractions from WT mouse forebrain.
(H) Quantification of the proportion of AKAP150 in the unpalmitoylated lower MW band and the mono- and di-palmitoylated higher MW bands across the subcellular fractions in (G).
(I) Quantification of the total proportion of palmitoylated AKAP150 (mono- plus di-) revealing significantly more palmitoylated AKAP150 in P2 versus S2 and TxP versus TxS (S2 0.26 ± 0.16, P2 0.63 ± 0.065, unpaired t test *p = 0.022; TxS 0.56 ± 0.059, TxP 0.73 ± 0.062, unpaired t test *p = 0.028; n = 3).
*p < 0.05, **p < 0.01, and ***p < 0.001 by unpaired t test. Data are reported as mean ± SEM; n = number of animals.
Previous biochemical and imaging studies indicate that AKAP79/150 associates with membrane lipids, including in lipid rafts, and other postsynaptic proteins, including PSD-95 and F-actin, and is localized not only in the PSD but also the extra-synaptic membrane (Carr et al., 1992; Colledge et al., 2000; Dell’Acqua et al., 1998; Gomez et al., 2002; Keith et al., 2012; Smith et al., 2006). To explore the effect of eliminating AKAP palmitoylation on its synaptic localization in vivo, we used differential centrifugation and detergent extraction (Grosshans et al., 2002; Sanderson et al., 2012, 2016; Smith et al., 2006) to isolate subcellular fractions from hippocampal lysates of 2- to 3-week-old mice followed by immunoblotting (Figure 1C). Intriguingly, we observed a selective decrease in AKAP150 CS protein compared with WT in the synaptosomal membrane fraction (P2) and a PSD-enriched fraction (TxP) derived from P2 by Triton X-100 detergent extraction (Figures 1C and 1D). An accompanying decrease in PKA-RII regulatory subunits was also seen in TxP for CS mice (Figure 1E). The distribution of PSD-95 across these fractions was not significantly different between WT and CS mice, with its highest levels detected in the TxP-PSD-enriched fraction as expected; however, we did observe a non-significant trend toward slightly increased PSD-95 levels in TxP for CS (Figure 1F). Overall, these fractionation data suggest that AKAP150CS is less associated with the PSD-enriched fraction than WT and thus that palmitoylation normally promotes AKAP150 localization in the PSD. Consistent with this idea, we combined subcellular fractionation with the APEGS palmitoylation assay in WT mice (Figure 1G) to reveal significant enrichment of palmitoylated AKAP150 in P2 relative to the S2 fractions and TxP relative to TxS fractions (Figures 1H and 1I). In particular, although unpalmitoylated AKAP150 predominates in whole extracts and the cytosolic-S2 fraction, mono- plus di-palmitoylated AKAP150 constitute the majority in synaptosomal-P2, perisynaptic-TxS, and PSD-enriched-TxP fractions, the latter of which contains the highest overall proportion of palmitoylated AKAP150 (Figures 1H and 1I).
AKAPCS Dendritic Spines Contain Smaller AKAP150 Nano-domains that Exhibit Reduced Overlap with the PSD
Because of the submicrometer dimensions of dendritic spines and organization of the PSD into even smaller nano-domains on the scale of ~100 nm (Nair et al., 2013; MacGillavry et al., 2013; Sinnen et al., 2017; Tang et al., 2016), we reasoned that any changes in AKAP150CS postsynaptic localization may be below the diffraction-limited resolution of standard confocal microscopy (~250 nm). Indeed, previous studies using standard microscopy revealed no differences in basal spine localization of GFP-tagged AKAP79 WT versus CS in transfected neurons (Keith et al., 2012). We therefore used a custom-built, two-color stimulated emission depletion (STED) nanoscope with a resolution of ~40–60 nm (Meyer et al., 2016) to assess the localization of AKAP150 relative to PSD-95 in dendritic spines of hippocampal neurons cultured from WT and CS mice (Figure 2). In agreement with our previous work on AKAP79CS-GFP, standard confocal imaging revealed that AKAP150CS and WT are both localized to dendritic spines and show substantial overlap with PSD-95; however, the improved resolution of STED revealed ~100–200 nm diameter AKAP150 clusters for both WT and CS that were not visible in confocal images and were located overlapping the PSD, closely surrounding the PSD, and also in distinct locations outside the PSD (Figures 2A and 2B). This ability of STED to resolve distinct extrasynaptic, perisynaptic, and PSD clusters of AKAP150 that are not visible in standard confocal imaging parallels findings for AMPARs using STED and stochastic optical reconstruction microscopy-photoactivated localization microscopy (STORM-PALM) that revealed previously unappreciated nano-domain organization (Nair et al., 2013; MacGillavry et al., 2013; Sinnen et al., 2017; Tang et al., 2016) (see also Figure 4). Using a custom, object-based image segmentation mask analysis method (Figure 2B) that we recently developed for intensity-based super-resolution imaging methods (i.e., STED; see STAR Methods), we found that the area (Figure 2C) and major axis length of individual AKAP150 objects (Figure 2D) in spines were both significantly reduced for the CS mutant compared with WT. Correspondingly, the total perimeter (Figure 2E) and area occupied by AKAP150 objects within spines (Figure 2F) were both significantly reduced for CS but with no changes in the average number of AKAP objects per spine (Figure 2G). Importantly, the proportional spatial overlap of AKAP150 and PSD-95 objects was also decreased for CS (Figure 2H) despite a small increase in total PSD area in spines (see Figure 4L). Collectively, these STED imaging data are in agreement with the fractionation data presented above and indicate that AKAP150CS localization in and around the PSD is reduced.
Figure 2. AKAP150CS Localization to the PSD Is Reduced.

(A and B) Confocal and STED imaging (A) and associated segmentation object masks (B) for 14–16 day in vitro (DIV) 14–16 hippocampal cultures from WT and CS mice stained for AKAP150 (red) and PSD-95 (green). STED images show enhanced resolution and provide better sub-synaptic visualization of AKAP150 localization relative to the PSD.
(C–F) Significant decrease in AKAP object area in AKAPCS cultures (C) (WT 0.01961 ± 0.0009 μm2, n = 102 spines; CS 0.01614 ± 0.0007 μm2, n = 106 spines; unpaired t test **p = 0.0043) that was accompanied by decreases in (D) AKAP object major-axis length (WT 0.205 ± 0.006 μm, CS 0.1874 ± 0.006 μm, unpaired t test *p = 0.0344), (E) total AKAP perimeter (WT 0.5585 ± 0.017 μm, CS 0.5034 ± 0.014 μm, *p = 0.0104), and (F) AKAP compartment area within spines (WT 0.9372 ± 0.019 μm2, CS 0.8662 ± 0.019 μm2, unpaired t test *p = 0.0104).
(G and H) No change is seen in AKAP object number per spine (G), but (H) AKAPCS PSD localization is reduced, as indicated by a decrease in AKAP and PSD-95 object overlap (WT 0.2971 ± 0.01, CS 0.2474 ± 0.01, **p = 0.0058).
*p < 0.05 and **p < 0.01 by unpaired t test. Data are reported as mean ± SEM.
AKAPCS Localization to REs Is Decreased
In previous work, we found that AKAP palmitoylation also controls targeting to REs. In addition, we observed that acute disruption of AKAP79/150 palmitoylation in rat hippocampal cultures resulted in enhanced basal RE fusion events in neuronal dendrites (Keith et al., 2012; Woolfrey et al., 2015). Accordingly, we assessed the co-localization of AKAP150 with REs marked by live-cell feeding with Alexa 488-labeled transferrin (TF-488) and also monitored basal exocytosis of transferrin receptor (TfR)-positive REs by expressing superecliptic pHluorin-tagged TfR (SEP-TfR) in WT and CS mouse dissociated hippocampal cultures (Figure S2). Consistent with previous work on human AKAP79, AKAP150 robustly co-localized with TF-488-positive puncta in WT mouse neurons but showed a significant decrease in RE localization in CS neurons (Figures S2A and S2B). Contrary to our previous findings showing that acute AKAP79CS overexpression increased basal RE exocytosis, basal RE exocytosis imaged with SEP-TfR was not significantly different in CS compared with WT mouse neurons, although a slight non-significant trend toward increased exocytosis was observed (Figures S2C and S2D). Collectively, these data suggest that RE exocytosis (as read out by TfR recycling) is largely normal in CS mice despite decreased AKAP150 localization to REs.
AKAPCS Mice Exhibit Enhanced CP-AMPAR-Mediated Basal Synaptic Transmission
Given that we observed reduced AKAP150 association with both REs and the PSD in CS mice, we wanted to explore how synaptic transmission and plasticity might be affected. To start, we characterized basal synaptic transmission at CA1 synapses in acute, ex vivo hippocampal slices from 2- to 3-week-old WT and CS mice by whole-cell voltage-clamp recording of AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs) and spontaneous excitatory postsynaptic currents (sEPSCs). Compared with WT, CS mice showed slightly enhanced mean mEPSC amplitude, slightly decreased mean mEPSC frequency (Figure 3A), and corresponding rightward shifts in the cumulative distributions of mEPSC amplitudes and inter-event intervals (Figure 3B). A similar decrease in sEPSC frequency and a non-significant trend toward increased sEPSC amplitude was observed in CS compared with WT (Figures 3C and 3D). Cultured hippocampal neurons from AKAPCS mice also exhibited slightly increased mEPSC amplitude (Figures S3A and S3B) but with no change in frequency (Figures S3A and S3C). Decreased mEPSC/sEPSC frequency could indicate a reduction in presynaptic release probability or a reduction in the overall number of synapses; however, analysis of CA1 dendritic spine numbers above revealed no differences between WT and CS (Figure S1C). We tested for changes in presynaptic release probability by measuring evoked AMPAR-mediated paired-pulse ratios (PPRs) at Schaffer collateral (SC) synapses. We observed no differences in PPR between WT and CS in either whole-cell −70 mV EPSC (Figure 3E) or extracellular field excitatory postsynaptic potential (fEPSP) recordings (Figure 3G), thus indicating normal presynaptic function in CS mice. Furthermore, input-output curves for evoked AMPAR EPSC amplitude (Figure 3F) and fEPSP slope (Figure 3H) were both similar for WT and CS mice.
Figure 3. AKAPCS Mice Exhibit Slightly Increased AMPAR mEPSC Amplitude and Decreased Frequency but Normal Evoked Basal Transmission at Hippocampal CA1 Synapses.

(A and B) Representative traces for mEPSC recordings with plots of mean amplitude and frequency (A) and cumulative distribution plots of mEPSC amplitude and inter-event interval (B) for CA1 neurons in acute hippocampal slices from WT and AKAPCS mice showing a slight increase in mEPSC amplitude and a slight decrease in mEPSC frequency (A: mEPSC amplitude: WT = 6.79 ± 0.371 pA n = 29 cells, CS = 7.883 ± 0.251 pA n = 35 cells, unpaired t test *p = 0.0145; mEPSC frequency: WT = 0.72 ± 0.059 Hz, CS = 0.57 ± 0.042, unpaired t test *p = 0.0475).
(C and D) Representative traces for sEPSC recording with plots of mean amplitude and frequency (C) and cumulative distribution plots of sEPSC amplitude and inter-event interval (D) for WT and CS mice showing a slight but not significant increase in sEPSC amplitude and a significant decrease in sEPSC frequency for CS mice (C: sEPSC frequency: WT 2.21 ± 0.297 Hz, n = 19 cells; CS 1.02 ± 0.0862 Hz, n = 24 cells; unpaired t test ***p = 0.0001).
(E–H) No changes in SC-CA1 evoked basal AMPAR transmission are observed for CS mice in (E and G) paired-pulse ratios or (F and H) input-output curves in either whole-cell EPSC or extracellular fEPSP recordings.
*p < 0.05 and ***p < 0.001 by unpaired t test. Data are reported as mean ± SEM.
A normal evoked SC-CA1 input-output relationship for CS mice indicates that basal AMPAR-mediated synaptic strength is largely unaffected despite somewhat decreased frequency of spontaneous transmission. However, the small increase in mEPSC amplitude could reflect a change in AMPAR subunit composition related to synaptic incorporation of higher conductance GluA2-lacking CP-AMPARs. Consistent with possible incorporation of CP-AMPARs at SC-CA1 synapses in CS mice, the ratio of evoked inward −70 mV AMPA peak current to outward +40 mV NMDA current (measured 50 ms after peak) was increased in CS mice relative to the corresponding ratio of peak outward +40 mV AMPA to NMDA current (Figure 4A), indicating the possible presence of inwardly rectifying CP-AMPARs in CS mice. Yet increased AMPA/NMDA ratios can arise not only from enhanced AMPAR function but also from decreased NMDAR function. However, the input-output relationship for evoked NMDAR EPSCs revealed no significant differences in basal NMDAR transmission between CS and WT mice, with if anything a trend toward increased NMDAR function in CS (Figure 4B). Taken together, these data suggest that the increases in mEPSC amplitude and AMPA/NMDA ratio observed in CS mice may be attributable to synaptic CP-AMPARs.
Figure 4. AKAPCS Mice Have Elevated Basal CP-AMPAR Activity at CA1 Synapses.

(A) Evoked SC-CA1 AMPA/NMDA EPSC ratios from WT and CS slices; AKAPCS mice show a substantial increase in −70 mV peak AMPA to +40 mV 50 ms after peak NMDA tail EPSC ratio and a smaller increase in the mixed AMPA and NMDA +40 mV peak to +40 mV 50 ms after peak NMDA tail EPSC ratio (−70 mV/+40 mV: WT 0.88 ± 0.080, n = 9 cells, CS 1.43 ± 0.15, n = 8 cells, unpaired t test **p = 0.0052; +40 mV: WT 1.26 ± 0.036, CS 1.46 ± 0.040, unpaired t test **p = 0.0017).
(B) No change in evoked NMDA +40 mV EPSC input-output (I-O) relationship in AKAPCS.
(C and D) Normalized AMPA EPSC I-V curve showing (C) decreased outward current at positive potentials (AMPA I-V at +40 mV: WT 0.73 ± 0.093, n = 10 cells; CS 0.45 ± 0.059, n = 9 cells; unpaired t test *p = 0.0251; normalized to −70 mV EPSC amplitude) and (D) increased −70 mV/+40 mV EPSC amplitude rectification index (RI: WT 1.30 ± 0.121, n = 21; CS 2.12 ± 0.187, n = 19; unpaired t test ***p = 0.0006) in AKAPCS slices.
(E) Inhibition of 70 mV AMPA EPSC amplitude in AKAPCS but not WT slices by 20 μM CP-AMPAR blocker NASPM (WT 0.065 ± 0.042, n = 8 cells, CS −0.39 ± 0.084, n = 5 cells; unpaired t test ***p = 0.0002; fold change baseline after NASPM application).
(F–I) STED imaging of cultured hippocampal neurons stained for surface GluA1 (sGluA1) and PSD-95 (F) showing for AKAPCS neurons (G) increased sGluA1 object area (WT 0.02202 ± 0.00014 μm2, n = 125 spines; CS 0.02835 ± 0.00015 μm2, n = 170 spines; unpaired t test **p = 0.0032) with (H) an increase in GluA1 object major-axis length (WT 0.19 ± 0.0067 μm, CS 0.2132 ± 0.0064 μm, *p = 0.0132) but with (I) no change in object number.
(J and K) AKAPCS spines also have increased total perimeter (J) (WT 0.5472 ± 0.02 μm, CS 0.6252 ± 0.02 μm, **p = 0.0070) and (K) area occupied by sGluA1 staining in spines (WT 0.4571 ± 0.019 μm2, CS 0.5111 ± 0.016 μm2, *p = 0.0359).
(L and M) The total area occupied by PSD-95 in spines is also increased in AKAPCS compared with WT (L) (WT 0.5056 ± 0.021 μm2, CS 0.5899 ± 0.016 μm2, **p = 0.0013) but with (M) no change in PSD-95 overlap with sGluA1.
*p < 0.05, **p < 0.01, and ***p < 0.001 by unpaired t test. Data are reported as mean ± SEM.
To directly test whether synaptic AMPAR subunit composition is different in CS mice, we used two approaches. First, we determined the current-voltage (I-V) relationship for AMPAR EPSCs over a range of holding potentials from −70 to +40 mV. CP-AMPARs exhibit inward rectification due to block of outward current by intracellular polyamines. As expected, WT mice displayed a linear AMPA EPSC I-V relationship, which is characteristic of GluA2-containing AMPARs. By contrast, SC-CA1 transmission in CS mice exhibited an inward-rectifying AMPA I-V relationship, which is indicative of GluA2-lacking CP-AMPARs (Figure 4C). Inward rectification in CS mice was also quantified as a significantly enhanced −70 mV/+40 mV AMPA EPSC rectification index (Figure 4D). Second, we applied NASPM, an extracellular polyamine, which selectively blocks inward current mediated by CP-AMPARs. Consistent with CS mice containing a greater number of synaptic CP-AMPARs, application of NASPM blocked ~40% of the inward AMPA EPSC in CS but not WT mice (Figure 4E). Furthermore, while mEPSC amplitude and frequency measured in WT mouse cultured hippocampal neurons were insensitive to the CP-AMPAR blocker IEM1460, the basal enhancement of mEPSC amplitude in CS cultured neurons was inhibited or reversed by IEM1460 application, with no impact on frequency (Figures S3D–S3H).
Finally, STED imaging and object-based segmentation analysis of surface GluA1 (sGluA1) and PSD-95 antibody staining (Figure 4F) revealed an increase in sGluA1 object area (Figure 4G) and major-axis length (Figure 4H) but with no change in the average number of sGluA1 objects per spine for CS compared with WT (Figure 4I). However, the total perimeter (Figure 4J) and area occupied by sGluA1 objects (Figure 4K) were both increased for CS. In addition, increased sGluA1 clustering in CS neurons was also accompanied by an increase in total area occupied by PSD-95 objects in spines (Figure 4L), perhaps explaining why the proportional overlap of sGluA1 with PSD-95 remained similar between CS and WT (Figure 4M). Overall, these sGluA1 STED imaging results are consistent with increased postsynaptic GluA1 expression in CS cultured neurons and increased basal CP-AMPAR activity measured by electrophysiology.
Previous work found that phosphorylation of GluA1 S845 by AKAP150-anchored PKA promotes and dephosphorylation by AKAP150-anchored CaN restricts CP-AMPAR synaptic incorporation (Lu et al., 2007; Sanderson et al., 2012, 2016, 2018). However, immunoblotting analysis of hippocampal subcellular fractions (Figure S4A) revealed no significant differences between WT and CS in either total GluA1 expression (Figure S4B) or pS845 levels, although non-significant trends toward increased pS845 were observed across all fractions in CS mice (Figure S4C). Overall, these data indicate that hippocampal neurons from CS mice have increased basal GluA1 CP-AMPAR synaptic activity; however, given the high single-channel conductance of these receptors, synaptic insertion of a relatively small number of S845 phosphorylated CP-AMPARs could account for this increased basal activity. Because the majority of AMPARs in CA1 are GluA1/2 heteromers (Lu et al., 2009), it would be very difficult to biochemically detect increased pS845 phosphorylation occurring in a small pool of CP-AMPARs in CS mice.
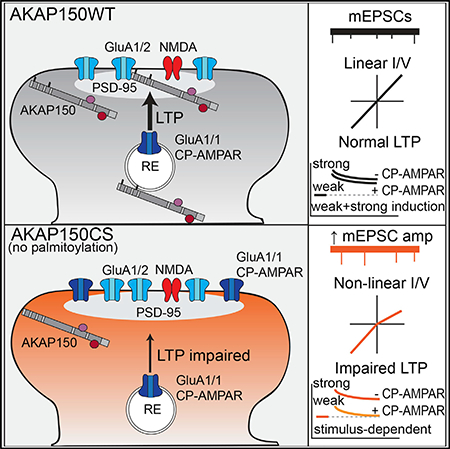
AKAP150 Palmitoylation Is Required for Expression of CP-AMPAR-Dependent but Not CP-AMPAR-Independent LTP
Our previous work found that AKAP150-anchored PKA and CaN modulate LTP and LTD at CA1 synapses through opposing each other in control of CP-AMPAR synaptic incorporation; however, the dependence of LTP on PKA signaling and AMPAR subunit composition is very flexible and developmentally plastic in mice between 2 and 8 weeks of age (Granger et al., 2013; Jensen et al., 2003; Kolleker et al., 2003; Lu et al., 2007; Sanderson et al., 2016; Zamanillo et al., 1999; Zhou et al., 2018). In addition, CP-AMPAR synaptic recruitment during LTP could be affected by the strength and type of induction stimulus, which is another major variable across previous studies (Adesnik and Nicoll, 2007; Gray et al., 2007; Jensen et al., 2003; Kolleker et al., 2003; Lu et al., 2007; Plant et al., 2006; Yang et al., 2010). Because of these factors, the contributions of GluA1 and CP-AMPARs to CA1 LTP remain unclear and controversial (Granger et al., 2013; Sanderson et al., 2016; Zhou et al., 2018). Therefore, we next examined how loss of AKAP150 palmitoylation affects LTP and LTD at CA1 synapses in 2- to 3-week-old mice. A standard 1 × 100 Hz, 1 s high-frequency stimulus (HFS) protocol elicited reliable LTP of fEPSP slope (~150%) in WT slices but failed to induce significant LTP in CS slices (Figures 5A and 5E). In contrast, LTD induced with prolonged low-frequency stimulation (LFS; 1 Hz, 900 pulses, 15 min) was comparable (~60%) at CA1 synapses in WT and CS mice (Figures 5B and 5F). To explore whether the LTP deficit in CS mice relates to altered CP-AMPAR regulation, we used two different common whole-cell pairing LTP induction protocols that we found differentially depend on CP-AMPARs in 2- to 3-week-old WT mice. In particular, we found that brief 2 × 100 Hz, 1 s stimulation, which is similar to HFS induction of LTP in fEPSP experiments, paired with 0 mV postsynaptic depolarization (Ahmad et al., 2012; Jurado et al., 2013) induced substantial LTP in WT (~200%) that was strongly impaired in CS slices and inhibited by NASPM in WT slices (Figures 5C and 5G). In contrast, LTP was similar and much greater in magnitude (~325%) for both CS and WT mice when induced with a stronger, prolonged pairing protocol (3 Hz, 90 s, 0 mV) (Adesnik and Nicoll, 2007; Jensen et al., 2003; Kolleker et al., 2003) that was largely insensitive to NASPM in WT slices (Figures 5D and 5H). These results indicate that the LTP deficits in CS mice are specifically related to impaired CP-AMPAR regulation and also suggest that high-conductance CP-AMPARs are more important for expression of the lower levels of LTP induced with weaker stimuli versus higher levels of LTP induced with stronger stimuli, which robustly recruit GluA2-containing AMPARs.
Figure 5. CP-AMPAR-Dependent LTP at CA1 Synapses Is Impaired in AKAPCS Mice.

(A and E) SC-CA1 fEPSP slope (normalized to baseline) recorded over time for WT and AKAPCS slices (A) and aggregate data for measurements of normalized fEPSP slope (E) (averaged over the last 10 min) showing robust 1×100 Hz 1 sec HFS induction of LTP in WT (~150%) that is significantly impaired in CS (A: ****p < 0.0001 by 2-way ANOVA over last 10 min; E: fEPSP slope for WT = 141.9 ± 4.55% n = 7 slices, CS = 110 ± 12.04% n = 7 slices, unpaired t test last 10 min *p = 0.028).
(B and F) SC-CA1 fEPSP slope (normalized to baseline) recorded over time for WT and AKAPCS slices (B) and aggregate data for measurements of normalizedfEPSP slope (F) (averaged over the last 10 min of recording) showing 1 Hz, 900 pulses (15 min) robust induction of LTD (~60%) in both WT and CS.
(C and G) Normalized EPSC amplitude (normalized to baseline) recorded over time (C) and aggregate data for measurements of normalized EPSC amplitude (G) (averaged over the last 10 min) showing CP-AMPAR dependent, NASPM-sensitive LTP induced by 2 × 100 Hz, 1 s HFS, 0 mV pairing in WT slices is impaired in AKAP CS slices (C: 2-way ANOVA with Tukey’s multiple comparisons test for last 10 min: WT NASPM versus WT ****p < 0.0001, WT versus CS ****p < 0.0001; G: WT = 204.9 ± 3.24% n = 5 cells, CS = 133.8 ± 9.41% n = 6 cells, WT NASPM = 119.6 ± 22.45% n = 5 cells; unpaired t test WT versus WT NASPM *p = 0.0113, WT versus CS *p = 0.0362).
(D and H) Normalized EPSC amplitude (normalized to baseline) recorded over time (D) and aggregate data for measurements of normalized EPSC amplitude (H) (averaged over the last 10 min) showing CP-AMPAR independent, NASPM insensitive LTP induced by 3 Hz, 90 s, 0 mV pairing in WT slices is normal in CS slices (H: WT = 337.4 ± 44.25% n = 6 cells, WT NASPM = 305.6 ± 51.8% n = 4 cells, CS = 389.1 ± 11.25% n = 5 cells). Data reported as mean ± SEM.
AKAPCS Mice Exhibit Enhanced, CP-AMPAR-Dependent De-depression after Prior Induction of LTD
Although the enhancement in basal AMPAR transmission is modest in CS mice, and a strong pairing induction stimulus can overcome the LTP deficit, we wanted to examine whether prior basal CP-AMPAR incorporation in CS mice was altering metaplasticity to prevent additional CP-AMPAR recruitment in response to HFS. Our previous studies found that AKAP-CaNdependent removal of CP-AMPARs from CA1 synapses is required during LTD (Sanderson et al., 2012, 2016). Thus, having demonstrated that LTD is comparable with WT in CS mice (Figure 5B), we wondered whether prior induction of LTD to remove a proportion of existing synaptic AMPARs might allow CP-AMPAR recruitment in response to subsequent LTP induction resulting in de-depression. With this in mind, we induced LTD with 1 Hz LFS and allowed its expression for 15 min before delivering 1 × 100 Hz HFS to induce LTP and de-depression. As seen in previous studies (Lee et al., 2000; Norris et al., 1996), HFS-induced de-depression in WT slices returned fEPSP responses back to pre-LFS baseline values within 30 min of induction (Figure 6A). In CS slices, not only did we observe HFS-induced de-depression, but this de-depression was also greater than that observed in WT. In addition, while NASPM had no significant impact in WT slices, it reduced de-depression in CS slices to WT levels (Figures 6A and 6B). Thus, the basal increase in CP-AMPAR synaptic activity in CS mice is altering the ability of CA1 synapses to undergo LTP, and prior removal of synaptic AMPARs by LTD can restore LTP responsiveness by allowing subsequent CP-AMPAR recruitment.
Figure 6. AKAPCS Mice Can Undergo CP-AMPAR-Dependent De-depression at CA1 Synapses.

(A and B) fEPSP slope (normalized to baseline) recorded over time (A) and aggregate data for measurements of normalized fEPSP slope (B) (averaged over last 10 min) showing that de-depression (induced by 1 Hz, 900 pulses LFS-LTD followed by 1 × 100 Hz, 1 s HFS-LTP 15 min later) is enhanced in CS mice (A: over last 10 min CS versus WT ****p < 0.0001 by 2-way ANOVA with Tukey’s multiple comparisons; B: WT 96.27 ± 8.537%, n = 7 slices, CS 111.8 ± 8.638%, n = 7 slices; unpaired t test CS versus WT ***p = 0.0010). CS but not WT de-depression is sensitive to CP-AMPAR blockade with NASPM (A: over last 10 min CS NASPM versus CS ***p < 0.001, WT NASPM versus CS ****p < 0.0001 by 2-way ANOVA with Tukey’s multiple comparisons; B: WT NASPM 85.42 ± 10.23%, n = 5 slices, CS NASPM 97.46 ± 8.487%, n = 8 slices; unpaired t tests WT NASPM versus WT p > 0.05 [n.s.], CS NASPMversus CS **p = 0.0016, WT NASPM versus CS NASPM *p = 0.0377). Data are reported as mean ± SEM.
DISCUSSION
Dynamic protein palmitoylation has emerged as a key regulator in the subcellular positioning of proteins in neurons to coordinate precise and specific signaling (Fukata et al., 2013; Globa and Bamji, 2017). Here, using biochemistry, super-resolution nanoscopy and electrophysiology we demonstrate the importance of palmitoylation of the postsynaptic scaffolding molecule AKAP150 in controlling basal AMPAR synaptic subunit composition to alter LTP.
Complete gene knockout is widely used to study the effect of disrupting protein function. However, for large, multivalent scaffold protein complexes that function as structural and signaling hubs, knockouts are problematic because of the disruption of multiple functions. In particular, AKAP150 knockout (KO) removes the opposing signaling functions of PKA and CaN, allowing compensation that makes mechanistic interpretations difficult. Accordingly, AKAP150-null mice exhibit different and in general more limited behavioral and synaptic phenotypes than AKAP150 knockin mice that are specifically deficient in either PKA (ΔPKA and D36) or CaN (ΔPIX) anchoring (Lu et al., 2007, 2008; Sanderson et al., 2012, 2016, 2018; Tunquist et al., 2008; Weisenhaus et al., 2010; Zhang et al., 2013). Thus, here we generated palmitoylation-deficient AKAPCS knockin mice to specifically address the role of palmitoylation in controlling AKAP150 postsynaptic targeting and AMPAR regulation.
AKAP150 Palmitoylation and PKA-CaN Anchoring in Control of Basal CP-AMPAR Incorporation
Importantly, we observed a very specific synaptic phenotype in AKAPCS animals that is distinct from, but overlapping with, phenotypes observed in either AKAP-PKA or AKAP-CaN anchoring-deficient mice. In particular, both CS and CaN anchoring-deficient ΔPIX mice (Sanderson et al., 2012, 2016) exhibit increased basal synaptic CP-AMPAR activity. However, although ΔPIX mice exhibit significantly enhanced GluA1 S845 phosphorylation and stronger EPSC inward rectification than CS mice (Sanderson et al., 2012), altered AMPAR subunit composition is only associated with increased mEPSC amplitude in CS mice. In addition, whereas blocking CP-AMPARs with IEM1460 in neurons cultured from WT mice had no impact on mEPSC activity, IEM1460 reduced basal mEPSC amplitude and frequency in ΔPIX mouse neurons to below WT levels (Sanderson et al., 2018). Yet in CS-cultured neurons IEM1460 only reduced elevated mEPSC activity back to WT levels. Thus, in ΔPIX mice the impact of CP-AMPARs on basal synaptic strength is offset by an accompanying, compensatory loss of GluA2-containing receptors, but in CS mice, while a smaller number of CP-AMPARs are added to synapses, little or no compensatory removal of GluA2-containing receptors is occurring. Overall, the impacts of loss of AKAP palmitoylation on basal AMPAR transmission are similar but clearly not identical to those resulting from loss of AKAP-CaN anchoring.
AKAP150 Palmitoylation and PKA-CaN Anchoring in CP-AMPAR Metaplasticity that Controls LTP and LTD Balance
In contrast, the CA1 LTP phenotypes in CS and ΔPIX mice are drastically different, with ΔPIX mice showing strongly enhanced (Sanderson et al., 2012) and CS mice exhibiting strongly impaired HFS-induced LTP. The relatively modest enhancement in basal AMPAR transmission in CS mice is unlikely to occlude LTP, and given our previous observations of enhanced CP-AMPAR-dependent LTP in ΔPIX mice, prior basal incorporation of CP-AMPARs alone cannot account for impaired LTP in CS mice. However, a key difference between the plasticity landscapes of these two knockin mice is the lack of LFS-induced LTD in ΔPIX but not CS mice. In ΔPIX mice, loss of AKAP-CaN anchoring impairs CP-AMPAR removal from synapses to alter metaplasticity at CA1 synapses in favor of LTP > LTD (Sanderson et al., 2012, 2016). In contrast, in CS mice LTD and CP-AMPAR synaptic removal mechanisms appear to be intact, pointing more toward a specific deficit in recruitment of additional CP-AMPARs to support LTP. Indeed, we were able to further link the LTP deficit in CS mice specifically to CP-AMPAR dysfunction by showing that it could be overcome by using a strong, prolonged whole-cell pairing induction stimulus that did not require CP-AMPAR recruitment in WT mice. In addition, we were able to establish that elevated basal CP-AMPAR activity in CS mice was contributing to the inability of HFS to recruit additional CP-AMPARs by showing that prior LTD induction to remove synaptic AMPARs allowed subsequent HFS to induce LTP/de-depression that was in part mediated by CP-AMPARs. Thus, overall, loss of AKAP150 palmitoylation increases basal CP-AMPAR synaptic incorporation but impairs additional recruitment to alter CA1 metaplasticity in favor of LTD > LTP.
Interestingly, prior characterization of PKA anchoring-deficient AKAP150 knockin mice also found deficits in LTP related to impaired CP-AMPAR recruitment, but only in adult (~8-week-old) and not juvenile (2- to 4-week-old) mice (Lu et al., 2007; Sanderson et al., 2016). In particular, although HFS-induced LTP at ~2 weeks of age was strongly inhibited by CP-AMPAR antagonists in WT mice, LTP was neither impaired nor sensitive to CP-AMPAR antagonists in ΔPKA mice (Sanderson et al., 2016). These studies, along with a number of other studies of CA1 LTP using GluA1-KO mice, S845A-knockin mice, and subunit replacement approaches, indicate the dependencies of LTP on PKA signaling, S845 phosphorylation, and AMPAR subunit composition are flexible and developmentally plastic in juvenile animals, (Granger et al., 2013; Jensen et al., 2003; Kolleker et al., 2003; Plant et al., 2006; Zamanillo et al., 1999; Adesnik and Nicoll, 2007; Lee et al., 2003, 2010; Yang et al., 2008, 2010). Thus, it is remarkable that the compensatory shift to HFS-LTP recruitment of GluA2-containing AMPARs that is observed in juvenile GluA1 KO, S845A, and AKAP150ΔPKA/D36 mice is not occurring in CS mice, in which the LTP deficit can be overcome only by prolonged whole-cell pairing that recruits GluA2-containing AMPARs even in WT mice. Importantly, our present findings demonstrating that CP-AMPAR recruitment depends strongly on LTP induction stimulus strength in general agree with previous observations made across several different ages (Gray et al., 2007; Jensen et al., 2003; Kolleker et al., 2003; Lu et al., 2007) and could explain discrepancies in previous studies of juvenile rodents that observed CP-AMPAR recruitment for LTP induced with comparatively weaker (Plant et al., 2006; Yang et al., 2008, 2010) but not stronger pairing protocols (Adesnik and Nicoll, 2007).
Interestingly, at this same early developmental age when LTP is normal in ΔPKA, D36, and S845A mice, LFS-LTD is impaired because AKAP-PKA anchoring and S845 phosphorylation are needed to promote transient recruitment of CP-AMPARs to CA1 synapses during LTD induction prior to their rapid removal by AKAP-anchored CaN (He et al., 2009; Lee et al., 2010; Lu et al., 2007; Sanderson et al., 2016). These LTD findings at CA1 synapses are in accordance with studies in other brain regions, including in the amygdala, ventral tegmentum, and nucleus accumbens, in which CP-AMPAR synaptic incorporation not only supports synaptic potentiation but can also prime synapses for LTD and de-potentiation (Clem and Huganir, 2010; Wolf, 2016). Accordingly, in CS mice basal CP-AMPAR incorporation may prime synapses to undergo normal LTD through AKAP-CaN-mediated removal with no need for additional CP-AMPAR recruitment. Consistent with effective synaptic removal of CP-AMPARs by LTD in CS mice, prior LFS induction of LTD allowed subsequent HFS induction of LTP and de-depression to recruit CP-AMPARs back to CA1 synapses.
AKAP Palmitoylation and Signaling in Multiple Locations during LTP and LTD
Our prior studies found that palmitoylation of human AKAP79 is required for its localization to dendritic REs (Keith et al., 2012; Woolfrey et al., 2015), a compartment that is known to deliver GluA1 to the plasma membrane in support of LTP (Hiester et al., 2017; Kennedy et al., 2010; Park et al., 2004). In addition, acute AKAP79CS overexpression in rat hippocampal neurons increased both basal RE exocytosis and synaptic CP-AMPAR activity. Here, although we also observed decreased AKAP150CS RE localization and increased basal CP-AMPAR activity in AKAPCS mice, we did not observe increased basal RE exocytosis. Thus, AKAPCS mice exhibit alterations in GluA1 CP-AMPAR regulation even in the absence of more widespread RE trafficking dysfunction. However, we found that palmitoylation is also required for normal AKAP150 association with the PSD, as shown by reduced co-localization and co-fractionation of AKAP150CS with PSD-95. Thus, impaired LTP in AKAPCS mice is likely related to decreased AKAP signaling in not only REs but also the PSD. In contrast, AKAP79/150 localization to the extrasynaptic plasma membrane, where AMPARs are endocytosed during LTD (Ashby et al., 2004; Beattie et al., 2000), is not affected by loss of palmitoylation. Thus, it is tempting to speculate that AKAP-PKA signaling that promotes CP-AMPAR synaptic incorporation during LTP requires AKAP localization to REs and the PSD to promote recycling and synaptic retention of receptors, while AKAP-CaN signaling that removes CP-AMPARs during LTD only requires extrasynaptic membrane targeting.
Accordingly, our prior work found that chemical LTP stimulation increased AKAP palmitoylation and localization to dendritic spines. Furthermore, overexpression of the AKAP79CS mutant or knockdown of its palmitoylating enzyme DHHC2 acutely interfered with a number of cellular correlates of LTP in cultured neurons including spine enlargement, RE exocytosis, GluA1 surface delivery, and mEPSC potentiation (Keith et al., 2012; Woolfrey et al., 2015). In contrast, chemical LTD stimulation decreased AKAP palmitoylation and localization to spines in coordination with spine shrinkage. Consistent with AKAP depalmitoylation favoring LTD > LTP as observed here in AKAPCS mice, AKAP79CS did not interfere with GluA1 endocytosis and was even more sensitive than WT to removal from spines by chemical LTD (Keith et al., 2012). In addition, overexpression of a constitutively lipidated AKAP79 mutant prevented both AKAP removal from spines and spine shrinkage following chemical LTD (Woolfrey et al., 2018). Thus, based also on our findings here ex Akap5CS, AKAP79/150 palmitoylation is required to support LTP but not LTD.
However, the observation that CS but not WT mice robustly recruit CP-AMPARs recently removed by LTD back to CA1 synapses during HFS-induced de-depression could reflect a loss of AKAP-CaN in REs, allowing enhanced GluA1 recycling and synaptic incorporation mediated by a pool of PKA other than that anchored to AKAP79/150 or possibly other kinases such as PKG, PKC, or CaMKII (Boehm et al., 2006; Kim et al., 2015; Opazo et al., 2010). Accordingly, loss of AKAP-CaN phosphatase signaling in REs, in addition to in the PSD, could also contribute to the increases in basal synaptic GluA1 surface expression and CP-AMPAR activity in AKAPCS mice by increasing receptors within the recycling pool and then also biasing PSD signaling toward receptor retention. All things considered, it is remarkable that such a specific perturbation of AKAP79/150 intracellular targeting caused by loss of palmitoylation has such a dramatic impact on synaptic plasticity, thus further underscoring how critical scaffold proteins and their organization of localized signaling pathways are for controlling neuronal function.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Polyclonal rabbit anti-AKAP 150 | Brandao et al., 2012 | RRID: AB_2532138 |
| Monoclonal mouse anti-PKA-RII b | BD Transduction Laboratories | Cat# 610625: RRID: AB_397957 |
| Monoclonal mouse anti-PSD95 | Millipore | Cat# MAB1596; RRID: AB_2092365 |
| Polyclonal rabbit anti-GluR1 | Millipore | Cat# ABN241; RRID: AB_2721164 |
| Polyclonal rabbit anti-GluR1, phosphoSer845 | Millipore | Cat# AB5849; RRID: AB_92079 |
| Anti-Rabbit IgG (H&L) Antibody (Goat) ATTO 647N Conjugated | Rockland | Cat# 611-156-122; RRID: AB_10893043 |
| Anti-Rabbit IgG (H&L) Antibody (Goat) ATTO 594 Conjugated | Rockland | Cat# 611-155-122; RRID: AB_10894686 |
| Anti-Mouse IgG (H&L) Antibody (Goat) ATTO 647N Conjugated | Rockland | Cat# 610-156-121; RRID: AB_10894200 |
| Anti-Mouse IgG (H&L) Antibody (Goat) ATTO 594 Conjugated | Rockland | Cat# 610-155-121; RRID: AB_10893162 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SUNBRIGHT Maleimide PEG | NOF America | Cat# Me-100MA (10 kD) |
| Tetrodotoxin (TTX) | Tocris Bioscience | Cat# 1078 |
| Picrotoxin | Tocris Bioscience | Cat# 1128 |
| QX-314 bromide | Tocris Bioscience | Cat# 1014 |
| NASPM trihydrochloride | Tocris Bioscience | Cat# 2766 |
| IEM 1460 | Tocris Bioscience | Cat# 1636 |
| DL-APV (AP5) | Tocris Bioscience | Cat# 0105 |
| NBQX disodium salt | Tocris Bioscience | Cat#1044 |
| Spermine tetrachloride | Tocris Bioscience | Cat# 0958 |
| Experimental Models: Organisms/Strains | ||
| AKAPCS mice (allele symbol: Akap5 < tm3.1Mdaq >; Allele synonyms: Akap5CS AKAP5CS, AKAP150CS) | This publication | RRID: MGI_6198520 |
| C57BL/6J mice | Jackson Laboratories | RRID: IMSR_JAX:000664 |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Slidebook | 3i- Intelligent Imaging Solutions | https://www.intelligentimaging.com/slidebook |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| pClamp/Clampfit | Molecular Devices | www.moldev.com |
| WinLTP | WinLTP Ltd. and The University of Bristol | http://www.winltp.com |
| Mosaic Suite (FIJI/ImageJ plugin) | Mosaic Group | http://mosaic.mpi-cbg.de/?q=downloads/imageJ |
| MATLAB | Mathworks | http://www.mathworks.com |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Dell’Acqua (mark.dellacqua@ucdenver.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of AKAP150 CS knockin mice
The Transgenic and Gene Targeting Core at the University of Colorado Anschutz Medical Campus constructed the Akap5CS targeting vector. The Akap5CS mutation introduced mutations of AKAP150 cysteines 36 and 123 to serines in the single coding exon of an Akap5 genomic DNA fragment via piggyBac (PB) transposon based method from a C57BL/6 BAC clone. In this targeting vector, the AKAP150 CS mutation was introduced by piggyBac method with a neomycin resistance cassette flanked by the 3′ and 5′ long-terminal repeat (LTR) of PB inserted within the Akap5 exon. The targeting construct was electroporated into a hybrid C57BL/6 129 embryonic stem (ES) cell line EC7.1 and G418-resistant clones were screened for homologous recombinants by PCR-based genotyping. The neomycin resistance cassette was then removed from the targeted locus by remobilizing the PB with transient expression of PB transposase. One positive clone was expanded, injected into blastocysts, and implanted into surrogate mothers. Chimeric F0 founders were born and bred to C57BL/6J to establish germ-line transmission. F1 mice heterozygous for the CS mutation were identified and then bred to yield F2 CS homozygous offspring. For PCR genotyping, DNA was extracted from tail snips using REDExtract-N-Amp Tissue PCR kit (Sigma-Aldrich) following manufacturer’s protocol. PCR with forward (5′- GGAGACC AGCGTTTCTGAGATT-3′) and reverse (5′- ATCTCCAAATCGTCTGCCTCTC-3′) primers amplified the mutated region of the coding sequence, giving a 461 bp fragment for both the wild-type (WT) allele and the CS allele. After PCR amplification, the samples were digested with HindIII for 90 min and then resolved on a DNA gel. For the WT allele, no fragment will result from cutting (461 bp fragment) while the CS allele results in two fragments (100, 360 bp). AKAP150CS mice were backcrossed to C57BL several generations but then maintained on a mixed C57BL/6J 129 background. Both male and female mice between the ages of post-natal day (P) 12–21 were used for experiments and analyzed together. Mixed litters of male and female neonatal day 1–3 mouse pups were used for cultures. All animal procedures were conducted in accordance with National Institutes of Health (NIH)–United States Public Health Service guidelines and with the approval of the University of Colorado, Denver, Institutional Animal Care and Use Committee.
Primary mouse hippocampal neuron culture
Mouse hippocampal neurons were cultured from postnatal day 1–3 mixed sex mice as previously described (Sanderson et al., 2012; 2018). Briefly, the hippocampus was dissected from postnatal day 1–3 AKAP150 WT or CS mice and dissociated in papain. Neurons were seeded at a density of 150,000–200,000 cells/well in 12 well dishes on 18 mm glass coverslips coated with poly-D-lysine and Laminin or 400,000–500,000 cells/well in 6 well dishes on 25 mm glass coverslips coated with poly-D-lysine and laminin (BD Biosciences). Cells were maintained at 37°C, 5% CO2 in Neurobasal-A medium supplemented with B27, Glutamax, and Pen/Strep for 14–16 days before processing.
METHOD DETAILS
Fractionation and immunoblotting of brain tissue
Subcellular fractionation and immunoblotting of WT and CS hippocampal or forebrain (cortex and hippocampus) lysates were performed as in (Sanderson et al., 2016; 2012; Smith et al., 2006; Grosshans et al., 2002). For immunoblotting, 15 μg of whole extract (WE), 10 μg of P2, 20 μg of S2, 5 μg of TxP, and 15 μg of TxS were resolved on Tris-SDS gels and transferred in 20% methanol to PVDF membranes. Membranes were incubated with primary antibodies for 2 hr as follows: rabbit anti-AKAP150 (1:1000) (Brandao et al., 2012), mouse anti-PKA-RIIβ (1:1000; BD Biosciences Transduction Laboratories), mouse anti-PSD-95 (1:1000; Millipore), rabbit anti-GluA1 (1:1000; Millipore), and rabbit anti-GluA1-S845 (1:1000; Millipore). Signal detection was performed using HRP-coupled secondary antibodies (Bio-Rad; 1:10,000) followed by ECL (West Pico or West Dura Chemiluminescent Substrate; Pierce). Chemiluminescence was imaged using an Alpha Innotech Fluorchem gel documentation system, and band intensities were analyzed using ImageJ (NIH). Band intensities were normalized to WT WE from the same blot.
APEGS palmitoylation assay
AKAP150 palmitoylation state was assessed using the APEGS (Acyl-PEG Exchange Gel-Shift) assay as previously described (Woolfrey et al., 2018; Yokoi et al., 2016). Forebrain whole extracts or subcellular fractions from above were tumbled in PBS buffer containing 4% SDS and 5 mM EDTA with 20 mM TCEP for 1 h at room temperature in the presence of protease inhibitors. Next, free thiols were blocked by incubation with 50 mM N-ethylmaleimide (NEM) overnight at room temperature. Following a chloroform-methanol precipitation (CMP), pellets were resuspended in 4% SDS PBS buffer and thioester bonds were cleaved with 1M Hydroxylamine (HAM, Sigma) for 1 h at room temperature with end over end rotation. After another CMP, free thiols were labeled with 10 kD polyethylene glycol moieties (SUNBRIGHT maleimide PEG, NOF America) for 1 h at RT with rotation. Following a final CMP, samples were re-suspended and boiled in sample buffer with 50 mM dithiothreitol and resolved via SDS-PAGE and western blotting with AKAP150 antibody.
Extracellular fEPSP recordings
For slice preparation, animals (P12-P21) were decapitated under anesthesia with isofluorane. The brain was removed into 4°C cutting solution (in mM: 3 KCl, 1.25 NaH2PO4, 12 MgSO4, 26 NaHCO3, 0.2 CaCl2, 220 sucrose, 10 glucose; all chemicals were purchased from Sigma-Aldrich.). Hippocampi were removed from the brain, and 400-μm-thick slices were made using a McIIwain tissue chopper. Slices were recovered at 29–31°C for >90 min in ACSF/cutting solution mixture (ACSF in mM: 126 NaCl, 5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 10 glucose, 2 N-acetyl cysteine). Following recovery, slices were transferred to a recording chamber and maintained at 29–31°C in ACSF as described above (without N-acetyl cysteine). A bipolar tungsten stimulating electrode was placed in the Schaffer collateral pathway to evoke fEPSPs recorded in CA1 stratum radiatum using a glass micropipette filled with ACSF. I-O curves were measured by evoking fEPSPs at various intensities until maximal response was determined by plotting initial fEPSP slope against stimulus intensity. For studies of LTP, LTD, and de-depression, the test stimulus intensity was set to evoke 40%–60% of the maximum slope. Both data acquisition and analysis was done using WinLTP.
Whole-cell electrophysiology
For whole-cell voltage-clamp electrophysiological recordings, 300 mm horizontal hippocampal slices were prepared as above (cutting solution in mM: 85 NaCl, 75 sucrose, 2.5 KCl, 1.3 NaH2PO4 monobasic, 24 NaHCO3, 0.5 CaCl2, 4 MgCl2, 25 D-Glucose) using a Vibratome. After 30 min at 31.5°C, slices were recovered at room temperature for >60 min in ACSF/cutting solution mixture (ACSF in mM: 126 NaCl, 2.5 KCl, 1 NaH2PO4 monobasic, 26.2 NaHCO3, 2.5 CaCl2, 1.3 MgSO4–7H20, 11 D-Glucose at ~290 mOsm). Slices were transferred to a recording chamber and maintained at 29.5°C and visualized using infrared–differential interference contrast microscopy. Pipettes had a resistance between 2 and 5 MΩ. CA1 at −70 mV and recorded from using an intracellular solution containing the following (in mM): 115 Cs-Methanesulfonate, 15 CsCl, 8 NaCl, 10 Tetraethylammonium-Cl, 0.2 EGTA, 2 Mg-ATP, 0.3 Na-GTP, 10 HEPES, 10 Na2-phosphocreatine, 1 MgCl2, pH 7.3 with CsOH at ~300 mOsm. AMPAR sEPSCs were isolated using 50 μm picrotoxin (Tocris) and mEPSCs were isolated with the addition of 0.5 μm TTX (Tocris) extracellularly. For hippocampal cultures, coverslips were transferred to ACSF containing 0.5 μm TTX and 50 mm picrotoxin or 0.5 μm TTX, 50 μm picrotoxin and 70 μM IEM1460 and then recorded from as above.
For Evoked EPSCs, a bipolar tungsten stimulating electrode was placed as in the field experiments and CA1 pyramidal cells were recorded from using an internal solution containing 5 mM QX-314 to prevent action potential firing. Baseline responses were established in whole-cell mode and then currents were evoked at holding potentials of −70 mV to assess inward AMPAR current and then +40 mV to assess outward AMPAR and NMDAR current. Traces (≥5) were averaged across recordings from a single neuron at each respective holding potential to calculate AMPA/NMDA ratios. AMPA currents were measured at the peak amplitude of the EPSC at both +40mV and −70mV divided by NMDA current at 50 ms after the onset of the EPSC at +40 mV. For NASPM sensitivity, ACSF containing 20 μM NASPM was washed on after establishing a baseline evoked response and the change in response was calculated as EPSC amplitude after NASPM /EPSC amplitude before NASPM.
For Evoked I-O curves, responses were established at various stimulus intensities and fixed multiplier setting. For PPR, baseline responses were established in whole-cell mode and then paired-pulses at various intervals were recorded at −70 mV to assess paired-pulse facilitation as a read out of presynaptic function.
For AMPA rectification measurements, AMPAR currents were isolated using 100 μM DL-APV (Tocris) and 50 μM Picrotoxin in extracellular solution and with 10 μM spermine and 5 mM QX-314 (Tocris) in the internal solution. Baseline responses were established in whole-cell mode and the currents were evoked at different holding potentials (−70, −40, −20, 0, +20, +40 mV). Rectification index was calculated by taking the −70 mV amplitude/+40 mV amplitude, resulting in a larger number for more rectifying channels/CP-AMPARs. For NMDA I-O measurements, NMDAR currents were isolated using 10 μM NBQX and 50 μM Picrotoxin. +40 mV responses were established at various stimulus intensities and fixed multiplier settings. For LTP experiments, slices were stimulated for 10–15 min at moderate stimulus intensity before going into whole-cell mode. Once a cell was patched, baseline was established within 5 min of breaking in. After a 3 min baseline measurement, cells were depolarized to 0 mV and then tetanized. Cells were then stepped back down to −70 mV and recorded for 50–60 min post-tetanus. Cells were monitored for membrane resistance and seal quality throughout. In NASPM LTP experiments, 20 μM NASPM was included in ACSF throughout. Whole-cell data were collected using a Digidata 1440 with Multiclamp 700B amplifier (Molecular Devices). Evoked experiments were conducted using a Model 2100 Isolated Pulse Stimulator (A-M Systems). All data were acquired with pCLAMP software and analyzed in Clampfit.
TF-488 feeding to label REs
DIV 14 neurons were transferred into Neurobasal with no additives and supplemented 0.1% BSA for 30 min at 37°C. Cells were incubated with Alexa 488 labeled transferrin (TF-488) (Invitrogen) for 30 min at 37°C and then processed for fixation and immunocytochemistry (rabbit anti-AKAP150 1:1000, followed by goat anti-rabbit-Alexa 568 1:1000). TF-488 (final concentration of 5 μg/well) was microcentrofuged at max speed for 1 min prior to application and only the supernatant was added to cells to prevent aggregation. Imaging was carried out on an Axio Observer microscope (Zeiss) with a 63 × Plan Apo/1.4 NA objective using 488 and 561 nm laser excitation and a CSU-XI spinning-disk confocal scan head (Yokogawa) coupled to an Evolve 512 EM-CCD camera (Photometrics) driven by SlideBook 6.0 (Intelligent Imaging Innovations). Z stacks of 13 optical sections (0.33 μm each) were acquired. Data were analyzed with SlideBook 6.0 using single optical sections of in-focus TF-488 signal. Masks were drawn over in-focus dendritic segments and only single-plane masks were analyzed using Pearson’s correlation for AKAP and TF-488 signals.
SEP-TfR imaging
Imaging of super-ecliptic pHluorin-tagged transferrin receptor was conducted essentially as previously described (Keith et al., 2012; Woolfrey et al., 2015). DIV 11–14 hippocampal neurons from WT and AKAP150CS mouse cultures were transfected (Lipofectamine 2000) with plasmids encoding SEP-TfR and mCherry (as a cell fill) and imaged 3 days later. Imaging was conducted on the spinning-disk confocal microscope detailed above. Prior to imaging, neurons were incubated in ACSF plus 1 mM MgCl2 for 30 min and were maintained during imaging at 33–35°C in a perfusion chamber (Warner Instruments). Baseline rates of SEP-TfR exocytic events (events defined as 2.5-fold above the median intensity of the dendrite) were determined by acquiring z stacks of 10 optical sections (1.0 μm spacing) every 6 s for 5 min.
Immunocytochemistry on mouse primary hippocampal neuron cultures for dendritic spine analysis
For dendritic spine counting in cultured hippocampal neurons, DIV12–14 neurons were transfected with a plasmid encoding GFP using Lipofectamine 2000 (Invitrogen) and fixed after two days of expression on DIV14–16. Neurons were washed with ACSF (in mM: 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 20 Glucose) x 2, then fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 in PBS, and blocked overnight in a 5% BSA/PBS solution. Primary anti-GFP antibodies were incubated for 2h at room temperature in 5% BSA/PBS. Cells were then washed in PBS and incubated in secondary antibody conjugated to Alexa 488 for 1h at room temperature. Coverslips were washed in PBS and mounted onto glass slides with Pro-Long Gold (Invitrogen). Images were obtained on a Zeiss Axiovert 200M microscope equipped with a 175W xenon lamp (Sutter), 63×Plan- Apo/1.4 NA objective, FITC/Alexa 488, Cy3/Texas Red and Cy5/Alexa 647 filter sets (Chroma), Coolsnap CCD camera, and Slidebook 5.0 software. Three-dimensional z stacks with 0.33 μm steps were collected. Spines were counted from 50–100 μm segments of secondary or higher-order dendrites in Slidebook 6.0 (3 individual neuron preps, 2–3 coverslips per prep per genotype).
Dendritic spine analysis by DiI labeling
Slices were prepared as for whole-cell electrophysiology (300 μm on a Vibratome). Slices were fixed in 4% PFA overnight at 4C, washed in PBS 3 × 15 min. After washing, sonicated DiI powder was collected on the tip of a needle and gently placed on the CA1 region of the hippocampus (Kim et al., 2007). DiI was allowed to incorporate overnight at room temperature. Slices were washed in PBS 3×15 min and then mounted onto glass slides using Vectashield (Vector Laboratories). Slices were imaged via spinning-disk confocal microscopy as detailed above. Spines were counted from for 50–100 μm segments of secondary or higher-order dendrites in ImageJ (NIH) (< 3 neurons per slice were counted, 2–3 slices/animal, 3 animals per genotype).
Surface GluA1 antibody labeling
DIV 14–16 neurons plated on #1.5 glass coverslips were transferred to ACSF with 1 mM Mg2+ for 30 min. Cells were transferred to ACSF for 30 min then rabbit anti-GluA1 antibody (Millipore 1:250) was live-fed for 15 min at 37°C before being washed in ice-cold ACSF 2× and fixed in 4% PFA. Cells were then processed for STED imaging by labeling with mouse anti-PSD-95 primary antibodies and fluorescent secondary antibody conjugates as described below.
Super-resolution stimulated emission depletion (STED) nanoscopy
Neurons plated on #1.5 glass coverslips were washed 2× ACSF then fixed with 4% paraformaldehyde for 10 min. Coverslips were washed 3×5 min with PBS with rotation and then permeabilized with 0.1% Triton. Neurons were next washed 3×5 min with PBS and then blocked overnight with filter-sterilized 5% BSA/PBS. Neurons were incubated with primary antibody in 5% BSA/PBS at room temperature (rabbit anti-AKAP150 1:1000, mouse anti-PSD-95 1:500), then washed 3×5 min PBS and incubated at room temperature for 1 h with secondary antibodies (goat anti-rabbit-Atto 647N 1:500 and goat anti-mouse-Atto 594 1:500; Rockland). Images were acquired on a custom built STED microscope (Meyer et al., 2016). Custom ordered 40 nm beads (Life Technologies) labeled with red and far-red dyes (proprietary) were used for resolution measurement and system alignment.
STED image analysis
The methodology of image segmentation and geometric analysis applied to STED images here will be described in more detail elsewhere along with its application to 3D-structured illumination microscopy (SIM) images (K.C.C. and M.L.D., unpublished data). Briefly, a Split-Bregman image segmentation algorithm, first described in (Paul et al., 2013) and subsequently incorporated into the MOSAIC image processing suite for ImageJ/FIJI (http://mosaic.mpi-cbg.de/) (Rizk et al., 2014) was utilized to delineate object boundaries from background-corrected STED images (using a histogram-based background estimator, also implemented as part of the MOSAIC suite). Binary masks generated in this process were then imported into MATLAB (Mathworks) where the geometric properties of the defined objects were calculated. Output metrics were then imported into Prism (GraphPad) for further analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data compilation and statistical analysis were performed in Prism (GraphPad) with significance value as α = 0.05. All data are reported as mean ± SEM. Prism provides exact p values unless p < 0.0001. The statistical tests used, p values and replicates with definition of n for all experiments can be found in the figure legends. No tests were used to estimate sample size. All experiments, except the initial APEGS assay in Figure 1B, were performed at least 3 separate times (or on at least 3 separate animals) to ensure rigor and reproducibility. Both male and female mice were used for electrophysiology experiments, and we observed no differences between sexes, therefore data from both sexes were pooled for all experiments.
Supplementary Material
Highlights.
AKAP150 palmitoylation controls localization to recycling endosomes and the PSD
AKAP150 palmitoylation limits basal synaptic incorporation of CP-AMPARs
CP-AMPAR synaptic incorporation during LTP depends on induction stimulus
AKAP150 palmitoylation is required for CP-AMPAR-dependent LTP
ACKNOWLEDGMENTS
We want to thank Emily S. Gibson for technical support. Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core, and AKAPCS mice were generated with assistance of the Gene Targeting and Transgenic Animal Cores, supported in part by Rocky Mountain Neurological Disorders Core (grant P30NS048154) and by the NIH/National Center for Advancing Translational Sciences (NCATS) Colorado Clinical and Translational Sciences Institute (CTSI) (grant UL1TR001082). The STED microscope was funded through a National Science Foundation (NSF) Major Research Instrumentation grant (DBI-1337573) and an NIH Shared Instrument grant (S10RR023381). AHA (16PRE29880006) and NIH/ National Institute of Neurological Disorders and Stroke (NINDS) (F31NS096815) predoctoral fellowships to A.M.P., NIH/NINDS grant R01NS040701 to M.L.D., and NIH/National Institute of Mental Health (NIMH) grant R00MH103531 to J.A. supported this work. The contents are the authors’ sole responsibility and do not necessarily represent official AHA or NIH views.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.085.
REFERENCES
- Adesnik H, and Nicoll RA (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci 27, 4598–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Su€dhof TC, and Malenka RC (2012). Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron 73, 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, and Chen L (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, and Henley JM (2004). Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J. Neurosci 24, 5172–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, and Malenka RC (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci 3, 1291–1300. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang M-G, Johnson RC, Esteban J, Huganir RL, and Malinow R (2006). Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51, 213–225. [DOI] [PubMed] [Google Scholar]
- Brandao KE, Dell’Acqua ML, and Levinson SR (2012). A-kinase anchoring protein 150 expression in a specific subset of TRPV1- and CaV 1.2-positive nociceptive rat dorsal root ganglion neurons. J. Comp. Neurol 520, 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigidi GS, Sun Y, Beccano-Kelly D, Pitman K, Mobasser M, Borgland SL, Milnerwood AJ, and Bamji SX (2014). Palmitoylation of d-catenin by DHHC5 mediates activity-induced synapse plasticity. Nat. Neurosci 17, 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, and Scott JD (1992). Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J. Biol. Chem 267, 16816–16823. [PubMed] [Google Scholar]
- Clem RL, and Huganir RL (2010). Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330, 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, and Scott JD (2000). Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27, 107–119. [DOI] [PubMed] [Google Scholar]
- Delint-Ramirez I, Willoughby D, Hammond GRV, Ayling LJ, and Cooper DMF (2011). Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J. Biol. Chem 286, 32962–32975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Acqua ML, Faux MC, Thorburn J, Thorburn A, and Scott JD (1998). Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 17, 2246–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Gustina AS, and Huganir RL (2014). PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini Ael.-D., Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, and Bredt DS (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi S-H, Wilson C, Nuriya M, Huganir RL, and Malinow R (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci 6, 136–143. [DOI] [PubMed] [Google Scholar]
- Fukata Y, and Fukata M (2010). Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci 11, 161–175. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, and Fukata M (2013). Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J. Cell Biol 202, 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globa AK, and Bamji SX (2017). Protein palmitoylation in the development and plasticity of neuronal connections. Curr. Opin. Neurobiol 45, 210–220. [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, and Dell’Acqua ML (2002). Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J. Neurosci 22, 7027–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, and Dell’Acqua ML (2005). Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol. Biol. Cell 16, 3574–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, and Nicoll RA (2013). LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493, 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Fink AE, Sariñana J, Vissel B, and O’Dell TJ (2007). Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol 98, 2488–2492. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, and Browning MD (2002). LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci 5, 27–33. [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, and Lee H-K (2009). Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc. Natl. Acad. Sci. U S A 106, 20033–20038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiester BG, Bourke AM, Sinnen BL, Cook SG, Gibson ES, Smith KR, and Kennedy MJ (2017). L-type voltage-gated Ca2+ channels regulate synaptic-activity-triggered recycling endosome fusion in neuronal dendrites. Cell Rep. 21, 2134–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang M-G, Ledoux J, Huganir RL, and Malinow R (2007). Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131, 160–173. [DOI] [PubMed] [Google Scholar]
- Huganir RL, and Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen V, Kaiser KMM, Borchardt T, Adelmann G, Rozov A, Burnashev N, Brix C, Frotscher M, Andersen P, Hvalby Ø, et al. (2003). A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J. Physiol 553, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Biou V, and Malenka RC (2010). A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat. Neurosci 13, 1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJM, Su€dhof TC, and Malenka RC (2013). LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, et al. (2008). Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay HY, Greene DL, Kang S, Kosenko A, and Hoshi N (2015). M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Invest 125, 3904–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, and Dell’acqua ML (2012). Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J. Neurosci 32, 7119–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, and Ehlers MD (2010). Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell 141, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BG, Dai H-N, McAtee M, Vicini S, and Bregman BS (2007). Labeling of dendritic spines with the carbocyanine dye DiI for confocal microscopic imaging in lightly fixed cortical slices. J. Neurosci. Methods 162, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, Arancio O, and Ziff EB (2015). Network compensation of cyclic GMPdependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc. Natl. Acad. Sci. U S A 112, 3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, Köhr G, Malinow R, Seeburg PH, and Osten P (2003). Glutamatergic plasticity by synaptic delivery of GluR-B(long)-containing AMPA receptors. Neuron 40, 1199–1212. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Barbarosie M, Kameyama K, Bear MF, and Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Takamiya K, Han J-S, Man H, Kim C-H, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, et al. (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Takamiya K, He K, Song L, and Huganir RL (2010). Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol 103, 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, and Hell JW (2007). Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, and Hell JW (2008). AKAP150-anchored PKA activity is important for LTD during its induction phase. J. Physiol 586, 4155–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, and Nicoll RA (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Song Y, Raghavachari S, and Blanpied TA (2013). Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron 78, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man H-Y, Sekine-Aizawa Y, and Huganir RL (2007). Regulation of alphaamino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. USA 104, 3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SA, Ozbay BN, Potcoava M, Salcedo E, Restrepo D, and Gibson EA (2016). Super-resolution imaging of ciliary microdomains in isolated olfactory sensory neurons using a custom two-color stimulated emission depletion microscope. J. Biomed. Opt 21, 66017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D, Hosy E, Petersen JD, Constals A, Giannone G, Choquet D, and Sibarita J-B (2013). Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci 33, 13204–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, et al. (2009). Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD95. J. Cell Biol 186, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, and Foster TC (1996). Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J. Neurosci 16, 5382–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, and Soderling TR (2006). Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem 281, 752–758. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, and Choquet D (2010). CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67, 239–252. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, and Ehlers MD (2004). Recycling endosomes supply AMPA receptors for LTP. Science 305, 1972–1975. [DOI] [PubMed] [Google Scholar]
- Paul G, Cardinale J, and Sbalzarini IF (2013). Coupling image restoration and segmentation: a generalized linear model/Bregman perspective. Int. J. Comput. Vis 104, 69–93. [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, and Isaac JTR (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci 9, 602–604. [DOI] [PubMed] [Google Scholar]
- Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T, Koval OM, Anderson ME, He K, Lee H-K, and Hell JW (2012). b2-Adrenergic receptor supports prolonged theta tetanus-induced LTP. J. Neurophysiol 107, 2703–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizk A, Paul G, Incardona P, Bugarski M, Mansouri M, Niemann A, Ziegler U, Berger P, and Sbalzarini IF (2014). Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat. Protoc 9, 586–596. [DOI] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, and Dell’Acqua ML (2009). Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J. Neurosci 29, 7929–7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Sprengel R, and Seeburg PH (2012). GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front. Mol. Neurosci 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, and Dell’Acqua ML (2012). AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J. Neurosci 32, 15036–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, and Dell’Acqua ML (2016). NMDA receptordependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89, 1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Scott JD, and Dell’Acqua ML (2018). Control of homeostatic synaptic plasticity by AKAP-anchored kinase and phosphatase regulation of Ca2+-permeable AMPA receptors. J. Neurosci 38, 2863–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Levental I, Mayor S, and Eggeling C (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, Dell’Acqua ML, and Kennedy MJ (2017). Optogenetic control of synaptic composition and function. Neuron 93, 646–660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, and Dell’Acqua ML (2006). cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J. Neurosci 26, 2391–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares C, Lee KFH, Nassrallah W, and Béïque J-C (2013). Differential subcellular targeting of glutamate receptor subtypes during homeostatic synaptic plasticity. J. Neurosci 33, 13547–13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubblefield EA, and Benke TA (2010). Distinct AMPA-type glutamatergic synapses in developing rat CA1 hippocampus. J. Neurophysiol 104, 1899–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, and Wolf ME (2005). Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci 25, 7342–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, and Schuman EM (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799. [DOI] [PubMed] [Google Scholar]
- Tang A-H, Chen H, Li TP, Metzbower SR, MacGillavry HD, and Blanpied TA (2016). A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 536, 210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, and Tsien RW (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Chiu S-L, Chen C-M, and Huganir RL (2012). Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron 73, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, and Scott JD (2008). Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc. Natl. Acad. Sci. U S A 105, 12557–12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, and McKnight GS (2010). Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS ONE 5, e10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME (2016). Synaptic mechanisms underlying persistent cocaine craving. Nat. Rev. Neurosci 17, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Sanderson JL, and Dell’Acqua ML (2015). The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J. Neurosci 35, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, O’Leary H, Goodell DJ, Robertson HR, Horne EA, Coultrap SJ, Dell’Acqua ML, and Bayer KU (2018). CaMKII regulates the depalmitoylation and synaptic removal of the scaffold protein AKAP79/150 to mediate structural long-term depression. J. Biol. Chem 293, 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang X-B, Frerking M, and Zhou Q (2008). Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc. Natl. Acad. Sci. U S A 105, 11388–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang X-B, and Zhou Q (2010). Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc. Natl. Acad. Sci. U S A 107, 11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, and Fukata M (2016). Identification of PSD-95 depalmitoylating enzymes. J. Neurosci 36, 6431–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, et al. (1999). Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 284, 1805–1811. [DOI] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, and Hell JW (2013). Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic b-adrenergic signaling. J. Biol. Chem 288, 17918–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, and Hell JW (2014). Capping of the N-terminus of PSD95 by calmodulin triggers its postsynaptic release. EMBO J. 33, 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu A, Xia S, Leung C, Qi J, Meng Y, Xie W, Park P, Collingridge GL, and Jia Z (2018). The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning.Nat. Neurosci 21, 50–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.