

Abstract
Medium sized carbocycles are particularly difficult to synthesize. Ring closing metathesis reactions (RCM) have recently been applied to construct eight-membered carbocycles, but trisubstituted double bonds in the eight-membered rings are more difficult to produce using RCM reactions. In this review, model examples and our own results are cited and the importance of the preparation of suitably designed precursors is discussed. Examples of RCM reactions used in the total synthesis of natural products are also outlined.
Keywords: ring closing metathesis, eight-membered carbocycles, synthesis
1. Introduction
Grubbs and his group developed ruthenium catalyst 1 in 1992 [1] and applied it to synthetic efforts [2,3]. Soon after, the first generation Grubbs catalyst 2 (Figure 1) was invented and became commercially available [4,5]. Molybdenum complex 5 (Schrock reagent) is known to work for the construction of trisubstituted double bonds; however, the complex is not so easy to handle [6,7]. Grubbs and his group further reported the commercially available so-called second-generation Grubbs catalyst 4 in 1999 [8]. Since then, ring closing metathesis (RCM) reactions have been used extensively by scientists, even in total syntheses [9,10,11,12,13,14,15]. The application of these catalysts began in the area of macrocyclic compounds, and later syntheses of more difficult 7-, 8-, 9- and 10-membered rings appeared in the literature [9,10]. In earlier work, RCM reactions were used in the synthesis of mostly heterocyclic compounds. We have been interested in these types of reactions, because terpenoids usually have trisubstituted double bonds in the carbocycles. However, the first generation Grubbs catalyst 2 was not applicable to the construction of the trisubstituted double bonds, but the second generation Grubbs catalyst 4 or imidazolinylidene catalyst 3 [16,17] can be used for such reactions. Thus, many chemists, as well as our group, started to use this commercially available catalyst. In this review, the synthesis of eight-membered carbocycles having disubstituted and the more difficult trisubstituted double bonds using RCM reactions will be presented [11,12,13,14]. A valuable review of enyne metathesis has appeared in the literature and therefore such syntheses are not included in this review [15].
Figure 1.

Structures of the reagents for RCM reactions.
2. Results and Discussion
2.1. Eight-membered carbocycles using RCM reactions (disubstituted double bonds)
It is well known that compounds possessing medium sized rings are difficult to prepare by conventional methods. Therefore, we have performed a preliminary check of the scope and limitations of the use of the Grubbs catalyst 2 for cyclization (Table 1). Cyclohexene 10b was obtained quantitatively and cycloheptene 11b was obtained in 40% yield, whereas cyclooctene 12b was procduced in 14% and cyclononene 13b in 4% yield, respectively. The larger ring compounds were not produced under these reaction conditions [18].
Table 1.
Cyclization to 6- to 11-membered compounds using catalyst 2 or 5.
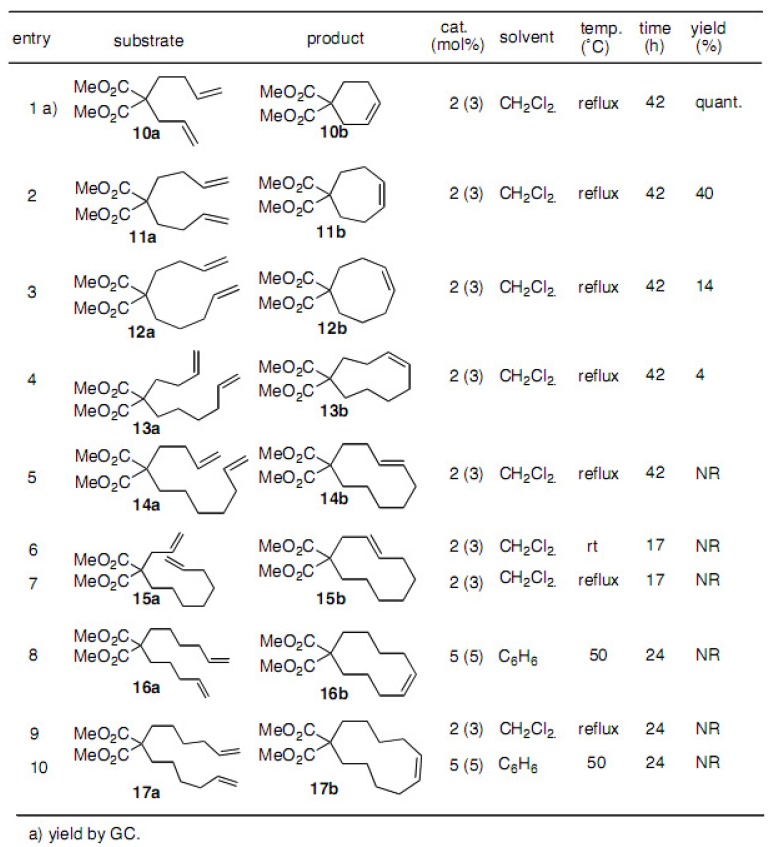 |
Grubbs reported the first application of his catalyst to the synthesis of eight-membered carbocycles using catalyst 1 (Scheme 1) [19]. Diallyl ether isomers had different reactivities towards the catalyst. The trans isomer afforded cyclooctene 7 in 60% yield, whereas a yield of 20% was afforded with the cis isomer 9. Both the substituents of the trans isomer 6 can adopt equatorial positions, but in the case of the cis isomer 8, one of them must be in the axial orientation.
Scheme 1.

Cyclization to the dioxacyclooctene ring.
The application of the first generation Grubbs catalyst 2 to the cyclization reaction of diene compound 18 was reported. The first generation catalyst 2 or Schrock catalyst 5 did not work in this type of reaction with or without the methyl group (Scheme 2) [20]. This substrate 18 (R=H) differs from 12a in its ester part. Grubbs did not describe the reaction conditions in detail. Successful cyclization (Table 1, entry 3) may be due to the longer reaction time. Although the Thorpe-Ingold effect is acting, it is not large enough to accelerate complete cyclization.
Scheme 2.

Attempt to cyclize a simple diene.
An attempt by Taylor was interesting (Scheme 3) [21]. The diene alcohol 20 fused with the five membered ring cyclized to cyclooctene compound 21 in 53% yield. However, the enol-lactone 22 failed to cyclize. The reason why this compound is not a good substrate is unclear.
Scheme 3.

Attempts to cyclize fused systems.
Fürstner reported the successful cyclization of 24 to azacyclooctene 25 using Nolan catalyst 3 (Scheme 4) [22]. The power of some catalysts was compared in this report.
Scheme 4.

Formation of azacyclooctene.
Construction of polyfunctional carbocycles were reported in 2002 (Scheme 5) [23]. Diene 26 bearing five functional oxygens produced the corresponding cyclooctene derivative 27 in 99% yield. It was a surprisingly high yield in the case of the first generation Grubbs catalyst 2.
Scheme 5.

Cyclization to polyfunctional cyclooctene.
Braddock reported that symmetrical diene 28 with a ketal protection cyclized to cyclooctene 29 in 89% yield, a comparable result to that shown in Scheme 5 [24]. However, the free substrate 30 produced only 19% yield of cyclooctene 31, as well as the one-carbon less cycloheptene derivative 32 in 33% yield. This result apparently arose from the isomerization of the double bond into the more substituted position followed by cyclization to cycloheptene. The researchers noticed that in some cases the double bonds can isomerize and such a phenomenon occurs. Moreover, at higher temperatures more isomerization was observed to occur (Scheme 6). This is discussed later in this review.
Scheme 6.

Cyclization to cyclooctene and cycloheptene.
Waldmann carried out an interesting experiment involving the competing cyclization to 6- or 8-membered compounds [25]. However, no cyclooctadiene 35 was obtained; instead cyclohexene 34 was formed in 60% yield (Scheme 7).
Scheme 7.

Competition to oxacyclooctadiene and oxacyclohexene.
As shown in Scheme 8, the double bond formed by the RCM reaction was isomerized to the conjugated position using catalyst 4. Therefore, they used RuClH(CO)(PPh3)3 (2%) as an isomerization catalyst after the RCM reaction to produce 37 in 70−74% yield [26].
Scheme 8.

Cyclization of diene to benzoazacyclooctene.
We have been studying the scope and limitations of the RCM reaction using simple dienes, as shown in Scheme 9 [27]. In this systematic work, we have found that the isomerization of the double bond occurs in the RCM reaction in the case of the second generation Grubbs catalyst 4 and at higher temperatures. Thus, compound 38 afforded a mixture of compounds 41 and 43, in the ratio of 86:14 with the first generation Grubbs catalyst 2. However, with the second generation Grubbs catalyst 4 the same substrate produced a 12% yield of a mixture of 41, 42, 43 and 44, in the ratio of 8:13:38:41. Interestingly, compound 39 afforded only compound 42 in 28% yield. Finally, compound 40 afforded a mixture of 41, 42, 43 and 44, and no cyclononene derivative was obtained.
Scheme 9.

Attempted cyclization of simple dienes to 7-, 8- and 9-membered carbocycles.
Further applications involving cyclization to cycloheptene derivative 46 and the ring opening to cyclooctene 47 was realized by Rodriguez (Scheme 10) [28,29]. An interesting review was published by Rodriguez [30].
Scheme 10.

Tandem RCM to 7-membered carbocycle and ring opening to fused cyclooctene.
A model study of the taxane skeleton was carried out for compound 48 (Scheme 11) [31]. Bicyclic acetate 49 was produced in 59% yield by the second generation Grubbs catalyst 4. The acetoxy group diastereoisomer did not cyclize, but rather afforded dimeric compounds. This is clearly due to the steric hindrance of the gem-dimethyl groups present in the cyclohexane ring.
Scheme 11.

Cyclization to cyclooctene derivative, a part of the taxane skeleton.
Prunet reported cyclization to fused cyclooctene systems (Scheme 12) [32,33]. The ease of cyclization was found to be strongly dependent on the protecting group. Mono TES protection of the diol 50 afforded only 6 and 7% yields of the corresponding diol 51 and its double bond isomer 52, respectively. However, the ketal 54 was formed in 93% yield using the molybdenum catalyst 5. Cyclic carbonate 55 produced the trans cyclooctene 56 for the first time in 30−40% yield with catalysts 2 or 5. The product was carefully analyzed and the cyclized compound had trans stereochemistry regarding the methyl group at the juncture position and the butyl group. The recovered substrate 57 had cis stereochemistry. These examples clearly indicate the importance of the conformation of the substrates; however, it is unclear at this stage what kind of conformation these substrates adopt. Nonetheless, it is noteworthy that ketal or cyclic carbonate forced both the double bond moieties close to each other, and was formally recognized as a type of Thorpe-Ingold effect. It remains a mystery as to why the trans cyclooctene 56 survived under these reaction conditions, although it isomerized to cis cyclooctene under the similar reaction conditions.
Scheme 12.

Effect of the protecting group in the cyclization to cyclooctene.
After the recognition of the double bond isomer 52 in Scheme 12, Prunet changed the substrate to compound 58 with the proper stereochemistry (Scheme 13) [34,35]. Thus, by use of catalyst 2, 3 and 4, the desired compound 59 was isolated in 65, 72 and 69% yields, respectively. The use of dichloroethane as a solvent gave good results. Ketal instead of cyclic carbonate afforded a quantitative result with this solvent. Compound 62, which bears the wrong stereochemistry at C8 for taxol, does not cyclize, even at higher temperatures.
Scheme 13.

Comparison of the catalysts and the solvents.
2.2. Olefin rearrangement
Olefin isomerization under the RCM reaction conditions is discussed briefly. Hoye noticed that olefin isomerization occurred with Grubbs catalyst 2 to give one-carbon less ketone 66 and proposed a mechanism for this reaction (Scheme 14) [36].
Scheme 14.

Olefin isomerization and the formation of a one-carbon less ketone.
Tori found that a similar isomerization and one-carbon less ketone 72 was formed in the reaction of compound 70 (Scheme 15) [37]. However, in addition to this ketone 72, ethyl ketone 71 was also produced in the ratio of 71:72 = 2:1. This process just involves a rearrangement of the double bond to the inner position and the tautomerization to the ketone.
Scheme 15.

Olefin isomerization and the formation of two ketones.
Prunet and Nolan observed the isomerization of the double bond in a series of synthetic works on the taxane skeleton [38]. As shown in Scheme 16, with catalyst 3 the more substituted olefin 75 was isolated at a higher temperature. The proposed mechanism involved the formation of the π-allyl complex 78 (oxidative addition) followed by hydride transfer to 75 (reductive elimination).
Scheme 16.

Olefin isomerization and the proposed mechanism.
The report by Wagener in 2003 proposes hydride and allyl mechanisms [39]. A model study was carried out to see how much isomerization occurs under metathesis conditions. The second generation Grubbs catalysis 4 induces isomerization at 50−60 ºC under concentrated conditions. Schmidt reported the possibility of poarticipation of a ruthenium hydride complex in the olefin isomerization [40].
2.3. Eight-membered carbocycles using the RCM reaction (trisubstituted double bond)
Until now, formation of the cyclooctene ring with disubstituted double bonds was discussed. Hereafter trisubstituted double bonds in cyclooctene will be discussed. Overman aimed at constructing a nine-membered heterocycle, most likely carried out in an NMR tube in C6D6. Surprisingly, the product obtained in 43% yield was the eight-membered compound 80 (Scheme 17) [41]. This, of course, arose from the isomerization of the vinyl group into the inner double bond followed by the RCM reaction. In other words, the cyclooctene ring is easier to prepare than the cyclononene ring presumably due to steric hindrance. The difference in steric energy was calculated by CONFLEX [42,43] and discussed later (vide infra).
Scheme 17.

Formation of the one-carbon less carbocycle with a trisubstituted double bond.
Fürstner succeeded with the RCM reaction to afford both diastereoisomers 82 and 84 in high yields using the molybdenum catalyst 5 (Scheme 18) [44]. Very interestingly, hexane was used as the solvent. This was the first report describing the production of an eight-membered carbocycle with a trisubstituted double bond applied to the synthesis of the terpenoids.
Scheme 18.

Synthesis of cyclooctene with a trisubstituted double bond.
The second generation Grubbs catalyst 4 was used for the construction of the trisubstituted cyclooctene ring by Granja (Scheme 19) [45]. The yield of 86 was 89% and this was a similar substrate to that employed by Fürstner.
Scheme 19.

Cyclooctene in the steroid-like ring system.
Wicha’s report is very interesting. Compound 87 was successfully cyclized to trisubstituted cyclooctene 88 in 95% yield (Scheme 20) [46]. However, its diastereoisomer 89 did not give cyclooctene, but rather cycloheptene 90. The formation of the dimer 91 is a rapid process in CH2Cl2 and the reaction of this dimer with the same catalyst 4 in benzene affords both cyclooctene 93 and cycloheptene 92. Once more, rearrangement occurred at a higher temperature. Wicha has presented an excellent review that includes their results [47].
Scheme 20.

Tri-substituted double bond in the cyclooctene ring.
An indirect approach to an eight-membered carbocycle has also been reported. For example, cycloheptene 95 was prepared by the RCM reaction followed by ring opening of the cylopropane ring to afford cyclooctenone 96 (Scheme 21) [48].
Scheme 21.

Tandem RCM and the ring opening of a three-membered ring.
A model study to produce eight-membered carbocycles was reported by Tori (Scheme 22) [49]. Compound 97 afforded only a trace amount of cyclooctene 98. The main product was dimer 99 obtained in 68% yield. In the case of compound 100 almost no cyclooctene 102 was produced, instead cycloheptene 101 was isolated in 27−64% yield, depending on the temperature and the reaction time.
Scheme 22.

Attempts to cyclize to cyclooctene with a tri-substituted double bond.
Since the cyclization of compound 97 and 100 did not give good results. The sp2 carbon was introduced next to the cyclohexane ring to force two diene partners close to each other. The formation of the cyclooctene ring 104 was realized in 41% yield; however, the competitive cyclization to the five membered ring 105 occurred in 33% yield. Interestingly, the isolated compound 104 had the OTES group in the β orientation. However, the configuration of the five membered compound 105 was unclear, although it was a single product (Scheme 23) [49].
Scheme 23.

Attempts to synthesize cyclooctene.
Further work toward the synthesis of sesterterpene YW3699 was described. Substrate 107 afforded three products 108−110 as shown in Scheme 24 [49]. Here, a normal RCM led to the formation of cyclooctene 108 and one-carbon less cycloheptene 109, as well as isomerized olefin 110.
Scheme 24.

The effects of the configuration of substrates.
It is very interesting to note that the diastereoisomer 111 cleanly cyclized to cyclooctene 112 in 61% yield (Scheme 25) [49]. The difference between these two diastereoisomers, 107 and 111, is attributed to the preferred conformation of the substrate. The introduction of the sp2 carbon at the C-2 position forces two olefin partners close to each other by the Thorpe-Ingold like effect. The stereochemistry was finally determined by the careful analysis of the products.
Scheme 25.

The effect of the configuration of substrates.
The four diastereoisomeric epoxides, 113a-116a, were treated with the second generation Grubbs catalyst 4 to give very interesting results (Scheme 26) [50]. Among the four, only one diastereoisomer 113a smoothly cyclized to cyclooctene derivative 113b (80% yield), while the other three resulted in just a trace amount of cyclooctene or decomposition. This is in a good agreement with the substrate 111. The difference in the reactivity must be due to the difference in the conformation, in which, the two ends of the olefins are in close proximity. Estimating how close they are to each other remains an unresolved issue.
Scheme 26.

The effect of the configuration of substrates.
We have calculated the energy minimum conformation using CONFLEX [42,43] for eunicellin diterpenoid derivatives. It is quite interesting that eight-membered carbocycle 80 has a higher energy than the nine-membered carbocycle 118 with a difference of 7 kcal/mol (Figure 2). The corresponding trans-isomers have higher steric energies at 22−14 kcal/mol. Therefore, the cyclization reaction does not depend on the product energy, but the transition state.
Figure 2.


Steric energies and conformations of 68, 80, and 117−119 calculated by CONFLEX.
The stable conformations of compounds 107 and 111' were also calculated. The steric energy for 107 is 69.4 kcal/mol and for 111' 71.0 kcal/mol. Compound 111' cyclized more effectively than 107. However, the most stable conformation for 111' was that shown in Figure 3, apparently the diene partners being distal from each other (olefins are indicated by red arrows). The cyclization does not depend on the ground state conformation, but the transition state.
Figure 3.

Steric energies and conformations calculated for 107 and 111' by CONFLEX.
It is also interesting that the steric energies for 82 and 84 were less than those of dienes 81 and 83, respectively (Figure 4). These figures indicate that cyclization should occur regardless of the ring junction. The diene partners are not very far away from each other in both cases.
Figure 4.

Steric energies and conformations calculated for 81−84 by CONFLEX.
The diene models of Prunet (butyl group was replaced with methyl) show that the diene partners are both in proximity, which is very interesting from the point that the substrate must be well designed and thus suitable for RCM reactions (Figure 5). They have also commented on the proximity between olefins by a brief calculation using Chem Draw [35].
Figure 5.

Steric energies and conformations calculated for 63' and 65' by CONFLEX.
3. Conclusions
In this review, cyclization reactions to cyclooctenes were briefly introduced, not only with trisubstituted double bonds but also with disubstituted double bonds. In a model study, only disubstituted eight-membered carbocycles are shown to be realized using catalyst 4. A large number of examples applied in this area have been reported. However, the success by Fürstner prompted us to attempt the synthesis of trisubstituted cyclooctene using the RCM reactions introduced above. In the literature, only the successful reactions are reported, and many examples of unsuccessful reactions and the corresponding data remain undisclosed. In the successful examples, the substrates have been eloquently designed to meet the requirements of the reactions. The diene partners are most likely allowed to interact in the reaction conditions. A type of Thorpe-Ingold effect appears to force both diene ends close to each other. The length of both chains bearing the olefin moiety should be designed to be equal, because the chance of interaction increases. In this context, examples 97 and 100 failed to cyclize; however, examples 107 and 111 succeeded. In these examples the introduction of the exomethylene group greatly aided the formation of the eight-membered carbocycle. It is again important to note that the substrate must be suitably designed to meet the requirement of cyclization.
Acknowledgments
We are grateful to all members in the laboratory for helpful discussions and enthusiastic effort.
References and Notes
- 1.Nguyen S.T., Johnson L.K., Grubbs R.H. Ring-opening metathesis polymerization (ROMP) of norbornene by a group VIII carbene complex in protic media. J. Am. Chem. Soc. 1992;114:3974–3975. [Google Scholar]
- 2.Nguyen S.T., Grubbs R.H. Syntheses and activities of new single-component, ruthenium-based olefin metathesis catalysis. J. Am. Chem. Soc. 1993;115:9858–9859. [Google Scholar]
- 3.Grubbs R.H., Miller S.J., Fu G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995;28:446–452. doi: 10.1021/ar00059a002. [DOI] [Google Scholar]
- 4.Schwab P., France M.B., Ziller J.W., Grubbs R.H. A series of well-defined metathesis catalysts-synthesis of [RuCl2(CHR)(PR3)2] and its reactions. Angew. Chem. Int. Ed. Engl. 1995;34:2039–2041. doi: 10.1002/anie.199520391. [DOI] [Google Scholar]
- 5.Schwab P., Grubbs R.H., Ziller J.W. Synthesis and applications of RuCl2(=CHR’)(PR3)2: The influence of the alkylidene moiety on metathesis activity. J. Am. Chem. Soc. 1996;118:100–110. [Google Scholar]
- 6.Schrock R.R., Murdzek J.S., Bazan G.C., Robbins J., DiMare M., O’Regan M. Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins. J. Am. Chem. Soc. 1990;112:3875–3886. [Google Scholar]
- 7.Schrock R.R. Olefin metathesis by molybdenum imino alkylidene catalysts. Tetrahedron. 1999;55:8141–8153. doi: 10.1016/S0040-4020(99)00304-X. [DOI] [Google Scholar]
- 8.Scholl M., Ding S., Lee C.W., Grubbs R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 9.Fürstner A., Langemann K. Macrocycles by ring-closing metathesis. Synthesis. 1997:792–803. [Google Scholar]
- 10.Maier M.E. Synthesis of medium-sized rings by the ring-closing metathesis reaction. Angew. Chem. Int. Ed. 2000;39:2073–2077. doi: 10.1002/1521-3773(20000616)39:12<2073::AID-ANIE2073>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Schuster M., Blechert S. Olefin metathesis in organic chemistry. Angew. Chem. Int. Ed. 1997;36:2036–2056. doi: 10.1002/anie.199720361. [DOI] [Google Scholar]
- 12.Armstrong S.K. Ring closing diene metathesis in organic synthesis. J. Chem. Soc., Perkin Trans. 1. 1998:371–388. doi: 10.1039/a703881j. [DOI] [Google Scholar]
- 13.Grubbs R.H., Chan S. Recent advances in olefin metathesis and its application in organic synthesis. Tetrahedron. 1998;54:4413–4450. doi: 10.1016/S0040-4020(97)10427-6. [DOI] [Google Scholar]
- 14.Fürstner A. Olefin metathesis and beyond. Angew. Chem. Int. Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]
- 15.Mori M. Recent progress on enyne metathesis: Its application to syntheses of natural products and related compounds. Materials. 2010;3:2087–2140. doi: 10.3390/ma3032087. [DOI] [Google Scholar]
- 16.Huang J., Stevens E.D., Nolan S.P., Peterson J.L. Olefin metathesis-active ruthenium complexes bearing a nucleophilic carbene ligand. J. Am. Chem. Soc. 1999;121:2674–2678. doi: 10.1021/ja9831352. [DOI] [Google Scholar]
- 17.Scholl M., Trnka T.M., Morgan J.P., Grubbs R.H. Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 1999;40:2247–2250. [Google Scholar]
- 18.Nakashima K. Dissertation, Tokushima Bunri University; 2006. Synthetic studies of medium- and macro-cyclic compounds using the olefin metathesis reaction and total synthesis and absolute configuration of several sphenolobane-type diterpenoids. [Google Scholar]
- 19.Miller S.J., Kim S.H., Chen Z.R., Grubbs R.H. Catalytic ring-closing metathesis of dienes: application to the synthesis of eight-membered rings. J. Am. Chem. Soc. 1995;117:2108–2109. [Google Scholar]
- 20.Kirkland T.A., Grubbs R.H. Effects of olefin substitution on the ring-closing metathesis of dienes. J. Org. Chem. 1997;62:7310–7318. doi: 10.1021/jo970877p. [DOI] [PubMed] [Google Scholar]
- 21.Edward S.D., Lewis T., Taylor R.J.K. Eight membered ether via diene metathesis: Synthetic approach to laureatin natural products. Tetrahedron Lett. 1999;40:4267–4270. doi: 10.1016/S0040-4039(99)00703-0. [DOI] [Google Scholar]
- 22.Ackermann L., Fürstner A., Weskamp T., Kohl F.J., Herrmann W.A. Ruthenium carbene complexes with imidazolin-2-ylidene ligands allow the formation of tetrasubstituted cycloalkenes by RCM. Tetrahedron Lett. 1999;40:4787–4790. [Google Scholar]
- 23.Marco-Contelles J., de Opazo E. Synthesis of enantiomerically pure, highly functionalized, medium-sized carbocycles from carbohydrates: Formal total synthesis of (+)-calystegine B2. J. Org. Chem. 2002;67:3705–3717. doi: 10.1021/jo0111107. [DOI] [PubMed] [Google Scholar]
- 24.Braddock D.C., Cansell G., Hermitage S.A., White A.J.P. An asymmetric synthesis of enantiopure chair and twist trans-cyclooctene isomers. Tetrahedron: Asymmetry. 2004;15:3123–3129. doi: 10.1016/j.tetasy.2004.07.036. [DOI] [Google Scholar]
- 25.Basu S., Waldmann H. Regioselectivity in the formation of small- and medium-sized cyclic ethers by diene-ene ring-closing metathesis. J. Org. Chem. 2006;71:3977–3979. doi: 10.1021/jo052367y. [DOI] [PubMed] [Google Scholar]
- 26.Dieltiens N., Stevens C.V., Masschelein K., Hennebel G., Van der Jeught S. Ring-closing metathesis and ring-closing metathesis-isomerization approach to 1-phosphonylated 2-benzazocines. Tetrahedron. 2008;64:4295–4303. doi: 10.1016/j.tet.2008.02.074. [DOI] [Google Scholar]
- 27.Tori M., Minami A., Nakashima K. Unpublished results Tokushima Bunri University; Japan: 2010. [Google Scholar]
- 28.Michaut A., Miranda-Garcia S., Menéndez J.C., Rodriguez J. Stereoselective synthesis of bicyclo[4.2.1]nonane skeletons by ring-closing metathesis: A new versatile methodology for the efficient assembly of functionalized cyclooctanoids. Org. Lett. 2004;6:3075–3078. doi: 10.1021/ol0489393. [DOI] [PubMed] [Google Scholar]
- 29.Michaut A., Miranda-Garcia S., Menéndez J.C., Rodriguez J. Stereoselective synthesis of bicyclo[4.2.1]nonanes - a temporary-bridge approach to cyclooctanoids. Eur. J. Org. Chem. 2008:4988–4998. [Google Scholar]
- 30.Michaut A., Rodriguez J. Selective construction of carbocyclic eight-membered rings by ring-closing metathesis of acyclic precursors. Angew. Chem. Int. Ed. 2006;45:5740–5750. doi: 10.1002/anie.200600787. [DOI] [PubMed] [Google Scholar]
- 31.Wenz M., Großbach D., Beitzel M., Blechert S. An approach towards enantiomerically pure taxoidic A,B-ring fragments. Synthesis. 1999:607–614. [Google Scholar]
- 32.Bourgeois D., Pancrazi A., Ricard L., Prunet J. Synthesis of highly functionalized cyclooctanes by ring-closing metathesis: unexpected formation of a trans isomer. Angew. Chem. Int. Ed. 2000;39:726–728. [PubMed] [Google Scholar]
- 33.Bourgeois D., Mahuteau J., Pancrazi A., Nolan S. P., Prunet J. Synthesis of BC ring-systems of taxol by ring-closing metathesis. Synthesis. 2000:869–882. [Google Scholar]
- 34.Schiltz S., Ma C., Ricard L., Prunet J. Synthesis of model BC bicycles of taxol using C10-C11 ring-closing metathesis strategy. J. Organomet. Chem. 2006;691:5438–5443. [Google Scholar]
- 35.Ma C., Schiltz S., Le Goff X.F., Prunet J. Ring-closing metathesis in the synthesis of BC ring-systems of taxol. Chem. Eur. J. 2008;14:7314–7323. doi: 10.1002/chem.200800774. [DOI] [PubMed] [Google Scholar]
- 36.Hoye T.R., Zhao H. Synthesis of a C(1)-C(14)-containing fragment of callipeltoside A. Org. Lett. 1999;1:169–171. doi: 10.1021/ol9906514. [DOI] [PubMed] [Google Scholar]
- 37.Nakashima K., Okamoto S., Sono M., Tori M. Isomerization reactions of allylic alcohols into ketones with the Grubbs reagent. Molecules. 2004;9:541–549. doi: 10.3390/90700541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourgeois D., Pancrazi A., Nolan S.P., Prunet J. The Cl2(PCy3)(IMes)Ru(=CHPh) catalyst: olefin metathesis versus olefin isomerization. J. Organomet. Chem. 2002;643/644:247–252. [Google Scholar]
- 39.Lehman S.E., Jr., Schwendeman J.E., O’Donnell P.M., Wagener K.B. Olefin isomerization promoted by olefin metathesis catalysts. Inorg. Chim. Acta. 2003;345:190–198. [Google Scholar]
- 40.Schmidt B. Catalysis at the interface of ruthenium carbene and ruthenium hydride chemistry: organometallic aspects and applications to organic synthesis. Eur. J. Org. Chem. 2004:1865–1880. doi: 10.1002/ejoc.200300714. [DOI] [Google Scholar]
- 41.Joe D., Overman L.E. An unexpected product arising from metal alkyldiene mediated ring-closing diene metathesis. Tetrahedron Lett. 1997;38:8635–8638. doi: 10.1016/S0040-4039(97)10373-2. [DOI] [Google Scholar]
- 42.Goto H., Osawa E. Corner flapping: A simple and fast algorithm for exhaustive generation of ring conformations. J. Am. Chem. Soc. 1989;111:89508951. [Google Scholar]
- 43.Goto H., Osawa E. An efficient algorithm for searching low-energy conformers of cyclic and acyclic molecules. J. Chem. Soc., Perkin Trans. 2. 1993:187–198. doi: 10.1039/p29930000187. [DOI] [Google Scholar]
- 44.Fürstner A., Langemann K. A concise total synthesis of dactylol via ring closing metathesis. J. Org. Chem. 1996;61:8746–8749. doi: 10.1021/jo961600c. [DOI] [PubMed] [Google Scholar]
- 45.Codesido E.M., Castedo L., Granja J.R. Access to [6.4.0]carbocyclic systems by tandem metathesis of dienynes. A step toward the synthesis of a preD3-D3 transition state analogue. Org. Lett. 2001;3:1483–1486. doi: 10.1021/ol0158011. [DOI] [PubMed] [Google Scholar]
- 46.Michalak K., Michalak M., Wicha J. Studies toward the total synthesis of di- and sesterterpenes with a dicyclopenta[a,d]cyclooctane skeleton. Construction of a versatile A/B ring building block via a ring-closing metathesis reaction and carbocationic rearrangement. Tetrahedron Lett. 2005;46:1149–1153. doi: 10.1016/j.tetlet.2004.12.077. [DOI] [Google Scholar]
- 47.Michalak K., Michalak M., Wicha J. Studies towards the total synthesis of di- and sesterterpenes with dicyclopenta[a,d]cyclooctane skeletons. Three-component approach to the A/B rings building block. Molecules. 2005;10:1084–1100. doi: 10.3390/10091084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lysenko I.L., Lee H.G., Cha J.K. Use of cyclopropanols as conformational constraints in RCM. Org. Lett. 2006;8:2671–2673. doi: 10.1021/ol0605631. [DOI] [PubMed] [Google Scholar]
- 49.Mizutani R., Miki T., Nakashima K., Sono M., Tori M. Trisubstituted double bond in the cyclooctene ring. Preparation using the RCM reaction. Heterocycles. 2009;78:2295–2314. doi: 10.3987/COM-09-11720. [DOI] [Google Scholar]
- 50.Mizutani R., Nakashima K., Saito Y., Sono M., Tori M. Studies toward the total synthesis of YW3699, a sesterterpenoid GPI biosynthesis inhibitor: Preparation of the tri-substituted cyclooctene ring using the RCM reaction. Tetrahedron Lett. 2009;50:2225–2227. doi: 10.1016/j.tetlet.2009.02.163. [DOI] [Google Scholar]