

Summary
Cell migration often involves the formation of sheet-like lamellipodia generated by branched actin filaments. The branches are initiated when Arp2/3 complex [1] is activated by WAVE regulatory complex (WRC) downstream of small GTPases of the Rac family [2]. Recent structural studies defined two independent Rac binding sites on WRC within the Sra-1/PIR121 subunit of the pentameric WRC [3, 4], but the functions of these sites in vivo have remained unknown. Here we dissect the mechanism of WRC activation and the in vivo relevance of distinct Rac binding sites on Sra-1, using CRISPR/Cas9-mediated gene disruption of Sra-1 and its paralog PIR121 in murine B16-F1 cells combined with Sra-1 mutant rescue. We show that the A site, positioned adjacent to the binding region of WAVE-WCA mediating actin and Arp2/3 complex binding, is the main site for allosteric activation of WRC. In contrast, the D site toward the C terminus is dispensable for WRC activation but required for optimal lamellipodium morphology and function. These results were confirmed in evolutionarily distant Dictyostelium cells. Moreover, the phenotype seen in D site mutants was recapitulated in Rac1 E31 and F37 mutants; we conclude these residues are important for Rac-D site interaction. Finally, constitutively activated WRC was able to induce lamellipodia even after both Rac interaction sites were lost, showing that Rac interaction is not essential for membrane recruitment. Our data establish that physical interaction with Rac is required for WRC activation, in particular through the A site, but is not mandatory for WRC accumulation in the lamellipodium.
Keywords: lamellipodium, protrusion, filopodium, migration, Arp2/3 complex, CRISPR/CAS9, Rho-GTPase
Graphical Abstract
Highlights
-
•
WRC harbors two Rac binding sites, A and D, with distinct functions in vivo
-
•
A site is the major WRC activation site
-
•
Rac-D site interaction optimizes WAVE-mediated actin filament assembly
-
•
Both sites are obligatory for WRC activation, but not its lamellipodial accumulation
Schaks et al. use gene disruption combined with re-expression of point mutants of the Sra-1/PIR121 subunit of mammalian WRC or the PIR ortholog in Dictyostelium to unravel differential functions of the two Rac interaction surfaces on WRC in vivo. These functions are crucial to both lamellipod/pseudopod protrusion and cell-migration efficiency.
Results and Discussion
Sra-1/PIR121 Double-KO B16-F1 Melanoma Cells Fail to Form Lamellipodia
Mammalian WRC (WAVE regulatory complex) displays various isoform combinations of its five essential subunits [5], which is why functional interference with individual subunits was hitherto mostly done using RNAi, revealing defects in cell-edge protrusion in organisms as evolutionarily distant as Drosophila, mouse, and human. Essential functions for individual subunits in the formation of lamellipodia and membrane ruffles were revealed [6, 7, 8, 9, 10] and complemented by genetic deletions in Dictyostelium [11, 12, 13, 14, 15] and mouse [16, 17, 18, 19]. Aside from knockouts (KOs) of individual, murine subunit isoforms such as WAVE1, WAVE2, or Abi-1 [16, 20], we currently lack a mammalian cell line permanently and entirely devoid of functional WRC. We thus engineered B16-F1-derived cell lines in which the two genes encoding Sra-1 and PIR121, termed CYFIP1 and CYFIP2 in the mouse, respectively, were stably disrupted using CRISPR/Cas9. Apart from confirming the essential function of WRC in lamellipodia formation and membrane ruffling, such a system should allow dissecting in vivo interactions between Sra-1/PIR121 and Rac recently established in vitro [3, 4]. Sra-1 and PIR121 are 87% identical at the amino acid level, and can both incorporate into WRC and share highly conserved, direct binding sites for Rac and the WASP homology 2, connector, acidic (WCA) module of WAVE, the actin- and Arp2/3-complex-binding end of WRC [3, 5, 7].
Simultaneous CRISPR/Cas9-mediated targeting of both CYFIP genes allowed establishing several clonal lines devoid of detectable Sra-1/PIR121 expression (Figures 1B and S1A). In analogy to disruption of the Dictyostelium ortholog Pir [15], Sra-1/PIR121 removal also diminished WAVE isoform expression, whereas it only partially reduced the expression of Nap1. The reasons for affecting just one posttranslationally modified Abi variant remain to be established (Figures 1B and S1A). The three clones analyzed further (3, 19, and 21) were completely devoid of lamellipodial protrusions, even upon strong stimulation of these structures using aluminum fluoride [21] (Figure S1B). Quantitation revealed lamellipodia formation in more than 90% of control cells, whereas not a single cell with lamellipodia could be discerned in respective KOs (n ≥ 344 for each clone; Figure S1D). This correlated with the absence of Arp2/3 complex accumulation at the cell periphery of KO lines (Figure S1F). KO cells also migrated at strongly reduced rates (by about 70%), indicating that migration speed in B16-F1 strongly depends on their ability to form lamellipodia (Figures S1C and S1E). An apparent increase of bi- or multinucleation upon Sra-1/PIR121 deletion indicated problems with cytokinesis, as observed previously for WRC subunit KOs [14, 15, 22], but this did not affect growth rates significantly (data not shown). Sra-1/PIR121 null cells could be specifically rescued to form lamellipodia (Figure 1A), express Nap1 and WAVE proteins at wild-type (WT) levels (Figure 1B), and migrate on laminin (Figure 1C; Video S1) by transfection (of clone 3) with EGFP-tagged Sra-1.
Figure 1.

Contribution of Distinct Rac Binding Sites in Sra-1 to Lamellipodia Formation
(A) Cell morphologies and lamellipodial phenotypes of B16-F1 control versus Sra-1/PIR121 KO cells (clone 3) transfected with EGFP or EGFP-tagged Sra-1, and stained for the actin cytoskeleton with phalloidin (scale bars, 20 μm).
(B) Cell lysates of B16-F1 cells, Sra-1/PIR121 KO cells (clone 3), as well as KO cells expressing EGFP-Sra-1 were subjected to western blotting to detect expression levels of WAVE complex components, as indicated.
(C) B16-F1 control cells, Sra-1/PIR121 KO clone 3, and the latter forming lamellipodia upon transfection with EGFP-tagged Sra-1 were analyzed for random migration speed (∗∗∗p ≤ 0.001; n.s. [not significant]: p > 0.05). Box and whisker plots represent data as follows: boxes correspond to 50% of data points (25%–75%), and whiskers correspond to 80% (10%–90%). Outliers are shown as dots, and lines and red numbers in boxes correspond to medians.
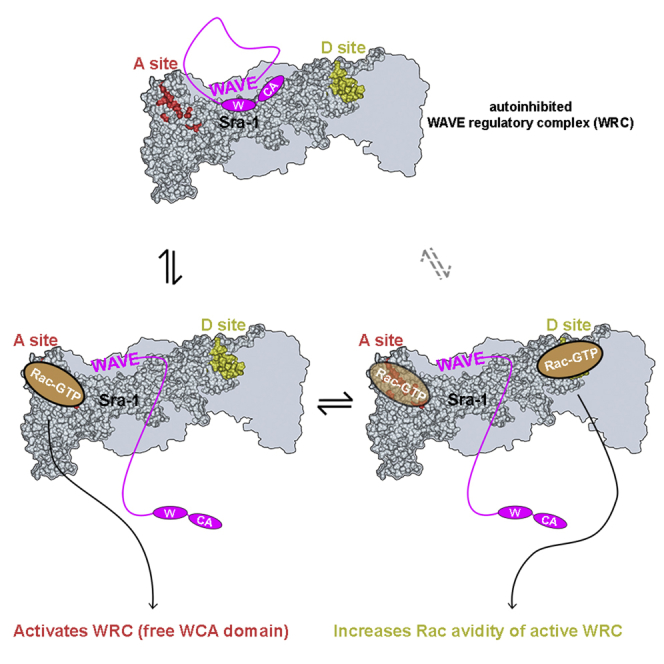
(D) Crystal structure of the WAVE complex (PDB: 3P8C [4]). From the view chosen, only WAVE (magenta), Sra-1 (green), and Nap1 (blue) are visible. Sra-1 possesses two binding sites for Rac (termed A site and D site) and sequesters the WH2 and C regions of WAVE. Rac binding to Sra-1 is thought to release interactions with the WH2 and C regions, thereby activating the WCA domain of WAVE.
(E) Sra-1/PIR121 KO cells (clone 3) were transfected with EGFP or various EGFP-Sra-1 constructs, lysed, and subjected to pull-downs with constitutively active Rac1 (Rac1-L61). Note strongly increased interaction of the WCA∗ mutant with Rac1, which was strongly and virtually entirely diminished upon additional mutation of the A and D site, respectively. Combinatorial mutation of both Rac binding sites in the WCA∗ background appeared to abolish detectable Rac1 interaction entirely. WCA∗: disrupted WH2 and C region contact sites (L697D/Y704D/L841A/F844A/W845A).
(F) Sra-1/PIR121 KO cells (clone 3) were transfected with the indicated EGFP-Sra-1 constructs and assayed for lamellipodia formation. Lamellipodial actin networks that were small, narrow, or displayed multiple ruffles were defined as “immature lamellipodia,” marked by arrowheads in cell images (right), as opposed to regular lamellipodia, marked by arrows (scale bar, 10 μm). Data in the bar chart are arithmetic means ± SEM from three independent experiments. Note that the A site mutation diminished lamellipodia formation in a fashion that could be restored by additional WCA∗ mutation of Sra-1. In the case of the D site, lamellipodial morphology was compromised in a fashion mostly independent from the WCA∗ mutation. The WIRS mutation had no detectable effect. To assess statistical significance of differences or confirm the absence of statistically relevant differences between experimental groups, a non-parametric, Mann-Whitney rank-sum test was performed in multiple, individual combinations of datasets. For each experimental group, we compared the number of cells with regular, i.e., “fully developed” lamellipodia, immature lamellipodia (see above), or the two groups combined, and hence all cells display either one of the lamellipodium-like structures. Selected combinations are as follows, with three p values representing aforementioned lamellipodial categories: WT-WIRS (n.s., n.s., n.s.); WT-C179R/R190D+WCA∗ (n.s., n.s., n.s.); WT-Y967A (∗∗, ∗∗, n.s.); WT-G971W (∗∗, ∗, n.s.); WT-Y967A+WCA∗ (∗∗, ∗∗, n.s.); Y967A-Y967A+WCA∗ (∗, n.s., n.s.); WT-WCA∗ (n.s., n.s., ∗∗). Statistical significance is expressed as ∗∗p ≤ 0.01, ∗p ≤ 0.05, and n.s. (not significant): p > 0.05. WIRS: Y923A/E1084A to mutate the WIRS-binding pocket; WCA∗: disrupted WH2 and C region contact sites (L697D/Y704D/L841A/F844A/W845A).
Phase contrast, time-lapse microscopy of B16-F1 wildtype, Sra-1/PIR121 KO clone #3 and the latter re-expressing EGFP-tagged Sra-1, as indicated, for restoration of lamellipodia-dependent migration. Time is in hours and minutes, scale bar is valid for all panels and equals 100μm.
The Rac-Binding A Site in Sra-1 Is Required for WAVE Complex Activation
Among multiple signals impacting on WRC function [3, 5, 23, 24], Rac appears obligatory for lamellipodia formation, at least in fibroblasts [25] (see also below), so we explored whether and under which conditions physical interactions between Rac and WRC, mediated by Sra-1, are essential for this process. Sra-1 possesses two Rac binding sites: the A site, located at the N terminus and homologous to the Rac binding site found in the WRC inhibitor CYRI [26], and the D site [3]. Both sites are required for Arp2/3 complex-mediated actin assembly in vitro [3]. Within WRC, Sra-1 sequesters the WCA domain of WAVE by binding to both W and C regions, thereby inhibiting the complex’s activity (see Figure 1D) [4]. Rac binding, in turn, competes with WCA sequestration, thereby releasing WCA for Arp2/3 complex activation. We compared the in vivo relevance of the A site and D site in Sra-1 (Figure 1D) by re-expressing WT Sra-1 and mutants deficient in Rac binding through either or both sites in Sra-1/PIR121 null cells. We also combined these Sra-1 variants with mutations causing disruption of Sra-1 binding to WAVE, termed WCA∗ (WCA activated), assumed to render WRC constitutively active [4]. WCA∗ dramatically augmented the interaction between Rac1 and the WAVE complex in Rac1 pull-down assays (Figure 1E), assuring that Rac binding to WRC competes with WCA domain sequestration. Additional mutation of the A site (C179R/R190D) strongly reduced binding to levels comparable to WT Sra-1. Mutation of the D site in the WCA∗ mutant background reduced binding to virtually undetectable levels, much like combinatorial mutation of both sites and in full accordance with published data [3]. We also confirmed that the strong increase of Rac binding to WCA∗ was specific for active, GTP-bound Rac1 (L61), as it was not detectable with N17-Rac (Figure S1G).
Next, we explored all these mutants upon transient transfection concerning their ability to rescue lamellipodia formation in Sra-1/PIR121-deficient cells migrating on laminin. Cells were categorized as forming or lacking lamellipodia as well as a group in which lamellipodia were not fully developed (immature lamellipodia; Figure 1F). In these conditions, >70% of cells were rescued in lamellipodia formation with WT Sra-1, although only half of those lamellipodia were fully developed. Notably, a mutant blocking the interaction between WRC and factors harboring the WRC-interacting receptor sequence (WIRS) [27, 28] did not display a statistically significant defect in lamellipodia formation (Figure 1F). This suggests that such interactions operate in specific regulation processes, but not as a general mechanism of lamellipodial WRC recruitment and/or activation. Returning to Rac binding, mutation of the A site caused an almost complete failure of restoration of lamellipodia formation (Figure 1F; for representative cell phenotypes, see Figure S1I). Both C179R and R190D mutants were tested individually and in combination [3, 4], with comparable results (Figure 1F). The rare examples of immature lamellipodia found were still weakly tipped by the rescue mutant (Figure S1K), consistent with the view that these structures are elicited by the rescue mutant displaying reduced but detectable Rac binding in pull-down assays (Figure S1H). Thus, in spite of the previously observed low affinity for Rac mediated by this site, in particular as compared to the D site [3], the A site is crucial for lamellipodia formation. To explore whether this was due to a failure of WRC activation or other reasons, we combined the A site mutation with disruption of WCA interaction (C179R/R190D+WCA∗). Importantly, this combinatorial Sra-1 mutant rescued lamellipodia formation with an efficiency indistinguishable from WT (Figure 1F). These data suggest that the A site is crucial for allosteric activation of WRC, but not needed in the absence of the inhibitory interaction of Sra-1 with WCA (Figures 1 and S1I). Consistent with lamellipodial phenotypes, A site+WCA∗ binds to Rac1 better than WT Sra-1 or the A site-alone mutant (Figures 1E and S1H). However, the increase of Rac binding caused by constitutive WRC activation (WCA∗) in the A site-mutated construct was much less robust than in the WT background, indicating a major contribution of the A site to allosteric activation (Figure S1H).
To test whether this function of the A site is conserved in evolution, we used an equivalent assay with Pir121-deficient Dictyostelium discoideum cells. Figure S2A emphasizes the degree of sequence conservation around A and D sites between both murine isogenes and the Dictyostelium ortholog. Pir121 KO in this organism causes defects in migration and loss of Scar/WAVE expression (Figures S2B and S2C) [12, 15], just as in B16-F1 cells (Figures S1A–S1F). These defects are correlated less with loss of lamellipod (also termed pseudopod) formation in Dictyostelium, because WASP can—inefficiently—produce pseudopods when WRC is lost [29, 30]. Yet, EGFP-tagged Pir121 WT prominently targeted to the cell periphery of Pir121 null cells and induced Arp2/3 complex-enriched structures, but A site mutants, A183R (not shown) or K193D/R194D (Figure 2A), failed to do so (also see Video S2). This phenotype was uncoupled from Scar/WAVE expression, which was fully restored by both EGFP-tagged WT and A site-mutated Pir121 (Figure S2B). Altogether, these data suggest the A site operates in WRC activation throughout evolution at least after the plant-animal split.
Figure 2.

Functional Comparison of Rac Binding Sites in Mouse and Dictyostelium Cells
(A) Subcellular localization of Scar/WAVE and Arp2/3 complexes. Wild-type and mutated Pir121-EGFP were expressed in PirA knockouts and visualized by confocal microscopy while migrating under agarose up a folate gradient. Top: wild-type Pir121-EGFP; middle: A site mutant (K193D/R194D); bottom: D site mutant (Y961A). Arp2/3 complex (red) is recruited to sites of WT Scar/WAVE complex (green) localization, but not upon expression of either PIR121 mutant. The D site mutant allows its recruitment to pseudopods, but there is no detectable Arp2/3 complex activation.
(B) Lifetimes of Scar/WAVE patches. Cells expressing PIR121-EGFP were allowed to migrate up folate gradients under agarose, and areas of local EGFP enrichment were observed by confocal microscopy. A site mutants showed no patches. Graph shows means ± SD; n > 25 cells; ∗∗∗p ≤ 0.001, non-parametric t test, Mann-Whitney.
(C) Frequency of Scar/WAVE patch generation. Cells were measured as in (B), but the rate of formation of patches was quantitated. Graph shows means ± SD; n > 25 cells; ∗∗∗p ≤ 0.001, non-parametric t test, Mann-Whitney.
(D) Reduced migration speed of cells expressing both A and D site mutants. The chemotactic speeds of cells expressing WT and mutant PIR121 were measured from the speed of cells allowed to migrate up folate gradients under agarose, and observed by differential interference contrast (DIC) microscopy. Data show means ± SD; n > 25 cells on 3 days; ∗∗∗p ≤ 0.001, one-way ANOVA, Dunnett’s multiple-comparison test.
(E) Random migration assay with B16-F1 Sra-1/PIR121 KO cells (clone 3) re-expressing the indicated Sra-1 variants, and analyzed as described in STAR Methods; cells with and without lamellipodia are displayed separately. n of cells analyzed was ≥130 per condition and specifically indicated for cells harboring lamellipodia. Lamellipodia-forming cells always migrated faster than respective controls, i.e., those cells lacking lamellipodia in each condition. Statistical significance is expressed as ∗∗∗p ≤ 0.001, ∗∗p ≤ 0.01, and ∗p ≤ 0.05; n.s. (not significant): p > 0.05.
(F and G) FRAP analysis of EGFP-Sra-1 variants expressed in Sra-1/PIR121 KO B16-F1 cells (clone 3). Data are arithmetic means with SEM of fluorescence intensities at acquired time points after bleaching, with intensities before each bleach individually normalized to 1. “n” equals the number of individual FRAP videos analyzed for each component (F). Half-times of recovery for each component were derived from curve fits (not shown) generated as described in STAR Methods. In (G), fitted data curves are displayed for comparison upon normalization of bleaching time point (0 s) to 0 and fluorescence recovery asymptote to 1. Constructs used were: WCA∗: disrupted WH2 and C region contact sites (L697D/Y704D/L841A/F844A/W845A); A site: C179R/R190D; D site: Y967A.
See also Figures S2 and S3 and Videos S2 and S4.
AiryScan imaging of cells to visualize WRC complex (green) and Arp2/3 complex (red) localization in wild type and PIR121 null cells expressing wild type and mutant (A or D site) Pir121-EGFP. Cells were imaged every 2 s, and resulting time-lapse movie is displayed at 10 frames/second.
The D Site in Sra-1 Is Dispensable for WRC Activation but Contributes to the Efficiency of Protrusion and Lamellipodial Actin Polymerization
Expression in Sra-1/PIR121 double null cells of mutants defective in Rac interaction through the D site (Y967A or G971W) (see [3]) caused comparably moderate effects. In particular, the frequency of lamellipodia formation was unchanged (Figure 1F). This was puzzling initially, as D site mutation abolished Rac binding almost completely (Figure 1E), consistent with previous data [3]. However, the majority of lamellipodia found in these cells appeared immature (see Figures 3A and S1I), indicating compromised WRC function in spite of its principal activation. A lack of an activation defect was experimentally confirmed by combining one of the D site mutants (i.e., Y967A) with WCA∗, as this failed to rescue important phenotypes, including lamellipodial morphology (Figure 3A) and protrusion velocity (Figures 3D and 3E).
Figure 3.

The D Site in Sra-1 Is Needed for Proper Lamellipodia Formation
(A) Representative Sra-1/PIR121 KO cells expressing different EGFP-Sra-1 variants, as indicated, and stained with phalloidin (scale bar, 10 μm).
(B) Quantification of lamellipodial width measurements.
(C) Quantification of filopodia formed anterior to lamellipodia.
(D) Representative kymographs of lamellipodial protrusion, induced by the indicated EGFP-Sra-1 variants.
(E) Quantification of lamellipodial protrusion velocity mediated upon expression of the respective Sra-1 variants.
(F) Representative Sra-1/PIR121 KO cells expressing different EGFP-Sra-1 variants and stained for the Arp2/3 complex subunit ArpC5A (scale bar, 10 μm).
(G) Quantification of ArpC5A intensity at the lamellipodium.
For quantifications in (B), (C), (E), and (G), data are displayed as described for Figure 1C, n = number of cells analyzed, and statistical significance is expressed as ∗∗∗p ≤ 0.001 and ∗∗p ≤ 0.01; n.s. (not significant): p > 0.05. See also Figure S3 and Video S3.
Again in analogy, D site-mutated Pir121 targeted to the cell periphery of Dictyostelium cells (Figure 2A; Video S2), unlike A site-mutated Pir121. However, as opposed to WT, activation of the Arp2/3 complex by D site-mutated Pir121 was compromised. Differences in D site mutant targeting were also illustrated by the evanescent nature of these Pir121 patches (Video S2). Consequently, the lifetimes of Pir121 Y961A patches at the cell periphery were strongly reduced from WT, and these short-lived patches were formed more frequently (Figures 2B and 2C). This was again consistent with reduced residency time at the lamellipodium tip of the mammalian D site mutant as revealed by fluorescence recovery after photobleaching (FRAP) (Figures 2F and 2G; see also below).
Furthermore, the immature lamellipodia seen in the mammalian system appeared particularly narrow (Figures 3A and 3B) and were tipped by filopodia, not typically seen in cells expressing WT Sra-1 (Figures 3A and 3C). These phenotypes were at best partially rescued by constitutive WAVE complex activation, as D site+WCA∗ behaved more like the D site mutant than WT (Figures 3B and 3C). Finally, lamellipodia protrusion was reduced in a statistically significant fashion with or without constitutive WRC activation, and less homogeneous (Video S3). In spite of all these phenotypes, D site+WCA∗-Sra-1 fully rescued Arp2/3 intensity values in lamellipodia, which were strongly reduced with the D site mutation alone (Figures 3F and 3G). Surprisingly, this rescue was not accompanied by rescued lamellipodial actin network polymerization (Figures 4A and 4B) or F-actin intensity (Figure 4C). Thus, most phenotypes caused by elimination of Rac binding through the D site cannot be bypassed by eliminating the necessity of WRC activation, except for efficient Arp2/3 complex incorporation (Figures 1F, 3F, and 3G). Continued Rac binding through the D site might thus favor a function mostly uncoupled from Arp2/3 complex activation (Figures 3F and 3G) yet promoting both lamellipodial actin filament assembly rates and quantities (Figures 4A–4C). To what extent this activity corresponds to the “distributive polymerase” activity recently ascribed to WAVE family proteins [31] remains an exciting question for future study.
Figure 4.

Further D Site-Associated Phenotypes and Analysis of the D Site Interaction Surface on Rac
(A and B) Determination of lamellipodial actin assembly rates. Sra-1/PIR121 KO cells (clone 3) were co-transfected with mCherry-Sra-1 variants (WT, D site mutant, and D site+WCA∗, as indicated) and EGFP-actin. Lamellipodial actin assembly rates were determined by photobleaching EGFP-actin within lamellipodial regions, and reading out network assembly rates as the sum of actin rearward flow during the fluorescence recovery period and simultaneous forward protrusion (A). Representative frames before and after bleaching (at 0 s) are shown in (B). Scale bar, 5 μm.
(C) Quantification of F-actin intensity levels in the lamellipodium obtained from phalloidin stainings.
Data and results of statistical analyses in (A) and (C) are displayed as described for quantifications in Figure 3.
(D) Rac1 docking onto the D site of Sra-1 based on the cryo-EM structure of Rac1 occupying the D site [3]. Amino acids mutated in this study (i.e., E31 and F37 in Rac1, as well as Y967 and G971 in Sra-1) are shown as sticks, except for G971, the position of which in the green helix is shown in red. Additional amino acids interacting with either E31/F37 of Rac1 or Y967 of Sra-1 are shown as lines.
(E) RAC1/2/3 genes were disrupted in B16-F1 cells using CRISPR/Cas9, and derived knockout clones were assayed for Rac expression. Antibodies employed are capable of detecting Rac1 or 3 and Rac2, respectively (see [25]). All lines including B16-F1 wild-type lack expression of hematopoietic Rac2, except for RAW macrophages (MΦ) used as control, and all KO clones except for clone 2 lack full-length Rac1/3 protein.
(F) Representative Rac1/2/3 KO clone (clone 1) and B16-F1 control cells plated on laminin-coated coverslips for analysis of the actin cytoskeleton by phalloidin staining. Note the complete absence of lamellipodia or lamellipodia-like structures upon elimination of Rac expression. Scale bar, 20 μm.
(G) Rac1/2/3 KO clone 1 was transfected with EGFP-tagged Rac1-L61 constructs as indicated, and plated on laminin-coated coverslips for analysis of cell morphology. Representative cells are shown. Note that mutation of E31 or F37 residues in Rac1-L61 resulted in the induction of compromised lamellipodia, highly reminiscent in phenotype of those observed upon expression of D site-mutated Sra-1 in Sra-1/PIR121 double KO cells. Scale bar, 10 μm.
High magnification, phase contrast video microscopy of individual Sra-1/PIR121 double KO B16-F1 melanoma cells (clone #3) transfected with EGFP-tagged versions (not shown) of wild type Sra-1 (WT), the D site mutant (Y967A) or the latter additionally activated through release of the WCA domain (Y967A+WCA∗). Note that cells positioned in center of each panel correspond to transfected ones. Time is in minutes and seconds; bar is valid for all panels and equals 20μm.
To solidify the view that all these defects can solely be explained by compromised Rac-D site interaction, additional experiments were performed. These included artificially mimicking an increase of endogenous Rac-GTP by AlF [21]. Among cells forming lamellipodia, only those expressing D site+WCA∗ were significantly shifted toward harboring fully developed versions of these structures, presumably because of unlimited access to the A site in these conditions (Figures S3B and S3C). In line with this, additional A site mutation (A+D site+WCA∗) caused complete failure to generate canonical lamellipodia irrespective of AlF treatment (Figures S3B and S3C; see also below). As a final, complementary experiment, full rescue of the D site phenotype could also be achieved by increasing the spatial proximity of WRC and active Rac through expressing D site+WCA∗ in fusion to constitutively active Rac1-L61 (Figures S3C and S3D). Albeit a bit more variable in localization than WT Sra-1 (Figure 1A and data not shown), the respective fusion protein was able to accumulate at the tips of fully developed lamellipodia and prominently induce their formation in Sra-1/PIR121 double KOs even without AlF treatment (Figure S3D). Overall, we conclude that lack of continuous WRC-Rac interactions and/or reduced avidity of WRC for Rac aside from deficiency in WRC activation generate the complexity of functional defects associated with D site mutation in Sra-1.
Both A and D Sites Are Relevant for Migration in Murine Melanoma and Dictyostelium Cells
We also analyzed the relative relevance of each binding site for the efficiency of migration. In Pir121 null Dictyostelium cells, only re-expression of WT Pir121 rescued migration speed in the null mutant to levels comparable to Ax3 control (Figures 2D and S2C; Video S4). In contrast, both A site and D site mutants improved the performance of the Pir121 KO line at best marginally. These data emphasize the importance of each of the two binding sites for migration, albeit for distinct reasons (Figures 2A–2C). In B16-F1 cells, analysis of migration was separated for cells with and without lamellipodia (Figure 2E), due to the large differences in migration efficiency between these two conditions (Figure S1). Sra-1/PIR121 KO cells forming lamellipodia by A site mutant-containing WRC migrated inefficiently, suggesting lamellipodia function in this case to be highly compromised (Figure 2E). A site+WCA∗ reverted migration efficiency to WT levels, clearly confirming that the defect in the A site mutant can be solely attributed to abrogated WRC activation. In contrast, D site mutant-mediated migration was much more efficient, but, as expected (see above), neither the D site mutant nor its constitutively active counterpart (D site+WCA∗) reached WT migration levels. These data imply that effects on dimension and dynamics of B16-F1 lamellipodia can quite directly be translated into rates of random cell migration.
Pseudopod formation in wild type parental strain Ax3, Pir121 knock out and cells expressing wild type and mutant (A and D site) Pir121-EGFP. Cells were imaged every 3 s, and time-lapse movie is shown at 10 frames/second.
Historical Rac Effector Loop Mutants Act through D Site-Mediated Interaction with WRC
To address the relevance of Rac-WRC interactions by complementary experiments, we generated Rac-free B16-F1 melanoma cells using CRISPR/Cas9-mediated disruption of all Rac genes, RAC1, 2, and 3 (Figure 4E). As expected [25], Rac null B16-F1 cells lacked lamellipodia entirely (Figure 4F and data not shown). Interestingly, two effector loop mutants of Rac1 described previously [32, 33], F37A and E31V, used here in the background of Rac1-L61, stimulated lamellipodia highly reminiscent in morphology of those seen above upon D site mutant rescue (Figures 3A, S1I, and S3B). Again, respective cells displayed jagged, concave, and narrow lamellipodia frequently tipped by filopodial bundles (Figure 4G). Due to the striking similarity in mutant phenotypes, we docked atomic models of Rac and WRC onto each other, guided by the published cryoelectron microscopy (cryo-EM) map of Rac occupying the D site in the WAVE complex [3], and found that Rac1 residues mutated in these two variants (F37 and E31) directly contact the D site of Sra-1 (Figure 4D). Thus, these data suggest the defects in lamellipodia formation previously described for these mutants [32, 33] to be caused by loss of Rac interaction with the D site of Sra-1 rather than p160ROCK [32], a now well-established key effector in RhoA signaling to myosin II-mediated contraction [34].
Physical Interactions between Rac and WRC Are Crucial, but Not Obligatory, for Lamellipodia Formation by Active WRC
As mentioned above, we also mutated both Rac interaction sites in Sra-1 simultaneously, which caused complete abolishment of detectable interaction with constitutively active Rac1 in pull-down assays (Figure 1E). Importantly, not a single cell (n = 202) expressing the A+D site mutant (C179R/R190D/Y967A) was recorded that displayed lamellipodia (Figure 1F). To distinguish between potential functions of these Rac interactions in activation of WRC versus its recruitment and/or maintenance at the lamellipodium tip membrane, we generated a potential, constitutively active WRC incapable of Rac interaction (A+D site+WCA∗). However, the frequency of lamellipodium formation induced was still low with this mutant, with the subfraction of cells (approximately 10%) classified as forming at best immature lamellipodia (Figures 1F and S1I). Although immature, these lamellipodia were still able to partially rescue the migration defect of Sra-1/PIR121 double null cells (Figure 2E). These data suggest that as opposed to activation, direct Rac-WRC interactions are not obligatory for recruitment and/or maintenance and turnover of WRC at the lamellipodium, and additional contributors to these processes must exist. To explore whether both the A site and D site contribute to the accumulation of WRC at the lamellipodium tip, independent of WRC activation, we moved on to assess the dwell time of EGFP-tagged Sra-1 variants by FRAP. WT Sra-1 showed a half-time of recovery at the lamellipodium tip of approximately 22 s (Figures 2F and 2G; for raw data, see Figure S3A), moderately slower than observed previously for WRC subunits expressed on top of endogenous proteins [35, 36]. However, the WCA∗ mutant displayed strongly delayed turnover of more than 3-fold (Figure 2E). To test to what extent this effect was possibly caused by direct interactions of activated WRC with Rac, both A site and D site mutants were explored in the context of WCA∗, and both were found to be reverted to turnover rates close to WT (24 and 28 s for A and D site mutant, respectively). The A site-alone mutant could not be tested in this context (Figure 1F), but turnover of the D site mutant was even accelerated as compared to WT (17 s; see Figures 2F and 2G). Collectively, these data suggest that Rac interactions with A and D sites display differential contributions to the allosteric activation of WRC in vivo, but both crucially contribute to the maintenance and stability of WRC following its activation at the lamellipodium tip.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Sra-1/PIR121 | [10] | pAB4955-B |
| Nap1 | [10] | pAB4953-B |
| pan-WAVE | this study | pAB5502 |
| Abi1 | this study, 6794 | pAB6794 |
| Pir121 (Dictyostelium) | [37] | N/A |
| Nap1 (Dictyostelium) | [13] | N/A |
| Scar (Dictyostelium) | [12] | N/A |
| Abi1 (Dictyostelium) | [22] | N/A |
| GFP | Roche | 11814460001; RRID: AB_390913 |
| Rac1/3 | Merck | 23A8; RRID: AB_309712 |
| Rac2 | [25] | mAB273-75-1 |
| ArpC5A | [38] | mAB323H3 |
| Goat anti-Mouse IgG (H+L) Alexa Fluor 594 | Invitrogen | A11032; RRID: AB_2534091 |
| Goat anti rabbit IgG (H+L) Dylight 800 | Invitrogen | SA535571; RRID: AB_2556775 |
| Rabbit anti Sheep IgG (H+L) Dylight 800 | Invitrogen | SA510060; RRID: AB_2556640 |
| Streptavidin Alexa 680 conjugate | Thermo Fisher Scientific | S32358 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa 488 phalloidin | Life Technologies | A12379 |
| ATTO-594 phalloidin | ATTO-TEC | AD594-81 |
| JetPrime | Polyplus | 114-07 |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare | RPN2236 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | MERCK | 11836170001 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | 78440 |
| Nutrient Mixture F-12 Ham | MERCK | N8641 |
| LoFlo Medium | FORMEDIUM | LF1001 |
| Laminin | Sigma-Aldrich | L2020 |
| Glutaraldehyde | Electron Microscopy Sciences | 16220 |
| Paraformaldehyde | Sigma-Aldrich | P6148 |
| GST-Rac1-L61 | [25] | N/A |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23227 |
| Experimental Models: Cell Lines | ||
| B16-F1 | ATCC | ATCC CRL-6323 |
| B16-F1 Sra-1/PIR121 KO#3 | This study | MS3 |
| B16-F1 Sra-1/PIR121 KO#19 | This study | MS19 |
| B16-F1 Sra-1/PIR121 KO#21 | This study | MS21 |
| B16-F1 Rac1/2/3 KO#1 | This study | MSr1 |
| B16-F1 Rac1/2/3 KO#2 | This study | MSr2 |
| B16-F1 Rac1/2/3 KO#3 | This study | MSr3 |
| B16-F1 Rac1/2/3 KO#5 | This study | MSr5 |
| B16-F1 Rac1/2/3 KO#7 | This study | MSr7 |
| B16-F1 Rac1/2/3 KO#8 | This study | MSr8 |
| B16-F1 Rac1/2/3 KO#9 | This study | MSr9 |
| B16-F1 Rac1/2/3 KO#10 | This study | MSr10 |
| B16-F1 Rac1/2/3 KO#11 | This study | MSr11 |
| Experimental Models: Organisms/Strains | ||
| Dictyostelium discoideum wild type | dictyBase [39] | Ax3 |
| PIR121 knock out (PIR121-), homologous recombination | This study | Sp3 |
| PIR121-/PIR121-EGFP (Dicty codon-optimized; REMI) | This study | Sp7 |
| PIR121-/PIR121K193D,R194D-EGFP, REMI | This study | Sp11 |
| PIR121-/PIR121 Y961A-EGFP, REMI | This study | Sp15 |
| PIR121-/PIR121 A183R-EGFP, REMI | This study | Sp17 |
| Oligonucleotides | ||
| Cas9-targeting sequence CYFIP1: GACAGAAATGCA TTTGTCAC |
This study | oligMS1 |
| Cas9-targeting sequence CYFIP2: GACAGGAATGCA TTTGTCAC |
This study | oligMS2 |
| Cas9-targeting sequence Rac1/2: ATGCAGGCCAT CAAGTGTG |
This study | oligMS3 |
| Cas9-targeting sequence Rac3: ATGCAGGCCATC AAGTGCG |
This study | oligMS4 |
| Mutagenesis primers, see Table S1 | This study | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro | Addgene | 48139 |
| pEGFP-C1 | Clontech | U55763.1 |
| pEGFP-C2-Sra-1-wt | This study | pMS1 |
| pEGFP-C2-Sra-1-C179R | This study | pMS2 |
| pEGFP-C2-Sra-1-R190D | This study | pMS3 |
| pEGFP-C2-Sra-1-C179R/R190D | This study | pMS4 |
| pEGFP-C2-Sra-1-Y967A | This study | pMS5 |
| pEGFP-C2-Sra-1-G971W | This study | pMS6 |
| pEGFP-C2-Sra-1-C179R/R190D/Y967A | This study | pMS7 |
| pEGFP-C2-Sra-1-C179R/R190D/WCA∗ | This study | pMS8 |
| pEGFP-C2-Sra-1-Y967A/WCA∗ | This study | pMS9 |
| pEGFP-C2-Sra-1-C179R/R190D/Y967A/WCA∗ | This study | pMS10 |
| pEGFP-C2-Sra-1-WIRS | This study | pMS11 |
| pmCherry-C2-Sra-1-WT | This study | pMS12 |
| pmCherry-C2-Sra-1-Y967A | This study | pMS13 |
| pmCherry-C2-Sra-1-Y967A/WCA∗ | This study | pMS14 |
| pGEX-2T-Rac1-Q61L | [40] | N/A |
| pEGFP-C1-Rac1-Q61L | This study | pMS15 |
| pEGFP-C1-Rac1-Q61L/E31V | This study | pMS16 |
| pEGFP-C1-Rac1-Q61L/F37A | This study | pMS17 |
| mRFPmars-ArpC4 | [14] | N/A |
| Pir121 Knock out vector | This study | pSP136 |
| Pir121Gfp, REMI vector | This study | pPT808 |
| Pir121K193D,R194D Gfp, REMI vector | This study | pPT810 |
| Pir121 Y961A Gfp, REMI vector | This study | pSP160 |
| Pir121 A183R Gfp, REMI vector | This study | pSP182 |
| Software and Algorithms | ||
| MetaMorph | Molecular Devices, Sunnyvale, CA, USA | https://de.moleculardevices.com/systems/metamorph-research-imaging/ |
| Prism 7 | Graphpad Software, La Jolla, USA | https://www.graphpad.com/scientific-software/prism/ |
| Sigma plot 12.0 | Systat Software, Erkrath, Germany | http://www.systat.de/SigmaPlot12_Produktseite.html |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Klemens Rottner (k.rottner@tu-braunschweig.de).
Experimental Model and Subject Details
B16-F1 cells were purchased from ATCC (CRL-6323, sex:male), and authenticated in a recent, official lab screen by local authorities to be mycoplasma-free, of murine origin and to lack any unexpected sequences such as commonly used antibiotic resistance genes or viral sequences. Dictystelium discoideum strain Ax3, haploid, mating type matA, derived from wild-type NC4 was obtained from Dictybase.
Method Details
Cell culture
B16-F1 cells and derived CYFIP1/2 double or Rac1/2/3 triple knockout clones were cultured in DMEM (4.5 g/L glucose; Invitrogen, Germany), supplemented with 10% FCS (PAA Laboratories, Austria), 2 mM glutamine (Thermo Fisher Scientific) and penicillin (50 Units/mL)/streptomycin (50 μg/mL) (Thermo Fisher Scientific). B16-F1 cells were routinely transfected in 35 mm dishes, using 0.5 μg DNA in total and 1 μL JetPrime for controls, and 1 μg DNA in total and 2 μL JetPrime for CYFIP1/2 and RAC1/2/3 knockout cells. After overnight transfection, cells were plated onto acid-washed, laminin-coated (25 μg/mL) coverslips for microscopy, or onto plastic dishes for obtaining samples for Western Blotting. Stimulation with aluminum fluoride as in Figures S1B, S3B, and S3C was performed in pre-warmed, full growth medium upon addition of AlCl3 and NaF to final concentrations of 50 μM and 30 mM, respectively [10, 21]. Treatments were carried out for 20 min and followed by fixation and staining as detailed below.
Dictyostelium discoideum strains were grown and maintained in Petri dishes or multiwell plates in filtered HL5 medium, including glucose, vitamins and microelements (ForMedium) with 100 μg/mL penicillin- streptomycin (Thermo Fisher Scientific). For transfections, 1 × 107 cells/mL were washed once with buffer (5 mM Na2HPO4, 5 mM KH2PO4 and 50 mM sucrose), followed by electroporation with 500 ng extrachromosomal plasmid at 500 V using ECM399 electroporator (BTX Harvard apparatus). Transfected cells were transferred into HL5 (100mm Petri-dishes), and 24 hr later, transformants selected and maintained using 50 μg/mL hygromycin.
DNA constructs
pEGFP-C1 and –C2 vectors were purchased from Clontech (Mountain View, CA, USA). The wild-type Sra-1 sequence, fused into pEGFP-C2 and used for all further mutants generated below corresponded to the splice variant CYFIP1a, sequence AJ567911 [10]. All Sra-1 mutations employing primers as listed in Table S1 were carried out by site-directed mutagenesis using Phusion DNA polymerase. mCherry-tagged Sra-1 constructs were generated by swapping EGFP with mCherry, kindly provided by Dr. Roger Tsien (University of California at San Diego, La Jolla, California, USA) using NheI/BsrGI restriction sites. For generating pEGFP-C1-Rac1-Q61L, the Rac1-Q61L fragment immobilized from pGEX-2T-Rac1-Q61L [40] with BamHI/EcoRI was ligated into pEGFP-C1 vector digested with BglII/EcoRI. Rac1 mutants were generated in analogy to Sra-1 (Table S1). For EGFP-tagged Sra-1-Y967A+WCA∗-Rac1-L61, Rac1-L61 was fused with an (GGS)6-linker to the C terminus of Sra-1-Y967A+WCA∗ using overlap extension PCR, and cloned into pEGFP-C2 vector using XhoI/SalI restriction sites. All constructs were sequence-verified.
Antibody generation
For generation of polyclonal antisera, recombinant Abi1 or the WCA-domain of WAVE2 were employed for immunization of rabbits (in duplicate) with five boosts (Eurogentec, Berlin, Germany). Sera from rabbits 6794 and 5502, respectively, were used for further experiments.
CRISPR/Cas9-mediated genome editing
B16-F1 cells lacking CYFIP1 and CYFIP2 genes were generated using CRISPR/Cas9 technology [41]. Specifically, both genes were simultaneously disrupted targeting a highly conserved DNA region in respective exon 2. Selected targeting sequences were cloned into pSpCas9(BB)-2A-Puro (Addgene plasmid ID: 48139), and equimolar amounts of resulting vectors targeting genomic regions of GACAGAAATGCATTTGTCAC (CYFIP1) or GACAGGAATGCATTTGTCAC (CYFIP2) transfected in combination. After puromycin selection for transfected cells (3 days), cells were extensively diluted and a few days later, macroscopically visible colonies picked, to obtain single cell-derived clones. Cell clones were screened for the absence of both gene products using a pan-CYFIP antibody, and defined as CYFIP1/2 double knockout clones following confirmation of the absence of any wild-type allele by sequencing of respective genomic regions.
For generation of Rac1/2/3 KO cells, B16-F1 cells were transfected with equimolar amounts of plasmids targeting ATGCAGGCCATCAAGTGTG (RAC1/2) and ATGCAGGCCATCAAGTGCG (RAC3) genomic regions. Derived cell clones were screened for the absence of Rac expression by Western Blotting.
Pir121 disruption, REMI, and site-directed mutagenesis in Dictyostelium
For disruption of the gene encoding Pir121 in Ax3, termed pirA [12], 5′ and 3′ arms of the knockout vector were PCR-amplified with primers provided in Table S1 and cloned upstream and downstream of a blasticidin cassette, respectively, into pAD70 vector [42]. The resulting vector was electroporated into Ax3 cells and pirA disruption confirmed by resistance to blasticidin (10 μg/mL), PCR and Western Blotting.
Constructs for Pir121-EGFP REMI (restriction enzyme-mediated integration) were made by amplifying Pir121 including the 5′ UTR promoter region with appropriate primers (Table S1), followed by cloning in fusion with EGFP into pDM1209 REMI vector. Appropriate residues in Rac binding sites were mutated by site directed mutagenesis using primers listed in Table S1. All constructs were verified by sequencing.
For genomic integration by REMI [43], clones were selected with G418 (10 μg/mL), and stability of Pir121 expression examined by Western Blotting after single cell expansion with bacteria for several generations.
Western Blotting
For preparation of whole cell lysates, cells were washed with PBS, lysed using lysis-buffer (2% SDS, 10% glycerol, 63 mM Tris-HCl pH 6,8) and sonicated to shear genomic DNA. An aliquot was used for protein measurement (BCA assay; Key Resources Table), and the remaining volume treated with β-mercaptoethanol and bromphenolblue to yield Laemmli buffer and boiled for 5 min. Western blotting was carried out using standard techniques with equal amounts of protein. Primary antibodies are listed in the Key Resources Table. HRP-conjugated secondary antibodies were purchased from Invitrogen. Chemiluminescence signals were obtained upon incubation with ECL Prime Western Blotting Detection Reagent (GE Healthcare), and were recorded with ECL Chemocam imager (Intas, Goettingen, Germany). Densitometric quantification of protein expression was performed using MetaMorph (Molecular Devices, Sunnyvale, CA, USA).
Dictyostelium cells were lysed in 1 x NuPAGE LDS sample buffer containing 20 mM DTT, HALT protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific), followed by boiling for 5 min. After SDS-PAGE, transfer onto nitrocellulose membranes (GE Healthcare Life Sciences) and blocking (5% non-fat milk), primary antibodies were used at a dilution of 1:1000. Fluorescently conjugated secondary antibodies (Thermo Fisher Scientific) were detected by Odyssey CLx Imaging system (LI-COR Biosciences).
Fluorescence staining and quantification
B16-F1 cells and Sra-1/PIR121 KO cell clones were seeded onto laminin-coated (25 μg/mL), 15 mm-diameter glass coverslips and allowed to adhere overnight. Next morning, cells were treated or not with aluminum fluoride, fixed with prewarmed, 4% paraformaldehyde (PFA) in PBS for 20 min, and permeabilized with 0.05% Triton X-100 in PBS for 1 min. For sole visualization of the actin cytoskeleton with phalloidin and in case of samples lacking EGFP expression, cells were fixed with 0.25% glutaraldehyde and 4% PFA followed by staining with Alexa 488-conjugated phalloidin (1:400). PFA-fixed cell samples following transfections with plasmids mediating expression of EGFP-tagged proteins were counterstained with ATTO-594-conjugated phalloidin (1:200). For stainings with ArpC5A antibodies, permeabilized cells were blocked with 5% horse serum and 1% BSA in PBS, followed by staining with monoclonal anti-ArpC5A antibody (Key Resources Table; undiluted hybridoma supernatant). Primary antibody was visualized with Alexa Fluor 594-coupled (1:100 dilution) anti-mouse IgG (see Key Resources Table). For fluorescence intensity measurements of ArpC5A- and phalloidin-stainings [41], respective lamellipodial regions were encircled using Metamorph software, and a larger, extracellular region defined as background. Average intensities of respective background regions were subsequently subtracted from pixel intensities derived from lamellipodial regions.
Time-lapse microscopy
For random migration assays, B16-F1 or Sra-1/PIR121 KO cells were seeded subconfluently into laminin-coated (25 μg/mL) μ-slide 4-well glass bottom microscopy chambers (Ibidi, Martinsried, Germany). After about 6 h, the chamber was mounted onto an inverted microscope (Axio observer, Zeiss, Jena, Germany), equipped with 37°C incubator and CO2 atmosphere. Phase-contrast movies were simultaneously acquired on multiple, randomly chosen positions using a 10 x /0.15 NA Plan Neofluar objective and a frame rate of 12 frames per hour for at least 10 hr. For analysis, cells were manually tracked using ImageJ (https://imagej.nih.gov/ij/). For the reconstitution experiment shown in Figure 1C, all B16-F1 control and Sra-1/PIR121 double KO cells (clone #3) were analyzed. Note that all B16-F1 control cells formed lamellipodia at some stage during the observation period. For the reconstitution experiment and in analogy to controls, only those double KO-cells transiently transfected with EGFP-Sra-1 that formed lamellipodia at some stage during the observation period were defined as successfully transfected and thus included into analysis. For the migration analyses of KO cells transfected with various Sra-1 constructs shown in Figure 2E, all cells were analyzed, but separated into cells forming lamellipodia at any time during the observation period (red) and those lacking lamellipodia throughout the observation period (blue).
For measuring protrusion velocities of lamellipodia induced by distinct Sra-1 mutants, live cell imaging was done with Sra-1/PIR121 KO #3 cells transfected with respective EGFP-tagged Sra-1 variants and migrating on laminin-coated glass coverslips (25 μg/mL). Cells were observed in an open heating chamber (Warner Instruments, Reading, UK) with a heater controller (Model TC-324 B, SN 1176) at 37°C. Cells were maintained in microscopy medium (F12 HAM HEPES-buffered medium, Sigma), including 10% FCS, 2 mM L-glutamine and 1% penicillin/streptomycin. Conventional video microscopy was performed on an inverted microscope (Axiovert 100TV, Zeiss) equipped with an HXP 120 lamp for epifluorescence illumination, a halogen lamp for phase-contrast imaging, a Coolsnap-HQ2 camera (Photometrics) and electronic shutters driven by MetaMorph software (Molecular Devices). Live cell images were obtained using a 63 x/1.4 NA Plan apochromatic oil objective. Lamellipodial protrusion velocities were determined based on kymographs obtained from time-lapse images, recorded for a time period of at least 4 min acquiring images every 5 s.
Microscopy and analysis of Dictyostelium cells
Cell migration was examined by under agarose folate chemotaxis assay [44]. Initially, 0.4% SeaKem GTG agarose was melted in Lo-Flo medium (ForMedium). After cooling of agarose, 10 μM folic acid was mixed and 5 mL poured into 50 mm glass bottom dish (MatTek Corporation) pre-treated with 1% BSA in Lo-Flo. After setting of the agarose, 5mm wide troughs were cut using a scalpel, and troughs filled with vegetative cells (2 × 106 cells/mL). Cells were allowed to migrate for 4 hr and routinely imaged with 10 x/0.3 NA or 60 x/1.4 NA DIC objectives on a Nikon ECLIPSE TE2000-R inverted microscope equipped with a monochromatic Retiga Exi cooled CCD camera. 10 x images were taken at 1 min intervals for 45 min, DIC images were captured every 3 s for 10 min. To determine localization and dynamics of Pir121-EGFP and mRFPmars-ArpC4, AiryScan superresolution imaging was performed on a Zeiss 880 inverted confocal microscope equipped with a 63 x/1.4 NA objective. All images were converted to tiff and processed by ImageJ (NIH). Chemotactic speed was calculated using home-made plug-in of ImageJ.
Fluorescence recovery after photobleaching
FRAP experiments were performed using an inverted Axio Observer microscope equipped with an automated stage, a DG4 light source (Sutter Instrument) for epifluorescence illumination, a VIS-LED for phase-contrast optics and a Coolsnap-HQ2 camera (Photometrics) driven by VisiView software (Visitron Systems, Puchheim, Germany). In addition, bleaching was performed using a 405 nm diode laser at 70–80 mW output power) controlled by the 2D-VisiFRAP Realtime Scanner (Visitron Systems, Puchheim, Germany).
For analyzing the dwell time in the lamellipodium, EGFP-Sra-1 variants were transfected into Sra-1/PIR121 KO cells (clone #3) and bleached within rectangular regions covering subfractions of lamellipodia, individually drawn for each experiment as indicated in the Figure (see Figure S3A). Movies were acquired at a rate of 2 s per frame. Intensity values in bleached regions were measured over time and background intensities obtained from close extracellular regions subtracted for each frame. Acquisition photobleaching was performed by correcting data to average intensities over time obtained from lateral, non-bleached lamellipodial tip regions. Data were curve-fitted with SigmaPlot 12.0 (Scientific Solutions SA, Pully-Lausanne, Switzerland) using single exponential equation: f = y0+a∗(1-exp(-b∗x)), and half times of recovery derived from fitted data [45].
For determination of actin assembly rates, Sra-1/PIR121 KO cells (clone #3) were cotransfected with mCherry-tagged Sra-1 constructs and EGFP-tagged actin, plated on laminin, and followed by bleaching of the latter and its analysis of recovery. Rates of lamellipodial actin network growth were directly obtained from measuring average widths of newly generated actin networks over time [35, 41].
GST-pull-down assays
For Rac1 pulldown assays, GST–Rac1-L61 was immobilized on glutathione–Sepharose beads in buffer B (50 mM Tris, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 0.1 mM DTT). Sra-1/PIR121 KO #3 cells were transfected with different EGFP-Sra-1 constructs and lysed two days after transfection with lysis buffer (1% Triton X-100, 140 mM KCl, 50 mM Tris/HCl pH 7.4/ 50 mM NaF, 10 mM Na4P2O7, 2 mM MgCl2 and Complete Mini, EDTA-free protease inhibitor [Roche]). Lysates were cleared and incubated with GST-Rac1-L61 beads for 60 min. Subsequently, beads were washed three times with lysis buffer lacking Triton X-100 and protease inhibitor, mixed with Laemmli buffer, boiled for 5 min and subjected to Western Blotting.
In silico interaction analysis
The structural model of Rac1 binding the WAVE complex at the D site was generated using HADDOCK (version 2.3) [46, 47, 48], coordinates of the WAVE complex (PDB: 3P8C [4]), a model of Rac1 (PDB: 3SBD [49]) and a deposited electron microscopy map (EMDB: EMD-6642 [3]). For the docking, residues within a distance of 5Å to residues of the other model were defined as active and within a distance of 8Å as passive (excluding active residues), after fitting the models into the map utilizing UCSF Chimera (version 1.12) [50]. Additional restrains were defined to fix issues with the magnesium ion bound to Rac1 wandering off during the second step of the docking. Based on the HADDOCK score, the best model from the biggest cluster was chosen as final model. The figure was generated using PyMOL software (Schrodinger, The PyMOL Molecular Graphics System, Version 2.0, 2015).
Quantification and Statistical Analysis
Quantitative experiments were performed at least in triplicates to avoid any potential impact of environmental influences or inadvertent error, but not routinely subjected to analyses blinded to the experimenter, for practical reasons. Effects on lamellipodial phenotypes derived from analyses on fixed samples and living cells were systematically obtained from sample sizes of hundreds and at least dozens of cells, respectively. Data points were not intentionally excluded from analyses at any time.
Statistical analyses of manually categorized lamellipodial phenotypes and distinct lamellipodial parameters such as width, protrusion rate and actin assembly rate were routinely performed using non-parametric statistical tests, such as Mann-Whitney Rank Sum Test, since the latter does not require compared datasets to be normally distributed. Experimental groups were explored for normal distribution using SigmaPlot 12.0 (Scientific Solutions SA, Pully-Lausanne, Switzerland). Sample sizes of statistical analyses are routinely provided in Figure legends, and n usually corresponded to cells analyzed, except for statistics done on Western band intensities. All statistics on mouse cell data were performed using SigmaPlot 12.0. For statistics of Dictyostelium data, Mann-Whitney was used in case of side-by-side comparisons of two experimental groups (Figures 2B and 2C). For comparison of multiple experimental groups, one-way ANOVA to assess principal statistical differences between groups followed by more informative Dunnett’s multiple comparison test were performed using Prism 7 software (Graphpad, La Jolla, USA).
Acknowledgments
This work was supported in part by Deutsche Forschungsgemeinschaft (DFG) grants GRK2223/1 (to K.R. and W.B.) and RO2414/3-2 (to K.R.), Cancer Research UK (grant CRUK-A20017 to R.H.I.), and intramural core grants (to T.E.S. and R.H.I.). We also thank Brigitte Denker for excellent technical assistance and Kai Städing for help with antibody generation.
Author Contributions
M.S., S.P.S., and F.K. performed wet experiments, and T.K. performed in silico structural analysis. P.T., A.S., and T.E.S. provided essential reagents. All authors analyzed and discussed the data. W.B., R.H.I., and K.R. supervised research; M.S., S.P.S., R.H.I., and K.R. interpreted all data and wrote the manuscript. All authors read and commented on the manuscript and/or amended parts of it.
Declaration of Interests
The authors declare no competing interests.
Published: November 1, 2018
Footnotes
Supplemental Information includes three figures, one table, and four videos and can be found with this article online at https://doi.org/10.1016/j.cub.2018.10.002.
Contributor Information
Robert H. Insall, Email: r.insall@beatson.gla.ac.uk.
Klemens Rottner, Email: k.rottner@tu-braunschweig.de.
Supplemental Information
References
- 1.Molinie N., Gautreau A. The Arp2/3 regulatory system and its deregulation in cancer. Physiol. Rev. 2018;98:215–238. doi: 10.1152/physrev.00006.2017. [DOI] [PubMed] [Google Scholar]
- 2.Rottner K., Faix J., Bogdan S., Linder S., Kerkhoff E. Actin assembly mechanisms at a glance. J. Cell Sci. 2017;130:3427–3435. doi: 10.1242/jcs.206433. [DOI] [PubMed] [Google Scholar]
- 3.Chen B., Chou H.T., Brautigam C.A., Xing W., Yang S., Henry L., Doolittle L.K., Walz T., Rosen M.K. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. eLife. 2017;6:e29795. doi: 10.7554/eLife.29795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Z., Borek D., Padrick S.B., Gomez T.S., Metlagel Z., Ismail A.M., Umetani J., Billadeau D.D., Otwinowski Z., Rosen M.K. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alekhina O., Burstein E., Billadeau D.D. Cellular functions of WASP family proteins at a glance. J. Cell Sci. 2017;130:2235–2241. doi: 10.1242/jcs.199570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derivery E., Fink J., Martin D., Houdusse A., Piel M., Stradal T.E., Louvard D., Gautreau A. Free Brick1 is a trimeric precursor in the assembly of a functional Wave complex. PLoS ONE. 2008;3:e2462. doi: 10.1371/journal.pone.0002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Innocenti M., Zucconi A., Disanza A., Frittoli E., Areces L.B., Steffen A., Stradal T.E., Di Fiore P.P., Carlier M.F., Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat. Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- 8.Kunda P., Craig G., Dominguez V., Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr. Biol. 2003;13:1867–1875. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Rogers S.L., Wiedemann U., Stuurman N., Vale R.D. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J. Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steffen A., Rottner K., Ehinger J., Innocenti M., Scita G., Wehland J., Stradal T.E. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bear J.E., Rawls J.F., Saxe C.L., III SCAR, a WASP-related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. J. Cell Biol. 1998;142:1325–1335. doi: 10.1083/jcb.142.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blagg S.L., Stewart M., Sambles C., Insall R.H. PIR121 regulates pseudopod dynamics and SCAR activity in Dictyostelium. Curr. Biol. 2003;13:1480–1487. doi: 10.1016/s0960-9822(03)00580-3. [DOI] [PubMed] [Google Scholar]
- 13.Ibarra N., Blagg S.L., Vazquez F., Insall R.H. Nap1 regulates Dictyostelium cell motility and adhesion through SCAR-dependent and -independent pathways. Curr. Biol. 2006;16:717–722. doi: 10.1016/j.cub.2006.02.068. [DOI] [PubMed] [Google Scholar]
- 14.King J.S., Veltman D.M., Georgiou M., Baum B., Insall R.H. SCAR/WAVE is activated at mitosis and drives myosin-independent cytokinesis. J. Cell Sci. 2010;123:2246–2255. doi: 10.1242/jcs.063735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Litschko C., Linkner J., Brühmann S., Stradal T.E.B., Reinl T., Jänsch L., Rottner K., Faix J. Differential functions of WAVE regulatory complex subunits in the regulation of actin-driven processes. Eur. J. Cell Biol. 2017;96:715–727. doi: 10.1016/j.ejcb.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Dubielecka P.M., Ladwein K.I., Xiong X., Migeotte I., Chorzalska A., Anderson K.V., Sawicki J.A., Rottner K., Stradal T.E., Kotula L. Essential role for Abi1 in embryonic survival and WAVE2 complex integrity. Proc. Natl. Acad. Sci. USA. 2011;108:7022–7027. doi: 10.1073/pnas.1016811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leithner A., Eichner A., Müller J., Reversat A., Brown M., Schwarz J., Merrin J., de Gorter D.J., Schur F., Bayerl J. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat. Cell Biol. 2016;18:1253–1259. doi: 10.1038/ncb3426. [DOI] [PubMed] [Google Scholar]
- 18.Soderling S.H., Langeberg L.K., Soderling J.A., Davee S.M., Simerly R., Raber J., Scott J.D. Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc. Natl. Acad. Sci. USA. 2003;100:1723–1728. doi: 10.1073/pnas.0438033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki D., Suetsugu S., Miki H., Kataoka Y., Nishikawa S., Fujiwara T., Yoshida N., Takenawa T. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–456. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 20.Suetsugu S., Yamazaki D., Kurisu S., Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev. Cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 21.Hahne P., Sechi A., Benesch S., Small J.V. Scar/WAVE is localised at the tips of protruding lamellipodia in living cells. FEBS Lett. 2001;492:215–220. doi: 10.1016/s0014-5793(01)02239-6. [DOI] [PubMed] [Google Scholar]
- 22.Pollitt A.Y., Insall R.H. Abi mutants in Dictyostelium reveal specific roles for the SCAR/WAVE complex in cytokinesis. Curr. Biol. 2008;18:203–210. doi: 10.1016/j.cub.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 23.Lebensohn A.M., Kirschner M.W. Activation of the WAVE complex by coincident signals controls actin assembly. Mol. Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oikawa T., Yamaguchi H., Itoh T., Kato M., Ijuin T., Yamazaki D., Suetsugu S., Takenawa T. PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat. Cell Biol. 2004;6:420–426. doi: 10.1038/ncb1125. [DOI] [PubMed] [Google Scholar]
- 25.Steffen A., Ladwein M., Dimchev G.A., Hein A., Schwenkmezger L., Arens S., Ladwein K.I., Margit Holleboom J., Schur F., Victor Small J. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J. Cell Sci. 2013;126:4572–4588. doi: 10.1242/jcs.118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fort L., Batista J.M., Thomason P.A., Spence H.J., Whitelaw J.A., Tweedy L., Greaves J., Martin K.J., Anderson K.I., Brown P. Fam49/CYRI interacts with Rac1 and locally suppresses protrusions. Nat Cell Biol. 2018;20:1159–1171. doi: 10.1038/s41556-018-0198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen B., Brinkmann K., Chen Z., Pak C.W., Liao Y., Shi S., Henry L., Grishin N.V., Bogdan S., Rosen M.K. The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell. 2014;156:195–207. doi: 10.1016/j.cell.2013.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squarr A.J., Brinkmann K., Chen B., Steinbacher T., Ebnet K., Rosen M.K., Bogdan S. Fat2 acts through the WAVE regulatory complex to drive collective cell migration during tissue rotation. J. Cell Biol. 2016;212:591–603. doi: 10.1083/jcb.201508081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritz-Laylin L.K., Riel-Mehan M., Chen B.C., Lord S.J., Goddard T.D., Ferrin T.E., Nicholson-Dykstra S.M., Higgs H., Johnson G.T., Betzig E., Mullins R.D. Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. eLife. 2017;6:e26990. doi: 10.7554/eLife.26990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veltman D.M., King J.S., Machesky L.M., Insall R.H. SCAR knockouts in Dictyostelium: WASP assumes SCAR’s position and upstream regulators in pseudopods. J. Cell Biol. 2012;198:501–508. doi: 10.1083/jcb.201205058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bieling P., Hansen S.D., Akin O., Li T.D., Hayden C.C., Fletcher D.A., Mullins R.D. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. EMBO J. 2018;37:102–121. doi: 10.15252/embj.201797039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamarche N., Tapon N., Stowers L., Burbelo P.D., Aspenström P., Bridges T., Chant J., Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- 33.Westwick J.K., Lambert Q.T., Clark G.J., Symons M., Van Aelst L., Pestell R.G., Der C.J. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol. Cell. Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellegrin S., Mellor H. Actin stress fibres. J. Cell Sci. 2007;120:3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- 35.Lai F.P., Szczodrak M., Block J., Faix J., Breitsprecher D., Mannherz H.G., Stradal T.E., Dunn G.A., Small J.V., Rottner K. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 2008;27:982–992. doi: 10.1038/emboj.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiner O.D., Marganski W.A., Wu L.F., Altschuler S.J., Kirschner M.W. An actin-based Wave generator organizes cell motility. PLoS Biol. 2007;5:e221. doi: 10.1371/journal.pbio.0050221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollitt A.Y., Blagg S.L., Ibarra N., Insall R.H. Cell motility and SCAR localisation in axenically growing Dictyostelium cells. Eur. J. Cell Biol. 2006;85:1091–1098. doi: 10.1016/j.ejcb.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Olazabal I.M., Caron E., May R.C., Schilling K., Knecht D.A., Machesky L.M. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr. Biol. 2002;12:1413–1418. doi: 10.1016/s0960-9822(02)01069-2. [DOI] [PubMed] [Google Scholar]
- 39.Chisholm R.L., Gaudet P., Just E.M., Pilcher K.E., Fey P., Merchant S.N., Kibbe W.A. dictyBase, the model organism database for Dictyostelium discoideum. Nucleic Acids Res. 2006;34:D423–D427. doi: 10.1093/nar/gkj090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 41.Kage F., Winterhoff M., Dimchev V., Mueller J., Thalheim T., Freise A., Brühmann S., Kollasser J., Block J., Dimchev G. FMNL formins boost lamellipodial force generation. Nat. Commun. 2017;8:14832. doi: 10.1038/ncomms14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidson A.J., Amato C., Thomason P.A., Insall R.H. WASP family proteins and formins compete in pseudopod- and bleb-based migration. J. Cell Biol. 2018;217:701–714. doi: 10.1083/jcb.201705160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuspa A., Loomis W.F. Tagging developmental genes in Dictyostelium by restriction enzyme-mediated integration of plasmid DNA. Proc. Natl. Acad. Sci. USA. 1992;89:8803–8807. doi: 10.1073/pnas.89.18.8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laevsky G., Knecht D.A. Cross-linking of actin filaments by myosin II is a major contributor to cortical integrity and cell motility in restrictive environments. J. Cell Sci. 2003;116:3761–3770. doi: 10.1242/jcs.00684. [DOI] [PubMed] [Google Scholar]
- 45.Koestler S.A., Steffen A., Nemethova M., Winterhoff M., Luo N., Holleboom J.M., Krupp J., Jacob S., Vinzenz M., Schur F. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol. Biol. Cell. 2013;24:2861–2875. doi: 10.1091/mbc.E12-12-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Vries S.J., van Dijk A.D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A.M. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 47.Dominguez C., Boelens R., Bonvin A.M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 48.van Zundert G.C.P., Melquiond A.S.J., Bonvin A.M.J.J. Integrative modeling of biomolecular complexes: HADDOCKing with cryo-electron microscopy data. Structure. 2015;23:949–960. doi: 10.1016/j.str.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Krauthammer M., Kong Y., Ha B.H., Evans P., Bacchiocchi A., McCusker J.P., Cheng E., Davis M.J., Goh G., Choi M. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phase contrast, time-lapse microscopy of B16-F1 wildtype, Sra-1/PIR121 KO clone #3 and the latter re-expressing EGFP-tagged Sra-1, as indicated, for restoration of lamellipodia-dependent migration. Time is in hours and minutes, scale bar is valid for all panels and equals 100μm.
AiryScan imaging of cells to visualize WRC complex (green) and Arp2/3 complex (red) localization in wild type and PIR121 null cells expressing wild type and mutant (A or D site) Pir121-EGFP. Cells were imaged every 2 s, and resulting time-lapse movie is displayed at 10 frames/second.
High magnification, phase contrast video microscopy of individual Sra-1/PIR121 double KO B16-F1 melanoma cells (clone #3) transfected with EGFP-tagged versions (not shown) of wild type Sra-1 (WT), the D site mutant (Y967A) or the latter additionally activated through release of the WCA domain (Y967A+WCA∗). Note that cells positioned in center of each panel correspond to transfected ones. Time is in minutes and seconds; bar is valid for all panels and equals 20μm.
Pseudopod formation in wild type parental strain Ax3, Pir121 knock out and cells expressing wild type and mutant (A and D site) Pir121-EGFP. Cells were imaged every 3 s, and time-lapse movie is shown at 10 frames/second.