

Abstract
A series of 1,3-oxazole, 1,3-thiazole, isomeric 1,2,4-oxadiazole, 1,3,4-oxadiazole, and 1,2,3,4-tetrazole heterocycles was synthesized. All the compounds shared as a common feature the presence of a 4-hydroxyphenyl substituent. The structures of the synthesized compounds were confirmed by MS, 1H-NMR, and elemental analysis. In vitro antimicrobial activity for all the newly synthesized compounds at concentrations of 200-25 μg/mL was evaluated against Gram+ve organisms such as methicillin-resistant Staphylococcus aureus (MRSA), Gram–ve organisms such as Escherichia coli (E. coli), and the fungal strain Aspergillus niger (A. niger) by the cup plate method. Ofloxacin and ketoconazole (10 μg/mL) were used as reference standards for antibacterial and antifungal activity, respectively. Compounds 15, 16, and 20 showed notable antibacterial and antifungal activities at higher concentrations (200 μg/mL), whereas 17-19 were found to display significant antibacterial or antifungal activity (25-50 μg/mL) against the Gram+ve, Gram–ve bacteria, or fungal cells used in the present study.
Keywords: 1,3-oxazole; oxadiazole congeners; 1,3-thiazole; tetrazole; antibacterial; antifungal
1. Introduction
Heterocycles containing nitrogen(s) and an oxygen or sulfur atom constitute an important class of compounds in the field of medicinal chemistry due to their interesting and diverse clinical applications [1]. For example, considerable evidence has accumulated during the past two decades demonstrating the various pharmacological effects of 1,3,4-oxadiazoles, which include antibacterial [1], antifungal [2], anthelmintic [3], antitubercular [4], anticancer [5], anti-HIV [6], antioxidant [7], analgesic [8], anti-inflammatory [9] and anticonvulsant [10] activities. In spite of the large number of antibiotics and chemotherapeutics currently available for medical usage, the spectre of increasing bacterial resistance has made it necessary to continue the search for new antimicrobial substances. To this end a large number of oxadiazole derivatives have been prepared, many of which have shown a wide spectrum of antimicrobial activity. Some oxadiazoles with different substituents, especially with a 4-hydroxy-phenyl moiety at different locations on the five-membered heterocyclic ring, produced fungicidal and bactericidal agents of various potencies [11,12]. These observations promoted us to develop, synthesize, and evaluate a novel series of diverse heterocycles of more simplified chemical structures that include 1,3-oxazole, 1,3-thiazole, isosteric oxadiazoles, and tetrazole heterocyclic ring systems, all bearing a 4-hydroxyphenyl moiety.
2. Results and Discussion
2.1. Synthesis
All novel heterocyclic compounds were synthesized by ring-closing reactions, with each derivative being prepared via a different synthetic pathway (Scheme 1). Key intermediates were the 4-substituted phenylmethyl ethers 9-14. Carbonyl compounds can be efficiently converted into 5-substituted oxazoles in a one-pot reaction with tosylmethyl isocyanide (TosMIC) [13]. Here, the oxazole compound 9 was obtained by reaction of equimolar quantities of TosMIC and the benzaldehyde precursor 1 with potassium carbonate in refluxing methanol (Scheme 1).
Scheme 1.

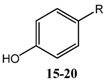
Synthesis of heterocyclic derivatives 15-20.
The most common and most versatile procedure for the formation of 1,3-thiazoles is the cyclocondensation of α-haloketones with appropriate thioamide derivatives [14]. In this study, the reaction of commercially available α-bromoketone 2 with thioacetamide under basic conditions furnished the 4-substituted heterocyclic derivative 10. The amide oxime 6 provided the starting point for the synthesis of the 3,5-disubstituted 1,2,4-oxadiazole 11, which was generated by reaction of hydroxylamine, liberated from its hydrochloride using potassium carbonate, with p-anisonitrile 3 in refluxing ethanol.
Dehydration, cyclization, and aromatization by subsequent treatment with acetic acid methyl ester in the presence of sodium hydride in tetrahydrofuran afforded the corresponding 5-methyl-1,2,4-oxadiazole compound 11 [15]. The 3-methyl-1,2,4-oxadiazole derivative 12 was prepared adapting the procedure described by Lin et al. [16]. Thus, reaction of the carboxamide 4 with N,N-dimethylacetamide dimethyl acetal and cyclization of the intermediate acylamidine with hydroxylamine provided 12 in good yield.
According to the procedure described by Ainsworth, synthesis of the 5-methyl-1,3,4-oxadiazole derivative 13 was achieved by treatment of the ester 5 with hydrazine to afford the hydrazide intermediate 7 followed by cyclization with triethyl orthoacetate [17]. In a direct one-pot reaction, primary amide 4 was converted with triazidochlorosilane into the 5-substituted tetrazole intermediate 8 [18]. Adapting the protocol of Begtrup and Larsen [19], compound 8 was methylated with methyl iodide in methanolic potassium hydroxide. The resulting mixture of isomeric 1-methyl- and 2-methyl-tetrazole derivatives was separated by column chromatography and showed a product ratio of 4:1 in favor of 14. The 1H- and 13C-NMR data for 14 were in full agreement with literature data [19,20,22]. Finally, proceeding from the different heterocyclic derivatives 9-14, methyl ether cleavage with BBr3 in dichloromethane provided the corresponding phenols 15-20 in high yields [21].
2.2. Biological Activity
The 1,3-oxazole derivative 15 and its bioisoster and the sulfur-containing 1,3-thiaziole derivative 16 exhibited low antimicrobial activity (MIC 200 μg/mL) compared to the reference substances ofloxacin and ketoconazole. Major improvement in antimicrobial activity was obtained with the next three compounds, the methyloxadiazole derivatives 17-19. Within this homogenous series, the oxadiazoles differ from each other solely in the position of the three heteroatoms of the five-membered aromatic ring with regard to the location of the methyl or 4-hydroxyphenyl moiety. The 1,2,4-oxadiazole derivative 17 having a 4-hydroxyphenyl at the 3-position of 5-methyl-1,2,4-oxadiazole exhibited a MIC of 25 μg/mL against S. aureus and A. niger, whereas 18 having the 4-hydroxyphenyl at the 5-position of 3-methyl-1,2,4-oxadiazole exhibited significant antimicrobial activity (MIC 25 μg/mL) against E. coli, and A. niger. The bioisosteric 5-methyl-1,3,4-oxadiazole 19 exhibited promising antibacterial activity (MIC 25 μg/mL) against all tested culture strains, indicating that changes in the relative positions of the heteroatoms inside the heterocycle rings influences the antimicrobial activity. Since a carboxylic acid group can bioisosterically be replaced by a tetrazole, the next compound 20 could be interpreted as a potential bioisoster of the corresponding benzoic acid methyl ester derivative, yet the 2-methyl-2H-tetrazole derivative 20 failed to exhibit high antimicrobial activity (MIC 200 μg/mL) against S. aureus, E. coli or A. niger (Table 1).
Table 1.
Antimicrobial activity of the title compounds.
| MIC µg/mL | ||||
|---|---|---|---|---|
| Compound | R | S. aureus | E. coli | A. niger |
| 15 | 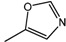 |
200 | 200 | 200 |
| 16 | 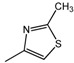 |
150 | 200 | 150 |
| 17 | 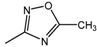 |
25 | 50 | 25 |
| 18 | 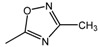 |
50 | 25 | 25 |
| 19 | 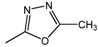 |
25 | 25 | 25 |
| 20 | 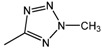 |
200 | 200 | 200 |
| Ofloxacin | 10 | 12.5 | -- | |
| Ketoconazole | -- | -- | 12.5 | |
3. Experimental
3.1. General Procedures
Melting points are uncorrected and were determined in open capillaries in a Büchi 512 Dr. Tottoli apparatus. 1H-NMR spectra were recorded on a Bruker WC 300 spectrometer with tetramethylsilane (TMS) as internal standard. Chemical shifts are reported in ppm downfield from internal tetramethylsilane as reference. 1H-NMR signals are reported in order: multiplicity (s, single; d, doublet; t, triplet; m, multiplet; *, exchangeable by D2O), number of protons, and approximate coupling constants expressed in Hertz. For compound 20, a 13C-NMR spectrum (100 MHz) was recorded on a Bruker DPX 400 Avance instrument, and chemical shifts that distinguish whether a methyl group is placed at the 1- or 2-position of the tetrazole moiety are reported in ppm downfield from internal TMS used as reference. Elemental analyses were performed on Perkin-Elmer 240B and 240C instruments. Analyses (C, H, N) indicated by the symbols of elements were within ±0.4% of the theoretical values. Chromatographic separations were done using a Chromatotron Model 7924 (Harrison Research) with 4 mm layers of silica gel 60 PF containing gypsum (Merck). EI-mass spectra were recorded using Finnigan MAT CH7A (70 eV), Finnigan MAT 711 (80 eV), or Kratos MS 25 RF (70 eV) instruments. +FAB-MS spectra were recorded on Finnigan MAT CH5DF instrument (xenon, DMSO)/glycerol). The MS spectra (data not shown) corresponded in all cases with the structures of the compounds depicted in the reaction schemes. The following abbreviations are used: Benz, benzoxathiazolyl; EtOH, ethanol; Et2O, diethyl ether; MeOH, methanol; Me2SO, dimethyl sulfoxide; Ox, oxadiazolyl; Ph, phenyl; Tet, tetrazolyl; Th, thiazolyl; THF, tetrahydrofuran.
Methyl 4-(1,3-oxazol-5-yl)phenyl ether (9): p-Toluenesulfonylmethyl isocyanide (0.27 g, 1,4 mmol) and 1 (0.19 g, 1.4 mmol) were dissolved in dry MeOH (30 mL). Next K2CO3 (0.19 g, 1.4 mmol) was added slowly over 30 min. After refluxing for 18 h the solvent was concentrated under vacuum and the residual aqueous layer was extracted with CH2Cl2. The organic extracts were combined, dried, and concentrated under reduced pressure. The oily product was purified by column chromatography (eluent: CH2Cl2) and crystallized at room temperature: Yield 61%; mp 61-63 °C; 1H-NMR δ ppm = 3.83 (s, 3H, H3CO), 7.02 (d, J = 8.6 Hz, 2H, Ph-3,5H), 7.52 (s, 1H, Ox-4H), 7.64 (d, J = 8.6 Hz, 2H, Ph-2,6H), 8.36 (s, 1H, Ox-2H); EI-MS m/z (%) 176 (M+, 21).
Methyl 4-(2-methyl-1,3-thiazol-4-yl)phenyl ether (10): 4-Methoxyphenacyl bromide (2, 1.49 g, 6.5 mmol), thioacetamide (0.48 g, 6.5 mmol), and K2CO3 (1.20 g, 8.7 mmol) were dissolved in dry N,N-dimethylformamide (30 mL). The mixture was stirred for 2 h at 100-120 °C, cooled, and the solvent was removed under reduced pressure. The solid residue was recrystallized from EtOH. Yield 79%; mp 59-60 °C; 1H-NMR δ ppm = 2.70 (s, 3H, Th-CH3), 3.79 (s, 3H, H3CO), 6.99 (d, J = 8.7 Hz, 2H, Ph-3,5H), 7.74 (s, 1H, Th-5H), 7.87 (d, J = 8.7 Hz, 2H, Ph-2,6H); EI-MS m/z (%) 205 (M+, 100).
Methyl 4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl ether (11): p-Anisonitrile (3, 6.66 g, 50 mmol), hydroxylamine hydrochloride (6.95 g, 100 mmol), and K2CO3 (10.34 g, 75 mmol) in dry EtOH (80 mL) were heated under reflux for 18 h. The mixture was cooled, filtered, and concentrated under reduced pressure. The crystalline residue was suspended in MeOH, and the final product 6 was isolated by filtration and dried. Powdered molecular sieves (4 Ǻ, 4 g) were added to a solution of 6 (1.66 g, 10 mmol) in dry THF (60 mL). After stirring for 30 min NaH (suspended in mineral oil, ω = 60%, 0.22 g, 5.5 mmol) was added, and the mixture was heated for 45 min at 60 °C. After cooling acetic acid methyl ester (1.48 g, 20 mmol) dissolved in dry THF was dropwise. Subsequently, the mixture was refluxed for 6 h under anhydrous conditions. Cooling, filtration, and removal of the solvent under reduced pressure resulted in a solid residue 11 which was recrystallized from EtOH. Yield 83%; mp 61-62 °C; 1H-NMR δ ppm = 2.64 (s, 3H, Ox-CH3), 3.83 (s, 3H, H3CO), 7.11 (d, J = 8.8 Hz, 2H, Ph-3,5H), 7.92-7.95 (m, 2H, Ph-2,6H); FAB+-MS m/z (%) 191 (M+ + H+, 100).
Methyl 4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl ether (12): A solution of 4-methoxybenzamide (4, 1.01 g, 6.7 mmol) in N,N-dimethylacetamide dimethyl acetal (15 mL) was stirred for 2.5 h at 120 °C under N2. The solvent was removed under reduced pressure and hydroxylamine hydrochloride (0.65 g, 9.4 mmol) dissolved in an aqueous NaOH solution (9.14 mL, c = 1 mol/L) was added. After addition of dioxane (10 mL) and acetic acid (12.5 mL) the mixture was stirred for 1.5 h at room temperature. Subsequently, the mixture was heated for 3 h at 90 °C, cooled, and a saturated solution of K2CO3 in H2O was added. Then, the mixture was concentrated under vacuum and the residual aqueous layer was extracted with CH2Cl2. The organic extracts were combined, dried, and concentrated under reduced pressure. The oily product 12 was purified by column chromatography (eluent: CH2Cl2) and crystallized at 4 °C. Yield 66%; mp 59-61 °C; 1H-NMR δ ppm = 2.39 (s, 3H, Ox-CH3), 3.87 (s, 3H, H3CO), 7.15-7.18 (m, 2H, Ph-3,5H), 8.03 (d, J = 8.8 Hz, 2H, Ph-2,6H); EI-MS m/z (%) 190 (M+, 92).
Methyl 4-(5-methyl-1,3,4-oxadiazol-2-yl)phenyl ether (13): A mixture of 4-methoxybenzoic acid methyl ester (5, 4.99 g, 30 mmol) and hydrazine monohydrate (3.00 g, 60 mmol) was heated at 120 °C for 3 h. Cooling and dilution with H2O (10 mL) afforded a precipitate of compound 7 which was filtered, washed with H2O, and dried in vacuo. Triethyl orthoacetate (10 mL) and 7 (1.66 g, 10 mmol) were heated at 120 °C for 3 h. Excess orthoacetate was evaporated, and the residue heated for further 2 h at 140 °C. The reaction was diluted with H2O (10 mL), saturated with K2CO3, and extracted with Et2O. Evaporation of the dried extracts afforded 13 as an oil. Yield 66%; mp 88°C; 1H-NMR δ ppm = 2.56 (s, 3H, Ox-CH3), 3.85 (s, 3H, H3CO), 7.91 (d, J = 8.8 Hz, 2H, Ph-3,5H), 7.91 (d, J = 8.7 Hz, 2H, Ph-2,6H); EI-MS m/z (%) 190 (M+, 93).
Methyl 4-(2-methyl-2H-tetrazol-5-yl)phenyl ether (14): A suspension of silicon(IV) chloride (9.35 g, 55 mmol) and sodium azide (7.15 g, 110 mmol) in dry acetonitrile (20 mL) was stirred for 1 h at ambient temperature. 4-Methoxybenzamide (4, 4.00 g, 27 mmol) was added dropwise. After stirring under for 3 h, the reaction mixture was poured into ice-cooled solution of K2CO3 in H2O, filtered, and acidified with aqueous HCl. The precipitated product 8 was filtered, washed with H2O, and dried in vacuo. A solution of 8 (1.86 g, 10.6 mmol) and KOH (2.81 g, 50 mmol) in dry MeOH (30 mL) was mixed at -25 °C with iodomethane (4.56 g, 32.1 mmol) in dry MeOH (5 mL). The temperature was raised to 20 °C during 1 h and the mixture refluxed for an additional hour. The solvent was removed and the crude product 14 purified by column chromatography (eluent: CH2Cl2). Yield 60%; mp 56-57 °C; 1H-NMR δ ppm = 3.83 (s, 3H, H3CO), 4.40 (s, 3H, NCH3), 7.10-7.13 (m, 2H, Ph-3,5H), 7.97-8.00 (m, 2H, Ph-2,6H); 13C-NMR (CDCl3) δ 39.7 (NCH3), 55.8 (H3CO), 114.7 (Ph-2,6C), 120.4 (Ph-4C), 128.7 (Ph-3,5C), 161.7 (Ph-1C), 165.6 (Tet-5C); EI-MS m/z (%) 190 (M+, 21).
3.2. General Procedure for Ether Cleavage
A solution of the corresponding ether (4 mmol) in dry CH2Cl2 (20 mL) was cooled to −80 °C under exceeding −60 °C. The reaction mixture was then allowed to warm to room temperature and stirred for additional 72 h. Subsequently, the mixture was cooled to −80 °C and MeOH (25 mL) was added dropwise. The organic layer was removed from the mixture under reduced pressure. After addition of a saturated K2CO3 solution in H2O to the aqueous layer, the crude product precipitated. It was isolated by filtration and recrystallized from EtOH.
4-(1,3-Oxazol-5-yl)phenol (15): From 9. Yield 41%; mp 43-45 °C; 1H-NMR δ ppm = 7.02 (d, J = 8.6 Hz, 2H, Ph-3,5H), 7.52 (s, 1H, Ox-4H), 7.64 (d, J = 8.6 Hz, 2H, Ph-2,6H), 8.36 (s, 1H, Ox-2H), 9.55 (s*, 1H, OH); EI-MS m/z (%) 161 (M+, 21). Anal. (C9H7NO2): C, H, N calcd. 67.07, 4.38, 8.69; found. 67.05, 4.26, 8.56.
4-(2-Methyl-1,3-thiazol-4-yl)phenol (16): From 10. Yield 70%; mp 151 °C; 1H-NMR δ ppm = 2.69 (s, 3H, Th-CH3), 6.81 (d, J = 8.6 Hz, 2H, Ph-3,5H), 7.64 (s, 1H, Th-5H), 7.55 (d, J = 8.6 Hz, 2H, Ph-2,6H), 10.45 (s*, 1H, OH); EI-MS m/z (%) 191 (M+, 100). Anal. (C10H9NOS): C, H, N calcd. 62.80, 4.74, 7.32; found. 62.31, 4.52, 7.02.
4-(5-Methyl-1,2,4-oxadiazol-3-yl)phenol (17): From 11. Yield 84%; mp 189-191 °C; 1H-NMR δ ppm = 2.62 (s, 3H, CH3), 6.89-6.92 (m, 2H, Ph-3,5H), 7.92 (d, J = 8.7, 2H, Ph-2,6H), 10.55 (s*, 1H, OH); EI-MS m/z (%) 176 (M+, 100). Anal. (C9H8N2O2): C, H, N calcd. 61.36, 4.58, 15.90; found. 61.34, 4.49, 15.48.
4-(3-Methyl-1,2,4-oxadiazol-5-yl)phenol (18): From 12. Yield 83%; mp 186 °C; 1H-NMR δ ppm = 2.37 (s, 3H, CH3), 6.94-6.98 (m, 2H, Ph-3,5H), 7.92 (d, J = 8.7 Hz, 2H, Ph-2,6H), 10.49 (s*, 1H, OH); EI-MS m/z (%) 176 (M+, 69). Anal. (C9H8N2O2): C, H, N calcd. 61.36, 4.58, 15.50; found. 61.33, 4.47, 15.43.
4-(5-Methyl-1,3,4-oxadiazol-2-yl)phenol (19): From 13. yYield 55%; mp 236 °C; 1H-NMR δ ppm = 2.56 (s, 3H, CH3), 6.98 (d, J = 8.6 Hz, 2H, Ph-3,5H), 7.85 (d, J = 8.6 Hz, 2H, Ph-2,6H), 10.26 (s*, 1H, OH); EI-MS m/z (%) 176 (M+, 100). Anal. (C9H8N2O2): C, H, N calcd. 61.36, 4.58, 15.50; found. 61.33, 4.44, 15.43.
4-(2-Methyl-2H-tetrazol-5-yl)phenyl ether (20): From 14. Yield 95%; mp 210-212 °C; 1H NMR δ ppm = 4.38 (s, 3H, NCH3), 6.92 (d, J = 8.6 Hz, 2H, Ph-3,5H), 7.87 (d, J = 8.5 Hz 2H, Ph-2,6H), 9.96 (s*, 1H, OH); EI-MS m/z (%) 176 (M+, 22). Anal. (C8H8N4O): C, H, N calcd. 54.54, 4.58, 31.80; found. 54.49, 4.41, 31.78.
3.3. Antimicrobial Activity
The quantitative in vitro antimicrobial study was carried on Muller-Hinton agar (Hi-media) plates (37 °C, 24 h) by the agar diffusion cup plate method [23]. The compounds (200-25 μg/mL) were screened for antimicrobial activity against the bacterial strains Staphylococcus aureus ATCC 25923 (S. aureus) (Gram+ve) and Escherchia coli ATCC 35218 (E. coli) (Gram-ve). Antifungal activity was tested on Sabouraud dextrose agar (Hi-media) plates (26 °C, 48-72 h) by the cup plate method against Aspergillus niger A733 (A. niger) also at a concentration level of 200-25 μg/mL. Ofloxacin and ketoconazole were used as standards for comparison of antibacterial and antifungal activity under the similar conditions. DMF was used as a solvent control for both antibacterial and antifungal activities, and the results are presented in minimal inhibition concentration (MIC) values (μg/mL) in Table 1.
4. Conclusions
The new compounds 15-20 presented here clearly differ in their corresponding antimicrobial activity depending on the type of the heterocycle. In the course of this study, particularly the derivatives possessing methyloxadiazole moieties 17-19 were identified as possessing moderate antibacterial activity against methicillin-resistant S. aureus (Gram positive) and E. coli (Gram negative) bacteria and antifungal activity against A. niger. These results, combined with the potential benefits or at least differences in pharmacokinetics make the titled oxadiazole congeners not only interesting simplified leads for the further chemical optimization of this class but also potentially interesting for future scope to study their mechanism of action and would be worthy of additional structure-activity relationship investigation.
Acknowledgments
The authors are thankful to Sigurd Elz, Faculty of Chemistry and Pharmacy, University of Regensburg, Germany, for spectral analysis.
Conflict of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 15-20 are available from the authors.
References and Notes
- 1.Jumat S., Nadia S., Ayad H., Hiba I.E.Y. Synthesis and antibacterial activity of some new 1,3,4-oxadiazole and 1,3,4-thiadiazole derivatives. J. Appl. Sci. Res. 2010;6:866–870. [Google Scholar]
- 2.Yang G.F., Liu Z.M., Qing X.H. Synthesis of 5,7-dimethyl-2-(5-substituted-1,3,4-oxadiazole-2-yl)-methylenethio-1,2,4-triazolo[1,5-a]pyrimidines as potential fungicides. Chin. Chem. Lett. 2001;12:877–880. [Google Scholar]
- 3.Patel K., Chandran J.E., Shah R., Vijaya J., Sreenivasa G.M. Synthesis, characterization and anthelmintic activity (Perituma posthuma) of new oxadiazole incorporated with imidazole and pyrazole. Int. J. Pharm. Bio. Sci. 2010;1:1–13. [Google Scholar]
- 4.Dewangan D., Pandey A., Sivakumar T., Rajavel R., Dubey R.D. Synthesis of some novel 2,5- disubstituted 1,3,4-oxadiazole and its analgesic, anti-inflammatory, anti-bacterial and anti-tubercular activity. Int. J. Chem. Tech. Res. 2010;2:1397–1412. [Google Scholar]
- 5.Holla B.S., Poojary K.N., Bhat K.S., Ashok M., Poojary B. Synthesis and anticancer activity studies on some 2-choloro-1,4-bis-(5-substituted-1,3,4-oxadiazole-2-ylmethyleneoxy)phenylene derivatives. Indian J. Chem. 2005;44B:1669–1673. [Google Scholar]
- 6.El-Sayeda W.A., El-Essawyb F.A., Alib O.M., Nasr B.S., Abdalla M.M., Abdel-Rahman A.A.-H. Anti-HIV activity of new substituted 1,3,4-oxadiazole derivatives and their acyclic nucleoside analogues. Z. Naturforsch. 2009;64C:773–778. doi: 10.1515/znc-2009-11-1203. [DOI] [PubMed] [Google Scholar]
- 7.Cena C., Bertinaria M., Boschi D., Giorgis M., Gasco A. Use of the furoxan (1,2,5-oxadiazole 2-oxide) system in the design of new NO-donor antioxidant hybrids. ARKIVOC. 2006;7:301–309. [Google Scholar]
- 8.Husain A., Ajmal M. Synthesis of novel 1,3,4-oxadiazole derivatives and their biological properties. Acta Pharm. 2009;59:223–233. doi: 10.2478/v10007-009-0011-1. [DOI] [PubMed] [Google Scholar]
- 9.Frank P.V., Girish K.S., Kalluraya B. Solvent-free microwave-assisted synthesis of oxadiazoles containing imidazole moiety. J. Chem. Sci. 2007;119:41–46. doi: 10.1007/s12039-007-0007-7. [DOI] [Google Scholar]
- 10.Zarghi A., Hamedi S., Tootooni F., Amini B., Sharifi B., Faizi M., Tabatabai S.A., Shafiee A. Synthesis and pharmacological evaluation of new 2-substituted-5-{2-[(2-halobenzyl) thio)phenyl}-1,3,4-oxadiazoles as anticonvulsant agents. Sci. Pharm. 2008;76:185–201. doi: 10.3797/scipharm.0803-10. [DOI] [Google Scholar]
- 11.Muhi-eldeen Z., Juma’a G., Al-kaissi E., Nouri L. Antimicrobial activity of some new oxadiazole derivatives. J. Chem. 2008;3:233–243. [Google Scholar]
- 12.Hussain S., Sharma J., Amir M. Synthesis and antimicrobial activities of 1,2,4-triazole and 1,3,4-thiadiazole derivatives of 5-amino-2-hydroxybenzoic acid. E. J.Chem. 2008;5:963–968. doi: 10.1155/2008/924734. [DOI] [Google Scholar]
- 13.van Leusen A.M., Hoogenboom B.E., Siderius H. A novel and efficient synthesis of oxazoles from tosylmethylisocyanide and carbonyl compounds. Tetrahedron Lett. 1972;23:2369–2372. [Google Scholar]
- 14.Vermin G. In: General Synthesis Methods for Thiazole and Thiazolium Salts. Metzger J.V., editor. Vol. 34. Interscience; New York, NY, USA: 1979. pp. 165–335. [Google Scholar]
- 15.Swain C.J., Baker R., Kneen C., Moseley J., Saunders J., Seward E.M., Stevenson G., Beer M., Stanton J., Walting K. Novel 5-HT3 antagonists. Indole oxadiazoles. J. Med. Chem. 1991;34:140–151. doi: 10.1021/jm00105a021. [DOI] [PubMed] [Google Scholar]
- 16.Lin Y.I., Lang S.A., Jr., Lovell M.F., Perkinson N.A. New synthesis of 1,2,4-triazoles and 1,2,4-oxadiazoles. J. Org. Chem. 1979;44:4160–4164. doi: 10.1021/jo01337a031. [DOI] [Google Scholar]
- 17.Anisworth C.J. The condensation of aryl carboxylic acid hydrazides with orthoesters. J. Am. Chem. Soc. 1955;77:1148–1152. doi: 10.1021/ja01610a019. [DOI] [Google Scholar]
- 18.El-Ahl A.A.S., Elmorsy S.S., Elbeheery A.H., Amer F.A. A novel approach for the synthesis of 5-substituted tetrazole derivatives from primary amides in mild one-step method. Tetrahedron Lett. 1997;38:1257–1260. [Google Scholar]
- 19.Begtrup M., Larsen P. Alkylation, acetylation and silylation of azoles. Acta Chem.Scand. 1990;44:1050–1057. doi: 10.3891/acta.chem.scand.44-1050. [DOI] [Google Scholar]
- 20.Fraser R.R., Haque K.E. Nuclear magnetic resonance and mass spectral properties of 5-aryltetrazoles. Can.J.Chem. 1968;46:2855–2859. doi: 10.1139/v68-471. [DOI] [Google Scholar]
- 21.Mcomie J.F.W., Watts M.L., West D.E. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron. 1969;24:2289–2292. [Google Scholar]
- 22.Begtrup M. 13C-NMR spectra of phenyl-substituted azoles: A conformational study. Acta Chem. Scand. 1973;27:3101–3110. doi: 10.3891/acta.chem.scand.27-3101. [DOI] [Google Scholar]
- 23.Barry A.L. The Antimicrobial Susceptibility Test: Principle and Practices. Edited by Illus lea and Febiger; Philadelphia, PA, USA: 1976. p. 180. [Biol. Abstr. 1977, 64, 25183]. [Google Scholar]