

Abstract
People with diabetes mellitus have increased infection risk. With diabetes, urinary tract infection (UTI) is more common and has worse outcomes. Here, we investigate how diabetes and insulin resistance impact the kidney’s innate defenses and urine sterility. We report that type 2 diabetic mice have increased UTI risk. Moreover, insulin-resistant prediabetic mice have increased UTI susceptibility, independent of hyperglycemia or glucosuria. To identify how insulin resistance affects renal antimicrobial defenses, we genetically deleted the insulin receptor in the kidney’s collecting tubules and intercalated cells. Intercalated cells, located within collecting tubules, contribute to epithelial defenses by acidifying the urine and secreting antimicrobial peptides (AMPs) into the urinary stream. Collecting duct and intercalated cell–specific insulin receptor deletion did not impact urine acidification, suppressed downstream insulin-mediated targets and AMP expression, and increased UTI susceptibility. Specifically, insulin receptor–mediated signaling regulates AMPs, including lipocalin 2 and ribonuclease 4, via phosphatidylinositol-3-kinase signaling. These data suggest that insulin signaling plays a critical role in renal antibacterial defenses.
Keywords: Immunology, Nephrology
Keywords: Insulin signaling, UTI/pyelonephritis
Introduction
Diabetes mellitus (DM) has become a global epidemic. According to the International Diabetes Federation, more than 415 million people worldwide have DM, and this number is expected to increase to 642 million by 2040 (1). In the United States, nearly half of the adult population has prediabetes or DM (2). DM is associated with many complications, including increased infection risk. With DM, one of the most common sites of infection is the urinary tract (3). In people with DM, urinary tract infection (UTI) is 10 times more common and more likely to progress to pyelonephritis and/or cause acute kidney injury (4). Complicated UTIs, including emphysematous pyelonephritis, renal abscess, and necrotizing papillitis, are also more common. The risk of bacteremia is 4 times greater and mortality rates are 5 times greater in DM patients with a UTI than in non–DM patients (5, 6). With DM, UTIs from multidrug-resistant microbes are more common, and antibiotics are increasingly insufficient to prevent UTI sequelae (7). Because of the growing incidence of DM and the increased UTI risk in people with DM, DM-associated UTI management has a substantial burden on medical costs (3). Understanding how the body shields the urothelium from pathogens in diabetic and nondiabetic states is vital to enhancing these defenses and developing novel UTI therapies.
Insulin resistance is the primary metabolic abnormality leading to the development of prediabetes and type 2 DM (T2DM). Insulin resistance is often subclinical and defined by inefficient insulin receptor (IR) signaling. Although insulin resistance at the receptor level is not fully defined, it does involve aberrant post-IR signaling and/or an overall change in the number of functional receptors (8, 9). Targeted IR deletion in tissues involved in energy homeostasis, including the pancreas, liver, muscle, and adipose tissue, promotes insulin resistance in vivo (10–13).
In the kidney, insulin is involved in a number of homeostatic responses including gluconeogenesis, regulation of renal blood flow, electrolyte/mineral balance, and vascular resistance (14). The IR is expressed throughout the glomerulus, nephron, and collecting duct. The IR is a tetrameric protein consisting of 2 extracellular α-subunits and 2 transmembrane β-subunits. When in a complex with insulin, the IR undergoes a conformational change that activates the β-subunit’s kinase activity and initiates a cascade of phosphorylation events leading to downstream target activation, including the phosphatidylinositol-3-kinase (PI3K/AKT) signaling pathway. AKT, also referred to as protein kinase B, is a key downstream insulin-mediated effector. PI3K/AKT is required for diverse cellular processes — including cellular proliferation, metabolism, protein synthesis, and protein trafficking (14–16).
Recently, we demonstrated that insulin and insulin-mediated PI3K/AKT activation contribute to renal host defenses by inducing antimicrobial peptide (AMP) production (17). In the kidney, AMPs are primarily produced by intercalated cells (ICs). ICs, which are located in the kidney’s collecting tubules, contribute to epithelial antibacterial defenses by acidifying the urine to inhibit the survival of invading uropathogens and producing innate immune proteins such as pattern recognition receptors, inflammatory cytokines, bacteriostatic iron binding proteins, and bactericidal AMPs (18, 19). Mice with reduced IC numbers develop substantially higher urinary and bladder bacterial burden when infected with uropathogenic E. coli (UPEC) (20, 21). During an ascending UTI (i.e., pyelonephritis), UPEC invades the collecting tubules and attaches to the luminal surface of ICs (20, 22). Thus, ICs are ideally positioned to defend the kidney from UPEC and have a key role in maintaining urine sterility.
Because people with DM have an increased propensity to develop UTIs, we hypothesized that DM impacts the immune functions of the kidney’s collecting tubules and ICs. Moreover, we hypothesized that insulin has an essential role in collecting duct and IC defenses by regulating PI3K/AKT activity and downstream AMP production. To test these hypotheses, we utilized prediabetic, T2DM, and cell-specific IR knockout models to investigate the role of insulin and IR-mediated signaling on renal antibacterial defenses. In doing so, we identified a crucial role for IR-mediated signaling in innate IC defenses and urine sterility.
Results
Type 2 diabetes mellitus and prediabetes increase UTI risk.
To verify that T2DM increases UTI susceptibility, we transurethrally infected 7-week-old female T2DM db/db mice and nondiabetic db/+ mice with UPEC (Figure 1). T2DM was identified by hyperinsulinemia, marked hyperglycemia (≥250 mg/dl), and glucosuria (Table 1) (23). Compared with nondiabetic controls, T2DM db/db mice had suppressed renal and bladder β-IR expression and AKT phosphorylation (Figure 1A and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI98595DS1). At 24 hours postinfection (HPI), urinary and bladder bacterial titers were enumerated. T2DM db/db mice had significantly greater UPEC burden compared with controls (Figure 1B), demonstrating that UPEC susceptibility increases with noncontrolled, hyperglycemic T2DM.
Figure 1. Type 2 DM and prediabetes increase UPEC susceptibility.

(A) Representative Western blots probed for β-IR, pAKT (ser473), and GAPDH in db/+ and db/db mice. Each lane depicts kidney β-IR expression and AKT phosphorylation from separate mice. (B) Female db/+ and db/db mice were transurethrally inoculated with UPEC. At 24 hours after infection, urinary and bladder UPEC burdens were enumerated. Each point depicts bacterial burden in a separate mouse. The horizontal line indicates the geometric mean of each group. (C) Representative Western blots probed for β-IR, pAKT (ser473), and GAPDH from control and TALLYHO mice. Each lane depicts kidney β-IR expression and AKT phosphorylation from separate mice. (D) Urinary and bladder UPEC burdens in female control and TALLYHO mice 24 hours after infection. Asterisks denote significance for the indicated pairwise comparison (Mann-Whitney U test). *P < 0.05.
Table 1. Fasting biometric data in diabetic, prediabetic, and control mice.
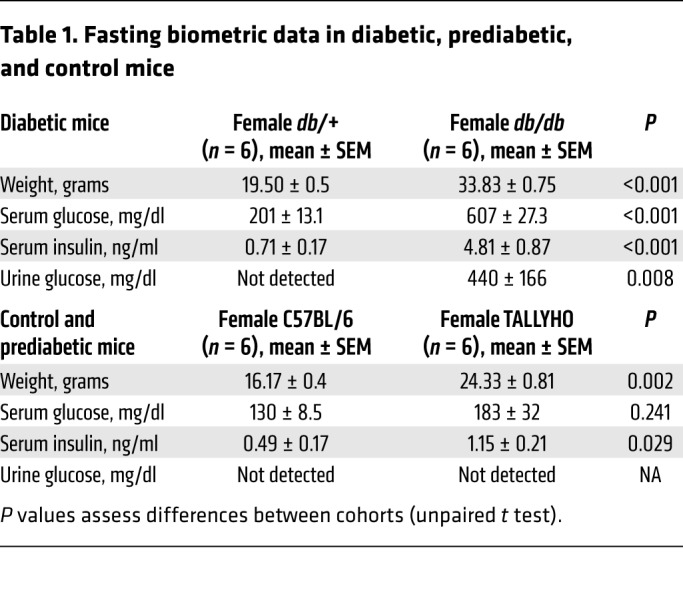
To assess the impact of insulin resistance on UTI risk, independent of hyperglycemia, we subjected 8-week-old C57BL/6J female mice and prediabetic TALLYHO mice to experimental UTI (24, 25). Prediabetes was defined by impaired glucose tolerance and/or increased insulin levels without marked hyperglycemia, glucosuria, or overt T2DM (Table 1) (23). Prediabetic TALLYHO mice also had suppressed renal β-IR expression and AKT activation (Figure 1C). Following experimental UTI, TALLYHO mice had a significantly greater urine UPEC burden compared with controls (Figure 1D). Although it did not reach statistical significance, bacterial bladder titers were also greater in TALLYHO mice compared with nondiabetic controls (Figure 1D). These results show that the hyperglycemic environment alone does not explain DM-associated UTI risk. Moreover, the results suggest that insulin resistance and IR-mediated signaling may play a key role in preventing UTI.
Generation and characterization of renal collecting duct and IC–specific IR knockout mice.
To evaluate the effects of insulin and IR-mediated signaling on urinary tract antibacterial defenses, we selectively deleted the IR in the kidney’s collecting duct and ICs. To delete the IR in the collecting ducts, mice homozygous for the floxed Insr gene (IRfl/fl), which possess loxP sites flanking exon 4, were crossed with kidney-specific (KSP) cadherin-Cre mice (Figure 2, A and B) (10, 26). Mice containing the floxed IR gene have been previously characterized (10). KSP-Cre+ IRfl/+ progeny were bred with IRfl/fl mice, generating KSP-Cre+ IRfl/fl offspring that lack the IR in the collecting ducts. To selectively delete the IR in the ICs, IRfl/fl mice were crossed with V-ATPase-Cre mice, which express Cre-recombinase under the IC-specific Atp6v1b1 promoter (Figure 2B) (27). Throughout this manuscript, mice in which the IR is selectively deleted are referenced as IRKO. Homozygous floxed littermates lacking the KSP-Cre or V-ATPase-Cre transgene served as controls (IRflox). IRKO mice exhibit normal phenotypes and development, normal renal histopathology (Supplemental Figures 2 and 3), normal serum chemistries and insulin levels, and no evidence of impaired glucose homeostasis (Table 2 and Supplemental Figure 4).
Figure 2. IR deletion in renal collecting ducts and ICs.

(A) The Insr lox allele before (IRflox) and after Cre-mediated exon 4 deletion (IRKO). The arrows labeled P2 and P3 indicate the PCR primer position. (B) Schematic of the breeding strategy used to delete the IR in renal collecting ducts and ICs using KSP-Cre and V-ATPase-Cre transgenic mice, respectively. (C) PCR shows a single 488-bp product indicative of the IR lox allele in kidneys and bladders of IRflox mice. In IRKO mouse kidneys, PCR generated the 488-bp product and a smaller 388-bp product. The 388-bp product confirms organ-specific Insr exon 4 deletion and the 488-bp product is attributed to other renal cell types in which Cre-recombination did not occur. Gapdh served as a loading control. Each lane identifies results from a separate mouse. (D) Relative Insr transcript expression in total kidney homogenate, dissected medulla, and FACS-isolated ICs from IRKO mice (white bars) compared with IRflox control mice (gray bars). Asterisks denote significance among genotypes (n = 5–6 mice/genotype; unpaired t test). *P < 0.05, **P < 0.01. (E) Representative Western blots probed for β-IR and GAPDH in dissected renal medulla and FACS-isolated ICs from IRflox and IRKO mice. (F) Western blots probed for pAKT (ser473) and GAPDH in dissected renal medulla from insulin-treated IRflox and IRKO mice. Each lane depicts kidney AKT phosphorylation from separate mice.
Table 2. Fasting biometric data in IRflox and IRKO mice.

PCR confirmed kidney-specific, Cre-mediated recombination in IRKO mice (Figure 2C). Real-time reverse transcription PCR (qRT-PCR) and Western blot showed reduced IR mRNA and protein expression in IRKO mouse kidneys relative to control littermates (Figure 2, D and E). We were unable to localize IR expression and/or deletion in the collecting duct with immunostaining. Thus, we incorporated the Cre-dependent tdTomato (tdT) fluorescent protein variant reporter strain into our breeding scheme (Figure 2B) (28). This approach allowed us to visualize Cre-mediated recombination and to isolate ICs by FACS (29). Western blot confirmed suppressed β-IR expression in FACS-isolated ICs (Figure 2E). Bladder IR expression was not affected (not shown). To confirm that IR deletion impacts downstream insulin-mediated effectors, IRKO and IRflox mice were starved and treated with increasing doses of insulin. Compared with controls, kidneys from insulin-treated IRKO mice showed suppressed PI3K/AKT activation (Figure 2F).
IR deletion in the kidney’s collecting ducts and ICs impairs host defense.
To evaluate if IR deletion affects UTI susceptibility, 6- to 8-week-old female IRflox and IRKO mice were transurethrally challenged with UPEC. At 24, 48, and 72 HPI, IRKO mice had greater UPEC urine and bladder burden (Figure 3). Similar burden differences were identified in the kidneys (Supplemental Figure 5). These results suggest that intact collecting duct IR-mediated signaling is critical for the kidney’s antibacterial defenses. Moreover, the results indicate that ICs play an essential role in maintaining urine sterility and that alterations in IC function impact lower urinary tract UPEC clearance.
Figure 3. IR deletion in renal collecting ducts and ICs increases UTI susceptibility.

Female IRKO mice and littermate IRflox controls were subjected to experimental UTI. At the indicated time points, urine was collected, bladders were harvested, and UPEC colonies were enumerated. Urinary (A and C) and bladder (B and D) UPEC burdens were combined from 3 independent experiments. The horizontal line indicates the geometric mean of each group. Asterisks denote significant P values for the indicated pairwise comparisons (Mann-Whitney U test). *P < 0.05, **P < 0.01. HPI, hours postinfection.
IR deletion does not impact IC populations or urine acidification.
Incorporation of the Cre-dependent tdT fluorescent protein reporter strain into our breeding scheme allowed us to FACS-isolate ICs and investigate the impact of IR deletion in an enriched cell population (Figure 4, A and B). Post FACS-sort enrichment checks demonstrated that tdT-expressing ICs were enriched from 1%–3% of total kidney cells to a 92%–97% pure population (Figure 4B) (29). IC enrichment was confirmed by IC-specific Atp6v1b1 transcript expression and V-ATPase β-1 subunit protein expression (Figure 4C) (30). To ensure that IR deletion does not impact IC populations, ICs were enumerated in IRKO mice expressing the tdT reporter (V-ATPase-Cre+ IRfl/fl tdT+/+). Compared with control V-ATPase-Cre+ tdT+/+ mice, IRKO mice express comparable IC numbers (Figure 4D).
Figure 4. IR deletion in the kidney’s ICs does not impact IC numbers or urine acidification.

(A) V-ATPase-Cre+ tdT+/+ mouse kidney labeled for aquaporin-2–positive principal cells (green), tdT+ ICs (red), and nuclei (blue). Original magnification, ×20 (left) and ×63 (right). Scale bars = 25 μm. (B) Representative FACS plots used to enrich tdT+ ICs with a sequential gating protocol. First, whole cells were gated (P1) and debris was excluded. From this population, only single cells were selected for further sorting by gating with the Trigger Pulse Width (P2). Single cells were then sorted by tdT expression. Rightmost plot is tdT+ postsort analysis showing enrichment from less than 2% in total kidney to greater than 90% after FACS. (C) Transcript expression of Atp6v1b1 and Slc5a2 in digested kidney, FACS-isolated tdT+ ICs, and FACS-isolated tdT– cells (n = 4 mice). Atp6v1b1 encodes the IC-specific β1 subunit of the V-ATPase hydrogen pump and Slc5a2 encodes the proximal tubule glucose transporter Sglt2. Asterisks denote significant P values for the pairwise comparisons (Kruskal-Wallis). Western blot confirms V-ATPase β1-subunit enrichment in tdT+ ICs (inset). (D) IC numbers in V-ATPase-Cre+ tdT+/+ (labeled tdT+) and IRKO kidneys enumerated by FACS (left of dashed line) and automated microscopic counting (right of dashed line). Graphs show the mean and SEM (n = 5–6 mice/genotype). (E) Ammonium chloride loading of IRflox and IRKO mice. Urine pH before (baseline) and after acid loading (NH4Cl). The horizontal line indicates the mean of each group. Asterisks indicate significant P values for the indicated pairwise comparison as determined by the paired 1-way ANOVA with Tukey’s test. *P < 0.05, **P < 0.01.
To determine whether IR deletion affects IC function via urine acidification, ammonium chloride was added to the drinking water of V-ATPase-Cre+ IRfl/fl tdT+/+ IRKO mice and IRflox littermate controls. The addition of ammonium chloride to the drinking water induces a metabolic acidosis as made evident by a decreased serum pH and subsequent urine acidification (31). Non–acid-loaded IRflox and IRKO mice (n = 8–10 mice/genotype) had similar serum pH (mean ± SEM: 7.45 ± 0.03 and 7.43 ± 0.04, respectively) and urine pH values (mean ± SEM: 5.78 ± 0.03 and 5.74 ± 0.05, respectively). After ammonium chloride loading, mean serum pH values decreased to 7.31 ± 0.03 in IRflox and 7.29 ± 0.02 in IRKO animals. IRflox and IRKO acidified their urine to a similar extent. Urine pH values decreased to 5.43 ± 0.07 in IRflox and 5.38 ± 0.09 in IRKO mice (Figure 4E). Together, these data suggest that IR deletion does not affect IC development or the ability of ICs to regulate urine acidification.
Preclinical studies suggest that ICs acidify the urine in response to transurethral UPEC challenge (20). Therefore, we collected serial urine samples from control and IRKO mice before and after experimental UTI. Following UPEC challenge, we did not identify significant changes in urine pH in IRflox or IRKO mice (Supplemental Figure 6).
IR deletion suppresses antimicrobial peptide expression.
To determine if IR deletion impacts IC AMP expression, we quantitated AMP transcript profiles in FACS-isolated ICs from V-ATPase-Cre+ tdT+/+ mice (Figure 5A). Next, we assessed relative AMP mRNA expression in isolated ICs from control and IRKO mice (Figure 5B). Compared with controls, ICs from IRKO mice had significantly decreased transcript expression of ribonuclease 4 (Rnase4) and lipocalin 2 (Lcn2, also referred to as NGAL). Western blot confirmed decreased RNase4 and Lcn2 peptide expression (Figure 5C). To ensure these expression changes were not related to the effects of tissue dissociation and/or FACS, we evaluated RNase4 and Lcn2 mRNA expression in kidney homogenates and measured urinary peptide concentrations. Rnase4 and Lcn2 transcripts were suppressed in noninfected and infected kidneys from V-ATPase-Cre IRKO mice (Figure 5D). Similarly, urinary RNase4 and Lcn2 peptide concentrations were suppressed in V-ATPase-Cre IRKO mice at baseline and after UPEC inoculation (Figure 5, E and F). Similar kidney mRNA and urinary peptide profiles were observed in KSP-Cre IRKO mice (Supplemental Figure 7). These findings suggest that IR-mediated signaling regulates AMP expression and that suppressed AMP production may increase UPEC susceptibility.
Figure 5. IR deletion in the kidney’s ICs suppresses antimicrobial peptide expression.

(A) Quantification of AMP mRNA expression in FACS-isolated ICs from noninfected V-ATPase-Cre+ tdT+/+ mice (n = 5). Transcript expression was determined by normalizing mRNA expression to serial dilutions of gene-specific plasmids. (B) Relative AMP mRNA expression in FACS-isolated ICs from noninfected V-ATPase-Cre+ tdT+/+ (striped bars) and IRKO mouse kidneys (white bars). Graphs show the mean and SEM (n = 6 mice/genotype). Asterisks denote significant P values for the pairwise comparisons (Mann-Whitney U test). (C) Representative Western blots on FACS-isolated ICs from V-ATPase-Cre+ tdT+/+ and IRKO mouse kidneys. (D) Relative total kidney AMP mRNA expression in IRflox (gray bars) and IRKO mice (white bars) at the indicated points postinfection (n = 7–8 mice/genotype). Relative transcript expression is normalized to IRflox expression at each time point. (E, F) Urinary AMP concentrations, normalized to urine creatinine (UCr), in IRflox (squares) and IRKO (triangles) mice at the indicated points postinfection. The horizontal line indicates the median of each group (n = 12–13 mice/genotype). (G) Isolated mouse urine from IRflox (squares) and IRKO (triangles) mice was incubated with and without anti–RNase4 antibody, anti–Lcn2 antibody, or an irrelevant antibody (IgG) prior to UPEC inoculation. The number of CFU was determined after a 90-minute incubation. The horizontal line indicates the median of each group (n = 7–9 mice/genotype). (E–G) Asterisks indicate significant P values for the indicated pairwise comparison (Kruskal-Wallis). (H) UPEC strains (UTI89 or MDR-UPEC) were incubated with serial dilutions of RNase4 peptide. After a 2-hour incubation, UPEC strains were plated on LB agar and colony counts were performed the following day. Repeat testing was performed on all bacterial isolates in triplicate. Data are mean CFU and SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that UPEC survival increases in urine with reduced AMP concentrations, we collected urine specimens from noninfected control and V-ATPase-Cre IRKO mice and inoculated them with UPEC. Compared with IRflox mice, UPEC survival increased in urine from isolated IRKO mice, suggesting that suppressed urinary AMP expression increases UPEC survival (Figure 5G). To validate the antimicrobial contributions of Lcn2 and RNase4, we added Lcn2 and RNase4 neutralizing antibodies to separate urine aliquots from IRKO and IRflox mice. In urine from IRflox controls, the addition of an antibody directed against RNase4 significantly suppressed UPEC killing. Although not reaching statistical significance, UPEC survival also increased in IRflox urine with the addition of an anti–Lcn2 antibody (Figure 5G). In contrast, adding Lcn2- or RNase4-neutralizing antibodies to IRKO urine did not impact UPEC survival. The addition of an irrelevant antibody had no effect. These data establish a role for Lcn2 and RNase4 in preventing UTI.
Ribonuclease 4 has antibacterial activity against UPEC.
Prior studies demonstrate that Lcn2 has antimicrobial activity against UPEC (20, 32). In contrast, RNase4’s antibacterial activity has not been investigated. To confirm RNase4’s antibacterial activity against UPEC, colony count reduction assays were performed by incubating multidrug-resistant UPEC (MDR-UPEC) and strain UTI89 with serial dilutions of recombinant RNase4 peptide. RNase4 exhibited potent bactericidal activity against both UPEC strains at micromolar concentrations (Figure 5H).
Insulin induces antimicrobial peptide expression via PI3K/AKT signaling.
To further define insulin’s effects on AMP expression, we cultured mouse outer medullary renal epithelial cells (mOMCD1), mouse inner medullary renal epithelial cells (mIMCD3), and human medullary renal epithelial cells in insulin-free media and treated them with recombinant insulin (100 nM). Insulin induced Lcn2 and RNase4 mRNA expression in all cell lines (Figure 6, A and B).
Figure 6. Insulin-mediated PI3K/AKT activation regulates AMP expression.

(A and B) Lcn2 and RNase4 mRNA expression in mOMCD1, mIMCD3, and human renal epithelial cells treated with vehicle control (gray bars) or insulin (white bars) for 24 hours. AMP expression is derived from 3 independent experiments performed in triplicate (n = 3). Columns represent mean and SEM. Asterisks denote significant P values for the indicated pairwise comparisons (Student’s t test). (C) Representative Western blots probed for pAKT (ser473), RNase4, and GAPDH from human renal epithelial cells treated with insulin with or without wortmannin (wort). (D) ELISA quantitated Lcn2 concentrations in renal epithelial cell culture media following insulin with or without wortmannin treatment (n = 3). Columns represent mean and SEM. Asterisks indicate significant P values for the indicated pairwise comparison as determined by 1-way ANOVA with Tukey’s test. (E) Relative Lcn2 and Rnase4 mRNA expression in noninfected kidneys from wortmannin (striped bars) or vehicle-treated (white bars) C57BL/6J mice (n = 6 mice/treatment). (F) One hour before UPEC challenge, female C57BL/6J mice were treated with intraperitoneal wortmannin (squares) or vehicle control (circles). At 6 HPI, urine was collected, bladders were harvested, and UPEC colonies were enumerated. The horizontal line indicates the geometric mean. (E, F) Asterisks denote significant P values for the pairwise comparisons (Mann-Whitney U test). (G) Representative Western blots confirm wortmannin suppression of renal AKT (ser473) phosphorylation. Each lane shows kidney pAKT activity from a separate mouse. *P < 0.05, **P < 0.01, ***P < 0.001.
Because IRKO mice have suppressed renal PI3K/AKT activity (Figure 2) and decreased AMP expression (Figure 5), we hypothesized that insulin induces AMP expression via PI3K/AKT. To confirm that insulin-induced Lcn2 and RNase4 expression are regulated by PI3K/AKT, human medullary epithelial cells were pretreated with wortmannin, a PI3K/AKT antagonist, and stimulated with insulin. Western blot showed insulin-induced RNase4 peptide production that was blocked by wortmannin (Figure 6C). Lcn2 was not routinely detected by Western blot in these cells. Thus, ELISA quantitated Lcn2 secretion into the culture media. Lcn2 concentrations increased nearly 7-fold after 24 hours of insulin treatment. Wortmannin blocked insulin-induced Lcn2 secretion (Figure 6D). These results verify that insulin induces renal epithelial Lcn2 and RNase4 expression through PI3K/AKT.
To confirm that PI3K/AKT regulates AMP expression in vivo, C57BL/6J mice were treated with intraperitoneal wortmannin or vehicle control. Compared with controls, kidneys from wortmannin-treated mice had decreased Lcn2 and Rnase4 transcript expression (Figure 6E). To assess the contributions of PI3K/AKT to urothelial defense, carrier and wortmannin-treated mice were subjected to UTI. After UPEC challenge, wortmannin-treated mice developed significantly greater UPEC burden (Figure 6F and Supplemental Figure 8). These findings confirm that PI3K/AKT regulates AMP expression and that it shields the urothelium from UPEC.
Youth with type 2 diabetes have suppressed urinary RNase4 and Lcn2 concentrations.
To investigate the effect of insulin resistance on human urinary AMP expression, we quantitated RNase4 and Lcn2 concentrations in urine specimens from healthy control and T2DM children and adolescents (Supplemental Table 1). ELISA demonstrated that T2DM youth had lower median urinary RNase4 and Lcn2 concentrations than healthy controls (Figure 7). Urinary RNase4 and Lcn2 concentrations negatively correlated with hemoglobin A1c (Spearman’s r = –0.362, P = 0.045 for RNase4; r = –0.359, P = 0.047 for Lcn2; Supplemental Figure 9). We did not observe a correlation between urinary AMP concentrations and the glomerular filtration rate or percentile of body mass index (not shown).
Figure 7. Urinary RNase4 and Lcn2 concentrations are suppressed in youth with type 2 diabetes.

(A) Urinary RNase4 and (B) Lcn2 concentrations, standardized to UCr, in healthy (n = 31) and T2DM children/adolescents (n = 31). Horizontal bars represent median urinary AMP/UCr concentrations. Asterisks denote significant P values for the indicated pairwise comparisons (Mann-Whitney U test). *P < 0.05.
Discussion
Although DM is recognized as a disease that increases infection susceptibility, the mechanisms that increase UTI risk are not established. Here, we investigate the impact of insulin and IR-mediated signaling on the kidney’s antibacterial defenses. Recent evidence suggests that epithelial cells in the kidney’s collecting duct activate host defenses to prevent lower urinary tract infection or an ascending pyelonephritis. Within the collecting tubules, ICs contribute to urine sterility by producing innate immune proteins, altering urine pH, and secreting bacteriostatic and bactericidal peptides and proteins into the urine (18, 22). This study provides new evidence that insulin and IR-mediated signaling have essential roles in regulating collecting duct and IC innate defenses. In doing so, our results identify unique mechanisms that may explain why certain patient populations, such as diabetics, have increased UTI risk. Our findings also provide evidence that the kidney’s collecting duct has a fundamental role in shielding the urothelium from UPEC.
Traditionally, DM-associated UTI has been attributed to glucosuria or hyperglycemia, which promote pathogen replication, facilitate UPEC binding to urothelial cells, inhibit neutrophil functions, and impair bladder emptying (6). In this regard, our data show that T2DM db/db mice, with marked hyperglycemia and glucosuria, have increased UTI susceptibility. Similarly, streptozotocin-induced T1DM hyperglycemic mice have increased UTI susceptibility, delayed UPEC clearance, and develop more severe infections compared with nondiabetic controls (33). Our data also demonstrate that insulin-resistant prediabetic TALLYHO mice have increased UPEC susceptibility, with urinary and bladder UPEC burden comparable to hyperglycemic db/db mice. These findings suggest that the hyperglycemic environment alone does not fully explain DM-associated UTI risk. They also indicate that insulin resistance may inhibit innate urothelial defenses and increase infection susceptibility.
Although the causes of insulin resistance are unclear, key mechanisms include disrupted IR signaling or decreased IR expression (8, 9). Published evidence shows that kidneys from insulin-resistant rats have suppressed medullary and collecting duct IR expression (34). Similarly, our data demonstrate that renal β-IR expression and downstream IR-mediated effectors, like PI3K/AKT, are suppressed in insulin-resistant db/db and TALLYHO mice. Suppressed β-IR expression and PI3K/AKT activity were also observed in db/db mouse bladders. Thus, decreased IR expression and suppressed insulin signaling could have a major impact on urothelial and renal epithelial innate immune functions, increasing UTI risk in prediabetes or T2DM.
To evaluate insulin’s effects on the kidney’s immune defenses, we deleted the IR in the collecting tubules and ICs. The IRKO model, which allows tissue-specific disruption of insulin signaling, provides a powerful approach to dissect insulin’s direct effects independent of lipotoxicity and abnormal glucose homeostasis (10, 13). Prior evidence suggests that insulin and IR signaling impact renal development, as explanted murine kidneys treated with a β-IR antisense oligodeoxynucleotide had a reduced nephron number and delayed growth (35). To determine whether IR deletion impacts IC development, we enumerated ICs in IRKO mice. IRKO mice have IC numbers comparable to those in controls. Histological lesions suggestive of abnormal renal organogenesis were not identified in IRKO mice. Similarly, when β-IR was selectively deleted in the kidney’s proximal tubule, thick ascending limb, or collecting duct, morphologic lesions indicative of abnormal kidney development were not identified. Instead, phenotypes were manifested by clinical or biochemical abnormalities (36–39).
Selective β-IR deletion in the kidney’s proximal tubule affects glucose homeostasis and gluconeogenesis (36, 37). Distal tubule IR deletion impacts sodium reabsorption and vascular resistance (38, 39). While IC β-IR deletion does not impact acid secretion or urine acidification in response to ammonium chloride loading, our data suggest that IR deletion suppresses the kidney’s innate defenses and increases UPEC susceptibility. Thus, intact insulin signaling is critical for activation of collecting duct epithelial defenses. Moreover, insulin resistance, in even a single cell type, affects host responses and markedly impacts phenotypic outcomes.
Although the collecting ducts’ contributions to host defense are only beginning to be recognized, evidence suggests that ICs participate in antimicrobial surveillance (18). In response to UPEC challenge, Paragas and colleagues demonstrated that ICs acidify the urine to inhibit bacterial replication (20). We did not observe urine acidification following experimental UTI. This occurrence may be a manifestation of the low UPEC titers we enumerated in mouse kidneys — perhaps a reflection of differences in the bacterial innoculum, UPEC strain, or experimental technique. In addition to acid secretion, additional evidence shows that ICs secrete AMPs into the urinary stream to combat invading pathogens. Some of these AMPs are produced at basal concentrations to prevent bacterial attachment whereas others are rapidly induced to facilitate pathogen clearance (18). To investigate how IR deletion impacts AMP expression, we profiled AMPs in IRKO mice. Compared with controls, Lcn2 and RNase4 mRNA and peptide expression were suppressed in kidneys, isolated ICs, and urine from IRKO mice.
Although only a limited number of AMPs have been characterized in the urinary tract, UTI risk increases when AMP production is disrupted (18, 40–42). When subjected to experimental UTI, Lcn2-KO mice develop higher bacterial bladder burden and are slower to eradicate UPEC than wild-type littermates (20, 32). Lcn2 binds the secreted siderophore enterochelin, which UPEC and other pathogens release to acquire iron. Without iron, UPEC is unable to replicate (20). Unlike Lcn2, RNase4’s antibacterial activity has not been previously defined. Here, we provide what we believe is the first evidence that RNase4, like other RNase A Superfamily members (i.e., RNase 6 and 7), has bactericidal activity against UPEC, including MDR-UPEC (43–45).
Additionally, we provide what we believe is the first data showing that RNase4 contributes to urine sterility while confirming that urinary Lcn2 inhibits UPEC survival. The addition of antibodies directed against RNase4 and Lcn2 to IRflox urine increased UPEC survival, linking UPEC survival to the antimicrobial activity of Lcn2 and RNase4. This phenomenon was more pronounced for RNase4 — perhaps reflecting RNase4’s potent antibacterial activity. UPEC survival was not impacted by the application of these antibodies to AMP-deficient IRKO urine. These data suggest that insulin regulates IC AMP expression, and that reduced urinary concentrations of Lcn2 and RNase4 predispose IRKO mice to UTI.
Prior evidence suggests that AMP expression is impacted by insulin bioavailability. Kidneys from T1DM and T2DM rodents have suppressed β-defensin 1 (BD-1) gene expression compared with nondiabetic controls (46, 47). In these rodents, DM-suppressed renal BD-1 mRNA and urinary BD-1 peptide expression are restored with insulin therapy. In other organ systems, insulin induces hepatic hepcidin production (48). Cohort studies show that people with T1DM have lower serum AMP concentrations of cathelicidin and BD-1 (49). Similarly, children with insulin-deficient T1DM have suppressed urinary RNase7 concentrations that increase with insulin therapy (17). Here, we collected random urine specimens from control and T2DM children and adolescents, and showed that T2DM youth have suppressed urinary RNase4 and Lcn2 concentrations. Urinary AMP concentrations negatively correlated with glycemic control. Collectively, these findings suggest that a prospective analysis of serial AMP concentrations is warranted in DM patients to assess how treatment, glycemic control, and long-standing DM impact AMP expression. These studies may identify novel AMP-based biomarkers for diabetic kidney disease and/or identify patients at risk for UTI or other DM-associated infections. High-risk patients with suppressed AMP profiles may benefit from close surveillance, UTI antibiotic prophylaxis, or ultimately novel strategies to activate insulin-signaling targets to boost endogenous AMP production.
Recently, we published in vitro data showing insulin induces RNase7 expression in human renal epithelial cells via PI3K/AKT signaling (17). Prior studies show that insulin upregulates Lcn2 production in omental adipose tissue explants. Insulin-induced Lcn2 production is inhibited by wortmannin (50). Here, we show that insulin induces RNase4 and Lcn2 expression via PI3K/AKT in vitro. Moreover, we demonstrate that pharmacologic PI3K/AKT inhibition suppresses in vivo AMP expression and decreases UPEC clearance. These results suggest that PI3K/AKT is an integral regulator of renal epithelial host defense and AMP expression. In addition, they suggest that decreased PI3K/AKT activation and downstream Lcn2 and RNase4 expression may increase UPEC susceptibility.
A major strength of the present study is the use of murine models to show that insulin resistance, independent of hyperglycemia/glucosuria, contributes to DM-associated UTI risk by suppressing renal AMP expression. The impact of insulin signaling on AMP expression is supported using in vitro culture models and human specimens. Still, there are limitations. We use PDM, T2DM, and genetically modified mice that are on nonrefluxing genetic backgrounds. As previously shown, it is difficult to establish pyelonephritis using nonrefluxing mice (51, 52). While our data demonstrate a role for collecting duct IR-mediated signaling in preventing lower UTI, we cannot definitively conclude that it has a role in preventing pyelonephritis. Additionally, while this study shows the key role for collecting duct IR-mediated signaling in UTI prevention, it does not provide mechanistic insight into the effects of insulin signaling in the lower urinary tract. As shown here and in our prior publication, IR is expressed in murine and human bladder (17). Finally, in regard to our human studies, youth in the T2DM cohort were receiving DM therapies. Consequently, this cross-sectional study cannot address the role of T2DM treatment and/or glycemic control on urinary AMP concentrations. Also, it does not assess whether suppressed urinary RNase4 or Lcn2 concentrations increase clinical UTI risk. A large prospective follow-up study may provide additional insight into these limitations.
To conclude, the present study identifies insulin and IR-mediated signaling as critical regulators of renal epithelial defenses. This study provides what we believe is the first evidence that IC-specific IR deletion and insulin resistance increase UTI risk by suppressing PI3K/AKT and downstream AMP expression. In doing so, they show that ICs protect the urothelium from uropathogens. Taken together, these findings elucidate a critical role for the IR-PI3K/AKT-AMP axis in IC antibacterial defenses and UTI prevention.
Methods
Mouse breeding and genotyping.
Mouse maintenance was performed in strict accordance with the Institutional Animal Care and Use Committee rules and regulations. Female TALLYHO, homozygous female db/db, and db/+ mice were purchased from Jackson Laboratory (stock 005314 and 00642). Because the diabetic trait in TALLYHO mice is polygenic and nondiabetic genetic controls do not exist, C57BL/6J mice were selected as controls as described in prior publications (24, 53).
To selectively delete the IR in the kidney’s collecting ducts or ICs, mice homozygous for the floxed Insr gene (Jackson Laboratory, stock 006955) were bred with KSP-Cre (Jackson Laboratory, stock 012237) or V-ATPase-Cre (provided by Raoul Nelson, University of Utah, Salt Lake City, UT, USA) mice, respectively, as outlined in Results. All mice were on a C57BL/6J genetic background. Animals were PCR genotyped according to previously published protocols and/or according to the Jackson Laboratory website (10, 27, 54). The primers used to detect Cre/LoxP recombination and the IR lox gene are outlined in Supplemental Table 2. All mice were maintained on standard rodent chow ad libitum with free access to water under controlled temperature and humidity with 12-hour light and dark cycles.
Mouse serum chemistries and insulin levels.
Fasting serum chemistries were measured using the Alfa Wassermann VetACE chemistry analyzer (Diamond Diagnostics) at The Ohio State University Comparative Pathology and Mouse Phenotyping Shared Resource. Plasma insulin levels were quantified using a mouse insulin ELISA (Crystal Chem Inc.). Serum glucose measurements were obtained using the AlphaTrak blood glucose monitoring system (Abbott Point of Care).
Experimental UTI.
Experimental UTI was performed as previously described (43, 55). Briefly, 6- to 8-week-old female mice were anesthetized and transurethrally inoculated with 107 CFU UPEC (strain UTI89) in 50 μl PBS. UTI89 was recovered from the urine of a patient with cystitis (56). At the indicated time points after inoculation, mice were reanesthetized and sacrificed via cervical dislocation. After sacrifice, organs were aseptically harvested and UPEC burdens were enumerated.
Mouse insulin dosing.
To investigate the effects of exogenous insulin therapy, IRflox and IRKO mice were starved for 6 hours. Mice were then intraperitoneally administered 0.5 U/kg insulin (Sigma-Aldrich), 1 U/kg insulin, or vehicle. One hour after injection, mice were anesthetized and sacrificed via cervical dislocation. After sacrifice, kidneys were aseptically harvested.
Mouse acid loading and pH measurements.
Metabolic acidosis was induced by providing IRKO and IRflox mice drinking water containing 0.28M ammonium chloride plus 2% sucrose (31). All animals had free access to standard rodent chow. Acid loading, water intake, food intake, and body weight were monitored daily. After 5 days of acid loading, urine and blood was collected. Blood pH was measured using an i-STAT G3+ cartridge (Abbott Point of Care). Urine was collected and urine pH was immediately measured using the Mettler Toledo SevenEasy pH meter.
Murine PI3K/AKT inhibition and experimental UTI.
Prior to UPEC inoculation, wortmannin (Sigma-Aldrich) was dissolved in DMSO (50–100 μl) and diluted 1:40 with sterile PBS. One hour before UPEC inoculation, mice were pretreated with intraperitoneal wortmannin (2 mg/mouse) or DMSO/PBS solution (vehicle). Experimental UTI, tissue isolation, and assessment of UPEC burden were performed as outlined above.
Fluorescence activated cell sorting.
tdT-expressing ICs were isolated by FACS as recently described with the following modifications (29). Kidneys were harvested and cell suspensions were prepared by TissueLyser II (Qiagen) homogenization followed by Collagenase IV (Thermo Fisher Scientific) digestion and straining through a 70-μm filter. ICs were isolated on a BD FACS Vantage with DiVa option cell sorter (Becton Dickinson) by sorting tdT-positive single events.
Immunofluorescent staining and IC counting.
Immunofluorescent staining in IRflox and IRKO mouse kidneys was performed as described (45). The kidney’s principal cells were labeled with a polyclonal goat aquaporin 2 antibody (catalog sc-9882, Santa Cruz Biotechnology) and tdT-positive ICs were labeled with a rabbit polyclonal RFP/tdTomato antibody (catalog 600-401-379, Rockland Immunochemicals Inc.). Alexa Fluor 488 AffiniPure Donkey Anti-Goat IgG (catalog 705-545-003, Jackson ImmunoResearch Laboratories) and Cy3 AffiniPure F(ab’)2 Fragment Donkey Anti-Rabbit IgG (catalog 711-166-152, Jackson ImmunoResearch Laboratories) served as the secondary antibodies. Using the BZ-9000 series fluorescence microscope cell counting program (Keyence), tdT-positive cells were enumerated in the cortex, outer medulla, and inner medulla and normalized to total cells, identified by nuclear DAPI staining.
Renal epithelial cells.
Mouse outer medullary cells (mOMCD1, provided by Snezana Petrovic, Wake Forest University, Winston Salem, NC, USA) and mouse inner medullary renal epithelial cells (mIMCD3, catalog CRL-2123, ATCC) were cultured in DMEM/F12 with 5% fetal bovine serum (57). Primary human renal epithelial cells were cultured in RenaLife culture medium with supplemental LifeFactors (Lifeline Cell Technology) at 37°C in 5% CO2 (17). Once reaching 80%–90% confluency, cells were cultured in insulin-free media overnight and then treated with the indicated drugs or vehicle for the specified times as previously described (17).
Quantitative real-time PCR.
RNA was isolated from murine tissues and cell culture lysates with TRIZol (Invitrogen). qRT-PCR was performed as previously described (43, 45). Primer sequences are shown in Supplemental Table 2. Relative expression changes were calculated using the 2–ΔΔCT method or normalized to gene-specific plasmid standards that were included with the indicated reactions (45).
SDS-PAGE Western blot and ELISA assay.
Protein extraction and Western blot on murine tissues and cell culture lysates were performed as previously described (17, 43). Primary antibodies included rabbit anti-RNase4 (catalog SAB1104141, Sigma-Aldrich), rabbit anti-Lcn2 (catalog HPA002695, Sigma-Aldrich), rabbit anti–β-IR (catalog sc-711, Santa Cruz), rabbit anti–phospho AKT (ser473; D9E; catalog 4060, Cell Signaling), and rabbit anti-GAPDH (catalog 2118S, Cell Signaling). Commercial ELISAs were performed to measure mouse RNase4 (MyBioSource), human RNase4 (Biomatik), and mouse and human Lcn2 (Abcam). Urinary RNase4 and Lcn2 concentrations were normalized to urine creatinine (Oxford Biomedical) (45).
Urine antimicrobial assay.
The antimicrobial activity of RNase4 and Lcn2 in murine urine was evaluated using colony count assays as previously described (17, 45). Briefly, 50 μl murine urine was inoculated with 5 μl 107 CFU/ml UPEC (strain UTI89). When indicated, 0.5 μg polyclonal anti–RNase4 antibody (catalog ab200717, Abcam), polyclonal anti–Lcn2 antibody (Abcam), or an IgG isotype antibody for an irrelevant peptide (catalog ab95316, Reg3a, Abcam) was added to each test isolate 30 minutes before UPEC inoculation. After 2 hours of incubation at 37°C, serial dilutions of the reaction mixtures were plated on lysogeny broth (LB) agar and the number of CFU was determined the following day.
Expression of recombinant mouse RNase4 peptide.
Using PCR, the murine Rnase4 mature peptide coding sequence was amplified from kidney cDNA and cloned into the E. coli expression vector pDEST17 (Invitrogen), which adds a 6-residue histidine tag at the N-terminus. Forward and reverse sequencing confirmed the absence of PCR errors. Full-length Rnase4-containing plasmids were transformed into E. coli BL21 AI cells (Invitrogen) to allow for l-arabinose inducible expression. Recombinant RNase4 protein was purified, refolded, and dialyzed as previously described (43, 44). Protein concentrations were determined by Bradford protein assay (Bio-Rad). The presence of the full-length RNase4 was visualized by SDS-PAGE followed by Coomassie staining.
Recombinant peptide antibacterial assay.
The antimicrobial activity of recombinant RNase4 was evaluated against UPEC using colony count reduction assays as previously described (44). UPEC (strain UTI89) or MDR-UPEC was incubated with serial dilutions of recombinant RNase4 for 2 hours at 37°C. The antimicrobial activity of RNase4 was analyzed by plating the incubation mixtures and determining CFU the following day. Repeat testing was performed on all bacterial isolates in triplicate. MDR-UPEC was isolated from a patient with cystitis and showed resistance to ampicillin, cefazolin, ceftriaxone, ceftazidime, ciprofloxacin, levofloxacin, and trimethoprim/sulfamethoxazole. MDR-UPEC showed intermediate resistance to cefepime, nitrofurantoin, and amoxacillin-clavulanate.
Human specimens.
Culture-negative urine specimens were collected from children and adolescents, aged 8–18 years, with a history of T2DM (58, 59). Patients were identified through the Nationwide Children’s Hospital diabetes or nephrology clinics. Exclusion criteria consisted of pregnancy, prisoners, institutionalized individuals, a known immunodeficiency, malignancy, immunosuppressant medications, indwelling hardware, comorbid urinary tract anomalies (including hydronephrosis, solitary kidney, pelvic kidney, renal dysplasia, cystic kidney disease, urinary tract obstruction, ureterocele, or neurogenic bladder), vesicoureteral reflux, chronic kidney disease stage 2–5, chronic antibiotic use, or antibiotic use within 1 week of enrollment. Healthy control patients were identified from these same locations and general pediatrics clinics.
Statistics.
Continuous differences among groups were evaluated for a normal distribution with the D’Agostino-Pearson Omnibus test, with normality defined as a P greater than 0.05. Comparisons on normally distributed data were performed by unpaired t test or by paired 2-tailed t test when appropriate; otherwise, the nonparametric Mann-Whitney U or Kruskal-Wallis test was used. Differences among groups with a P less than 0.05 were considered significant. Data from in vitro experiments were normally distributed and are presented as mean ± SEM. Treated and control cells were compared by 1-way ANOVA with the Tukey procedure. Data were analyzed using GraphPad Prism 6.0 (GraphPad Software).
Study approval.
All experimental procedures involving mice were approved by The Research Institute at Nationwide Children’s Hospital Institutional Laboratory Animal Care and Use Committee (protocol AR16-00043). For human subject research, The Nationwide Children’s Hospital Institutional Review Board approved this study (IRB14-00376). Written informed consent was obtained from all patients. For subjects younger than 18 years of age, written parental/guardian consent and patient assent were obtained.
Author contributions
JDS supervised the project and contributed to the design and interpretation of all experiments. TE contributed to the design and interpretation of all experiments, and performed FACS-IC isolation and Western blot analysis. MJM characterized the IRKO mouse, and performed all in vivo experiments, qRT-PCR, immunostaining, ELISA, and urine assays. MJM, BL, and KB performed experimental UTI. ALS and KB designed and performed the mouse acid loading experiments. JM, BB, LS, and ALS contributed to the design of in vivo and/or in vitro studies. JM and KB managed the mouse colony and breeding. CJ purified recombinant RNase4 peptide and CM performed the peptide antimicrobial assays and collected human urine specimens. JDS, MJM, and TE wrote the manuscript with input from all coauthors.
Supplementary Material
Acknowledgments
We acknowledge Krista La Perle (The Ohio State University School of Veterinary Medicine) for reviewing the renal histopathology. The Ohio State University Comparative Pathology and Mouse Phenotyping Shared Resource is supported by the NIH National Cancer Institute (P30 CA016058). This work is supported by the American Society of Nephrology’s Norman Siegel Research Scholarship and the NIH National Institute of Diabetes and Digestive and Kidney Diseases Diabetes Complications Consortium Pilot and Feasibility Program (DK076169) and R01 DK114035 (to JDS). BB is supported by the NIH National Institute of Diabetes and Digestive and Kidney Diseases (K08 DK102594).
Version 1. 11/12/2018
Electronic publication
Version 2. 12/03/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(12):5634–5646.https://doi.org/10.1172/JCI98595.
See the related Commentary at Why are diabetics prone to kidney infections?.
Contributor Information
Tad Eichler, Email: tad.eichler@nationwidechildrens.org.
Kristin Bender, Email: kristin.bender@nationwidechildrens.org.
Jackie Metheny, Email: jd_metheny@hotmail.com.
Birong Li, Email: rollin.li@nationwidechildrens.org.
Claudia Mosquera, Email: Claudia.MosqueraVasquez@nationwidechildrens.org.
Cindy James, Email: james.456@osu.edu.
Laura Schwartz, Email: laura.schwartz@nationwidechildrens.org.
Brian Becknell, Email: brian.becknell2@nationwidechildrens.org.
John David Spencer, Email: john.spencer@nationwidechildrens.org.
References
- 1. IDF Diabetes Atlas. 7th ed. International Diabetes Federation. 2015. http://diabetesatlas.org/IDF_Diabetes_Atlas_8e_interactive_EN/ Accessed October 18, 2018.
- 2. National Diabetes Statistics Report, 2017. Centers for Disease Control Prevention. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf Accessed October 18, 2018.
- 3.Korbel L, Spencer JD. Diabetes mellitus and infection: an evaluation of hospital utilization and management costs in the United States. J Diabetes Complicat. 2015;29(2):192–195. doi: 10.1016/j.jdiacomp.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiu PF, Huang CH, Liou HH, Wu CL, Wang SC, Chang CC. Long-term renal outcomes of episodic urinary tract infection in diabetic patients. J Diabetes Complicat. 2013;27(1):41–43. doi: 10.1016/j.jdiacomp.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Muller LM, et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. 2005;41(3):281–288. doi: 10.1086/431587. [DOI] [PubMed] [Google Scholar]
- 6.Geerlings SE. Urinary tract infections in patients with diabetes mellitus: epidemiology, pathogenesis and treatment. Int J Antimicrob Agents. 2008;31 Suppl 1:S54–S57. doi: 10.1016/j.ijantimicag.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 7.Nitzan O, Elias M, Chazan B, Saliba W. Urinary tract infections in patients with type 2 diabetes mellitus: review of prevalence, diagnosis, and management. Diabetes Metab Syndr Obes. 2015;8:129–136. doi: 10.2147/DMSO.S51792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6(1):a009191. doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 10.Brüning JC, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2(5):559–569. doi: 10.1016/S1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 11.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329–339. doi: 10.1016/S0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 12.Blüher M, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3(1):25–38. doi: 10.1016/S1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- 13.Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6(1):87–97. doi: 10.1016/S1097-2765(05)00015-8. [DOI] [PubMed] [Google Scholar]
- 14.Hale LJ, Coward RJ. Insulin signalling to the kidney in health and disease. Clin Sci. 2013;124(6):351–370. doi: 10.1042/CS20120378. [DOI] [PubMed] [Google Scholar]
- 15.Butlen D, Vadrot S, Roseau S, Morel F. Insulin receptors along the rat nephron: [125I] insulin binding in microdissected glomeruli and tubules. Pflugers Arch. 1988;412(6):604–612. doi: 10.1007/BF00583761. [DOI] [PubMed] [Google Scholar]
- 16.Sechi LA, De Carli S, Bartoli E. In situ characterization of renal insulin receptors in the rat. J Recept Res. 1994;14(6-8):347–356. doi: 10.3109/10799899409101509. [DOI] [PubMed] [Google Scholar]
- 17.Eichler TE, et al. Insulin and the phosphatidylinositol 3-kinase signaling pathway regulate Ribonuclease 7 expression in the human urinary tract. Kidney Int. 2016;90(3):568–579. doi: 10.1016/j.kint.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becknell B, Schwaderer A, Hains DS, Spencer JD. Amplifying renal immunity: the role of antimicrobial peptides in pyelonephritis. Nat Rev Nephrol. 2015;11(11):642–655. doi: 10.1038/nrneph.2015.105. [DOI] [PubMed] [Google Scholar]
- 19.Chassin C, Tourneur E, Bens M, Vandewalle A. A role for collecting duct epithelial cells in renal antibacterial defences. Cell Microbiol. 2011;13(8):1107–1113. doi: 10.1111/j.1462-5822.2011.01614.x. [DOI] [PubMed] [Google Scholar]
- 20.Paragas N, et al. α-Intercalated cells defend the urinary system from bacterial infection. J Clin Invest. 2014;124(7):2963–2976. doi: 10.1172/JCI71630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hains DS, et al. Carbonic anhydrase 2 deficiency leads to increased pyelonephritis susceptibility. Am J Physiol Renal Physiol. 2014;307(7):F869–F880. doi: 10.1152/ajprenal.00344.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chassin C, et al. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol. 2006;177(7):4773–4784. doi: 10.4049/jimmunol.177.7.4773. [DOI] [PubMed] [Google Scholar]
- 23.Islam MS, Venkatesan V. Experimentally-induced animal models of prediabetes and insulin resistance: a review. Acta Pol Pharm. 2016;73(4):827–834. [PubMed] [Google Scholar]
- 24.Kim JH, et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J Endocrinol. 2006;191(2):437–446. doi: 10.1677/joe.1.06647. [DOI] [PubMed] [Google Scholar]
- 25.Kim JH, Stewart TP, Zhang W, Kim HY, Nishina PM, Naggert JK. Type 2 diabetes mouse model TallyHo carries an obesity gene on chromosome 6 that exaggerates dietary obesity. Physiol Genomics. 2005;22(2):171–181. doi: 10.1152/physiolgenomics.00197.2004. [DOI] [PubMed] [Google Scholar]
- 26.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13(7):1837–1846. doi: 10.1097/01.ASN.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- 27.Miller RL, et al. The V-ATPase B1-subunit promoter drives expression of Cre recombinase in intercalated cells of the kidney. Kidney Int. 2009;75(4):435–439. doi: 10.1038/ki.2008.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veeneman JM, de Jong PE, Huisman RM, Reijngoud DJ. Re: Adey et al. Reduced synthesis of muscle proteins in chronic renal failure. Am J Physiol Endocrinol Metab 278: E219-E225, 2000. Am J Physiol Endocrinol Metab. 2001;280(1):E197–E198. doi: 10.1152/ajpendo.2001.280.1.E197. [DOI] [PubMed] [Google Scholar]
- 30.Brown D, Hirsch S, Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature. 1988;331(6157):622–624. doi: 10.1038/331622a0. [DOI] [PubMed] [Google Scholar]
- 31.Nowik M, Kampik NB, Mihailova M, Eladari D, Wagner CA. Induction of metabolic acidosis with ammonium chloride (NH4Cl) in mice and rats--species differences and technical considerations. Cell Physiol Biochem. 2010;26(6):1059–1072. doi: 10.1159/000323984. [DOI] [PubMed] [Google Scholar]
- 32.Steigedal M, et al. Lipocalin 2 imparts selective pressure on bacterial growth in the bladder and is elevated in women with urinary tract infection. J Immunol. 2014;193(12):6081–6089. doi: 10.4049/jimmunol.1401528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosen DA, Hung CS, Kline KA, Hultgren SJ. Streptozocin-induced diabetic mouse model of urinary tract infection. Infect Immun. 2008;76(9):4290–4298. doi: 10.1128/IAI.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiwari S, Halagappa VK, Riazi S, Hu X, Ecelbarger CA. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol. 2007;18(10):2661–2671. doi: 10.1681/ASN.2006121410. [DOI] [PubMed] [Google Scholar]
- 35.Liu ZZ, Kumar A, Ota K, Wallner EI, Kanwar YS. Developmental regulation and the role of insulin and insulin receptor in metanephrogenesis. Proc Natl Acad Sci USA. 1997;94(13):6758–6763. doi: 10.1073/pnas.94.13.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiwari S, et al. Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. J Am Soc Nephrol. 2013;24(8):1209–1214. doi: 10.1681/ASN.2012060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandey G, et al. Reduced insulin receptor expression enhances proximal tubule gluconeogenesis. J Cell Biochem. 2017;118(2):276–285. doi: 10.1002/jcb.25632. [DOI] [PubMed] [Google Scholar]
- 38.Li L, et al. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am J Physiol Renal Physiol. 2013;304(3):F279–F288. doi: 10.1152/ajprenal.00161.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiwari S, et al. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc Natl Acad Sci U S A. 2008;105(17):6469–6474. doi: 10.1073/pnas.0711283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chromek M, et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12(6):636–641. doi: 10.1038/nm1407. [DOI] [PubMed] [Google Scholar]
- 41.Houamel D, et al. Hepcidin as a major component of renal antibacterial defenses against uropathogenic Escherichia coli. J Am Soc Nephrol. 2016;27(3):835–846. doi: 10.1681/ASN.2014101035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison G, Kilanowski F, Davidson D, Dorin J. Characterization of the mouse beta defensin 1, Defb1, mutant mouse model. Infect Immun. 2002;70(6):3053–3060. doi: 10.1128/IAI.70.6.3053-3060.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becknell B, et al. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int. 2015;87(1):151–161. doi: 10.1038/ki.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spencer JD, et al. Ribonuclease 7, an antimicrobial peptide upregulated during infection, contributes to microbial defense of the human urinary tract. Kidney Int. 2013;83(4):615–625. doi: 10.1038/ki.2012.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spencer JD, et al. Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int. 2011;80(2):174–180. doi: 10.1038/ki.2011.109. [DOI] [PubMed] [Google Scholar]
- 46.Froy O, Hananel A, Chapnik N, Madar Z. Differential effect of insulin treatment on decreased levels of beta-defensins and Toll-like receptors in diabetic rats. Mol Immunol. 2007;44(5):796–802. doi: 10.1016/j.molimm.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Hiratsuka T, et al. Structural analysis of human beta-defensin-1 and its significance in urinary tract infection. Nephron. 2000;85(1):34–40. doi: 10.1159/000045627. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, Li H, Jiang X, Shi W, Shen Z, Li M. Hepcidin is directly regulated by insulin and plays an important role in iron overload in streptozotocin-induced diabetic rats. Diabetes. 2014;63(5):1506–1518. doi: 10.2337/db13-1195. [DOI] [PubMed] [Google Scholar]
- 49.Brauner H, et al. Markers of innate immune activity in patients with type 1 and type 2 diabetes mellitus and the effect of the anti-oxidant coenzyme Q10 on inflammatory activity. Clin Exp Immunol. 2014;177(2):478–482. doi: 10.1111/cei.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan BK, et al. Ex vivo and in vivo regulation of lipocalin-2, a novel adipokine, by insulin. Diabetes Care. 2009;32(1):129–131. doi: 10.2337/dc08-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bowen SE, Watt CL, Murawski IJ, Gupta IR, Abraham SN. Interplay between vesicoureteric reflux and kidney infection in the development of reflux nephropathy in mice. Dis Model Mech. 2013;6(4):934–941. doi: 10.1242/dmm.011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun. 1998;66(6):2798–2802. doi: 10.1128/iai.66.6.2798-2802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JH, et al. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics. 2001;74(3):273–286. doi: 10.1006/geno.2001.6569. [DOI] [PubMed] [Google Scholar]
- 54.Shao X, Johnson JE, Richardson JA, Hiesberger T, Igarashi P. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13(7):1824–1836. doi: 10.1097/01.ASN.0000016443.50138.CD. [DOI] [PubMed] [Google Scholar]
- 55.Spencer JD, et al. Expression and significance of the HIP/PAP and RegIIIγ antimicrobial peptides during mammalian urinary tract infection. PLoS One. 2015;10(12):e0144024. doi: 10.1371/journal.pone.0144024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A. 2000;97(16):8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun X, et al. Deletion of the pH sensor GPR4 decreases renal acid excretion. J Am Soc Nephrol. 2010;21(10):1745–1755. doi: 10.1681/ASN.2009050477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.American Diabetes Association. 2. Classification and diagnosis of diabetes. Diabetes Care. 2017;40(Suppl 1):S11–S24. doi: 10.2337/dc17-S005. [DOI] [PubMed] [Google Scholar]
- 59.Copeland KC, et al. Management of newly diagnosed type 2 Diabetes Mellitus (T2DM) in children and adolescents. Pediatrics. 2013;131(2):364–382. doi: 10.1542/peds.2012-3494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.