

Abstract
Sugar- and lipid-derived aldehydes are reactive carbonyl species (RCS) frequently used as surrogate markers of oxidative stress in obesity. A pathogenic role for RCS in metabolic diseases of obesity remains controversial, however, partly because of their highly diffuse and broad reactivity and the lack of specific RCS-scavenging therapies. Naturally occurring histidine dipeptides (e.g., anserine and carnosine) show RCS reactivity, but their therapeutic potential in humans is limited by serum carnosinases. Here, we present the rational design, characterization, and pharmacological evaluation of carnosinol, i.e., (2S)-2-(3-amino propanoylamino)-3-(1H-imidazol-5-yl)propanol, a derivative of carnosine with high oral bioavailability that is resistant to carnosinases. Carnosinol displayed a suitable ADMET (absorption, distribution, metabolism, excretion, and toxicity) profile and was determined to have the greatest potency and selectivity toward α,β-unsaturated aldehydes (e.g., 4-hydroxynonenal, HNE, ACR) among all others reported thus far. In rodent models of diet-induced obesity and metabolic syndrome, carnosinol dose-dependently attenuated HNE adduct formation in liver and skeletal muscle, while simultaneously mitigating inflammation, dyslipidemia, insulin resistance, and steatohepatitis. These improvements in metabolic parameters with carnosinol were not due to changes in energy expenditure, physical activity, adiposity, or body weight. Collectively, our findings illustrate a pathogenic role for RCS in obesity-related metabolic disorders and provide validation for a promising new class of carbonyl-scavenging therapeutic compounds rationally derived from carnosine.
Keywords: Endocrinology, Metabolism
Keywords: Diabetes, Drug therapy, Obesity
Introduction
Overnutrition from fatty acids and complex carbohydrates is known to cause oxidative stress from multiple enzymatic and nonenzymatic sources due to the high caloric content and the prevalence of these macronutrients in the Western diet. A pathological role for oxidative stress in obesity has been clearly established through extensive clinical and experimental studies. Reactive sugar- and lipid-derived aldehydes (reactive carbonyl species [RCS]) are spontaneously formed during the preparation of high-fat/high-sugar–containing foods under high heat (1) and are formed in vivo as a byproduct of oxidative stress. These RCS alter proteins via the covalent modifications of cysteine, arginine, lysine, and histidine and form adducts with phospholipids and DNA. Glucose-derived oxoaldehydes such as methylglyoxal (MGO) accumulate in oxidative tissues and react with protein functional groups, forming advanced glycation end-products (AGEs) (2, 3). AGEs are a major cause of chronic inflammation, cardiovascular disease, diabetes, and even some cancers, all of which are associated with obesity (4–8). In addition to reducing sugars and sugar-derived breakdown products, lipid-derived aldehydes are another significant source of carbonyl stress in vivo. Individuals who consume diets rich in vegetable and corn oil have very high endogenous levels of n-6 polyunsaturated fatty acids (PUFAs). The α,β-unsaturated carbonyls derived from n-6 PUFA oxidation have particularly diverse biological effects. Of these, 4-hydroxynonenal (HNE) and acrolein (ACR) have been the most widely studied (9). These aldehydes rapidly form adducts with proteins and phospholipids (10), and chronic exposure to high levels of these aldehydes is toxic (11). The accumulation of HNE has been extensively documented in blood and tissue samples from obese/diabetic patients, typically as a marker of oxidative stress (12–16). However, emerging studies suggest that these reactive aldehydes are more than simply by-products of oxidative stress. Rather, the carbonyl-modifying activity imposed by these reactive species may have a distinct pathogenic role in obesity-related disorders such as insulin resistance, chronic inflammation and fibrosis, dyslipidemia, and liver disease (15, 17–23). Thus, novel compounds that mitigate the production or enhance the removal of RCS remain compelling therapies for cardiovascular and metabolic diseases associated with obesity.
l-carnosine is a naturally occurring dipeptide (β-alanyl-histidine), which, along with its analogs, is a potent endogenous scavenger of RCS and highly concentrated (mM) in muscle and nervous tissues (24). The utility of l-carnosine as a pharmacological agent has been demonstrated in rodent models of metabolic syndrome and cardiovascular disease (25–27) and by its widespread use as eyedrop therapy in patients with ocular diseases (28). Major obstacles exist with respect to the clinical applicability of l-carnosine as an oral drug therapy, however. The largest of these is that high serum and tissue carnosinase activity in humans abrogates the bioavailability of circulating carnosine by rapid hydrolysis of the peptide bond (24).
Here, we present the rational design, characterization, and pharmacological evaluation of carnosinol, i.e., (2S)-2-(3-amino propanoylamino)-3-(1H-imidazol-5-yl)propanol, a reduced derivative of l-carnosine that is impervious to metabolism by carnosinase. Carnosinol displayed selectivity for reaction with RCS in vitro and in vivo, oral bioavailability and long duration in vivo, and negligible toxicity in human cell cultures and animal models. In rodent models of diet-induced obesity, carnosinol dose-dependently reduced systemic carbonyl stress, normalized glycemic control and many inflammatory parameters, and mitigated steatohepatitis. Collectively, these findings illustrate a distinct pathological role of RCS in metabolic diseases of obesity and validate the use of a novel RCS-scavenging l-carnosine derivative to treat these diseases.
Results
Rational design of carnosinol
In addition to the common properties that characterize a drug-like molecule (i.e., chemical and metabolic stability, bioavailability, and safety), our a priori rationale was that an RCS-sequestering therapeutic compound should: (a) be stable in plasma; (b) effectively scavenge (i.e., trap) circulating RCS; and (c) be highly reactive and selective toward damaging RCS (29). l-carnosine fulfils some of the above-mentioned requirements (30). However, it lacks an important basic requirement to be a suitable RCS-sequestering agent, because it is unstable in the circulation as a result of the hydrolytic action of carnosinases.
Hence, the rational design of improved l-carnosine derivatives should be focused on molecules that, besides maintaining or even enhancing quenching activity and selectivity, are endowed with plasma stability and oral bioavailability. To this end, the ideal derivative should: (a) maintain or better optimize its quenching activity, at least toward HNE, while preserving its selectivity; (b) maintain the active transport by human H+/peptide cotransporter-1 (hPepT1); and (c) eliminate recognition by human serum carnosinase (30, 31). As depicted in Figure 1A, this can be pursued by modifying the carboxyl group, which (a) is not involved in the quenching mechanism, even though its complete deletion has a detrimental effect, as seen in carcinine; (b) has a crucial role in carnosinase-1 (CN1) binding (Figure 1B); and (c) has a marginal role in hPepT1 transport (Figure 1C). Reduction of the carboxyl group has only a modest impact on the PepT1 interaction (Figure 1C), since both the carboxyl and hydroxyl functions elicit comparable and (weak) H-bonds with surrounding backbone atoms, as also confirmed by very similar interaction energies (–19.056 vs. –17.934 kcal/mol). In contrast, the carnosine carboxyl group is engaged in a pivotal ion pair with Arg350 in CN1 (Figure 1B), and its reduction to the hydroxyl function has a dramatic impact on the stability of the CN1-carnosinol complex, as confirmed by the reported drop in the interaction energy (–22.770 vs. –14.862 kcal/mol).
Figure 1. In silico models and biochemistry of carnosinol metabolism.

(A) Structure-activity relationships for l-carnosine and comparison with carnosinol. (B) Putative complex between carnosinol and CN1, highlighting the missing ion pair with Arg350 when compared with the corresponding complex for l-carnosine. (C) Putative complex between carnosinol and hPepT1, revealing that the inserted hydroxyl group elicits H-bonds similar to those already observed for l-carnosine.
Given these modeling results, the carboxyl reduction to yield carnosinol should meet all the above requirements and should even enhance the quenching activity, since it optimizes some physicochemical parameters that influence the quenching mechanism (e.g., flexibility and lipophilicity). Besides a greater flexibility, physicochemical analysis reveals that carnosinol is also more lipophilic than carnosine, as seen in experimental log P and log D7.4 values, a property that would benefit the molecular engagement by lipophilic RCS (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI94307DS1). Furthermore, carboxyl reduction did not significantly affect the ionization constant or the chelating activity toward transition metal ions such as Cu2+, which are roughly comparable to those of carnosine (chelating activity of Cu2+ [pKCuHL] = 12.73, [pKCuL] = 5.49). Quantum mechanical simulations showed that carnosinol also possesses favorable stereoelectronic parameters (Supplemental Table 1).
Carnosinol is a selective and potent RCS-sequestering agent
We first performed in vitro testing of the sequestering activity of carnosinol toward the most widely studied RCS involved in oxidative-based diseases and belonging to the chemical classes of α,β-unsaturated aldehydes, i.e., 4-hydroxynonenal (HNE), ACR dialdehydes, i.e., malondialdehyde (MDA) and glyoxal (GO), and ketoaldehydes, i.e., MGO.
Sequestering of HNE.
The sequestering activity of carnosinol toward HNE and ACR was first evaluated and compared with l-carnosine and for other small molecules known to possess similar properties (32). We directly compared carnosinol reactivity toward ACR and HNE with the reactivity of l-carnosine toward these aldehydes. Both compounds effectively reduced free aldehyde levels via conjugate formation over time, but interestingly, carnosinol showed a more rapid depletion of both ACR and HNE during the incubation compared with l-carnosine (Supplemental Table 2). We then determined selectivity by comparing carnosinol reactivity with HNE and the biogenic aldehyde pyridoxal (Table 1). The data are reported as consumption percentages (Q% ± SD) of HNE after 24 hours and consider a quencher/RCS ratio of 1:1 for HNE and 10:1 for pyridoxal (30). Carnosinol was the most reactive compound with respect to l-carnosine and other well-known RCS-sequestering agents such as aminoguanidine and pyridoxamine. We found carnosinol to be less effective only by comparison with hydralazine, a commonly prescribed antihypertensive medication with nonselective RCS reactivity, as shown by its effects with pyridoxal. In contrast, we found that carnosinol did not react with pyridoxal, and hence it can be considered a selective RCS-sequestering agent, like the parent compound l-carnosine.
Table 1. Comparison of carnosinol reactivity with HNE and the biogenic aldehyde pyridoxal, reported as consumption percentages (Q% ± SD).

The mechanism of carnosinol reaction toward HNE was fully elucidated on the basis of the reaction products identified and characterized by mass spectrometry (MS) experiments (Supplemental Figure 1). The proposed reaction mechanism (Figure 2A) is similar to that already clarified for l-carnosine and based on the formation of an imine derivative (CI) that catalyses the Michael adduct (CII) (Figure 2B).
Figure 2. Carnosinol is a selective sequestering agent of HNE.

(A) Proposed reaction mechanism of carnosinol (m/z = 213) with HNE (MW = 156 kDa). Carnosinol, like l-carnosine, reacts with HNE through a 2-step mechanism. The reaction starts with the formation of a reversible Schiff base (an α,β-unsaturated imine, CI) to yield the macrocyclic adduct through an intramolecular Michael addition, which hydrolyzes to form the stable hemiacetal derivative CII. (B) Mass spectrum of the reaction mixture of carnosinol with HNE incubated for 24 hours at 37°C, characterized by the peaks at m/z = 213, attributed to the carnosinol; m/z = 351, attributed to the Schiff base (CI); and m/z = 369, attributed to the Michael adduct (CII).
The increased potency of carnosinol compared with that of l-carnosine can be explained by the favorable stereoelectronic parameters as predicted by quantum mechanical simulations (Supplemental Table 1) and also by the ability of the hydroxyl group to form a hemiacetal intermediate with HNE, which, in addition to the amino group of β-alanine, further catalyzes the formation of the Michael adduct.
The quenching ability of carnosinol to inhibit HNE induced protein covalent adducts, and its comparison with the known quenchers was then evaluated in a MS-based assay that was recently set up in our laboratory (33). The assay consists of determining the ability of the tested compounds to inhibit the formation of HNE-induced adducts on a target protein, human ubiquitin, whose carbonylation specifically involves Lys6 and His68. Shown in Figure 3 is the MS spectrum of ubiquitin incubated in the absence and presence of HNE, inducing the formation of an HNE Michael adduct characterized by a molecular weight (MW) shifted by 156 kDa with respect to native ubiquitin. Carnosinol dose-dependently inhibited ubiquitin carbonylation. The greater activity with respect to hydralazine when measured via HPLC as compared with MS can be easily explained by considering that hydralazine forms a reversible Schiff base with HNE, as already reported (30), that cannot compete with the formation of Michael adducts on ubiquitin. By contrast, the more stable carnosinol-HNE adduct is not shifted in the presence of ubiquitin; thus, hydralazine was less efficient when tested by MS. The percentage of inhibition of HNE-induced ubiquitin carbonylation for carnosinol and some selected reference compounds are also reported in Table 1. Note again that hydralazine depletes pyridoxal levels completely, a feature of this drug that undoubtedly contributes to the adverse risk profiles related to vitamin B deficiency.
Figure 3. Carnosinol inhibits HNE-induced ubiquitin carbonylation.

(A) MS spectrum of ubiquitin control sample focusing on the z = 11 multicharged ion peak at m/z = 779; (B) 11+ peaks obtained upon incubation of ubiquitin with HNE, corresponding to unmodified (m/z = 779) and HNE-modified (m/z = 793) forms of the protein. (C and D) 11+ peaks obtained upon incubation of ubiquitin with HNE and carnosinol at 2 different molar ratios: HNE/carnosinol 1:0.5 mol/mol (C) and HNE/carnosinol 1:1 mol/mol (D), showing reduced intensity for the peak corresponding to the modified protein (m/z = 793).
We then used both HPLC and MS to test carnosinol sequestering activity toward MGO and MDA (Supplemental Figures 2 and 3 and Table 2). We found carnosinol to be more effective as a MGO-sequestering agent than l-carnosine and of the same order of potency of well-established scavengers such as aminoguanidine and hydralazine. An adduct characterized by a methylglyoxal-lysine dimer –like (MOLD-like) structure was identified by MS as the major reaction product between carnosinol and MGO (Supplemental Figure 2). The sequestering activity toward MDA was found superimposable with respect to carnosine, and the N-propenal derivative of carnosinol was identified as the reaction product (Supplemental Figure 3).
Table 2. HPLC and MS testing of carnosinol sequestering activity toward MGO and MDA.
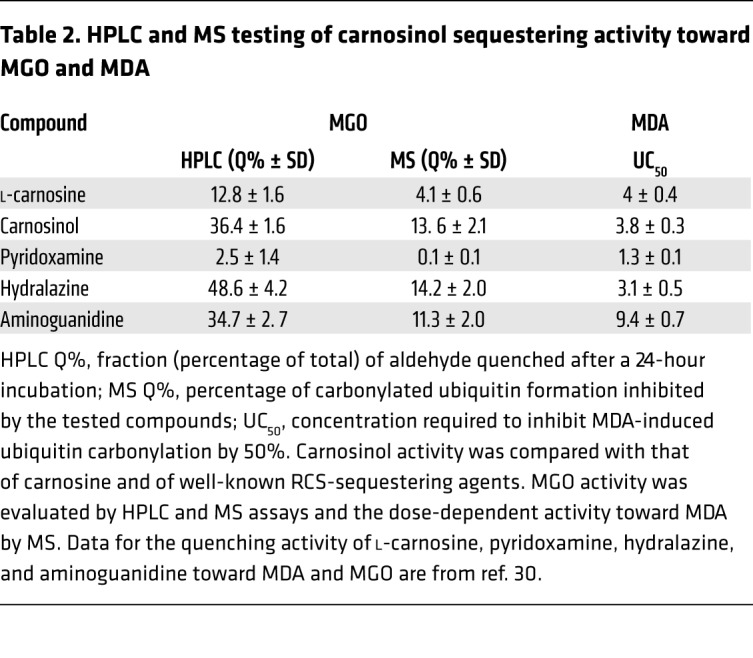
Taken together, the data indicate that carnosinol is selective and very reactive toward biogenic sugar- and lipid-derived aldehydes and the most potent RCS-sequestering agent among those commercially available.
ADMET studies
Carnosinol was evaluated in in vitro ADMET studies as well as in exploratory pharmacokinetic (PK) and toxicological studies (additional information is available in the supplemental material). Carnosinol was found to be stable in human serum for at least 60 minutes, as well as in rat and human liver preparations, thus indicating that it is not hydrolyzed by carnosinases. In vitro experiments indicated a good safety profile of the compound (no inhibition of the most important isoenzymes of cytochrome P450, no interaction with hERG K+ channel, no cell toxicity up to 100 μM concentration in a human hepatoma cell line). We then confirmed the safety profile of carnosinol in rats in both single-and repeat-dose oral toxicity studies. PK studies in rats showed a moderately good oral bioavailability that gave a significant exposure in plasma after oral administration at a dose of 45 mg/kg.
The data reported above demonstrate that carnosinol fulfils the optimization requirements we took into account in the rational design.
Dose-dependent effects of carnosinol on systemic inflammation and metabolic derangements in fructose-fed rats
Prior studies by our group and others have demonstrated that oral administration of l-carnosine and its derivatives effectively reduces atherogenesis (27, 34, 35), dyslipidemia, and renal dysfunction (25) in rodent models of cardiometabolic disease. To determine whether carnosinol retained or surpassed the efficacy of l-carnosine, we tested the effect of low-dose (10 mg/kg/day) and high-dose (45 mg/kg/day in the drinking water) carnosinol on cardiometabolic and inflammatory parameters using a short-term intervention (3 weeks) in rats fed a high-fructose (HF) diet (60% in the food pellet) (Figure 4A). An additional group of rats was treated for an equivalent period with a clinically relevant dose of rosiglitazone, the PPAR agonist widely prescribed for the treatment of metabolic syndrome and type 2 diabetes and which we recently determined to be a very effective therapy in this model (36). HF feeding led to significant gains in body weight (Figure 4B and Supplemental Table 3) compared with control-fed rats, and drug treatment did not cause significant differences in body weight gains. Total food and water intake was similar between the groups. Systolic blood pressure was substantially higher in fructose-fed rats, and this was dose-dependently reduced by carnosinol to levels comparable to those seen with rosiglitazone (Supplemental Table 3). This improvement in blood pressure with carnosinol mirrors the effect of l-carnosine supplementation on these parameters in similar models (25, 26).
Figure 4. Dose-dependent mitigation of inflammation and metabolic disease parameters by carnosinol in HF diet–fed rats.

(A) Study design for HF diet–induced metabolic disease in a rat model. The effect of either a HF diet alone or in combination with low-dose (10 mg/kg) or high-dose (45 mg/kg) carnosinol or rosiglitazone in the drinking water on overall changes in body weight (B) and the levels of serum AGEs (C), TNF-α (D), IL-6 (E), C-reactive protein (F), triglycerides (G), cholesterol (H), glucose (I), and insulin (J). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (K), along with liver triglycerides (L) and liver cholesterol (M) levels, are also shown. †P < 0.01 versus control diet; §P < 0.01 versus HF diet alone; *P < 0.01 versus HF plus carnosinol (10 mg/kg); 1-way ANOVA using a Newman-Keuls post hoc test for multiple comparisons (n = 6).
Next, we sought to examine the effect of carnosinol on markers of systemic oxidative and inflammatory stress. Notably, carnosinol dose-dependently reduced serum AGEs (Figure 4C), urinary 8-isoprostanes (Table 3), and proinflammatory cytokines (Figure 4, D–F) in fructose-fed rats. Importantly, the beneficial effects of carnosinol treatment on oxidative and inflammatory stress were paralleled by improvements in metabolic parameters. Carnosinol treatment led to dose-dependent reductions in serum triglycerides and cholesterol (Figure 4, G and H), improved glycemic control (Figure 4, I and J), and mitigated liver toxicity (Figure 4K) and steatosis (Table 3) induced by fructose feeding. All improvements in systemic metabolic and inflammatory parameters with carnosinol treatment were paralleled by significant reductions in plasma (Figure 4, L and M), kidney, and liver of HNE adducts (Supplemental Figure 4). Liver and renal fibrosis were not significantly affected by fructose feeding or drug treatment (data not shown), although the urinary creatinine clearance rate decreased with fructose feeding (Table 3), and this effect was blunted with carnosinol and rosiglitazone treatment.
Table 3. Hepatic and renal parameters in a HF-fed rat model.

Carnosinol improves glycemic control and muscle insulin sensitivity in mouse models of severe carbonyl stress and diet-induced obesity
Glutathione peroxidase 4 (GPx4), also named phospholipid hydroperoxide glutathione peroxidase, is the only known selenoenzyme that exclusively neutralizes lipid peroxides in membranes and lipoproteins (37). Homozygous null GPx4 mice die around gestational day 7, underscoring the critical role for this enzyme in development. GPx4-haploinsufficient (GPx4+/–) mice have approximately 40% of the WT GPx4 enzyme levels in oxidative tissues and are highly susceptible to environmental stressors due to enhanced lipid peroxidation and protein carbonylation (38, 39). Recently, we observed that GPx4+/– mice displayed exacerbated metabolic derangements and cardiomyopathy when fed an n-6 PUFA–enriched, high-fat/high-sucrose (HFHS) diet (15). These cardiometabolic disorders in GPx4+/– mice were accompanied by extensive carbonyl stress in liver and heart, and cardiac mitochondria in obese GPx4+/– mice had decreased fatty acid–supported respiration and increased ROS production compared with obese WT mice. These results are consistent with mitochondrial localization of GPx4 and its known role in protecting mitochondria from oxidative stress (40). Importantly, diabetic patients have higher levels of HNE adducts and lower GPx4 enzyme levels in their myocardial tissue compared with nondiabetic patients (15), a finding that corroborates previous observations concerning HNE adducts in diabetic patients (12–14, 16). Thus, GPx4+/– mice are ideal for the pharmacological evaluation of carnosinol, because much of the underlying stress-induced pathology of these mice is due to oxylipid-derived aldehydes.
To determine whether carnosinol is effective at mitigating obesity-related metabolic disorders, we administered the compound at the high dose (45 mg/kg/day in the drinking water) in a cohort of HFHS diet–induced obese WT mice. Given our previous findings in GPx4+/– mice on a HFHS diet, a cohort of GPx4+/– mice was used in parallel with WT mice to allow for assessment of carnosinol in a clinically relevant, translational model of severe carbonyl stress. Drug was administered to mice starting after 8 weeks on the HFHS diet, and a control group was fed normal chow for the duration of the study (Supplemental Figure 5A). As expected, the HFHS diet increased body weights and adiposity in both WT and GPx4+/– mice (Supplemental Figure 5, B and C), and carnosinol had no effect on these parameters, although fasting serum triglyceride and cholesterol levels were decreased in the carnosinol-treated mice (Supplemental Table 4). We detected no effect of carnosinol on whole-body energy expenditure (Supplemental Figure 6) or food or water intake (data not shown) in mice compared with those on a HFHS diet alone. Carnosinol treatment led to enhanced glucose disposal following oral glucose challenge in obese WT, but not GPx4+/–, mice (Figure 5, A–C). This improved glucose disposal may be attributed in part to increased skeletal muscle insulin sensitivity, as carnosinol normalized insulin-stimulated 2-deoxyglucose (2-DOG) uptake in extensor digitorum longus (EDL) to levels similar to those in control-fed lean mice (Figure 5D), although this effect was not seen in soleus (Figure 5E). We observed no improvement in insulin sensitivity with carnosinol in soleus tissue from obese WT mice, although we observed a modest improvement in obese GPx4+/– soleus (Figure 5F).
Figure 5. Therapeutic effect of carnosinol on metabolic homeostasis and carbonyl stress in mouse models of diet-induced obesity.

The effect of a HFHS diet with and without carnosinol treatment is shown for glucose tolerance (A–C, n = 8) and insulin sensitivity of EDL (D) and soleus muscle (E) tissue upon termination of the study for each group (n = 6). Representative immunoblots for HNE adducts in whole-tissue homogenates prepared from mixed gastrocnemius skeletal muscle (F) and pancreas (H) tissue (n = 3 mice per group), along with the corresponding densitometric analysis (G and I). †P < 0.01 versus control diet within each respective genotype. A 2-way ANOVA followed by Tukey’s multiple comparisons test was used to test for the main effect of the treatment within each genotype.
Reactive aldehyde derivatives of lipid peroxidation have recently come into focus as novel redox signaling agents that, paradoxically, have beneficial and pathological roles, depending on the concentration and tissues and cells affected (11, 41). Protein-HNE adducts are known to be increased in skeletal muscle of type 2 diabetic and obese/insulin-resistant patients, and these adducts are associated with the severity of insulin resistance (42). Experimental models have reported that lipid peroxidation in skeletal muscle blunts insulin signaling and glucose uptake in skeletal muscle via HNE adduct formation (43, 44). Lipid peroxidation and HNE adduct formation have also emerged as potential causal factors in hyperinsulinemia and the eventual loss of pancreatic β cell function in models of obesity/overnutrition (45–47), which is further evidence of a system-wide pathological role for reactive aldehydes in metabolic syndrome. Here, HNE adduct formation in mixed gastrocnemius skeletal muscle (Figure 5, F and G) and pancreas (Figure 5, H and I) increased with a HFHS diet, particularly in GPx4+/– mice. Carnosinol effectively mitigated the accumulation of these adducts in both WT and GPx4+/– mice on a HFHS diet, suggesting that this may be one aspect of the mechanism by which carnosinol improves systemic glycemic control in obese mice.
Liver inflammation and fibrosis are mitigated by carnosinol in mouse models of severe carbonyl stress– and diet-induced obesity
Nonalcoholic steatohepatitis (NASH) is among a cluster of obesity-related pathologies and is closely linked with insulin resistance. NASH is distinct from fatty liver disease, in that intralobular inflammation and fibrosis are present in addition to the steatosis (48). The fibrosis component of this disease has been specifically identified by numerous studies to be the most likely to predict adverse outcomes in patients (49). Thus, therapeutic strategies that specifically target liver inflammation and fibrosis in obese patients will be highly valued by clinicians (50). A number of studies have implicated RCS as having a causal role in NASH, due in large part to the known effect of RCS on activation of the proinflammatory receptor for AGEs (RAGE) pathway (22, 51). In the recent study from our group, obese GPx4+/– mice were found to have elevated RAGE expression in their heart tissue, and this corresponded to greater cardiac inflammatory cytokine expression and fibrosis (15). In the present study, RAGE expression in liver was unchanged in WT and GPx4+/– mice on a HFHS diet, but carnosinol significantly decreased RAGE expression in both groups (Figure 6A). Expression of the proinflammatory cytokines TNF-α and IL-6 was significantly higher in the livers of HFHS-fed mice, and carnosinol mitigated the expression of TNF-α, but not IL-6, in this tissue (Figure 6, B and C).
Figure 6. Carnosinol effect on liver inflammation and steatosis in mouse models of diet-induced obesity.

Expression of the proinflammatory genes RAGE (A), TNF-α (B), and IL-6 (C) in the livers of mice from each treatment group was determined by quantitative reverse transcription PCR (qRT-PCR). Representative images of liver histology showing H&E staining (D), oil red O staining of triglycerides (E), and Picrosirius red staining under polarized light for collagen/fibrosis (F) in mice from each treatment group. Original magnification, ×100; scale bars: 10 μm. Both insoluble (G) and soluble (H) forms of liver hydroxyproline were quantified, along with expression of collagen 1a1 (Col1a1), determined by qRT-PCR (I). Quantified data are shown as the mean ± SEM (n = 6/group). †P < 0.01 versus control diet for each respective genotype. A 2-way ANOVA followed by Tukey’s multiple comparisons test was used to test for the main effect of treatment within each genotype.
To further examine the effect of carnosinol on liver pathology in obesity, sections of liver tissue were fixed and stained with oil red O and Picrosirius red to label triglycerides and collagen, respectively. As in the previous study, we observed that the HFHS diet increased liver triglyceride content in WT and GPx4+/– mice, with substantially greater lipid deposition occurring in the obese GPx4+/– mice (Figure 6, D and E). Interestingly, we found that the increase in liver triglyceride deposition with a HFHS diet was not accompanied by changes in serum triglycerides (Supplemental Table 4), although this could be attributable to necropsy being performed while the mice were in a fasted state. Although total triglyceride content in liver did not significantly change with carnosinol (data not shown), we observed a shift from predominantly macro- to microvesicular steatosis with carnosinol treatment in obese WT and GPx4+/– mice (Figure 6E). Carnosinol also blunted fibrosis in the liver as indicated by Picrosirius red staining (Figure 6F). In order to determine whether a HFHS diet and/or carnosinol treatment alters collagen cross-linking in the tissue, we measured the ratio of insoluble to soluble hydroxyproline quantity in liver extracts. A HFHS diet led to an increase in the insoluble hydroxyproline fraction in WT and GPx4+/– livers (Figure 6G), and carnosinol normalized these levels. Indeed, carnosinol treatment was associated with significantly lower levels of collagen overall, including soluble hydroxyproline content (Figure 6H), regardless of diet or genotype. This decrease in collagen content caused by carnosinol treatment in obese WT and GPx4+/– mice does not appear to be due to decreased collagen gene expression, as we detected no effect of carnosinol on collagen-1 (Figure 6I) or collagen-4 (not shown) mRNA levels.
Pharmacodynamics and reactivity of carnosinol in mouse models of obesity and in human serum
In order to ascribe the beneficial effects of carnosinol in our obese mouse models to its aldehyde-scavenging capacity in vivo, we developed a high-resolution liquid chromatography-MS–based (LC-MS-based) approach to measure free carnosinol and carnosinol-aldehyde adducts in biological material. Pooled samples of tissue from skeletal muscle, pancreas, liver, kidney, and adipose tissue were analyzed using this method, and free carnosinol was found to be detectable only in liver, kidney, and adipose tissue (Supplemental Table 5). We found that carnosinol levels in liver and kidney were lower in the HFHS-fed GPx4+/– mice than in the WT mice.
Importantly, the lower concentration of free carnosinol in these mice corresponded with a higher concentration of carnosinol-ACR adducts in the liver as compared with concentrations in HFHS-fed WT mice treated with carnosinol (Figure 7A). Specifically, a signal at 269.16082 (i.e., the expected m/z value for the carnosinol-ACR Michael adduct) was detectable at the same retention time observed in a reference sample prepared by spiking an aliquot of carnosinol-ACR adduct in rat liver homogenate at a final concentration of 0.5 μM (Figure 7A, bottom). The narrow mass range considered for extracting the chromatograms in Figure 7A (i.e., 1 ppm mass tolerance) and the reproducible retention time if compared with a spiked sample are conclusive evidence of the formation of a carnosinol metabolite after supplementation. Tandem mass spectra of the signal at 269.16 from traces in Figure 7A were able to confirm the structure of the putative adduct (Supplemental Figure 7). No signal of adducts with other reactive carbonyl species (e.g., HNE, HHE, malondialdehyde) were detectable in control or carnosine-supplemented animals. This may be due to the instability of carnosinol-HNE adducts in liver homogenate, which we determined in separate experiments. Only 63.24% ± 3.47% of an initial amount of 5 μM carnosinol-HNE conjugate was detectable after a 2-hour incubation in liver homogenate, with an estimated half-life of 3 hours according to a first-order decay model. We did not observe this instability with the carnosinol-ACR adduct in liver homogenate.
Figure 7. Pharmacodynamics and aldehyde-scavenging capacity of carnosinol.

LC-MS analyses were performed on pooled, prepared liver homogenates from WT and GPx4+/– mice from each of the treatment groups. (A) Single-ion chromatograms of the carnosinol-ACR adduct (269.16082 m/z, ± 1 ppm). The bottom trace in A is the reference standard used in this analysis and was prepared by spiking rat liver homogenate with 0.5 μM carnosinol-ACR adduct. (B) LC-MS analysis showing the single-ion chromatogram of carnosinol-HNE adducts in human serum spiked with carnosinol and HNE (top), carnosinol only (middle), and serum only. Values shown in the green boxes denote the corresponding concentration of these adducts.
To further characterize carnosinol reactivity and the stability of aldehyde adducts, we tested the HNE-scavenging capacity of carnosinol in human serum by spiking the serum with a known concentration of preformed carnosinol and HNE as well as with carnosinol alone. Using our high-resolution LC-MS approach, we found that not only was carnosinol-HNE detectable, but unlike in tissues, the adduct was highly stable in human serum, with more than 85% of the carnosinol-HNE adduct remaining after a 2-hour incubation (Figure 7B and Supplemental Figure 8). Moreover, carnosinol can form adducts with trace amounts of HNE that are already present in human serum.
Discussion
An association between sugar- and lipid-derived RCS and obesity has been known for many years. The extent to which carbonyl stress plays a causal role in the metabolic disorders of obesity has remained unclear, however. In the present study, we sought to investigate the pathological role of RCS in obesity with a rational design and pharmacological evaluation of carnosinol, a chemical analog of l-carnosine with high oral bioavailability and a RCS-scavenging capacity. Following the design and characterization of carnosinol, we performed a comprehensive set of in vitro and in vivo studies profiling its effects across a range of experimental models, including rodent models of diet-induced obesity and metabolic syndrome. Our findings support the hypothesis that sugar- and lipid-derived RCS have a causal role in metabolic disorders associated with obesity. Moreover, we show that carnosinol represents a promising lead compound in a new class of RCS-scavenging agents derived from the histidyl dipeptide l-carnosine.
Recently, several important studies involving experimental and clinical models have illustrated the unique pathological role of lipid-derived aldehydes, particularly HNE, in the development of cardiometabolic disease. A study in healthy lean men determined that 1 week of a high-fat/high-carbohydrate (i.e., hypercaloric) diet induced systemic insulin resistance and glucose intolerance. Importantly, the authors showed that HNE modification of glucose transporter-4 (GLUT4) in adipocytes, on a residue near the glucose transport channel, played a role in diminished glucose uptake following the hypercaloric diet (20). Other studies have shown lipid peroxidation and HNE to be mediators of chronic inflammation and insulin resistance in adipose tissue (12, 19, 52) and skeletal muscle (42, 43) in obesity. From a mechanistic standpoint, the most important outstanding question involves the temporality and tissue dependency of RCS. Specifically, it is still unclear how long the oxidative stress must persist after the onset of caloric overload in order for RCS pathogenicity to emerge. Moreover, it is still not clear whether RCS are equally toxic and pathogenic in every tissue. Considering the critical role of the liver, skeletal muscle, and adipose tissue in glucose disposal and intermediary metabolism, these organs represent the most obvious target for examination. For example, a likely explanation for the decrease in serum and liver triglycerides with carnosinol treatment in the rodent models is that this effect is secondary to the improvement in insulin sensitivity and glycemic control. It is also noteworthy that in the carnosinol-treated mice, adipose tissue retains the highest concentration of the drug, behind liver and kidney tissue. Although adipose tissue was not a major focus of this study, much work over the past decade has documented the presence and pathogenicity of RCS in adipose tissue as it pertains to metabolic syndrome (12, 19, 52, 53). Thus, the effect of carnosinol on RCS in adipose tissue, and the resulting impact on metabolic parameters, may have played a significant role in the outcomes of the present study.
Though it is clear that lipid peroxidation and RCS do indeed have deleterious effects in the context of obesity, there are also complex time-, concentration-, and tissue-dependent factors to consider. Studies in pancreatic β cells have shown that short-term exposure to low levels of lipid peroxidation and subsequent 4-hydroxyalkenal formation stimulates an adaptive response mediated by PPARδ, which causes increased glucose-stimulated insulin secretion (46, 47). In a previous study, we found that numerous enzymes involved in redox buffering and fatty acid metabolism are enhanced in rat myocardium in parallel with lipid peroxidation following 12 weeks of a HFHS diet (54). Such a “hormetic” effect of oxidative stress has been documented by other groups using similar obese models (55). Certainly, much remains to be determined about the precise mechanisms and factors involved in lipid peroxidation and subsequent RCS in obesity.
A major determinant of lipid peroxidation now known to be intimately involved in regulating disease pathology is the expression and activity of the selenoenzyme GPx4. As 1 of only 3 antioxidant enzymes essential for development (56), GPx4 has recently been the target of intense scrutiny by investigators. In particular, a critical role for GPx4 in regulating ferroptosis and subsequent organ failure has been reported (57–61). Furthermore, genetic variants of gpx4 that result in diminished activity and/or enzyme content are associated with obesity and cardiovascular disease in humans (62–64). We previously reported that GPx4-deficient (GPx4+/–) male mice acquire severe insulin resistance, steatohepatitis, and cardiomyopathy on a HFHS diet and that diabetic patients have diminished GPx4 content and elevated HNE adduct levels in their heart tissue compared with levels in age-matched nondiabetic patients (15). In the present study, we used male GPx4+/– mice as a model to pharmacologically assess carnosinol in a context of enhanced lipid peroxidation–induced by obesity. As shown in Figure 5, GPx4+/– mice had greater levels of HNE adducts in skeletal muscle and pancreas following a HFHS diet compared with levels in WT mice. Carnosinol was effective at mitigating HNE adducts in these organs from both WT and GPx4+/– mice but was only fully effective at mitigating insulin sensitivity and glucose tolerance in WT mice. A reason for this may be that the dose of carnosinol used in this study was insufficient for a complete therapeutic effect in the GPx4+/– mice. Evidence supporting this conclusion is that there was a greater concentration of the carnosinol-ACR adduct in HFHS-fed GPx4+/– livers (Figure 7A), which corresponded with a lower concentration of free carnosinol in the livers and kidneys of HFHS-fed GPx4+/– mice (Supplemental Table 5). These findings are consistent with a greater aldehyde load in the GPx4+/– mice on a HFHS diet and a correspondingly greater demand for carnosinol in these mice as compared with WT mice on a HFHS diet. Taken together, we have uncovered compelling evidence that the therapeutic effect of carnosinol is mediated, at least in part, by its RCS-scavenging capacity.
Other compounds that scavenge RCS have shown significant therapeutic potential in preclinical studies using cardiovascular, renal, and metabolic disease models. Of all those tested to date, aminoguanidine and members of the B vitamin family (pyridoxamine, thiamine) have shown the most promising therapeutic effects (29, 65). However, in randomized, controlled trials these compounds have yielded largely disappointing results (with the exception of pyridoxamine), probably because of their promiscuous reactivity to all aldehydes (e.g., pyridoxal). Such off-target effects, particularly in the case of aminoguanidine (66), have made existing RCS-scavenging therapies unsuitable for mainstream clinical practice (67). These findings underscore the complexity of carbonyl biochemistry in physiological systems and illustrate that, in principle, agents that scavenge or block RCS have clinical potential. Thus, the development of novel RCS-scavenging therapies is warranted.
Recently, interventional studies based on a daily dose of l-carnosine (2 g/day for 12 weeks) in overweight and obese subjects resulted in the preservation of insulin sensitivity and insulin secretion and normalized glucose tolerance compared with the placebo group. Furthermore, l-carnosine supplementation reduced 2-hour insulin levels after oral glucose tolerance tests (OGTTs) in a subgroup of individuals with impaired glucose tolerance (68). Although l-carnosine was not detected in serum, it was easily detectable in the urine as a conjugate with ACR (69). The in vivo RCS-scavenging capacity of l-carnosine was further confirmed by 2 independent studies reporting the presence of covalent adducts between carnosine and lipid peroxidation by-products (e.g., ACR, HNE, HHE) in the urine of human volunteers (70, 71).
Given the findings of these translational studies, it would seem that orally supplemented l-carnosine, despite its rapid serum degradation by carnosinases, provides a promising therapeutic effect in obese subjects, one that may be linked to a RCS-scavenging mechanism. However, several studies indicate that the beneficial effect of l-carnosine is strictly associated with its serum stability and bioavailability. An allelic variant of CN1 that results in increased enzyme activity (and, consequently, lower serum l-carnosine levels) is linked to diabetic nephropathy (72). Transgenic db/db mice overexpressing human CN1 and having reduced serum carnosine levels exhibit higher fasting plasma glucose and HbA1c levels, to the extent that the glucosuria in these mice causes a significant reduction of body weight (73). It can be inferred from these studies that serum l-carnosine levels are directly linked to glycemic control and that human CN1 presents an attractive drug target for this patient population.
Our group envisioned 2 approaches that could circumvent the challenge posed by endogenous carnosinases: (a) inhibition of these enzymes during simultaneous oral l-carnosine therapy; and (b) design of stable carnosine peptide mimetics resistant to carnosinases. The former approach was not pursued because of a high potential for toxicity, as CN1 is critical for neurotransmitter production, histidine metabolism, and other vital functions. To address the latter approach, we previously synthesized and characterized the carnosinase-resistant enantiomer d-carnosine and found that this compound exhibited a significant RCS-scavenging effect in vitro and in vivo, but had poor intestinal absorption due to a low affinity for and transport by PepT1 (74). Other groups have successfully made carnosine derivatives that are resistant to carnosinase and initially showed therapeutic potential, but failed in preclinical testing largely because of decreased absorption (75, 76). An octylester derivative of d-carnosine showed enhanced intestinal absorption and therapeutic effects in a mouse model of cardiometabolic disease (34), but the translational applicability of this compound is low because of potency issues that likely stem from dosing limitations resulting from diminished absorption. Carnosinol is the most promising l-carnosine derivative that has been synthesized at this point. It is easily transportable by PepT1 and not metabolized by CN1 (Figure 1), it maintains the outstanding safety profile of l-carnosine, and it was determined to be more reactive toward HNE, MGO, and ACR compared with l-carnosine (Tables 1 and 2 and Supplemental Table 2). As such, it represents a promising lead compound for counteracting RCS, particularly lipid peroxidation–derived α,β-unsaturated aldehydes, which have recently come to the forefront as primary driving forces in chronic disease.
Conclusions and translational perspective.
The present study supports the hypothesis that sugar- and lipid-derived RCS have a causal role in metabolic disorders associated with obesity and further demonstrates that carnosinol represents a very promising lead compound in a new class of RCS-scavenging agents derived from the histidyl dipeptide l-carnosine.
Methods
A more detailed description of the materials and methods used in this study can be found in the supplemental material.
Materials and reagents.
Solvents for HPLC and LC-MS and all analytical-grade chemicals were purchased from Sigma-Aldrich. l-carnosine and carnosinol were gifts of Renato Canevotti and Stefania Gagliardia (Flamma S.p.A.). For molecular and biochemical endpoints, all reagents were from Sigma-Aldrich unless otherwise noted above.
ADMET studies.
ADMET studies were performed by Nikem Research following standard experimental protocols. PK studies were performed using fasted CD male rats obtained from Charles River Laboratories. Additional details and descriptions of the ADMET studies are provided in the supplemental material.
Rodent models of obesity and metabolic syndrome.
Both rat and mouse models of diet-induced obesity were used in this study. Eight-week-old male Sprague-Dawley rats (Harlan Laboratories) weighing 200 ± 20 g were used for this study. Rats were housed under constant environmental conditions and were fed standard laboratory rat chow or a 60% HF diet (Mucedola S.R.L.) and tap water ad libitum. In particular, the control diet contained 60% corn starch (carbohydrates), 20% casein (protein), 0.3% methionine, 5% lard (fat), 8% cellulose, 5% mineral mixture and 1% vitamin mixture, and zinc carbonate 0.004%. The fructose diet contained all the ingredients except corn starch, which was replaced by an equal quantity of fructose. Animals were acclimatized for a period of at least 7 days before use in the study. Subsequently, rats were randomly divided into 5 groups. The control group received a standard rat chow diet for 6 weeks, whereas the other 4 groups of mice were given a fructose-enriched diet for 6 weeks. Three weeks after starting the fructose diet, two groups were treated with carnosinol at two doses: 10 and 45 mg/kg/day. An additional group was treated with 10 mg/kg/day rosiglitazone (GlaxoSmithKlein), and the fifth group continued with the fructose-enriched diet alone (HF group). Rosiglitazone was orally administered to rats by gastric gavage during the last 3 weeks of the study, while carnosinol was dissolved in the water. During all the experiments, rats had ad libitum access to food and water.
For mouse models of diet-induced obesity, C57BL6/J female mice (The Jackson Laboratory) were crossed with male GPx4+/– mice, and the pups were genotyped by PCR using previously described primers (15). At 8 to 12 weeks of age, WT and GPx4+/– male age-matched littermates were randomly assigned to groups and individually housed. Mice were fed either a control (TD110367) or a HFHS (TD110365) diet from Harlan-Teklad Laboratories ad libitum for 25 weeks. The composition of this diet was a special formulation consisting of mixed saturated and n-6 PUFA (44.6% kcal/g fat), with a high-sucrose (34% kcal/g) content (54). After 8 weeks of the HFHS diet, half of the mice in the HFHS diet cohort (WT and GPx4+/–) were administered carnosinol (45 mg/kg/day) in their drinking water until study termination at 20 weeks. This dose was calculated on the basis of a series of days on which water consumption was meticulously recorded, and the dose of carnosinol was maintained and adjusted according to the body weight of each mouse throughout the duration of the study.
Statistics.
Data from our in vitro analyses are presented as the mean ± SD. Physiological and biochemical data for the rodent models are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism, version 7 (GraphPad Software). For the fructose-fed rat model, 1-way ANOVA was performed on continuous variables followed by a Newman-Keuls post hoc test comparing all groups, with a P value of less than 0.05 considered statistically significant. For the HFHS mouse model experiments, a 2-way ANOVA was used to assess genotype (WT vs. GPx4+/–) and treatment (control diet vs. HFHS vs. HFHS and carnosinol), following by a Tukey’s multiple comparisons test. This allowed us to compare the main effects of the treatment within each genotype and to ascertain whether there were interactions between the genotype and the treatment. A P value of less than 0.05 was considered statistically significant.
Study approval.
All in vivo studies were performed in accordance with Italian law (D.L.vo 116/92). All aspects of this study involving the care and use of laboratory animals received IACUC approval from the University of Milan in accordance with Italian law (D. L.vo 116/92) and with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) in the United States. Studies in rat models were approved by the animal ethics committee of the University of Milan and communicated to the Italian Ministry of Health, corresponding to article seven of D.L. 116/92. Studies using mouse models were performed with the approval of the IACUC of East Carolina University, in compliance with NIH guidelines for the care and use of laboratory animals.
Author contributions
GV, GA, and EJA made equivalent intellectual contributions to this manuscript and are the guarantors of all the data contained herein. GV (in silico modeling) and GA (bioanalysis and testing) were principally responsible for the design of carnosinol. EJA was principally responsible for the overall study design, experimental model development, and pharmacological testing of carnosinol and for writing the manuscript. LAK, KF, TBM, LR, EG, LC, M. Colzani, DDM, GR, and M. Carini designed and conducted experiments, analyzed data, and assisted with manuscript preparation. LR and EG designed and optimized the assay for LC-MS carnosinol-aldehyde adduct determination. RC and SG contributed to carnosinol synthesis.
Supplementary Material
Acknowledgments
Carnosinol was provided by Flamma S.p.A. This project was supported by funds from the Ministry of Education, Universities, and Research of Italy (MIUR) (PRIN 2009); the Regione Lombardia – MIUR (L.297–Art. 12/BioTech DM27909 and decree no. 6737 July, 2, 2009); and the NIH (R01HL122863 and R21AG057006, to EJA). The authors would like to thank Cherese Beatty (East Carolina University), who assisted with animal experiments and tissue histology. GV gratefully acknowledges Concetta De Stefano (University of Messina, Messina, Italy) for assistance in determining the chelation constants.
Version 1. 09/18/2018
In-Press Preview
Version 2. 10/22/2018
Electronic publication
Version 3. 12/03/2018
Print issue publication
Funding Statement
Portions of this project were funded by the Ministry of Education, University and Research (MIUR), Italy
Footnotes
Conflict of interest: GA, GV, and M. Carini of the University of Milan and Flamma S.p.A. are co-inventors on the patent “Amino alcohol derivatives and their therapeutic activities” (patent nos. EP 2519507B1 and US 8623900B2), which includes carnosinol. RC and SG are employees of Flamma S.p.A., the manufacturer of carnosinol.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(12):5280–5293.https://doi.org/10.1172/JCI94307.
See the related Commentary at Therapeutic potential of carbonyl-scavenging carnosine derivative in metabolic disorders.
Contributor Information
Ethan J. Anderson, Email: ethan-anderson@uiowa.edu.
Giulio Vistoli, Email: giulio.vistoli@unimi.it.
Lalage A. Katunga, Email: Lalage.a.katunga@health.slu.edu.
Katsuhiko Funai, Email: kfunai@health.utah.edu.
Luca Regazzoni, Email: luca.regazzoni@unimi.it.
T. Blake Monroe, Email: blake-monroe@uiowa.edu.
Ettore Gilardoni, Email: ettore.gilardoni@unimi.it.
Luca Cannizzaro, Email: luca.m.cannizzaro@gmail.com.
Mara Colzani, Email: mara.colzani@gmail.com.
Danilo De Maddis, Email: demaddis.danilo@gmail.com.
Giuseppe Rossoni, Email: giuseppe.rossoni@unimi.it.
Renato Canevotti, Email: r.canevotti@flamma.it.
Stefania Gagliardi, Email: sgagliardi57@gmail.com.
Marina Carini, Email: marina.carini@unimi.it.
Giancarlo Aldini, Email: giancarlo.aldini@unimi.it.
References
- 1.Uribarri J, et al. Dietary advanced glycation end products and their role in health and disease. Adv Nutr. 2015;6(4):461–473. doi: 10.3945/an.115.008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt AM, Yan SD, Stern DM. The dark side of glucose. Nat Med. 1995;1(10):1002–1004. doi: 10.1038/nm1095-1002. [DOI] [PubMed] [Google Scholar]
- 3.Cho SJ, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem. 2007;14(15):1653–1671. doi: 10.2174/092986707780830989. [DOI] [PubMed] [Google Scholar]
- 4.Palimeri S, Palioura E, Diamanti-Kandarakis E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: recommendations for dietary management. Diabetes Metab Syndr Obes. 2015;8:415–426. doi: 10.2147/DMSO.S63089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Puyvelde K, Mets T, Njemini R, Beyer I, Bautmans I. Effect of advanced glycation end product intake on inflammation and aging: a systematic review. Nutr Rev. 2014;72(10):638–650. doi: 10.1111/nure.12141. [DOI] [PubMed] [Google Scholar]
- 6.Hu H, Jiang H, Ren H, Hu X, Wang X, Han C. AGEs and chronic subclinical inflammation in diabetes: disorders of immune system. Diabetes Metab Res Rev. 2015;31(2):127–137. doi: 10.1002/dmrr.2560. [DOI] [PubMed] [Google Scholar]
- 7.Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol Metab Clin North Am. 2013;42(4):697–719. doi: 10.1016/j.ecl.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Turner DP. Advanced glycation end-products: a biological consequence of lifestyle contributing to cancer disparity. Cancer Res. 2015;75(10):1925–1929. doi: 10.1158/0008-5472.CAN-15-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11(1):81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 10.Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. 2009;47(5):469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 11.Negre-Salvayre A, et al. Pathological aspects of lipid peroxidation. Free Radic Res. 2010;44(10):1125–1171. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 12.Frohnert BI, et al. Increased adipose protein carbonylation in human obesity. Obesity (Silver Spring) 2011;19(9):1735–1741. doi: 10.1038/oby.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toyokuni S, et al. Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid Redox Signal. 2000;2(4):681–685. doi: 10.1089/ars.2000.2.4-681. [DOI] [PubMed] [Google Scholar]
- 14.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54(20):1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katunga LA, et al. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol Metab. 2015;4(6):493–506. doi: 10.1016/j.molmet.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell AP, et al. Lipid peroxidation in skeletal muscle of obese as compared with endurance-trained humans: a case of good vs. bad lipids? FEBS Lett. 2003;551(1-3):104–106. doi: 10.1016/S0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126(1):50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen G, et al. Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic Biol Med. 2013;65:978–987. doi: 10.1016/j.freeradbiomed.2013.08.163. [DOI] [PubMed] [Google Scholar]
- 19.Frohnert BI, Long EK, Hahn WS, Bernlohr DA. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes. 2014;63(1):89–100. doi: 10.2337/db13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boden G, et al. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7(304):304re7. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uribarri J, et al. Elevated serum advanced glycation endproducts in obese indicate risk for the metabolic syndrome: a link between healthy and unhealthy obesity? J Clin Endocrinol Metab. 2015;100(5):1957–1966. doi: 10.1210/jc.2014-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung C, et al. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J Hepatol. 2014;60(4):832–838. doi: 10.1016/j.jhep.2013.11.033. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, et al. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS One. 2013;8(8):e70663. doi: 10.1371/journal.pone.0070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boldyrev AA, Aldini G, Derave W. Physiology and pathophysiology of carnosine. Physiol Rev. 2013;93(4):1803–1845. doi: 10.1152/physrev.00039.2012. [DOI] [PubMed] [Google Scholar]
- 25.Aldini G, et al. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med. 2011;15(6):1339–1354. doi: 10.1111/j.1582-4934.2010.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai K, et al. Role of L-carnosine in the control of blood glucose, blood pressure, thermogenesis, and lipolysis by autonomic nerves in rats: involvement of the circadian clock and histamine. Amino Acids. 2012;43(1):97–109. doi: 10.1007/s00726-012-1251-9. [DOI] [PubMed] [Google Scholar]
- 27.Barski OA, et al. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2013;33(6):1162–1170. doi: 10.1161/ATVBAHA.112.300572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babizhayev MA, Micans P, Guiotto A, Kasus-Jacobi A. N-acetylcarnosine lubricant eyedrops possess all-in-one universal antioxidant protective effects of L-carnosine in aqueous and lipid membrane environments, aldehyde scavenging, and transglycation activities inherent to cataracts: a clinical study of the new vision-saving drug N-acetylcarnosine eyedrop therapy in a database population of over 50,500 patients. Am J Ther. 2009;16(6):517–533. doi: 10.1097/MJT.0b013e318195e327. [DOI] [PubMed] [Google Scholar]
- 29.Aldini G, Carini M, Yeum KJ, Vistoli G. Novel molecular approaches for improving enzymatic and nonenzymatic detoxification of 4-hydroxynonenal: toward the discovery of a novel class of bioactive compounds. Free Radic Biol Med. 2014;69:145–156. doi: 10.1016/j.freeradbiomed.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Colzani M, De Maddis D, Casali G, Carini M, Vistoli G, Aldini G. Reactivity, selectivity, and reaction mechanisms of aminoguanidine, hydralazine, pyridoxamine, and carnosine as sequestering agents of reactive carbonyl species: a comparative study. ChemMedChem. 2016;11(16):1778–1789. doi: 10.1002/cmdc.201500552. [DOI] [PubMed] [Google Scholar]
- 31.Vistoli G, et al. Computational approaches in the rational design of improved carbonyl quenchers: focus on histidine containing dipeptides. Future Med Chem. 2016;8(14):1721–1737. doi: 10.4155/fmc-2016-0088. [DOI] [PubMed] [Google Scholar]
- 32.Aldini G, et al. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic Res. 2013;47(Suppl 1):93–137. doi: 10.3109/10715762.2013.792926. [DOI] [PubMed] [Google Scholar]
- 33.Colzani M, et al. A novel high resolution MS approach for the screening of 4-hydroxy-trans-2-nonenal sequestering agents. J Pharm Biomed Anal. 2014;91:108–118. doi: 10.1016/j.jpba.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Menini S, et al. D-Carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br J Pharmacol. 2012;166(4):1344–1356. doi: 10.1111/j.1476-5381.2012.01834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown BE, et al. Supplementation with carnosine decreases plasma triglycerides and modulates atherosclerotic plaque composition in diabetic apo E(-/-) mice. Atherosclerosis. 2014;232(2):403–409. doi: 10.1016/j.atherosclerosis.2013.11.068. [DOI] [PubMed] [Google Scholar]
- 36.Cannizzaro L, et al. Regulatory landscape of AGE-RAGE-oxidative stress axis and its modulation by PPARγ activation in high fructose diet-induced metabolic syndrome. Nutr Metab (Lond) 2017;14:5. doi: 10.1186/s12986-016-0149-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34(2):145–169. doi: 10.1016/S0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 38.Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34(4):496–502. doi: 10.1016/S0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 39.Liang H, et al. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic Biol Med. 2009;47(3):312–320. doi: 10.1016/j.freeradbiomed.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai H, Koumura T, Nakajima R, Nomura K, Nakagawa Y. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371(Pt 3):799–809. doi: 10.1042/BJ20021342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riahi Y, Cohen G, Shamni O, Sasson S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab. 2010;299(6):E879–E886. doi: 10.1152/ajpendo.00508.2010. [DOI] [PubMed] [Google Scholar]
- 42.Ingram KH, et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. J Clin Endocrinol Metab. 2012;97(7):E1182–E1186. doi: 10.1210/jc.2011-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillon NJ, Croze ML, Vella RE, Soulère L, Lagarde M, Soulage CO. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology. 2012;153(5):2099–2111. doi: 10.1210/en.2011-1957. [DOI] [PubMed] [Google Scholar]
- 44.Prasannarong M, Santos FR, Hooshmand P, Hooshmand P, Giovannini FJ, Henriksen EJ. The lipid peroxidation end-product and oxidant 4-hydroxynonenal induces insulin resistance in rat slow-twitch skeletal muscle. Arch Physiol Biochem. 2014;120(1):22–28. doi: 10.3109/13813455.2013.834937. [DOI] [PubMed] [Google Scholar]
- 45.Koulajian K, et al. Overexpression of glutathione peroxidase 4 prevents β-cell dysfunction induced by prolonged elevation of lipids in vivo. Am J Physiol Endocrinol Metab. 2013;305(2):E254–E262. doi: 10.1152/ajpendo.00481.2012. [DOI] [PubMed] [Google Scholar]
- 46.Cohen G, et al. Role of lipid peroxidation and PPAR-δ in amplifying glucose-stimulated insulin secretion. Diabetes. 2011;60(11):2830–2842. doi: 10.2337/db11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen G, et al. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia. 2015;58(6):1333–1343. doi: 10.1007/s00125-015-3566-z. [DOI] [PubMed] [Google Scholar]
- 48.Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–496. doi: 10.1146/annurev-pathol-012615-044224. [DOI] [PubMed] [Google Scholar]
- 49.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43(8):617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 50.Rinella ME, Sanyal AJ. Management of NAFLD: a stage-based approach. Nat Rev Gastroenterol Hepatol. 2016;13(4):196–205. doi: 10.1038/nrgastro.2016.3. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi M, et al. Involvement of the TAGE-RAGE system in non-alcoholic steatohepatitis: Novel treatment strategies. World J Hepatol. 2014;6(12):880–893. doi: 10.4254/wjh.v6.i12.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimsrud PA, Picklo MJ, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol Cell Proteomics. 2007;6(4):624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Ruskovska T, Bernlohr DA. Oxidative stress and protein carbonylation in adipose tissue - implications for insulin resistance and diabetes mellitus. J Proteomics. 2013;92:323–334. doi: 10.1016/j.jprot.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher-Wellman KH, et al. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J Physiol (Lond) 2013;591(14):3471–3486. doi: 10.1113/jphysiol.2013.254193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rindler PM, Plafker SM, Szweda LI, Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J Biol Chem. 2013;288(3):1979–1990. doi: 10.1074/jbc.M112.412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conrad M. Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochim Biophys Acta. 2009;1790(11):1575–1585. doi: 10.1016/j.bbagen.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlson BA, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31. doi: 10.1016/j.redox.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seiler A, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Wortmann M, et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ Res. 2013;113(4):408–417. doi: 10.1161/CIRCRESAHA.113.279984. [DOI] [PubMed] [Google Scholar]
- 62.Rupérez AI, et al. Association of genetic polymorphisms for glutathione peroxidase genes with obesity in Spanish children. J Nutrigenet Nutrigenomics. 2014;7(3):130–142. doi: 10.1159/000368833. [DOI] [PubMed] [Google Scholar]
- 63.Polonikov AV, et al. The C718T polymorphism in the 3′-untranslated region of glutathione peroxidase-4 gene is a predictor of cerebral stroke in patients with essential hypertension. Hypertens Res. 2012;35(5):507–512. doi: 10.1038/hr.2011.213. [DOI] [PubMed] [Google Scholar]
- 64.Crosley LK, Bashir S, Nicol F, Arthur JR, Hesketh JE, Sneddon AA. The single-nucleotide polymorphism (GPX4c718t) in the glutathione peroxidase 4 gene influences endothelial cell function: interaction with selenium and fatty acids. Mol Nutr Food Res. 2013;57(12):2185–2194. doi: 10.1002/mnfr.201300216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engelen L, Stehouwer CD, Schalkwijk CG. Current therapeutic interventions in the glycation pathway: evidence from clinical studies. Diabetes Obes Metab. 2013;15(8):677–689. doi: 10.1111/dom.12058. [DOI] [PubMed] [Google Scholar]
- 66.Tilton RG, et al. Prevention of diabetic vascular dysfunction by guanidines. Inhibition of nitric oxide synthase versus advanced glycation end-product formation. Diabetes. 1993;42(2):221–232. doi: 10.2337/diab.42.2.221. [DOI] [PubMed] [Google Scholar]
- 67.Turgut F, Bolton WK. Potential new therapeutic agents for diabetic kidney disease. Am J Kidney Dis. 2010;55(5):928–940. doi: 10.1053/j.ajkd.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 68.de Courten B, et al. Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity (Silver Spring) 2016;24(5):1027–1034. doi: 10.1002/oby.21434. [DOI] [PubMed] [Google Scholar]
- 69.Regazzoni L, et al. A carnosine intervention study in overweight human volunteers: bioavailability and reactive carbonyl species sequestering effect. Sci Rep. 2016;6:27224. doi: 10.1038/srep27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bispo VS, de Arruda Campos IP, Di Mascio P, Medeiros MH. Structural elucidation of a carnosine-acrolein adduct and its quantification in human urine samples. Sci Rep. 2016;6:19348. doi: 10.1038/srep19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baba SP, et al. Role of aldose reductase in the metabolism and detoxification of carnosine-acrolein conjugates. J Biol Chem. 2013;288(39):28163–28179. doi: 10.1074/jbc.M113.504753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janssen B, et al. Carnosine as a protective factor in diabetic nephropathy: association with a leucine repeat of the carnosinase gene CNDP1. Diabetes. 2005;54(8):2320–2327. doi: 10.2337/diabetes.54.8.2320. [DOI] [PubMed] [Google Scholar]
- 73.Sauerhöfer S, et al. L-carnosine, a substrate of carnosinase-1, influences glucose metabolism. Diabetes. 2007;56(10):2425–2432. doi: 10.2337/db07-0177. [DOI] [PubMed] [Google Scholar]
- 74.Orioli M, et al. Design, synthesis, ADME properties, and pharmacological activities of β-alanyl-D-histidine (D-carnosine) prodrugs with improved bioavailability. ChemMedChem. 2011;6(7):1269–1282. doi: 10.1002/cmdc.201100042. [DOI] [PubMed] [Google Scholar]
- 75.Stvolinsky SL, et al. Biological activity of novel synthetic derivatives of carnosine. Cell Mol Neurobiol. 2010;30(3):395–404. doi: 10.1007/s10571-009-9462-7. [DOI] [PubMed] [Google Scholar]
- 76.Bertinaria M, et al. Synthesis, physicochemical characterization, and biological activities of new carnosine derivatives stable in human serum as potential neuroprotective agents. J Med Chem. 2011;54(2):611–621. doi: 10.1021/jm101394n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.