

Abstract
We report the molecular, cellular, and clinical features of 5 patients from 3 kindreds with biallelic mutations in the autosomal LIG1 gene encoding DNA ligase 1. The patients exhibited hypogammaglobulinemia, lymphopenia, increased proportions of circulating γδT cells, and erythrocyte macrocytosis. Clinical severity ranged from a mild antibody deficiency to a combined immunodeficiency requiring hematopoietic stem cell transplantation. Using engineered LIG1-deficient cell lines, we demonstrated chemical and radiation defects associated with the mutant alleles, which variably impaired the DNA repair pathway. We further showed that these LIG1 mutant alleles are amorphic or hypomorphic, and exhibited variably decreased enzymatic activities, which lead to premature release of unligated adenylated DNA. The variability of the LIG1 genotypes in the patients was consistent with that of their immunological and clinical phenotypes. These data suggest that different forms of autosomal recessive, partial DNA ligase 1 deficiency underlie an immunodeficiency of variable severity.
Keywords: Genetics, Immunology
Keywords: B cells, Genetic diseases, T cell development
Introduction
The DNA ligase family is an evolutionarily conserved group of proteins found in both prokaryotic and eukaryotic cells that have previously been implicated in rare immune defects. Substrate dependence divides this family into 2 groups, with prokaryotes utilizing mostly NAD+-dependent ligases and eukaryotes using ATP-dependent ligases. Vertebrate cells encode 3 ATP-dependent DNA ligases (1). Ligase 4 (LIG4) is responsible for nonhomologous end-joining repair after double-stranded breaks, and is essential in B and T cells for VDJ recombination (2). Compound heterozygous and homozygous mutations in LIG4 have previously been associated with T-B-NK+ severe combined immunodeficiency (SCID) although phenotypic variability has been reported (3, 4). Ligase 3 (LIG3), expressed in vertebrates but with homologues in many lower eukaryotes (5), is the only ligase expressed in mitochondria; this ligase has been implicated in both single-strand (6, 7) and double-strand (8) break repair in mitochondria and in the nucleus. No cases of primary immune deficiency (PID) caused by LIG3 deficiency have been reported to date. DNA ligase 2 is thought to be a proteolytic fragment of LIG3, and appears to have the functions of this ligase (9). Ligase 1 (LIG1), while not essential for viability in a murine B cell line (10), plays a key role in joining of Okazaki fragments during DNA replication and for repair of single-stranded DNA damage during base excision repair (11). Recent studies found a role for LIG1 in double-strand break repair through the alternative-NHEJ pathway (12, 13), although current understanding of this pathway is incomplete. When catalyzing repair, LIG1 completely surrounds and partially unwinds DNA, interacting with the minor groove both upstream and downstream of the nick (14). Enzymatic ligation involves a 3-step process involving the initial adenylylation of the active site lysine (K568), subsequent transfer of the AMP to the 5′-PO4 of the DNA, and finally nucleophilic attack of the 3′-OH to seal the nick and release AMP (15).
Deleterious mutations in LIG1 have been described in only one patient 25 years ago, who was originally suspected of having Bloom’s syndrome based on clinical phenotype (16). This patient, who died at age 19 of pneumonia, exhibited hypogammaglobulinemia, red cell macrocytosis, sun sensitivity, lymphocytic liver infiltrates, and short stature (16). The variants identified in this patient were subsequently shown to severely impact LIG1 catalytic activity. One of these variants, E566K, disrupts the ATP-binding site of the protein and thus abolished enzymatic activity (17, 18). The other, R771W, falls next to a DNA-binding motif and was demonstrated to retain only a fraction of wild-type (WT) activity (18). As expected, a fibroblast cell line from this patient was shown to be susceptible to DNA damage, caused by both ionizing and UV irradiation, in addition to several alkylating agents (19, 20).
Recently in 2 independent centers, we identified 2 unrelated patients presenting with hypogammaglobulinemia in early childhood who were shown to have the same compound heterozygous mutations in DNA ligase 1. Further exploration of PID cohorts led to the identification of 3 additional patients with biallelic LIG1 mutations in a single kindred, prompting study of the role of LIG1 in the pathogenesis of this group of immunodeficient patients. Here we describe the molecular, cellular, and clinical heterogeneity of inherited LIG1 deficiency.
Results
Clinical phenotypes and identification of LIG1 variants.
Patients 1 and 2 (P1 and P2, Figure 1A) are from unrelated kindreds A and B, and were identified as children with early-onset hypogammaglobulinemia, lymphopenia, and macrocytic red blood cells (Table 1). P1 also had an early history of diarrhea which resolved, and P2 demonstrated early susceptibility to viral infections. Both had normal growth and neurodevelopment and showed absence of dysmorphia or other clinical complications (Table 2). In both patients, a transcobalamin II deficiency was first suspected due to the striking erythrocyte macrocytosis. Vitamin B12 and folate levels were normal in both, and supplementation in P1 had no effect; no mutations in TCN2 were found. Lacking a specific genetic diagnosis, each was diagnosed with a form of common variable immune deficiency (CVID), and has been treated with replacement immunoglobulin. Further study revealed that both patients had reduced absolute counts of CD3+ T cells but increased proportions of γδ T cells. We investigated both patients and their parents using whole-exome sequencing (WES) followed by selected primary immune deficiency disease panels. Two rare LIG1 variants were identified in both patients, T415Mfs*10 and R641L (Figure 1, B and C; Table 2). The presence of the same 2 variants in 2 unrelated White patients from different geographic locations prompted exploration of identity-by-descent (21). However, no evidence of a familial relationship between the 2 families was found. In addition, using the 1000 Genomes Project SNPs, both sets of parents were found to be either homozygous or heterozygous for known but different haplotypes surrounding the shared mutations, suggesting that these are common haplotypes in the population and not a founder effect.
Figure 1. LIG1 mutations and functions.

(A) Pedigrees for 3 kindreds with recessive mutations in LIG1. Patients 1 and 2 (P1 and P2) are from unrelated parents and both have the LIG1 mutations T415Mfs*10 and R641L. Patients 3 and 4 (P3 and P4) are brothers whose parents share grandmothers; patient 5 (P5) is a maternal cousin; these subjects have homozygous missense variants, P529L and R771W. The parents of P5 also share the same grandmother. (B) Sanger sequencing of P1 and a control, also P3 and a control. (Note that P2 has the same mutations as P1 and that P3 has the same genotype as P4 and P5.) (C) Schematic of LIG1 protein structure and approximate locations of all the identified mutations, in this and prior reports (16, 17). (D) Mechanism of LIG1-catalyzed DNA ligation. LIG1 consumes ATP and forms a covalent bond between AMP and K568. AMP is then transferred to the 5′-phosphate of a nicked DNA substrate. Attack by the 3′-hydroxyl seals the nick and releases AMP. All 3 chemical steps require Mg2+ as an essential cofactor.
Table 1. Demographic and immunological phenotypes of LIG1-deficient patients.

Table 2. Clinical information.

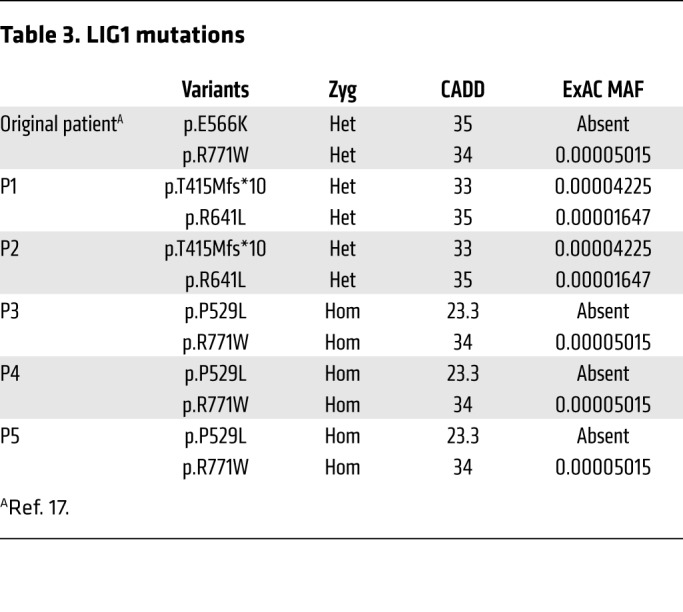
Patients 3 and 4 (P3 and P4, kindred C, Figure 1A) are brothers born to consanguineous parents who share grandmothers, originating from the Sudan. Both were diagnosed as infants with SCID, complicated by multicystic dysplastic kidneys and severe anemia requiring blood transfusions. They had profound hypogammaglobulinemia, lymphopenia with low B and T cells, but normal NK cell numbers (Tables 1 and 2). P3 and P4 also had red cell macrocytosis and an elevated proportion of γδT cells. Both patients were treated with hematopoietic stem cell transplantation. A female maternal cousin of the brothers, who shares the same grandparents and the same genotype as P3 and P4, has chronic respiratory tract infections and early growth failure (P5 in Figure 1A). Although she also has macrocytosis, low numbers of B cells, and is hypogammaglobulinemic, she is not so profoundly T cell lymphopenic and is maintained on immune globulin replacement and antibiotics. Neither dysmorphia or neurodevelopmental defects is present (Tables 1 and 2). Due to the known consanguinity, we hypothesized that these children suffer from an autosomal recessive disorder. Analysis of WES revealed that all 3 patients have 2 homozygous missense variants in LIG1, P529L and R771W, (Figure 1, B and C; Table 3). The latter variant was identified in the first patient in the original report (17).
Table 3. LIG1 mutations.
Population genetics of LIG1 variants.
The T415Mfs*10, P529L, and R641L LIG1 variants identified in the 3 kindreds were not found in any other patients in an in-house database (JLC laboratory) of 3,595 patients with immune deficiency. A heterozygous copy of the R771W substitution was identified in a single patient in this large cohort, but a second mutation was not present. The P529L and E566K variants have not been reported in public databases (Exome Aggregation Consortium [ExAC], Broad Institute; 1000 Genomes Project, Phase 3, http://www.1000genomes.org/) whereas the T415Mfs*10, R641L, and R771W variants occur at frequencies of 3 × 10–5, 2 × 10–5, and 4 × 10–5, respectively. Consequently, the highest probability of being homozygous for these alleles in worldwide populations is estimated to be 1.6 × 109, but in compound heterozygous states it is compatible with an estimated occurrence of CVID (1:20,000 to 1:50,000; Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI99629DS1). Combined annotation-dependent depletion (CADD) scores for the T415Mfs*10, R641L, and R771W variants are above the mutation significance cutoff (MSC) for LIG1 (Table 3) (22), indicating that they have a high likelihood of contributing to disease pathogenesis. Assuming an autosomal recessive model, no deleterious polymorphism (frameshift or loss of splice site) with a MAF higher than 10–4 in LIG1 has been reported in the worldwide population (Supplemental Figure 1). We also noted that all 5 mutations occur at highly conserved residues across many species (Supplemental Figure 2). Collectively, the genetic data suggested that P1 and P2 from kindreds A and B have compound heterozygous, and P3, P4, and P5 from kindred C have homozygous, rare, and likely deleterious LIG1 variants.
Defective repair response to DNA damage in LIG1–/– and mutant cells.
To further examine the functional consequences of the LIG1 mutants identified in our patients, LIG1–/– cell lines were constructed in HEK-293T cells with the CRISPR-Cas9 DNA-editing system, by independently engineering 2 pools of cells with 2 sgRNAs targeting exon 4 or exon 7 of the LIG1 gene (Figure 2A). Following subcloning of these 2 pools, we identified several clones with loss of LIG1 protein expression in both exon 4 and exon 7 targeted regions (Figure 2B). DNA sequencing of these clones showed biallelic LIG1 frameshift mutations around the targeted sites, as shown for colony 37, targeting exon 7, in Figure 2C. The LIG1–/– cells from colony 37 were then used to generate stable cell lines, as well as cell lines singly reexpressing WT LIG1, or the individual LIG1 mutant alleles observed in patients. The expression of each mutant was validated by both Sanger sequencing (not shown) and protein blotting (Figure 2D). The P529L, R641L, and R771W variants were expressed at similar levels as WT LIG1, but the truncated protein produced by the T415Mfs*10 mutation was at a much lower level, as was the previously identified patient’s mutation, E566K. We then examined whether the LIG1–/– HEK-293T cells also displayed sensitivity to DNA damage caused by ethyl methanesulfonate (EMS). This reagent produces alkylated bases which require base excision repair, involving single-strand breaks as an intermediate (20, 23). As expected, LIG1–/– clones 37 and 54 displayed similar loss of viability when exposed to EMS, as compared with native HEK-293T cells (Figure 3A). Showing that the loss of ligase function led to cell death, clone 37 LIG1–/– cells could be rescued from loss of viability by stable transfection with WT protein (Figure 3B). Cells with other mutations of interest, R641L and R771W, were also impaired, with intermediate loss of viability. We also γ-irradiated LIG1–/– cells and measured the number of γH2AX foci per nuclei by immunofluorescence as a measure of early DNA damage as described. LIG1–/– HEK-293T cells showed an increase in γH2AX foci after γ-irradiation, as compared with WT HEK-293T cells (Figure 3, C and D), in agreement with previous studies (24), but again, cells were also rescued following stable expression of WT protein. Note that LIG1–/– cells were generated using 2 different sgRNAs targeting different upstream exons, and both cell lines exhibited similar EMS damage (Figure 3A) and an increase in γH2AX foci with irradiation, both repaired by WT LIG1 (Figure 3C), thereby limiting potential off-target effects of CRISPR-Cas9 DNA editing.
Figure 2. Production of LIG1–/– and mutant cells.

(A) Schematic of LIG1 showing location of the gRNA targets. Targets were chosen in exon 4 (red) and exon 7 (blue), which are both upstream of the catalytic core. (B) Immunoblot using the rabbit polyclonal anti-LIG1 on cell lysates from CRISPR-Cas9–treated colonies. One colony from each exon (red and blue boxes) was chosen for experiments. (C) Sanger sequencing of exon 4 colony demonstrated the homozygous deletion of 32 bp and introduction of a premature stop codon; the exon 7 colony 37 showed homozygous deletion of 26 bp and also a premature stop codon. Plasmids containing WT LIG1 or the mutants found in LIG1 from patients (P529L, E566K, R771W, R641L, or T415Mfs*10) were transduced into colony 37 LIG1–/– HEK-293T cells using retroviral vectors. (D) Immunoblots of total cell lysates extracts (TCEs; performed 2 times) demonstrated absence of expression of LIG1 protein in the LIG1 knockout (clone 37) and restoration of LIG1 expression by transduced cells, as examined by rabbit polyclonal anti-LIG1. Immunoprecipitation with anti-FLAG demonstrated both FLAG and LIG1 polypeptide in these cells, validating the expression of truncated LIG1 in the T415Mfs*10 mutant. Each mutation was validated with Sanger sequencing (not shown).
Figure 3. Defective repair of LIG1–/– HEK-293T and mutant cells.

(A) LIG1–/– clones 37 and 54 were more sensitive to EMS, as compared with HEK-293T cells but similar to each other (2 experiments, 6 replicates; mean ± SD; P = 0.03, P = 0.04). (B) EMS sensitivity of clone 37 cells was rescued by complementation with WT LIG1, but only partially with R641L or R771W (3 experiments, 6 replicates; mean ± SD). (C) Responses of HEK-293T cells to γ radiation: WT HEK-293T cells, clone 37 LIG1–/–, and LIG1–/– + WT LIG1 were irradiated (25 Gy), incubated for 3 hours, and inspected for number of γH2AX foci per nuclei (3 experiments). (D) γH2AX foci/nucleus for HEK-293T cells, LIG1–/–, and LIG1–/– cells complemented with WT LIG1, before and after irradiation with 25 Gy. LIG1–/– cells demonstrated increased foci compared with HEK-293T cells (P = 0.0001), with substantial rescue with WT protein: LIG1–/– versus LIG1 –/– + WT, now P = 0.05 (3 experiments; 50 nuclei/condition counted per experiment; mean ± SD). (E) HEK-293T cells, LIG1–/– cells, and LIG–/– cells transfected with WT LIG1, incubated with media alone or 0.5 mM EMS, and examined by comet assay. Data are mean ± SD and percentage of cellular DNA in the comet tail for 79 to 86 cells/condition; performed 3 times. (F) Comparing DNA damage in 0.5 mM EMS-treated HEK-293T cells to LIG1–/–, T415Mfs*10, R771W, E566K cells, P < 0.0001, to R641L cells, P = 0.69. Adding the P529L variant was similar to adding WT LIG1 (differences, P = 0.9). (G) After 16 hours EMS, LIG1–/–, and mutant cells were washed, replated in fresh media, and tested for the differences in DNA damage at intervals. Mean percent shown for each. At 8 hours, differences emerged: HEK-293T versus LIG1–/–, P = 0.007; LIG1–/– versus T415Mfs*10, P = not significant; LIG1–/– versus R641L, P = 0.02; LIG1–/– versus R771W, P = 0.04 (50–88 colonies/condition).
Using comet assays, we then tested populations of LIG1–/– cells or LIG1–/– cells transduced with WT LIG1 or the mutations identified in patients, after exposure to various concentrations of EMS, to compare DNA breaks. As expected, the LIG1–/– cells demonstrated significantly more DNA damage when exposed to an optimum concentration of EMS as compared with native HEK-293T cells (P = 0.0001), but when transduced with WT LIG1, again showed complete reversal of this sensitivity (Figure 3E). In fact, LIG1–/– cells reconstituted with WT LIG1 demonstrated significantly less DNA damage than even native cells, potentially due to the more than 2-fold increase in LIG1 protein content present in these transduced cells (Supplemental Figure 6). In addition, the LIG1 variants identified as particularly deleterious (the truncated mutation T415Mfs*10 and mutants E566K and R771W) again showed significantly more damage in this assay, as compared with baseline HEK-293T cells (P = 0.0001), while the variant R641L showed damage similar to that of HEK-293T baseline cells (P = 0.69). Mutants with the P529L variant were also similar to LIG1–/– cells rescued with WT LIG1 (differences P = 0.9) (Figure 3F).
To additionally define the differences in the LIG1 mutations observed in our patients, we used the comet assay in a kinetic experiment, again exposing cells to EMS for 16 hours, but then washing cells and replating them in fresh media to examine the comparative ability of mutants to repair DNA damage over 2, 4, 8, 12, or 16 hours. Again, as expected, LIG1–/– cells transduced with WT LIG1 demonstrated significantly less DNA damage as compared with all mutant cells, starting as early as 4 hours into the recovery period (P = 0.001), showing that absence of LIG1 in HEK-293T cells is associated with the disruption of normal repair responses in these cells. LIG1–/– cells were also similar to the truncation mutant T415Mfs*10, whereas the R641L mutant was similar to the R771W mutant at all time intervals (P not significant in each case). However, the R641L and R771W mutants displayed somewhat improved recovery as compared with LIG1–/– cells, demonstrating less DNA damage after 8 hours (differences between LIG1–/– and R641L and R771W cells, P = 0.02 and P = 0.04, respectively.)
LIG1 variants impair protein structure and function.
The defective response of LIG1–/– cells to DNA damage prompted us to examine how the observed mutations affect protein structure. The T415Mfs*10 variant introduces a premature stop codon in the LIG1 transcript, causing truncation of the protein and loss of its entire catalytic core. Thus, this allele is predicted to lead to an inactive enzyme. The R641L and R771W missense variants occur at residues adjacent to separate DNA-binding motifs (Figure 1C), suggesting that they may interfere with the ability of LIG1 to interact with DNA. The crystal structure of LIG1 bound to 5′-adenylated nicked DNA (14) revealed that R641 is located in a hairpin loop that interacts with the minor groove of nicked DNA via interactions with amino acids Q636, K642, R643, and K644. R641 itself interacts with the negatively charged D600 via an ionic bond (Supplemental Figure 3). Replacement of this arginine with a hydrophobic leucine is predicted to abolish this interaction, destabilizing the adjacent DNA-binding region, and consequently decreasing the efficiency of the enzyme. Similarly, replacement of R771 with a hydrophobic tryptophan may lead to inefficient binding of LIG1 to nicked DNA. In agreement with this assessment, the R771W variant has been shown to severely abrogate catalytic activity of LIG1 (16, 18). We suspected that the presence of this variant in the homozygous state in P3 and P4 would be enough to cause LIG1 deficiency. In support of this, the P529L missense variant, also homozygous in P3 and P4, is not adjacent to any DNA-binding or functional motifs (Figure 1C). Therefore, with the exception of the P529L variant, the missense mutations observed in our patients are predicted to be disruptive to normal catalytic activity of LIG1.
Impaired LIG1 catalytic activity.
Based on the predicted structural defects of these variants, we hypothesized that the T415Mfs*10, E566K, R641L, and R771W mutations lead to defective enzymatic ligation. To test this, we transfected LIG1–/– HEK-293T cells with LIG1-pRetro plasmids containing each of the observed mutations and then purified the LIG1 protein. Protein concentrations were normalized (Figure 4A) and each protein assayed for ligase activity using a synthetic nicked 28 bp oligonucleotide duplex (Figure 4B). The ligated 28 bp product was separated from unreacted DNA on a polyacrylamide gel. Band intensity was measured and normalized to WT activity. Disruption of the active site, K568, and the AMP-binding site, E566, leads to inactive enzymes, whereas 2 missense variants (R409H and V753M) reported at high frequency in ExAC (and therefore likely tolerated) retain over 75% of WT activity (Figure 4, C and D). The R771W missense mutation exhibits only 4.5% of WT activity, in agreement with a previous report (16). As expected, the P529L substitution does not affect activity of the enzyme. The T415Mfs*10 frameshift lacks the catalytic core of the protein, and is, as expected, functionally inactive or unstable, whereas the R641L substitution demonstrates an estimated 7% of WT activity. Because R641 is located in a hairpin loop that binds DNA (Supplemental Figure 3), we also explored the effects of damaging mutations in nearby residues by alanine scanning mutagenesis (Figure 5). Disruption of the salt bridge with R641 by the D600A mutation has similar deleterious effects on catalytic activity. In contrast, single replacement of other nearby residues does not cause a similar decrease in ligase activity (Figure 5). Thus, the R641L mutation disrupts an essential feature of LIG1 structure that is responsible for normal repair of nicked DNA (Supplemental Figure 3). Together, these enzymatic studies demonstrate that the mutations observed in the patients, apart from the P529L variant, are severely deleterious and have a significant impact on the repair of damaged DNA.
Figure 4. Measurement of enzymatic activity of LIG1 mutants.

(A) Normalized expression of purified LIG1 protein from HEK-293T cells, containing patient or control mutations. Proteins were separated by SDS-PAGE and detected using rabbit polyclonal anti-LIG1 and anti-Flag antibodies as shown. (B) Schematic of 28 bp synthetic nicked oligonucleotide complex used for the enzymatic assay. (C) Representative ligation assay. Ligation forms a 28 bp ligated product. (D) Quantification of enzymatic activity normalized to WT LIG1 activity (%). (3 experiments; data are mean ± SD.) Controls: GFP and FNT= Flag, a nuclear localization signal, and thioredoxin.
Figure 5. Exploration of the DNA-binding loop containing Arg-641 by alanine scanning mutagenesis.

Residues adjacent to R641 in the DNA-binding loop were mutated to alanine (A) purified from HEK-293T cells, and then assessed for enzymatic activity. (A) Representative ligation assay. (B) Enzymatic activity is normalized to WT LIG1 activity (%), and the corresponding immunoblot is shown. Data are mean ± SD and are representative of 3 experiments using polyclonal anti-LIG1. Controls: WT LIG1; FNT= Flag, a nuclear localization signal, and thioredoxin.
Comparing biochemical effects of LIG1 missense mutations.
We further investigated the specific biochemical defects in the LIG1 patient alleles using recombinant enzyme, as previously described for the structure and mechanism of WT Δ232 LIG1 (14, 25). The WT, P529L, R641L, and R771W variants were expressed in E. coli, purified to homogeneity, and analyzed by SDS-PAGE (Figure 6A). The active concentration of each enzyme was determined by titrating increasing LIG1 against a fixed DNA concentration in the absence of ATP (Supplemental Figure 4). The mutant enzymes had a similar active fraction as the WT LIG1 and the corrected concentrations were used to compare the steady-state kinetics of ligation for the mutant enzymes (see Figure 6B for representative initial rates determination). The dependence on the DNA substrate reveals the maximal turnover rate constant was approximately 3-fold slower for the R641L and R771W mutants and the Km values were slightly elevated (Figure 6C, Table 4). Catalytic efficiency (kcat/Km) for the R641L and R771W mutants was significantly reduced (5- and 11-fold, respectively; Table 3). We next assessed the dependence of ligation on the concentration of Mg2+ (Figure 6D). Surprisingly, we observed a 4- to 5-fold decrease in Mg2+ affinity for both the R641L and R771W mutants (Table 3). Additionally, we noted a buildup of adenylylated DNA in reactions containing either R641L or R771W at low magnesium concentrations (Figure 7A). This behavior is indicative of abortive ligation, where LIG1 performs adenylyl transfer but dissociates from the adenylylated DNA before it can complete ligation (Figure 7B). When ATP is available, LIG1 rapidly undergoes self-adenylylation, rendering it incapable of rebinding the aborted intermediate. To quantify this abortive ligation, reactions were performed at physiologically relevant ATP and Mg2+ concentrations. Under these conditions, R641L and R771W underwent abortive ligation approximately 50% of the time, compared with less than 1% for WT and the P529L variant (Figure 7C). Interestingly, this deficiency at a physiological Mg2+ concentration could be substantially rescued by elevating the concentration of free Mg2+ to 5 mM.
Figure 6. Steady-state kinetic analysis of LIG1 mutants.

(A) Recombinant WT Δ232 LIG1 and mutants were purified to homogeneity and 1 μg protein analyzed by SDS-PAGE with Coomassie stain. M, marker. (B) Representative initial rates for ligation measured for R641L with varying amounts of Mg2+. The dependence on nicked DNA substrate (C) and Mg2+ cofactor (D) were determined for WT and mutant proteins (mean ± SD; n ≥ 3) and fit to the Michaelis-Menten equation (best fit parameters are summarized in Table 3).
Table 4. Kinetic parameters for ligation catalyzed by LIG1.

Figure 7. R641L and R771W LIG1 mutants generate abortive ligation intermediates at physiological Mg2+ concentration.

(A) Representative denaturing polyacrylamide gel analyzing multiple-turnover ligation with 5 nM LIG1, 500 nM DNA, and 1 mM free Mg2+. (B) Schematic for abortive ligation by LIG1. After catalyzing adenylyl transfer, LIG1 can either catalyze the nick-sealing step or dissociate from the adenylylated DNA intermediate. (C) Fraction abortive ligation was calculated for WT and mutant enzymes at both 1 mM and 5 mM free Mg2+ (mean ± SD; n ≥ 3). ND, not detected. See Supplemental Figure 5 for the initial rates for formation of ligated product and adenylated DNA.
LIG1 expression in patients’ leukocytes.
LIG1 mRNA expression in cells of patients, parents, and family members was determined by reverse transcription quantitative PCR (RT-qPCR) in PBMCs and EBV-immortalized B cells from P1 in kindred A (Figure 8, A and B). Levels of the LIG1 transcript were not decreased in P1 when compared with the parents, brother, sister, or controls. In contrast, immunoblots of LIG1 protein from EBV-immortalized B cells in kindreds A and B revealed decreased expression levels of full-length LIG1 in individuals with the T415Mfs*10 variant (Figure 8, C–F). This is in agreement with the observation that HEK-293T cells containing the T415Mfs*10 mutation also have very weak expression of a truncated LIG1 protein (Figure 2D). Therefore, cells harboring the frameshift mutation have approximately 50% the steady-state level of full-length LIG1 that must come from the other allele.
Figure 8. Protein and mRNA expression in B cells.

Reverse transcription qPCR was used to measure LIG1 mRNA levels in PBMCs (A) and EBV-B cells (B) from P1, family members, and 5 controls in 3 replicates. (C and D) EBV-B cell lysates were separated by SDS-PAGE and the expression of LIG1 was detected by chemiluminescence using rabbit monoclonal anti-LIG1 (recognizes aa 1–100) for P1 and family members (C) or P2 and family members with 1 control (D). (E) Quantification of LIG1 protein expression in blot C for P1, mother (R641L/WT), father (T415Mfs*10/WT), brother (T415Mfs*10/WT), and sister (WT/WT). (F) Quantification of LIG1 protein in blot D for P2, mother (R641L/WT) and father (T415Mfs*10/WT), as compared with a control (C) (immunoblotting, n = 3; data are mean ± SD).
Defective repair responses to DNA damage in B and T cells.
B lymphoblastoid cell lines from members of kindred A and B were then examined for DNA damage caused by EMS. B cells with the same compound heterozygous LIG1 mutations from both families demonstrated loss of viability in this survival assay, whereas heterozygous members had variably reduced survival in comparison to transformed cells of controls (Figure 9, A and B). We also examined the effects of radiation on peripheral blood T cells from P1 and his family members, using peripheral blood PBMCs exposed to 10 Gy radiation, then fixed and permeabilized at 1, 8, 24, and 43 hours. CD3+ T cell populations were examined by FACS using phosphorylated histone H2AX (γH2AX) as a marker of early DNA damage (26) as described (27). T cells from P1 demonstrated increased γH2AX whereas cells from individuals with heterozygous variants and controls showed intermediate and normal staining, respectively (Figure 9C). These data indicate that both B and T cells demonstrate a decreased capacity to respond to chemical and radiation-induced DNA damage.
Figure 9. Defective lymphocyte repair responses to EMS and radiation.

Lymphoblastoid B cells of subjects from kindred A (A) and kindred B (B) were cultured in media with increasing concentrations of ethyl methanesulfonate (EMS) to examine DNA sensitivity, determining cell survival (3 experiments; 6 replicates of cell cultures for each condition; mean ± SD). (C) The effects of radiation on peripheral blood T cells from P1 and family members were determined after exposure to 10 Gy radiation. After permeabilization, CD3+ T cell populations were examined by flow cytometer for γHAX foci, and the mean fluorescence intensity (MFI) was determined as described (43).
IgH rearrangement and somatic hypermutation.
As patients P1 and P2 presented with early-onset hypogammaglobulinemia, we determined the effect of LIG1 deficiency on IgH rearrangement and somatic hypermutation. For this, we generated cDNA from PBMC RNA from P1, family members, and controls, and sequenced the VDJ-VH3 region by TOPO-TA cloning. While the average CDR3 length was similar for the tested individuals (not shown), the isolated VH3 clones from both P1 and his mother had a much lower percentage of mutated clones than controls (Figure 10A). In addition, of the mutated clones, P1 demonstrated a significantly decreased extent of mutations and fewer mutations per clone as compared with his parents, siblings, or controls (Figure 10B).
Figure 10. Effect of LIG1 deficiency on somatic hypermutation.

VH3 clones from 80 colonies per subject from P1 and family members (mother, father, sister, and brother) were amplified from cDNA from PBMC RNA, using primers specific for the VDJ VH3 region; individual clones were sequenced following TA-TOPO cloning. (A) The percentage of VH3 sequences in these clones for each member of the family that contained 2 or more mutations is shown, in comparison to similarly isolated clones of 2 healthy controls. (B) For clones that contained at least 2 mutations in the VH3 sequences, we then determined the number of mutations in each clone for each member of the family again, as compared with 2 healthy controls. *P = 0.05; ***P = 0.001.
Discussion
We identified autosomal recessive partial LIG1 deficiency as the cause of immunodeficiency in 5 patients from 3 different kindreds, of different ages and ethnicities, and with a wide spectrum of immunological and clinical manifestations, ranging from an early-onset Ig production defect to severe combined B and T cell deficiency requiring hematopoietic stem cell transplantation. For P1 and P2, the complete loss of catalytic activity from the T415Mfs*10 mutation is coupled with a second mutation, R641L, that disrupts an important DNA binding loop but retains a modest level of enzyme activity, perhaps accounting for the more moderate clinical phenotype in these subjects. Finding 2 identical compound heterozygous mutations in unrelated White subjects P1 and P2, from geographically dispersed locations, was surprising; however, exploration of familial relationship found no evidence of a founder effect, indicating that these mutations arose independently in these families. Although there is also no familial relationship, or even ethnic similarity between kindred C and the originally described patient, P3 and P4 harbor the same R771W substitution found in the earlier patient (16). This mutation was also found to be heterozygous in 1 other individual in our internal cohort of 3,595 immune-deficient patients, but is reported at approximately 1 in 20,000 individuals in ExAC, possibly explaining its occurrence in the 2 unrelated families.
For kindred C, the homozygous R771W mutant is likely the cause of disease in these patients, as the LIG1 protein resulting from the P529L variant, also found in this family, has normal catalytic activity and efficiency. Additionally, the R771W mutation studied in a fibroblast cell line derived from the original patient had an estimated 3%–5% of WT enzyme activity, delayed joining of Okazaki fragments, and caused hypersensitivity to DNA-damaging agents (17, 18, 24, 28, 29). However, in other studies, mice with homozygous R771W mutations were viable and had preserved hematopoietic lineages but early growth failure and splenomegaly due to extensive extramedullary hematopoiesis. As the mice aged, the growth failure resolved but an increased number of spontaneous tumors were observed (30). In our studies both the R771W and R641L mutant proteins exhibited dramatic increases in abortive ligation, which would block other DNA ligases and require increased contributions from aprataxin (APTX), which repairs the abortive ligation adduct (31) or flap displacement synthesis (FEN1) (25). We predict that deficiencies in flap processing and/or aprataxin could increase the severity of these LIG1 mutations and contribute to the clinical variability seen in patients harboring identical LIG1 alleles, but note that no potentially deleterious mutations in these pathways were identified in our subjects. A notable feature of the R771W and R641L LIG1 mutant enzymes is their sensitivity to Mg2+ concentration, requiring elevated Mg2+ for efficient ligation (Table 3). The weakened Mg2+ binding and elevated abortive ligation leading to the accumulation of adenylylated DNA intermediates seen in vitro could contribute to the cellular phenotypes observed in patients and suggests possible worsening of the enzyme defect in any clinical state of magnesium deficiency.
All of the patients, including the originally reported subject (16), share a somewhat unique clinical feature: red cell macrocytosis, similar to lethally irradiated mice rescued with LIG1–/– hematopoietic cells (29). Although the exact mechanism is not clear, impaired DNA synthesis is known to lead to macrocytic anemias (32). Thus, LIG1 deficiency may lead to increased erythrocyte size through failure to produce DNA quickly enough during replication. A more detailed exploration of Okazaki fragment repair and cell-cycle dynamics in erythrocyte progenitors would provide more insight into this mechanism.
Patients P1 and P2 presented with hypogammaglobulinemia presumably through impaired B lymphocyte development. B cells from subjects in kindreds A and B who carry the T415Mfs*10 mutation had reduced LIG1 enzyme levels. EBV-transformed lymphoblastoid B cells from P1 and P2 also demonstrated increased sensitivity to chemical damage. We also examined the VH3 region for both numbers and extent of mutations. Mutation extent was significantly reduced in P1, suggesting that LIG1 has a greater role during somatic hypermutation than previously known. We also observed T cell lymphopenia and increased numbers of γδ T cells, especially as a fraction of CD3+ T cells, in all the patients, and showed that peripheral T cells from P1 demonstrated increased γH2AX signal following γ-irradiation. This finding was confirmed by γ-irradiation of LIG1–/– cells, which could be rescued by complementation with WT LIG1. While the mechanism of γδ T cell development is also not well understood (33, 34), a defect in DNA replication and repair caused by LIG1 mutations may lead to ineffective commitment of T cell precursors to the αβ lineage or inefficient recombination of the αβ locus. Increased circulating γδ T cells have not been reported in LIG4 deficiency (4), but increases in this population have been noted both in patients with other DNA repair defects, including those with ataxia telangiectasia (35), and in patients with rare hypomorphic mutations in RAG1 (36, 37).
These studies confirm one of the main lessons learned from the study of PIDs, namely, that defects in the same gene may give rise to a number of different clinical phenotypes (38). While P1 and P2 had primarily an early-onset B cell defect, targeted genetic studies in other hypogammaglobulinemic subjects have also uncovered a number of pathogenic variants, including hypomorphic mutations in other genes associated with SCID, including RAG1, RAG2, ADA, and DCLRE1C (Artemis) (38–42). While severe defects of LIG4 abrogate nonhomologous end-joining and prevent T and B cell development, hypomorphic heterozygous mutations led to panhypogammaglobulinemia and lymphopenia in another case (27). As hypomorphic mutations in numerous genes that control overall lymphoid growth and development (IKZF1, NFKB1, STAT3, CTLA4, PIK3CD, and LRBA) also may appear clinically as B cell defects (43, 44), unbiased approaches such as whole-exome and whole-genome sequencing are likely to uncover a number of additional unexpected genetic causes of B cell failure.
Methods
Whole-exome sequencing.
Genomic DNA from peripheral blood mononuclear cells from P1 and family members was extracted and sheared with a Covaris S2 Ultrasonicator. After generation of an adaptor-ligated library (Illumina), exome capture was performed with SureSelect Human All Exon 37, 50, or 71 Mb kits (Agilent Technologies). Massively parallel WES was performed on a HiSeq 2000 or 2500 (Illumina), which generates 72- or 100-base reads. We used the Genome Analysis Software Kit (GATK) best-practice pipeline to analyze our WES data (45). Reads were aligned to the human reference genome (hg19) using the Maximum Exact Matches algorithm in Burrows-Wheeler Aligner (46). PCR duplicates were removed using Picard tools (http://picard.sourceforge.net/). The GATK base quality score recalibrator was applied to correct sequencing artifacts. GATK HaplotypeCaller was used to identify variant calls. Variant annotation was performed with SnpEff (http://snpeff.sourceforge.net/). Exomes were annotated with ExAC r0.3.1 (Exome Aggregation Consortium, Broad Institute) and the 1000 Genomes Project, Phase 3 (http://www.1000genomes.org/). DNA was prepared from blood from P2 and parents at National Jewish Health, and analyzed by next generation sequencing (Illumina MiSeq) using libraries prepared for a custom PID gene panel (345 genes, Agilent Sure Select QXT Target Enrichment System).
Peripheral blood cells and generation and culture of EBV-B cell lines.
PBMCs from patients and controls were isolated using Ficoll-Paque gradients under IRB-approved protocols and with informed consent. EBV generated from the blood of cotton-top tamarin monkey peripheral blood lymphocytes (MilliporeSigma, B95-8) was used to generate lymphoblastoid cells lines which were cultured in RPMI 1640 with 10% or 20% FBS, 2 mM l-glutamine, 1% HEPES buffer, and 1% antibiotic/antimycotic at 37°C.
Antibodies.
The antibodies used for immunoblot analyses were rabbit monoclonal anti-LIG1 (Abcam, ab177946, which recognizes an LIG1 epitope within aa 1–100), or polyclonal rabbit anti-LIG1 (Proteintech Group, 18051-1-AP), anti-Flag (Cell Signaling, 14793S), and anti-actin (Santa Cruz, sc-8432). The antibody used for detection of γH2AX foci by immunofluorescence was anti-γH2AX (Ser139) (Biolegend, 613402), followed by goat anti–mouse Alex Fluor 647 (Thermo Fisher Scientific, A32728.) Antibodies used for flow cytometry of peripheral blood T cells were anti–γH2AX-Alexa Fluor 647 and anti–CD3-APC-eFluor 780 (eBioscience, 47-0038-41) (27).
Generation of LIG1–/– cells using CRISPR-Cas9.
HEK-293T cells (ATCC CRL-1573) cultured in DMEM with 10% FBS, 1% HEPES, and 1% antibiotic/antimycotic were transfected with pSpCas9(BB)-2A-GFP (Addgene, PX458) plasmids using Superfect (Qiagen). The Cas9 plasmids each contained a single gRNA target specific for LIG1 exon 4 (5′-GGGATTCCGAAGCGTCGCAC-3′) or exon 7 (5′-CTCTGAAACGCTTTCCGTCG-3′). sgRNAs were designed with no predicted off-target sites. After 24 hours, GFP+ cells were single-cell sorted into 96-well plates and allowed to grow for 2–3 weeks. Single-cell colonies were expanded and Sanger sequenced to confirm nonsense mutations or frameshift mutations leading to early termination using primers for exon 4 (forward: 5′-GGAATCCAAGTGGGGAGAAAGG-3′; reverse: 5′-AGAGGCCACCCCTGAGTTAG-3′) or exon 7 (forward: 5′-AGCAGGAAAGGAAGAAGGGTC-3′; reverse: 5′-ATATCTTCCATCTGCAGGGTCAG-3′). Immunoblot was used to confirm absence of LIG1 expression in knockout lines.
Generation of stable LIG1 mutant cell lines.
pRetro-Flag plasmids were prepared containing Flag-tagged WT or mutant LIG1 genes. LIG1 patient or control mutations were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent, 200521). Plasmids containing VSV-g, Gag-pol, and WT or mutated LIG1 were transfected into HEK-293T LIG1–/– cells and incubated for 24 hours, after which the culture media was replaced. After an additional 24 hours, supernatants were collected, centrifuged, and filtered through a 0.2-μm syringe filter. Filtered virus was added to 50–100,000 LIG1–/– HEK-293T cells and 4 μg/ml polybrene transfection reagent (EMD Millipore, TR-1003-G). Plates were spun for 45 minutes at 450 g and 37°C, then incubated for 24 hours, after which the culture media was replaced. After an additional 24 hours, 5 μg/ml blasticidin was added to each condition, and selective media was replaced every 3 days. When all cells in the nontransfected condition died, cells from transfected conditions were diluted to 20 cells/ml and plated in 10-cm dishes. Once visible colonies were present in the dishes, 10 colonies per condition were expanded. Immunoblot was used to confirm presence of LIG1 protein and Sanger sequencing was used to confirm specific LIG1 mutations in each line.
Chemical DNA damage.
To examine DNA sensitivity to chemical damage, 4 × 105 lymphoblastoid B cells of kindred A and B were cultured in RMPI1640 media with 10% fetal calf serum and antibiotics, with increasing concentrations (0, 0.2, 0.5, 1, 2, 3 mM) of ethyl methanesulfonate (EMS, Alpha Aeser, A12938) a monofunctional alkylating agent that generates alkylated nucleobases in DNA. B cells in triplicate wells were incubated at 37°C and 5% CO2 for 4 days. Cell survival was determined by adding 40 μl Alamar-Blue reagent (Bio-Rad, BUS012B) to each well, and determining the fluorescence of duplicate 100-μL aliquots in 96-well plates, at excitation wavelength 530–560 nm and emission wavelength 590 nm (POLARstar Omega, BMG Labtech). EMS sensitivity was also examined for ligase-deficient and mutant HEK-293T cells. For this, 200 cells were plated in 2 ml DMEM media in 6-cm plates, allowed to adhere overnight, and then 0, 20, 50, 100, and 200 μM EMS in 4 ml media was added. Plates with cells forming adherent colonies in each condition with 6 replicates in each condition were then washed with 0.9% NaCl, fixed for 1 hour with 2 ml 10% formalin in saline, and then with 2 ml 0.01% Crystal Violet solution. After washing, the plates were inverted, dried, and the colonies counted as described (47). EMS sensitivity was also tested using a comet assay, which allows examination of strand breaks in single eukaryotic cells (48). For this, 2 × 106 cells (HEK-293T cells, LIG1–/–, LIG1–/– cells transduced with either WT LIG1 or the different mutants found in patients) were plated in 60-mm plates, grown to confluency overnight, and then incubated in media containing 0.2, 0.5, or 1.0 mM EMS, or media alone for controls, to test optimum concentrations. After 4, 8, or 16 hours to determine optimum times, cells were immobilized on slides with agarose (CometSlide, Trevigen) lysed with alkali, subjected to electrophoresis, and then stained with SYBR Gold according to manufacturer’s instructions. Cells were examined with a fluorescence microscope (Axioimager Z2M) and the images analyzed using OpenComet Image J (www.opencomet.org). To examine the kinetics of DNA repair, cells were incubated with 0.5 mM EMS (the optimum concentration) for 16 hours (the optimum time), then washed in PBS and replated in fresh media. Cells were then examined by the comet assay again at 0, 4, 8, 12, and 16 hours to compare DNA damage, expressing differences as the mean percentage of DNA in the comet tail in each cell line.
Radiation damage: γH2AX staining.
HEK-293T cells were grown overnight on glass coverslips and then irradiated using a Cesium irradiator. After incubation for 1–5 hours, cells were fixed with 2% paraformaldehyde (PFA) for 5 minutes followed by 4% PFA for 5 minutes and then permeabilized with FACS Permeabilizing Solution 2 (BD Biosciences, 347692) for 20 minutes. Permeabilized cells were blocked using PBS with 2% FBS for 30 minutes and then stained with an anti-γΗ2AX antibody for 90 minutes. Following this, an anti-mouse secondary antibody conjugated with Alexa Fluor 542 was added for 40 minutes. Lastly, cell nuclei were stained with DAPI for 5 minutes. Coverslips were attached to glass slides using FluorSave reagent (EMD Millipore, 345789) and stored at 4°C. γH2AX signal for 50 nuclei per condition was detected using a Zeiss LSM880 confocal microscope and images were analyzed using ImageJ (www.rsbweb.nih.gov/ij/), Zen (Zeiss), and Photoshop (Adobe). Three separate experiments were performed. To examine the effects of radiation on PBMCs, cells were exposed to 10 Gy radiation, fixed after 1, 6, 24, and 40 hours using the Fix&Perm permeabilization kit (Invitrogen/Life Technologies), and then examined by FACS for fluorescence-labeled γH2AX foci in CD3+ T cells, as previously described (27).
LIG1enzymatic activity examined by alanine scanning mutagenesis. pRetro-Flag plasmids containing the genes for WT or mutant LIG1 with alanine replacements were transfected into knockout LIG1 HEK-293T cells using X-tremeGENE HP DNA transfection reagent (MilliporeSigma, 6366244001). After 72 hours, the cells were harvested and lysed on ice with standard lysis buffer. Protein concentration was measured by Bradford assay and equal concentrations of total protein were mixed with anti-Flag magnetic beads (MilliporeSigma, M8823) and incubated overnight. Beads were washed and LIG1 protein was eluted using 3× Flag peptide solution (MilliporeSigma, F4799). Purified LIG1 protein was used in a modified enzymatic activity assay (25). The ligation assay was performed in 50 mM MOPS (pH 7.5), 300 nM nicked DNA substrate, 20 mM MgCl2, 0.1 mM ATP, 1.0 mM DTT, 150 mM NaCl, 0.05 mg/ml BSA. The substrate was annealed by mixing equimolar amounts of the oligonucleotides 5′-CCGAATCAGTCCGACGACGCATCAGCAC, 5′-GTGCTGATGCGTC, and 5′P-GTCGGACTGATTCGG-Cy3 (synthesized by Integrated DNA Technologies) in buffer comprised of 10 mM NaMES (pH 6.5) and 50 mM NaCl, heating to 90°C for 2 minutes, and cooling in sequential steps as follows: 70°C for 2 minutes, 50°C for 2 minutes, 40°C for 2 minutes, 30°C for 2 minutes, 10°C for 2 minutes, and 4°C for 2 minutes. Purified LIG1 (1 μL) was added to ligation buffer (20 μL) and the mixture was incubated for 30 minutes at 37°C. Ligation reactions were quenched by cooling to 4°C followed by addition of 20 μL loading buffer (urea/EDTA). Samples were incubated at 95°C for 5 minutes, rapidly cooled on ice, and separated on denaturing urea acrylamide gels. The Cy3 label was detected using Typhoon Trio Imager (GE Healthcare). Fluorescence intensity was analyzed using ImageJ and the relative activity of each mutant was calculated by dividing by the activity of the WT enzyme under the same conditions.
Generation of recombinant human LIG1 in E. coli.
The plasmid used for expression of the minimal catalytic domain of human LIG1 (232–919) has been previously described (14). Mutant Δ232 LIG1 vectors were constructed using site-directed mutagenesis with synthetic primers, and the mutagenesis was confirmed by sequencing of the coding region. The Δ232 LIG1 constructs were transformed and expressed in BL21(DE3) Rosetta 2 cells. Cells were grown in a shaker at 37°C in Terrific Broth media (Research Products International) supplemented with auto-induction components (49) until they reached an OD600nm of approximately 1. The shaker was then turned down to 16°C and expression proceeded for 16–18 hrs. Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris-Cl at pH 7.5, 10% glycerol, 300 mM NaCl, 5 mM BME, and 0.1% IGEPAL CA-630; Sigma-Aldrich), and frozen at –80°C.
After thawing, protease inhibitors (0.5 mM PMSF, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin A) were added to cells. Cells were lysed by passage through an EmulsiFlex-C3 (Avestin) with 20 kpsi pressure. After centrifugation of the lysate, polyethyleneimine was added to the soluble extract to a final concentration of 0.2%. Following a second centrifugation step, soluble extract was bound to a HisTrap column (GE Healthcare) equilibrated with buffer 1 (20 mM HEPES at pH 7.5, 0.1% IGEPAL CA-630, 500 mM NaCl, 20 mM imidazole). The column was washed with buffer 1, followed by buffer 2 (buffer 1 with 50 mM imidazole). The protein was eluted from the column using a 0%–100% gradient of buffer 3 (20 mM HEPES at pH 7.5, 0.1% IGEPAL CA-630, 100 mM NaCl, 50 mM imidazole) to buffer 4 (buffer 3 with 300 mM imidazole). The His-tag was cleaved using PreScission protease, and a mix of 1 mM ATP and 10 mM MgCl2 was added to the pooled fractions to ensure that Δ232 LIG1 was fully adenylylated. Proteins were further purified with HiTrap Q (GE Healthcare) and HiTrap Blue (GE Healthcare) columns and dialyzed into storage buffer (25 mM at Tris-Cl pH 7.5, 150 mM NaCl, 1 mM DTT, and 0.1 mM EDTA). Protein concentrations were estimated using absorbance at 280 nm. Active enzyme concentrations were determined using single-turnover ligation reactions, in which increasing concentrations of LIG1 were incubated with a fixed concentration of the fluorescein-labeled nicked 28-mer substrate (25).
Gel-based ligation assay.
Ligation assays were carried out at 37°C in a standard reaction buffer consisting of 50 mM NaMOPS at pH 7.5, 1 mM dithiothreitol, 0.05 mg/ml BSA, and sufficient NaCl to maintain a constant ionic strength of 150 mM. Concentrations of ATP, MgCl2, 28-mer substrate, and LIG1 were varied as indicated below. Reactions were quenched in a 50 mM EDTA/90% formamide solution, heated to 95°C, and separated on a 15% (wt/vol) polyacrylamide gel containing 8M urea. The fluorescein-labeled oligonucleotide was detected using a Typhoon Trio+ imager (GE Healthcare), and the gel images were analyzed using ImageQuant TL software (GE Healthcare) as previously described (25). Reported values for all ligation assays are the average of at least 3 independent experiments.
Multiple-turnover ligation assays.
Steady-state DNA dependence was determined in the standard reaction buffer with 200 μM ATP, 20 mM MgCl2, 0.1 nM WT or mutant Δ232 LIG1, and varying amounts of the 28-mer substrate. Steady-state Mg2+ dependence was determined with 100 μM ATP, 500 nM 28-mer substrate, 1 nM enzyme, and varying amounts of MgCl2. Free magnesium concentrations were calculated using the dissociation constants for the ATP-Mg2+ complex (25). Initial rates for all steady-state reactions were determined by linear regression, and the rates were fit with the Michaelis-Menten equation: (Vinit/[E]) = Vmax[S]/(Km + [S]).
For reactions in which an accumulation of both intermediate and product was observed, the initial rates of the formation of each species were determined individually. Ligation efficiency for the Mg2+ dependence was determined by dividing the initial rates of product formation by the sum of the rates of product and intermediate accumulation. To investigate the abortive ligation burden under physiological conditions, 5 nM enzyme was added to a reaction mix containing the standard reaction buffer with 3 mM MgCl2, 2 mM ATP, and 100 nM 28-mer DNA substrate. Initial rates were determined as stated above, and the fraction of abortive ligation was determined by dividing the rate of intermediate accumulation by the sum of the rates of product and intermediate formation: Fraction abortive ligation = (Vintermediate)/(Vproduct + Vintermediate).
PBMC and EBV cell lysis and immunoblotting.
EBV-B cells, PBMCs, or HEK-293T cells were lysed on ice with lysis buffer with Halt Protease Inhibitor and phenylmethylsulfonyl fluoride (both from Thermo Fisher Scientific). Protein lysates were quantified using the Pierce BCA protein quantification assay (Thermo Fisher Scientific). Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Immunoreactive proteins were detected with HRP-coupled secondary antibodies via chemiluminescence. Densitometry of blots was measured using ImageJ.
Gene expression IgH rearrangement and somatic hypermutation in B cells.
RNA was extracted from PBMCs or EBV-immortalized B cells with the RNeasy extraction kit (Qiagen) and reverse-transcribed with a Superscript III first-strand synthesis kit (Invitrogen). LIG1 mRNA was quantified by RT-qPCR in a TaqMan Gene Expression Assay, with normalization against GAPDH probes. For BCR repertoire screening, cDNA from PBMCs from P1, family members, and controls was amplified using primers for the VDJ-VH3 region (forward: 5′-GGGGTACCATGGAGTTTGGGCTGAG-3′; reverse: 5′-GGAATTCTCACAGGAGACGAG-3′) (44). Single clones were selected using TOPO-TA cloning (Thermo Fisher Scientific, K4500J10) and transformed into TOP10 competent cells. Eighty colonies from each individual were selected and amplified using M13 primers (forward: 5′-GTAAAACGACGGCCAG-3′; reverse: 5′-CAGGAAACAGCTATGAC-3′). PCR products were Sanger sequenced (Macrogen USA) and aligned using IgBlast (NCBI). Clones were considered mutated if they contained at least 2 mutated bases in the FWR1, CDR1, FWR2, CDR2, or FWR3 regions.
Statistics.
Data are mean ± SD. Comparing different sets of mutant or native cells under baseline or damaging conditions, the Mann-Whitney nonparametric test was used; for group analysis, the Kruskal-Wallis ANOVA was used. We defined significance as P less than 0.05 and used Prism GraphPad for all analyses.
Study approval.
Human studies were approved by the Institutional Review Boards of the Icahn School of Medicine at Mount Sinai, Great Ormond Street and/or the Weill Cornell Medical Center at Qatar, or the Boston Children’s Hospital. Written informed consent was obtained from the parents of the patients.
Author contributions
PM, BB, JKA, EWG, and AC performed the massively parallel sequencing, whole-exome evaluation, and mutation analyses. PM produced the LIG1–/– cells and mutants and the radiation damage assay on B cells. JC performed structural analyses, comet assays, and damage assays with PC. PM and JC performed functional studies on LIG1–/– cells and mutants. JC performed the alanine scanning studies. PM and LR performed Western blotting and cell cultures. TJJ and PJO produced recombinant enzymes and all functional studies of the LIG1 mutants, effects of magnesium, and provided expertise on LIG1 structural modeling. LDN and KF performed the γH2AX radiation damage study on CD3+ T cells. CCR, PM, AW, JKA, PRR, ME, EWG, TBO, and CD recruited patients and gathered the detailed clinical information needed for this study. CCR conceived the study. PM, CCR, TJJ, PJO, JLC, and EWG wrote the manuscript, which was critically reviewed by all the authors.
Supplementary Material
Acknowledgments
Funding for this study was provided by NIH grants AI-061093, AI-086037, and AI-48693; the David S. Gottesman Immunology Chair at the Icahn School of Medicine at Mount Sinai (to CCR); and in part by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases of the NIH. EWG acknowledges support from Grifols, the Eugene F. and Easton M. Crawford Charitable Lead Unitrust, and the Joanne Siegel Fund. This work was further supported by the Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM) Paris Descartes University, Howard Hughes Medical Institute, and the French National Research Agency (ANR 14-CE15-0009-01). PM acknowledges support from the Medical Sciences Training program (T32 GM 007280) and the Jeffrey Model Foundation.TJJ acknowledges partial support from the Cellular Biotechnology Training Program supported by the National Institute of General Medical Sciences of the NIH (T32 GM008353). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Version 1. 11/05/2018
Electronic publication
Version 2. 12/03/2018
Print issue publication
Funding Statement
National Institutes of Health grants, AI-061093, AI-086037, AI-48693, and David S. Gottesman Immunology Chair at the Icahn School of Medicine at Mount Sinai (CCR), and in part by the Division of Intramural Research of the NIAID of the NIH. EWG acknowledges support from Grifols, the Eugene F. and Easton M. Crawford Charitable Lead Unitrust, and the Joanne Siegel Fund. The work was further supported by the Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM) Paris Descartes University, Howard Hughes Medical Institute, and the French National Research Agency (ANR 14-CE15-0009-01). PM acknowledges partial support from the Medical Sciences Training program, T32007280 and T.J.J. acknowledges partial support from the Cellular Biotechnology Training Program supported by the NIGMS of the National Institutes of Health (GM008353). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
as listed here
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(12):5489–5504.https://doi.org/10.1172/JCI99629.
Contributor Information
Patrick Maffucci, Email: Patrick.Maffucci@mssm.edu.
Jose Chavez, Email: jose.chavez@mssm.edu.
Thomas J. Jurkiw, Email: tjurkiw@umich.edu.
Patrick J. O’Brien, Email: pjobrien@umich.edu.
Jordan K. Abbott, Email: abbottj@njhealth.org.
Paul R. Reynolds, Email: reynoldsp@njhealth.org.
Luigi D. Notarangelo, Email: luigi.notarangelo2@nih.gov.
Kerstin Felgentreff, Email: Kerstin.Felgentreff@gmx.de.
Patricia Cortes, Email: pcortes@med.cuny.edu.
Bertrand Boisson, Email: bebo283@rockefeller.edu.
Lin Radigan, Email: lin.radigan@mssm.edu.
Aurélie Cobat, Email: aurelie.cobat@inserm.fr.
Chitra Dinakar, Email: cdinakar@stanford.edu.
Mohammad Ehlayel, Email: mehlayel@gmail.com.
Tawfeg Ben-Omran, Email: tawben11@hotmail.com.
Erwin W. Gelfand, Email: gelfande@njhealth.org.
Jean-Laurent Casanova, Email: Jean-Laurent.Casanova@mail.rockefeller.edu.
Charlotte Cunningham-Rundles, Email: Charlotte.Cunningham-Rundles@mssm.edu.
References
- 1.Martin, MacNeill SA. ATP-dependent DNA ligases. Genome Biol. 2002;3(4):REVIEWS3005. doi: 10.1186/gb-2002-3-4-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2(4):477–484. doi: 10.1016/S1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 3.van der Burg M, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116(1):137–145. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altmann T, Gennery AR. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. 2016;11(1):137. doi: 10.1186/s13023-016-0520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simsek D, Jasin M. DNA ligase III: a spotty presence in eukaryotes, but an essential function where tested. Cell Cycle. 2011;10(21):3636–3644. doi: 10.4161/cc.10.21.18094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okano S, Lan L, Tomkinson AE, Yasui A. Translocation of XRCC1 and DNA ligase IIIalpha from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005;33(1):422–429. doi: 10.1093/nar/gki190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell. 2007;27(2):311–323. doi: 10.1016/j.molcel.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65(10):4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 9.Tomkinson AE, Mackey ZB. Structure and function of mammalian DNA ligases. Mutat Res. 1998;407(1):1–9. doi: 10.1016/S0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 10.Han L, Masani S, Hsieh CL, Yu K. DNA ligase I is not essential for mammalian cell viability. Cell Rep. 2014;7(2):316–320. doi: 10.1016/j.celrep.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timson DJ, Singleton MR, Wigley DB. DNA ligases in the repair and replication of DNA. Mutat Res. 2000;460(3-4):301–318. doi: 10.1016/S0921-8777(00)00033-1. [DOI] [PubMed] [Google Scholar]
- 12.Masani S, Han L, Meek K, Yu K. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc Natl Acad Sci U S A. 2016;113(5):1261–1266. doi: 10.1073/pnas.1521630113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu G, et al. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative joining. Proc Natl Acad Sci U S A. 2016;113(5):1256–1260. doi: 10.1073/pnas.1521597113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432(7016):473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 15.Lehman IR. DNA ligase: structure, mechanism, and function. Science. 1974;186(4166):790–797. doi: 10.1126/science.186.4166.790. [DOI] [PubMed] [Google Scholar]
- 16.Webster AD, Barnes DE, Arlett CF, Lehmann AR, Lindahl T. Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene. Lancet. 1992;339(8808):1508–1509. doi: 10.1016/0140-6736(92)91266-B. [DOI] [PubMed] [Google Scholar]
- 17.Barnes DE, Tomkinson AE, Lehmann AR, Webster AD, Lindahl T. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell. 1992;69(3):495–503. doi: 10.1016/0092-8674(92)90450-Q. [DOI] [PubMed] [Google Scholar]
- 18.Prigent C, Satoh MS, Daly G, Barnes DE, Lindahl T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol. 1994;14(1):310–317. doi: 10.1128/MCB.14.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teo IA, Arlett CF, Harcourt SA, Priestley A, Broughton BC. Multiple hypersensitivity to mutagens in a cell strain (46BR) derived from a patient with immuno-deficiencies. Mutat Res. 1983;107(2):371–386. doi: 10.1016/0027-5107(83)90177-X. [DOI] [PubMed] [Google Scholar]
- 20.Teo IA, et al. A biochemical defect in the repair of alkylated DNA in cells from an immunodeficient patient (46BR) Carcinogenesis. 1983;4(5):559–564. doi: 10.1093/carcin/4.5.559. [DOI] [PubMed] [Google Scholar]
- 21.Browning SR, Browning BL. Identity by descent between distant relatives: detection and applications. Annu Rev Genet. 2012;46:617–633. doi: 10.1146/annurev-genet-110711-155534. [DOI] [PubMed] [Google Scholar]
- 22.Itan Y, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindahl T. Recognition and processing of damaged DNA. J Cell Sci Suppl. 1995;19:73–77. doi: 10.1242/jcs.1995.supplement_19.10. [DOI] [PubMed] [Google Scholar]
- 24.Soza S, et al. DNA ligase I deficiency leads to replication-dependent DNA damage and impacts cell morphology without blocking cell cycle progression. Mol Cell Biol. 2009;29(8):2032–2041. doi: 10.1128/MCB.01730-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor MR, Conrad JA, Wahl D, O’Brien PJ. Kinetic mechanism of human DNA ligase I reveals magnesium-dependent changes in the rate-limiting step that compromise ligation efficiency. J Biol Chem. 2011;286(26):23054–23062. doi: 10.1074/jbc.M111.248831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul K, et al. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PLoS ONE. 2013;8(3):e59505. doi: 10.1371/journal.pone.0059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felgentreff K, et al. Ligase-4 deficiency causes distinctive immune abnormalities in asymptomatic individuals. J Clin Immunol. 2016;36(4):341–353. doi: 10.1007/s10875-016-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lönn U, Lönn S, Nylen U, Winblad G. Altered formation of DNA in human cells treated with inhibitors of DNA topoisomerase II (etoposide and teniposide) Cancer Res. 1989;49(22):6202–6207. [PubMed] [Google Scholar]
- 29.Bentley DJ, et al. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J Cell Sci. 2002;115(Pt 7):1551–1561. doi: 10.1242/jcs.115.7.1551. [DOI] [PubMed] [Google Scholar]
- 30.Harrison C, Ketchen AM, Redhead NJ, O’Sullivan MJ, Melton DW. Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res. 2002;62(14):4065–4074. [PubMed] [Google Scholar]
- 31.Schellenberg MJ, Tumbale PP, Williams RS. Molecular underpinnings of Aprataxin RNA/DNA deadenylase function and dysfunction in neurological disease. Prog Biophys Mol Biol. 2015;117(2-3):157–165. doi: 10.1016/j.pbiomolbio.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslinia F, Mazza JJ, Yale SH. Megaloblastic anemia and other causes of macrocytosis. Clin Med Res. 2006;4(3):236–241. doi: 10.3121/cmr.4.3.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibata K. Close link between development and function of gamma-delta T cells. Microbiol Immunol. 2012;56(4):217–227. doi: 10.1111/j.1348-0421.2012.00435.x. [DOI] [PubMed] [Google Scholar]
- 34.Xiong N, Raulet DH. Development and selection of gammadelta T cells. Immunol Rev. 2007;215(1):115–31. doi: 10.1111/j.1600-065X.2006.00478.x. [DOI] [PubMed] [Google Scholar]
- 35.Carbonari M, et al. Relative increase of T cells expressing the gamma/delta rather than the alpha/beta receptor in ataxia-telangiectasia. N Engl J Med. 1990;322(2):73–76. doi: 10.1056/NEJM199001113220201. [DOI] [PubMed] [Google Scholar]
- 36.Ehl S, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. 2005;115(11):3140–3148. doi: 10.1172/JCI25221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Villartay JP, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115(11):3291–3299. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picard C, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38(1):96–128. doi: 10.1007/s10875-017-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchbinder D, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. 2015;35(2):119–124. doi: 10.1007/s10875-014-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volk T, et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum Mol Genet. 2015;24(25):7361–7372. doi: 10.1093/hmg/ddv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shovlin CL, et al. Adult presentation of adenosine deaminase deficiency. Lancet. 1993;341(8858):1471. doi: 10.1016/0140-6736(93)90910-9. [DOI] [PubMed] [Google Scholar]
- 42.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16(4):234–246. doi: 10.1038/nri.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maffucci P, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. 2016;7:220. doi: 10.3389/fimmu.2016.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuehn HS, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. 2016;374(11):1032–1043. doi: 10.1056/NEJMoa1512234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 48.Fairbairn DW, Olive PL, O’Neill KL. The comet assay: a comprehensive review. Mutat Res. 1995;339(1):37–59. doi: 10.1016/0165-1110(94)00013-3. [DOI] [PubMed] [Google Scholar]
- 49.Studier FW. Stable expression clones and auto-induction for protein production in E. coli. Methods Mol Biol. 2014;1091:17–32. doi: 10.1007/978-1-62703-691-7_2. [DOI] [PubMed] [Google Scholar]
- 50.Shearer WT, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003;112(5):973–980. doi: 10.1016/j.jaci.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Garcillán B, et al. γδ T Lymphocytes in the diagnosis of human T cell receptor immunodeficiencies. Front Immunol. 2015;6:20. doi: 10.3389/fimmu.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.