

Abstract
Nonalcoholic fatty liver disease (NAFLD) arises from mitochondrial dysfunction under sustained imbalance between energy intake and expenditure, but the underlying mechanisms controlling mitochondrial respiration have not been entirely understood. Heterotrimeric G proteins converge with activated GPCRs to modulate cell-signaling pathways to maintain metabolic homeostasis. Here, we investigated the regulatory role of G protein α12 (Gα12) on hepatic lipid metabolism and whole-body energy expenditure in mice. Fasting increased Gα12 levels in mouse liver. Gα12 ablation markedly augmented fasting-induced hepatic fat accumulation. cDNA microarray analysis from Gna12-KO liver revealed that the Gα12-signaling pathway regulated sirtuin 1 (SIRT1) and PPARα, which are responsible for mitochondrial respiration. Defective induction of SIRT1 upon fasting was observed in the liver of Gna12-KO mice, which was reversed by lentivirus-mediated Gα12 overexpression in hepatocytes. Mechanistically, Gα12 stabilized SIRT1 protein through transcriptional induction of ubiquitin-specific peptidase 22 (USP22) via HIF-1α increase. Gα12 levels were markedly diminished in liver biopsies from NAFLD patients. Consistently, Gna12-KO mice fed a high-fat diet displayed greater susceptibility to diet-induced liver steatosis and obesity due to decrease in energy expenditure. Our results demonstrate that Gα12 regulates SIRT1-dependent mitochondrial respiration through HIF-1α–dependent USP22 induction, identifying Gα12 as an upstream molecule that contributes to the regulation of mitochondrial energy expenditure.
Keywords: Hepatology, Metabolism
Keywords: Fatty acid oxidation, G-proteins, Obesity
Introduction
The liver plays a major role in maintaining whole-body energy balance by regulating lipid metabolism (1, 2). Upon changes in nutrient availability following food intake, hepatic lipid metabolism is tightly controlled through fine-tuning regulation of both fatty acid (FA) oxidation and lipogenesis, which is an essential process for the maintenance of metabolic homeostasis under physiological and pathological conditions. When this equilibrium is disturbed by excess caloric supply and impaired energy expenditure due to mitochondrial dysfunction, ectopic lipid is accumulated within hepatocytes, favoring hepatic steatosis as an early risk factor for the development of nonalcoholic fatty liver disease (NAFLD) (2–4).
Both prolonged fasting and Western dietary intake share a common metabolic feature in terms of increased concentrations of FA serving as a major fuel source. In the liver under starvation conditions, when the glycogen stores are depleted with the inhibition of lipogenesis, FAs mobilized from adipose tissues are oxidized primarily in mitochondria to produce ketone bodies and/or reesterified into triglyceride (TG) for storage. In contrast, impaired mitochondrial FA oxidation in the liver is frequently observed along with increased de novo synthesis of FA in pathologic situations, such as insulin resistance and obesity, indicating that mitochondrial capacity to oxidize FA plays a key role in modulating lipid metabolism. Thus, identification of the signaling node or nodes regulating mitochondrial FA oxidation is warranted for the treatment of NAFLD. However, the pathways that control mitochondrial FA utilization in response to varying physiologic conditions are not entirely defined yet.
G proteins represent major molecular switches that converge varying cell-surface signals from activated GPCRs upon diverse extracellular stimuli. Over 800 different genes encode GPCRs in humans, whereas only approximately 20 genes encode G proteins, implying a converging role of G proteins for signal transduction. The G protein α subunit, a component of heterotrimeric G proteins, can be classified largely into Gs, Gi/o, Gq, and G12. Although the roles of Gs, Gi/o, and Gq have been well characterized, G12 family members were identified relatively recently, and their functions have been uncovered at a slower pace (5). Gα12 is ubiquitously expressed in metabolic organs, including the liver (6). In particular, Gα12 has drawn considerable interest in the field of cancer biology due to its triggering effect on cell growth and oncogenic transformation (7, 8). Moreover, Gα12 was overexpressed in highly proliferating cancer cells (8–11). Interestingly, a considerable portion of endogenous Gα12, but not other Gα subunits, is physically associated with mitochondria (12), raising the possibility that Gα12 is associated with mitochondrial function (e.g., mitochondrial energy metabolism) more directly than other Gα proteins. Given that mitochondrial activity favors cancer cell growth (13), it is presumed that Gα12 may also contribute to energy metabolism in normal cells under pathophysiological conditions. However, the metabolic impact of Gα12 signaling in cellular energy balance has remained unexplored, although recent studies have investigated the roles of a few other G proteins in lipid and/or glucose metabolism.
Sirtuin 1 (SIRT1), a NAD+-dependent protein deacetylase, plays a role in the regulation of the transcriptional network in various metabolic processes, especially FA oxidation (14, 15). However, the upstream regulator linking cell-surface signaling and SIRT1 is incompletely understood. In the present study, cDNA microarray analysis using the liver of Gna12-KO mice enabled us to define SIRT1, PPARα, and PPARγ coactivator 1α (PGC1α) as the “core partners” for the regulation of genes responsible for mitochondrial respiration controlled by Gα12; ablation or knockdown of the Gα12 gene suppressed SIRT1 induction by fasting and its downstream mitochondrial target genes associated with FA oxidation. Consistently, Gna12-KO mice subjected to fasting showed increased TG accumulation in the liver compared with WT mice, and this change was normalized by hepatocyte-specific Gα12 overexpression. Mechanistically, we revealed that Gα12 promotes SIRT1 stability by inducing ubiquitin-specific peptidase 22 (USP22) through HIF-1α, unraveling the regulatory role of Gα12 in SIRT1 expression. Furthermore, we found that high-fat diet–fed (HFD-fed) Gna12-KO mice were prone to hepatic steatosis and obesity due to decreases in energy expenditure. In line with this, we observed that Gα12 levels were markedly diminished in patients with either simple steatosis or nonalcoholic steatohepatitis (NASH) as compared with individuals without steatosis. Our findings show that Gα12 signaling controls lipid metabolism through the regulation of the HIF-1α/USP22/SIRT1 axis, revealing its regulatory role in energy expenditure.
Results
Ablation of Gna12 augments fasting-induced liver steatosis in mice.
A sustained fasting condition promotes liver steatosis, as FA derived mainly from adipose tissues are being accumulated (16). To investigate whether Gα12 levels change depending on nutritional status, we first assessed the effect of fasting on Gα12 in mouse liver. Of note, fasting of WT mice for 24 to 48 hours markedly enhanced Gα12 expression in the liver (Figure 1, A and B), which is suggestive of the role of Gα12 signaling in lipid metabolism. To better understand the metabolic impact of Gα12 on the physiological adaptation to fasting, we then analyzed the lipid profiles in the liver of Gna12-KO mice subjected to fasting for 24 hours. Gna12-KO mice displayed a significant increase in liver fat accumulation compared with WT mice, as revealed by both histochemical and biochemical analyses for lipids (Figure 1, C–E). In contrast, serum TG and cholesterol levels were lower in fasted Gα12-KO mice presumably due to diminished fat secretion from hepatocytes (Figure 1F). Referring to the published literature, the distribution of genotypes from the offspring of Gna12+/– intercrosses was Mendelian, and mice with either heterozygous or homozygous deletion of Gna12 were fertile without apparent morphological or behavioral abnormalities (17). In order to provide insight into the physiological relevance of the Gα12-signaling pathway in our experimental model, male mice heterozygous for Gna12 deficiency (Gna12 Het mice) (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI97831DS1) were additionally subjected to fasting for 24 hours together with WT and Gna12-KO mice to compare hepatic lipid profiles between genotypes. As expected, the partial effect of heterozygous deletion of Gna12 was corroborated in the context of hepatic lipid metabolism, as assessed by oil red O staining of liver sections and TG measurements (Supplemental Figure 1B). These results indicate that Gα12 signaling may be adaptively increased under fasting conditions, whereas a deficiency in Gα12 renders the liver more susceptible to fat accumulation.
Figure 1. Association of Gα12 signaling with fasting-induced liver steatosis.

(A) qRT-PCR assays for Gna12 in the liver from 10-week-old mice fed ad libitum or fasted for 24 hours (n = 4–6/group). Rel., relative. (B) Immunoblotting for Gα12 in liver homogenates from WT mice fed ND ad libitum or fasted for indicated times. Blots were run in parallel using the same samples. (C) Representative gross appearance of liver tissues from the mice shown in A (n = 3/group). (D) Representative H&E staining (left; n = 5/group) and oil red O staining (right; n = 3/group) of the liver sections. Scale bars: 100 μm. (E) Hepatic TG contents (n = 5/group). (F) Serum TG and total cholesterol levels (n = 5/group). Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (A) or ANOVA, followed by LSD post hoc tests (E and F).
Gα12 regulation of SIRT1 contributes to FA oxidation in mitochondria via the PPARα network.
In an effort to find the molecules regulated by the Gα12 pathway, we performed cDNA microarray analyses using Gna12-KO liver tissue. First, our analysis of the PANTHER Gene Ontology (GO) term demonstrated that the “metabolic process” pathway was notably altered in Gna12-KO livers (Figure 2A). Similarly, our additional GO analysis for identical data sets using the DAVID bioinformatics program verified that ablation of Gna12 caused downregulation of 4 major signaling pathways: DNA metabolism, lipid biosynthesis, amine catabolism, and DNA repair (Figure 2B). Since Gna12-KO mice did not show obvious growth retardation or any other developmental defects, which may reflect abnormal DNA metabolism (18), we focused on lipid metabolism, particularly alterations in the expression of clusters of genes involved in FA oxidation, with the aim of understanding the basis of altered lipid profiles observed in Gna12-KO mice. Thorough analysis of the microarray results enabled us to find PPARα target genes as one of the major pathways suppressed by Gna12 KO (Figure 2C). In the analysis of the gene network using the STRING database, SIRT1, PPARα, and PGC1α as “core partners” were found to be closely interconnected with a subset of genes affected by Gna12 deficiency (Figure 2D). Of those linked to the core network, the genes associated with lipid catabolism, acyl-CoA metabolism, ketogenesis, and peroxisomal oxidation processes were all markedly suppressed.
Figure 2. Gα12 regulation of mitochondrial respiration via SIRT1/PPARα network.

(A) PANTHER pathway analysis in the cDNA microarrays performed using RNA samples extracted from the livers of 8-week-old male WT or Gna12-KO mice that had been fasted overnight before sacrifice (n = 3/group). Percentage of number of genes that belong to respective pathway categories over total number of genes analyzed is shown. (B) GO analysis of major signaling pathways in cDNA microarrays using the DAVID bioinformatics database. (C) Heatmap of the genes associated with energy metabolism in the same cDNA microarrays used for A. The log2 ratios of Gna12-KO/WT were presented using heatmap (blue, underexpression; red, overexpression). (D) Core network analysis associated with the SIRT1/PPARα pathway. PPARα-associated genes affected by Gna12 KO are represented as colored circles and assigned to specific subcategories. Genes upregulated (red circles) or downregulated (blue circles) in the microarrays are shown for each subcategory. Line thickness represents the strength of evidence provided by the STRING database.
We then narrowed our focus to the regulatory potential of Gα12 on SIRT1 and found that SIRT1 levels were distinctly reduced in livers deficient in Gα12, whereas other isoforms associated with mitochondrial function (i.e., SIRT3 and SIRT5) were not (or minimally if at all) affected (Figure 3A) (19). Similar results were obtained in the experiments using primary hepatocytes (Figure 3B). Consistently, infection of HepG2 cells with an adenoviral construct encoding for a constitutively active mutant of Gα12 (Ad-Gα12QL) increased SIRT1 levels, whereas shRNA-mediated stable knockdown of the Gα12 gene in AML12 cells showed the opposite effect (Figure 3B). Carnitine palmitoyl transferase-1 (CPT1) and PGC1α levels were also diminished in the liver or in primary hepatocytes (Figure 3C), indicating that Gna12 ablation might cause a decrease in mitochondrial lipid oxidation. Among the members existing in the core network controlled by SIRT1, attention was paid to PPARα because it is a transcription factor that globally regulates genes associated with FA oxidation in physiologic situations (20). PPARα target gene transcripts responsible for FA oxidation were substantially downregulated (Figure 3D), which was consistent with the inhibition of SIRT1 and PGC1α. In line with this, the oxygen consumption rate (OCR) in mitochondrial fractions prepared from the liver tissue (Figure 3E) and palmitate oxidation in primary hepatocytes were also decreased (Figure 3F). Our results corroborate the role of Gα12 in the regulation of FA oxidation, which is controlled by PPARα target gene products in conjunction with SIRT1.
Figure 3. Gα12 regulation of SIRT1-dependent mitochondrial respiration in the liver.

(A) SIRT1 inhibition by Gna12 KO. Immunoblottings for SIRT1, SIRT3, and SIRT5 were performed using liver homogenates from 14-week-old WT or Gna12-KO mice fed ND (upper panel). Lower panel shows quantification (n = 3/group). (B) Effects of Gα12 modulation on SIRT1 levels. Immunoblottings for SIRT1 were performed (upper) and quantified (lower) using primary hepatocytes from WT or Gna12-KO mice (left, n = 3/group), HepG2 cells infected with Ad-Gα12QL or control (Ad-Con) (middle, n = 4/group), or AML12 cells stably expressing sh-Gα12 or control (sh-Luci) (right, n = 3/group). (C) Immunoblottings for CPT1 and PGC1α in liver or primary hepatocytes from WT or Gna12-KO mice (upper) and their respective quantifications (lower, n = 3/group each). (D) qRT-PCR assays for PPARα target genes responsible for FA oxidation in the liver or primary hepatocytes (n = 3–11/group). (E) OCR in mitochondria. OCR was measured using the mitochondrial fraction prepared from liver tissues of WT or Gna12-KO mice (n = 3/group). Analyzed OCR was normalized to the protein concentrations for each set of samples determined by the Bradford method. (F) Palmitate oxidation in primary hepatocytes. [3H]-palmitate oxidation rate was determined using primary hepatocytes from WT or Gna12-KO mice, and 5 × 105 cells per well were cultured in 12-well plates. Data shown are from 1 representative experiment of 2 independent experiments (n = 3 mice/group). Each dot represents an individual pool of primary hepatocytes isolated from each mouse. Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (A–F). For A–C, the blots in each panel were run in parallel using the same samples, and β-actin was used as a normalization control for densitometric analysis. For D, box-and-whisker plots show median (horizontal lines within boxes), 5%–95% (ends of the boxes), and range of minimum to maximum.
Gna12 ablation suppresses SIRT1 along with enhanced fat accumulation under fasting conditions.
During the period of calorie restriction or fasting, metabolic adaptations occur in various organs by changing a large subset of genes necessary for maintaining energy homeostasis. Given that SIRT1 is induced in the fasting state as a core regulator of lipid metabolism (21, 22), we examined the effect of Gna12 ablation on adaptive change in SIRT1 under fasting conditions. While fasting of WT animals for 24 hours markedly increased SIRT1 and CPT1 levels in the liver, Gna12 KO completely prevented this effect (Figure 4A). In Gna12 Het mice, the protein levels were partially diminished, strengthening the functional relevance of Gα12 signaling in our experimental model (Supplemental Figure 1C). In addition, the fasting-inducible transcript levels of Acadl and Acadm were diminished in the liver of Gna12-KO mice (Figure 4B). Moreover, Gna12 KO lowered basal or fasting-inducible SIRT1 expression in skeletal muscle and brown adipose tissue; although the fasting effect on SIRT1 in white adipose tissue seemed to be relatively mild, the inhibitory effect of Gna12 KO on SIRT1 was also observed in this tissue (Supplemental Figure 2). To further evaluate the regulatory role of Gα12 in lipid metabolism, WT mice were hydrodynamically injected with a plasmid (50 μg) encoding shRNA-Gα12 (sh-Gα12) or shRNA-nontargeting control luciferase (sh-Luci) via the tail vein for knockdown of Gα12 in the liver (10). As expected, mice injected with sh-Gα12 plasmid exhibited diminished SIRT1 and CPT1 expression in association with increased liver TG content upon fasting compared with mice injected with the sh-Luci plasmid (Figure 4, C and D). Next, we employed an albumin promoter–driven lentiviral Gα12 delivery system to validate the link between Gα12 and SIRT1 and to exclude off-target effects. Enforced expression of Gα12 specifically in hepatocytes caused recovery of SIRT1 and CPT1 expression in the liver of Gna12-KO mice under fasting conditions (Figure 4E). Similarly, hepatic lipid accumulation in the animals was notably attenuated by the lentiviral gene delivery (Figure 4F). These results indicate that Gα12 regulates levels of SIRT1 and, consequently, its downstream molecules responsible for mitochondrial FA oxidation.
Figure 4. Lack of fasting induction of SIRT1 by Gna12 KO.

(A) Abrogation of SIRT1 and CPT1 induction upon fasting by Gna12 KO. Immunoblottings for SIRT1 and CPT1 were performed and quantified on the liver homogenates from 12-week-old mice fed ad libitum, followed by fasting and refeeding for 24 hours (n = 4–5/group). (B) qRT-PCR assays for Acadl and Acadm in the liver (n = 5/group). (C) Effect of hepatic Gα12 gene knockdown on fasting induction of SIRT1. Immunoblottings for SIRT1 and CPT1 (center) in the liver homogenates and SIRT1 quantification (far right). Mice at 8 weeks of age were subjected to hydrodynamic injection with the plasmid-expressing sh-Gα12 or control (sh-Luci) (n = 4–6/group) (left). Third panel shows qRT-PCR assay for Gna12 in the liver (n = 4/group). (D) Representative H&E staining (left) and hepatic TG contents (right) from the same mice as in C (n = 4–6/group). Scale bars: 100 μm. (E) Effect of hepatocyte-specific Gα12 overexpression on fasting induction of SIRT1. Eight-week-old WT or Gna12-KO mice were injected with Lv-Gα12alb (or control) via the tail vein (left). Immunoblottings for SIRT1 and CPT1 were done on the liver homogenates (center) and SIRT1 quantification (right). Mice were subjected to fasting as in A. (n = 4/group). Third panel shows qRT-PCR assay for Gna12 in the liver (n = 7–10/group). (F) Representative H&E staining (left) and hepatic TG contents (right) from mice as described in E (n = 4–6/group). For E and F, only fasted groups were analyzed for ease of data presentation. Scale bars: 100 μm. Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (C and E, mRNA levels) or ANOVA followed by LSD (A and D) or Bonferroni’s (B, C, E, and F) post hoc tests. For A as well as C and E (protein levels), the blots in each panel were run in parallel using the same samples and β-actin was used as a normalization control for densitometric analysis.
HIF-1α–mediated USP22 induction contributes to SIRT1 upregulation by Gα12.
Nutritional status modulates SIRT1 levels through transcriptional and/or posttranslational mechanisms (23). Hepatic NAD+ and NADH contents reciprocally modulate SIRT1 transcript levels (24). In our study, however, Gna12-KO mice showed no changes in Sirt1 mRNA, NAD+, and NADH in the liver (Supplemental Figure 3, A and B), raising the idea that Gα12 may posttranslationally regulate SIRT1. Consistently, pyruvate and lactate levels, which affect NAD+/NADH and SIRT1 de novo synthesis (21), were not changed (Supplemental Figure 3B). In an effort to find the molecule or molecules responsible for SIRT1 regulation, we checked the effect of Gα12 on the stability of SIRT1 and found that Ad-Gα12QL infection not only attenuated the intensities of ubiquitinated SIRT1, but enhanced SIRT1 stability in HepG2 cells, as fortified by the outcome of an experiment using cycloheximide (Figure 5A). These results support the concept that Gα12 regulation of SIRT1 may result from modulation of protein ubiquitination.
Figure 5. Gα12 regulation of SIRT1 via HIF-1α–mediated induction of USP22.

(A) Inhibition of SIRT1 ubiquitination and degradation by Gα12. SIRT1 immunoprecipitates from HepG2 cells infected with Ad-Gα12QL (or Ad-Con) were immunoblotted for ubiquitin (left) and quantified (middle, n = 3). In another experiment, HepG2 cells were treated with 10 μM cycloheximide for indicated times (right, n = 3). (B) Effect of USP22 gene silencing on inhibition of SIRT1 ubiquitination by Gα12. (C) qRT-PCR assays for Usp22 in the liver (left, n = 5/group) or primary hepatocytes (right, n = 4/group). (D) Luciferase reporter assays for USP22 promoter activity in Gα12-overexpressed AML12 cells. The result shown is combined from 3 independent experiments (n = 6–8 replicates/group for each experiment). Box-and-whisker plot shows median (horizontal lines within boxes), 5%–95% percentile (ends of the boxes), and range of minimum to maximum values (whiskers). Each dot represents an outlying value. Mut1 or Mut2, promoter-reporter constructs with deletion of respective HIF-1α response element sites. (E) Increase in SIRT1 level by Gα12 overexpression through HIF-1α/USP22 axis (right, n = 4/group). (F) Effect of HIF-1α or USP22 gene silencing on SIRT1 induction by Gα12. (G) Effect of RhoA/Rock pathway inhibition on HIF-1α induction by Gα12. (H) Effect of Gα12 overexpression in the liver on HIF-1α/USP22/SIRT1 axis. Immunoblottings were done on the liver homogenates obtained from mice as in Figure 4E. (I) Effect of Gα12 overexpression in hepatocytes on HIF-1α/USP22/SIRT1 axis. Immunoblottings were done on mouse primary hepatocytes infected with Ad-Gα12QL (or Ad-Con) and quantified (n = 3/group). Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (A, C, D, and E) or ANOVA followed by Bonferroni’s post hoc test (I). For A, B, and E–I, blots in each panel were run in parallel using the same samples and β-actin was used as a normalization control for densitometric analysis.
Based on the report that USP22 deubiquitinates SIRT1 for stabilization (25), we determined whether Gα12 signaling regulates SIRT1 ubiquitination via USP22. The effect of Gα12 overexpression on SIRT1 ubiquitination was assessed in HepG2 cells deficient in USP22 (siRNA knockdown). As expected, USP22 silencing prevented Ad-Gα12QL from lowering the intensities of ubiquitinated SIRT1 (Figure 5B). In line with this, Gna12-KO mice displayed a decrease in Usp22 mRNA in the liver or primary hepatocytes (Figure 5C), demonstrating that Gα12 signaling regulates SIRT1 ubiquitination through USP22. To find putative transcription factor or factors for USP22 expression downstream from Gα12, we next used the PROMO analysis program and predicted HIF-1α as a candidate interacting with DNA-binding sites located in the promoter region of Usp22 (Figure 5D). In luciferase reporter assays using a construct containing the –2.2 kb region of Usp22 and its hypoxia regulatory element mutant constructs, the 2 DNA-binding sites located at –539/–535 bp and –287/–283 bp were functionally active (Figure 5D). In parallel, Ad-Gα12QL infection augmented SIRT1 levels in HepG2 cells, and this event depended on HIF-1α or USP22, as evidenced by the results of siRNA knockdown experiments (Figure 5, E and F). In line with several published reports (26–28), inhibition of the RhoA/Rock pathway attenuated the Gα12 overexpression effect on HIF-1α expression (Figure 5G). Consistently, hepatocyte-specific lentiviral delivery of Gα12 in Gna12-KO mice facilitated upregulation of HIF-1α, USP22, and SIRT1 in the liver (Figure 5H), as corroborated in the experiments using primary hepatocytes from WT or Gna12-KO mice subjected to Ad-Gα12QL infection (Figure 5I).
To verify the signaling proposed in this study, we performed a hydrodynamic injection of either human USP22 overexpression plasmid or control vector (mock) into Gna12-KO mice via tail vein; Gna12-KO mice injected with USP22 plasmid displayed enhanced SIRT1 expression along with attenuated liver TG accumulation upon fasting, compared with Gna12-KO mice injected with mock vector (Figure 6, A and B). To strengthen our contention that SIRT1 levels decreased by Gna12-KO may contribute to hepatic steatosis, we examined the effect of SIRT1 overexpression on changes in fat accumulation in the liver of Gna12-KO mice under fasting conditions. As expected, Gna12-KO mice exhibited decreased hepatic SIRT1 and CPT1 levels compared with WT controls under fasting conditions, which was reversed by SIRT1 overexpression (Figure 6C). Likewise, hepatic lipid accumulation augmented by Gna12 KO was significantly attenuated by SIRT1 overexpression (oil red O staining of liver sections and hepatic TG assays) (Figure 6D). To confirm the role of SIRT1 in the Gα12-signaling pathway in vitro, we additionally measured OCR in AML12 cells stably expressing sh-Gα12 or control (sh-Luci); Gα12 knockdown notably suppressed mitochondrial OCR (i.e., basal, ATP linked, and maximal respiration), which returned to control levels by SIRT1 overexpression (Figure 6E). Similarly, overexpression of SIRT1 sufficiently rescued the phenotype of Gna12-KO hepatocytes, as proven by diminished lipid accumulation after palmitate treatment (Supplemental Figure 4A). We additionally attempted to examine the effect of Ad-SIRT1 infection on mitochondrial FA oxidation in Gna12-KO primary hepatocytes; only a slight increase was found in this experiment, presumably due to insufficient SIRT1 overexpression (and CPT1 also) in Gna12-KO hepatocytes as compared with WT cells (Supplemental Figure 4B). Taken together, these results provide strong evidence that Gα12 signaling facilitates USP22 expression through HIF-1α and that induced USP22 stabilizes SIRT1 protein.
Figure 6. Rescue of metabolic phenotype of Gna12 KO by overexpression of USP22 or SIRT1.

(A) Effect of hepatic USP22 overexpression on SIRT1 induction by fasting. Immunoblotting for SIRT1 and USP22 (center) in the liver homogenates and SIRT1 quantification (far right). WT and Gna12-KO mice at 12 weeks of age were hydrodynamically injected with the plasmid expressing USP22 or control vector (Mock) (n = 3–5/group) (left). Third panel shows densitometric analysis for USP22 in the liver (n = 3–5/group). (B) Representative oil red O staining (left) and hepatic TG contents (right) (n = 3–5/group). Scale bars: 100 μm. (C) Effect of hepatic SIRT1 overexpression on CPT1 induction by fasting. Immunoblotting for SIRT1 and CPT1 (center) in the liver homogenates and their respective quantifications (right) (n = 3–4/group). WT and Gna12-KO mice at 15 weeks of age were injected with the adenovirus carrying mouse SIRT1 (Ad-SIRT1, 2.8 × 109 PFU/mouse) or GFP control (Ad-Con)via the tail vein (left). (D) Representative oil red O staining (left) and TG contents (right) in liver tissues (n = 3–4/group). Original magnification, ×20. (E) Effect of SIRT1 overexpression on OCR in AML12 cells. OCR was measured in AML12-sh-Gα12 (or AML12-sh-Luci) cells infected with Ad-SIRT1 (or Ad-Con) in the presence of oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (1 μM), or rotenone plus antimycin A (0.5 μM each). Results represent 4 independent experiments (n = 6–8 replicates/group for each experiment). Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (A, USP22) or ANOVA followed by LSD (A [SIRT1], C, and E) or Bonferroni’s (B and D) post hoc tests. For A–D, only fasted groups were analyzed for ease of data presentation. For A and C, blots in each panel were run in parallel using the same samples and β-actin was used as a normalization control for densitometric analysis.
HFD feeding renders Gna12-KO mice highly susceptible to liver steatosis.
To understand the role of Gα12 in energy metabolism in the setting of metabolic excess, we examined Gα12 levels in the livers of both human subjects with NAFLD and obese animal models. In cohort no. 1, NAFLD patients with either steatosis or steatohepatitis exhibited apparent, but not statistically significant, decreases in hepatic GNA12 mRNA levels as compared with those in normal subjects (Figure 7A). To strengthen the clinical relevance of our finding, we additionally assessed Gα12 protein levels using a separate set of human liver specimens with varying degrees of hepatic steatosis (cohort no. 2). Of note, Gα12 protein levels were markedly lowered in livers of patients having either simple steatosis or NASH as compared with individuals without steatosis (Figure 7A). However, GNA12 mRNA and its protein levels tended to slightly decrease in the livers of HFD-fed mice (Figure 7B). In primary hepatocytes from HFD-fed mice, Gα12 protein levels were notably decreased as compared with those of normal chow diet–fed (ND-fed) control (Figure 7B).
Figure 7. Augmented liver steatosis and decreased FA oxidation in Gna12-KO mice fed HFD.

(A) qRT-PCR assays for GNA12 (left, n = 6–15/group for cohort no. 1), immunoblotting for Gα12 (middle, n = 2–5/group for cohort no. 2), and quantification of Gα12 levels (right, cohort no. 2) in human liver specimens obtained from NAFLD patients or normal subjects. Each dot represents an individual subject. (B) qRT-PCR assays and immunoblottings for Gα12 in mouse liver. Gna12 mRNA levels were analyzed in the liver of mice fed HFD (or ND) for 8 weeks (n = 3 or 12/group for set no. 1) or 9 weeks (n = 8 or 16/group for set no. 2) or ob/ob or db/db mice (n = 5/group). For immunoblottings, Gα12 levels were assessed in the liver or primary hepatocytes from mice as in set no. 2 and quantified (n = 3/group). (C) Representative H&E staining (left) or oil red O (right) staining of the liver tissue from WT or Gna12-KO mice fed HFD for 16 weeks (n = 3/group). Scale bars: 100 μm. (D) Hepatic TG contents in WT or Gna12-KO mice as in C (n = 7/group). (E) Immunoblottings for SIRT1 in the liver (upper panel), skeletal muscle (middle panel), or white adipose tissue (lower panel) from the same mice as in C. (F) qRT-PCR assays for transcripts associated with FA oxidation in the liver (n = 6–14/group), skeletal muscle (n = 6–10/group), or white adipose tissue (n = 4–12/group) from the same mice as in C. Values represent mean ± SEM. Data were analyzed by 2-tailed Student’s t test (A, B, D, and F). For A, B, and E, blots were run in parallel using the same samples and β-actin was used as a normalization control for densitometric analysis. For A and F, box-and-whisker plots show median (horizontal lines within boxes), 5%–95% percentile (ends of the boxes), and range of minimum to maximum values (whiskers).
Next, we monitored the effect of Gna12 KO on liver steatosis and changes in the expression of genes responsible for FA oxidation. HFD-fed Gna12-KO mice displayed profound fat accumulation in the liver (Figure 7C). Consistently, hepatic TG contents as well as serum LDL cholesterol levels were significantly elevated (Figure 7D and Table 1). Of note, serum liver enzyme activities (e.g., alanine transaminase [ALT], aspartate transaminase [AST], and lactate dehydrogenase [LDH]) and other serum lipid parameters (e.g. total cholesterol, HDL cholesterol, TG, and free FA contents) were decreased in HFD-fed Gna12-KO mice (Table 1), presumably due to decreased production of inflammatory mediators in other cell types (29). In line with this, an additional lipidomic analysis from HFD-fed Gna12-KO mice showed decreases in ceramide and/or sphingolipid contents in plasma (Supplemental Figure 5), supporting our view that overall inflammatory responses diminished in whole-body Gna12-KO mice. Several lines of evidence clearly demonstrate that the JNK pathway plays a role in inflammation, contributing to metabolic disease, including obesity and insulin resistance (30–32). Based on the notion that Gα12 signaling controls JNK activity (33, 34), we examined whether Gα12 gene knockdown attenuates palmitate-induced apoptosis. As expected, AML12 cells deficient in Gα12 (AML12-sh-Gα12) displayed a significant decrease in cytotoxicity upon palmitate treatment (MTT assays) (Supplemental Figure 6A). In parallel with this, cleaved caspase-3 and phosphorylated JNK levels were lowered (Supplemental Figure 6B). Palmitate treatment inhibited Akt phosphorylation (i.e., cell viability marker) to a lesser degree in Gα12 gene knockdown cells than in control cells (Supplemental Figure 6B). These outcomes support the possibility that decreased JNK activity might account for attenuated liver injury in Gna12-KO mice fed on HFD.
Table 1. Serum metabolic profiles in WT and Gna12-KO mice fed either ND or HFD for 16 weeks.
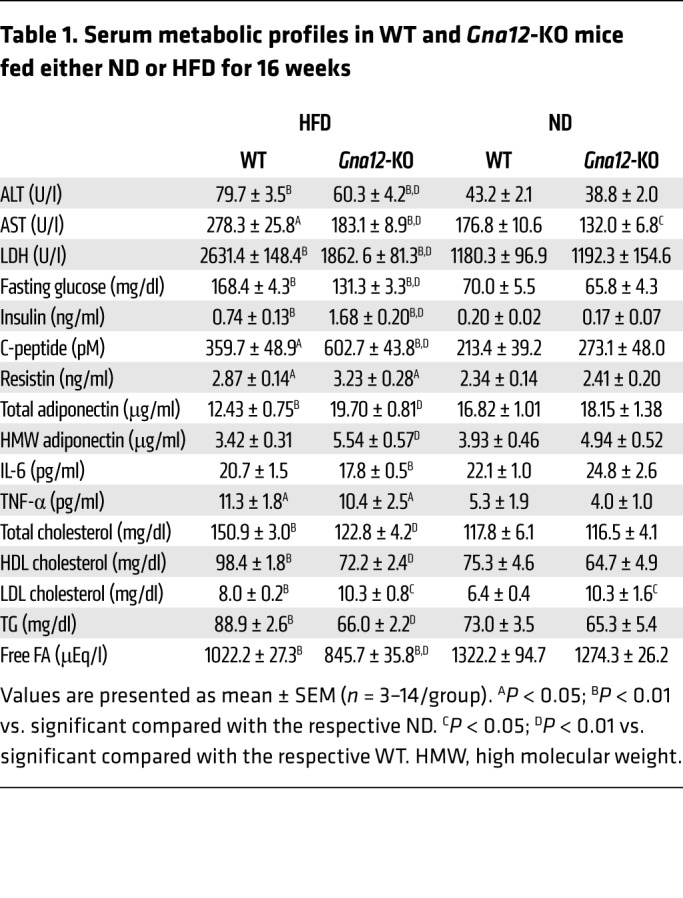
The energy metabolizing capacity in organs is governed by a highly dynamic transcriptional network. Based on our finding from microarray analysis that Gα12 regulates the PPARα target gene network, which includes SIRT1 (Figure 2), we measured SIRT1 levels in metabolic tissues from WT and Gna12-KO mice fed HFD. Hepatic SIRT1 levels were markedly lowered in Gna12-KO mice without significant differences in Sirt1 mRNA, NAD+, and NADH contents (Figure 7E and Supplemental Figure 3). The transcript levels of lipid oxidation genes were notably suppressed in the livers of HFD-fed Gna12-KO mice (Figure 7F), whereas those of lipogenic genes were minimally or moderately enhanced, presumably due to adaptive changes (Supplemental Figure 7). Similar results were observed in skeletal muscle and white adipose tissue (Figure 7, E and F), strengthening the concept that a deficiency in Gα12 exacerbates HFD-induced hepatic steatosis as a consequence of decreases in mitochondrial lipid oxidation.
Gna12 KO does not interfere with glucose metabolism and insulin sensitivity.
Since liver steatosis is strongly associated with insulin resistance, which contributes to the adverse consequences of metabolic syndrome (2–4), we further assessed glucose tolerance and insulin sensitivity using the animal model to see whether Gα12 also controls glucose metabolism. In glucose- or insulin-tolerance tests, time courses of blood glucose levels were slightly different between HFD-fed Gna12-KO mice and the corresponding WT mice (Supplemental Figure 8, A and B). In the hyperinsulinemic-euglycemic clamp experiment, the glucose infusion rate required to maintain euglycemia during the clamp was rather weakly enhanced at early times in HFD-fed Gna12-KO mice, although this trend was lost at later steady state (Supplemental Figure 8C). The glucose-production rate in the liver at either the basal state or under clamped conditions was not changed in the animals (Supplemental Figure 8D). In addition, there were no differences in whole-body glucose flux comprising glucose uptake, glycolysis, and glycogen synthesis (Supplemental Figure 8E). However, it is noteworthy that Gna12-KO mice fed HFD exhibited lower fasting glucose with hyperinsulinemia compared with WT mice (Table 1). Based on the recent study demonstrating that JNK activation in pancreatic β cells deregulates glucose-stimulated insulin secretion (35), we hypothesized that Gα12 gene deletion might affect JNK-dependent signaling pathways in β cells, since we used a whole-body gene-ablation model. Therefore, we assessed the effect of Gα12 overexpression on insulin secretion from Min6 cells (a mouse insulinoma-derived cell line displaying characteristics of pancreatic β cells). As expected, Gα12 overexpression suppressed insulin secretion with JNK activation, which was prevented by JNK inhibitor treatment (Supplemental Figure 8F). Additionally, we assessed insulin degrading enzyme (IDE) in the liver, where approximately two-thirds of circulating insulin is degraded in a physiological process called insulin clearance (36), and found that IDE levels in the liver were not different between genotypes (Supplemental Figure 8, G and H). Together, these results support the idea that suppressed JNK signaling and antiinflammatory response due to whole-body Gα12 gene deletion contribute to a mild effect on glucose homeostasis distinct from deregulation of lipid metabolism.
Gα12 ablation augments diet-induced obesity due to decreased energy expenditure.
Next, we monitored the impact of Gna12 KO on obesity development and whole-body energy expenditure. WT and Gna12-KO mice fed ND showed no difference in body weight gain (Figure 7A) and had normal phenotype (data not shown). When maintained on HFD for 16 weeks ad libitum, Gna12-KO mice developed obesity at an accelerated rate as compared with the WT controls (Figure 8A). Food intake, fecal output, and excreted fecal lipid were all comparable to each other (Figure 8A). Of note, a deficiency in the Gα12 gene fortified the effect of HFD feeding on lean mass and fat mass gains (Figure 8B). In HFD-fed Gna12-KO mice, epididymal fat weight was increased along with adipocyte enlargement (Figure 8C). In parallel, serum leptin levels were doubled in the animals (Figure 8D).
Figure 8. Changes in whole-body energy metabolism and adiposity by a deficiency of Gα12.

(A) Effect of Gna12 KO on body weight gains, food intake, fecal output, and fecal lipid content in mice fed HFD. Body weight (n = 8–14/group) and daily food intake (n = 9–10/group) of WT or Gna12-KO mice fed HFD were monitored once every week for 16 weeks. Fecal output and fecal lipid content were measured during the 13th week (n = 9–10/group). (B) Adiposity in Gna12-KO mice fed HFD. Fat mass was assessed by weighing total epididymal, mesenteric, inguinal, perirenal fat pads, and brown adipose tissue. Lean body mass was assessed by subtracting fat mass from total body mass (left, n = 9–10/group). (C) Epididymal fat pad weight (left, n = 9–10/group) and representative H&E staining (right, n = 3/group) of white adipose tissue from WT or Gna12-KO mice fed HFD for 16 weeks. Scale bars: 100 μm. (D) ELISA assays for serum leptin (n = 12–14/group). (E) Energy expenditure and respiratory quotient profiles. Metabolic profiles were measured in WT or Gna12-KO mice fed HFD for 4 weeks using comprehensive animal metabolic monitoring system (CLAMS) (n = 11–12/group). (F) Whole-body oxygen consumption. Oxygen consumption was measured in mice as described in E (n = 11–12/group). (G) Body temperature and locomotor activity. Resting rectal body temperature was measured in WT or Gna12-KO mice fed HFD for 12 weeks (left, n = 5–6/group). Locomotor activities were monitored using CLAMS in mice as described in E (right, n = 11–12/group). Values represent mean ± SEM. Data were analyzed by ANOVA followed by Bonferroni’s (A) post hoc test or 2-tailed Student’s t test (B–F). For A, F, and G, box-and-whisker plots show median (horizontal lines within boxes), 5%–95% percentile (ends of the boxes), and range of minimum to maximum values (whiskers).
Next, we measured energy expenditure of HFD-fed Gna12-KO mice using a monitoring system of animal metabolism. Gna12-KO mice fed HFD showed decreases in total energy expenditure and total OCR compared with the corresponding WT controls (Figure 8, E and F). Respiratory quotients were not significantly different between genotypes (Figure 8E). Body temperature was markedly lower, with no change in locomotor activities, in Gna12-KO mice than in WT mice (Figure 8G). In an effort to assess whether brown adipose tissue is involved in lowering body temperature, as observed in Gna12-KO mice, we examined levels of uncoupling protein 1 (UCP1), an uncoupling protein responsible for thermogenesis, in the tissues of mice fed either ND or HFD and found no change in UCP1 expression in the brown adipose tissue despite a compensatory increase in its transcript level (Supplemental Figure 9, A and B). Histologic morphology and tissue weights were comparable between genotypes (Supplemental Figure 9C). These results suggest that UCP1-dependent thermogenesis in brown adipose tissue may have a marginal role in lowering body temperature in the animals. Overall, our results demonstrate that Gna12-KO mice are more susceptible to diet-induced obesity as a consequence of decrease in energy expenditure.
Adenosine signaling may affect Gα12 regulation of the USP22/SIRT1 axis.
Several lines of evidence indicate that adenosine signaling has been clinically implicated as a therapeutic target for various pathophysiologic situations, including cardiovascular disease, ischemia-reperfusion, and inflammatory disease (37–41). Adenosine functions as a biological ligand through binding to distinct corresponding GPCRs (i.e., A1, A2a, A2b, and A3) (37, 41). To assess the possible link between adenosine and Gα12 signaling, SIRT1 levels were measured in WT or Gα12-deficient mouse embryonic fibroblasts (MEF) treated with each agonist for the receptors in our supplementary experiment. Interestingly, treatment of WT cells with each agonist notably increased SIRT1 levels, which were abrogated by a deficiency of Gα12 (Supplemental Figure 10A). Similar outcomes were obtained using AML12 cells stably expressing shRNA directed against Gα12 (sh-Gα12) (Supplemental Figure 10B). In addition, primary hepatocytes exposed to each adenosine receptor agonist displayed marked increases of SIRT1 and USP22 (Supplemental Figure 10C).
Adenosine concentrations in the extracellular region vary upon metabolic stimuli. Consistently, fasting significantly enhanced serum adenosine concentrations in mice (Supplemental Figure 10D). No change was observed in the liver homogenates. Thus, elevated circulating adenosine in conjunction with increase of Gα12 would amplify GPCR-mediated SIRT1 induction to maintain systemic energy homeostasis.
Discussion
Gα12 belongs to the group of heterotrimeric G proteins that control various cellular responses, including growth, motility, proliferation, and transdifferentiation (7–11). So far, the impact of Gα12 on cellular energy metabolism has not been investigated. Our results revealed the role of Gα12 signaling in mitochondrial respiration for the control of lipid oxidation and the underlying basis of its regulation of SIRT1, as mediated by HIF-1α–dependent transcriptional induction of USP22. Since Gα12 and SIRT1 are ubiquitously expressed in most metabolic tissues (6), our results support the notion that Gα12 signaling plays a role in overall FA metabolism and, consequently, whole-body energy expenditure.
Moreover, we verified that fasting conditions increased the level of Gα12 in the liver in parallel with fat accumulation and that Gα12 ablation exacerbated fasting-induced liver steatosis along with decreasing circulating fat. These findings raised the contention that Gα12 signaling is essential for metabolic processing of fat in the liver and thus its homeostatic balance between liver and systemic lipid metabolism. Our study also showed that the primary mechanism by which Gα12 controls lipid metabolism engages the SIRT1/PPARα/PGC1α axis, as fortified by the results of cDNA microarray and gene network analyses for WT and Gna12-KO mouse liver. Our results showing that a deficiency of the Gα12 gene deregulates PPARα target gene expression and thereby increases susceptibility to fasting-induced liver steatosis with diminished FA oxidation are in line with previous reports demonstrating a link between SIRT1 and PPARα (42, 43). Considering that multifaceted metabolic adaptations that involve SIRT1 induction are observed under fasting conditions, it is highly likely that hepatic Gα12 levels are enhanced as an adaptive response to fasting. Indeed, our results confirmed the lack of fasting induction of Sirt1 by ablation of Gα12 and the consequent exacerbation of liver steatosis. By the same token, overexpression of either Gα12 or SIRT1 in the liver by viral gene transfer reversed these effects. Thus, it is highly likely that Gα12 regulates lipid metabolism in a SIRT1-dependent pathway.
As an extended effort to verify the proposed molecular basis in obesity models, we further examined the role of Gα12 in liver steatosis in a diet-induced obesity model and found that HFD-fed Gna12-KO mice displayed massive lipid accumulation in the liver due to suppression of the genes involved in mitochondrial respiration and FA oxidation downstream of SIRT1. Moreover, such outcomes were verified in other tissues, including skeletal muscle and white adipose tissue, strengthening the concept that Gα12 signaling may be responsible for whole-body energy metabolism. In a previous study, however, a certain amount of Gα12 was found to be localized in mitochondria, negatively modulating its motility, respiration, and membrane potential (12). In addition, active mutants of Gα12 inhibited phosphorylation of Bcl-2, causing mitochondrial fragmentation and membrane permeabilization (12), which represents distinctive features in comparison with our current findings. Considering the notion that the preservation of functional capacity of healthy mitochondria contributes to homeostatic maintenance of FA oxidation, it is also likely that Gα12 has distinct effects on mitochondria in a SIRT1-independent pathway in accordance with its subcellular distribution (44, 45). Detailed molecular insight into the role of Gα12 at mitochondria needs to be further explored.
In our results, Gα12 expression, particularly as seen in protein levels, was notably diminished in subjects with either simple steatosis or NASH as compared with those without steatosis. Contrary to the findings from human liver specimens, Gα12 levels in the liver of HFD-fed WT mice showed mild decreases compared with those of ND-fed control despite a notable decrease in primary hepatocytes. Given that Gα12 plays a key role in inflammatory and immune responses (29, 46), it is presumed that Gα12 levels might be altered in a subset of nonparenchymal cells (e.g., inflammatory cells or fibroblasts) that reside within inflamed fatty liver. Similarly, our previous study also demonstrates that Gα12 levels were upregulated in hepatic stellate cells in fibrotic liver (47). Thus, we carefully raise the possibility that the severity of inflammatory response and/or fibrotic change affects Gα12 in the liver, although it is quite challenging to compare the degree of inflammation among different species and/or experimental models.
It is well established that JNK activation contributes to chronic inflammation and consequent metabolic disorders (30–32). In our previous report and others, the Gα12 pathway activates JNK via RhoA (33, 34), which may explain diminished overall inflammatory responses, as indicated by decreases in liver injury markers (i.e., ALT and AST) and inflammatory lipid mediators (i.e., sphingolipids and ceramides) in whole-body Gna12-KO mice. Similarly, the levels of proinflammatory cytokines (e.g., IL-6 and TNF-α) secreted predominantly by inflamed adipose tissue were lower in Gna12-KO mice. Hence, diminished inflammation might cause mild changes, if any, in glucose tolerance distinct from exacerbation in hepatic steatosis and obesity. Previous studies have provided the pathogenic role of SIRT1 deficiency in insulin resistance and hyperglycemia (48–50). However, our results showed unaltered glucose metabolism and insulin sensitivity in HFD-fed Gna12-KO mice, presumably due to diminished inflammatory response. In addition, our finding that Gna12-KO mice exhibited hyperinsulinemia together with lowered fasting glucose levels might result from suppressed JNK signaling in Gα12-deficient pancreatic β cells. Also, we do not necessarily exclude the possibility that Gα12 signaling regulates other pathway(s) affecting glucose metabolism.
The observation that Gα12 stabilizes SIRT1 by decreasing its ubiquitination supports the hypothesis that Gα12 posttranslationally regulates SIRT1. Persistent activation of JNK1 facilitates SIRT1 degradation (51). Since the Gα12 pathway positively controls JNK activity (34), the effect of Gα12 on SIRT1 deubiquitination depends on a pathway independent of JNK signaling. USP belongs to the members of the deubiquitinase family, controlling target protein stability via inhibition of ubiquitin-mediated proteosomal degradation. Based on the previous observation that USP22 deubiquitinates SIRT1 for stabilization (25), we were tempted to determine whether Gα12 regulation of SIRT1 engages USP22. Our results demonstrated, for what we believe is the first time, that Gα12 transcriptionally activates the USP22 gene via HIF-1α, leading to inhibition of ubiquitin-mediated SIRT1 degradation, as corroborated by the results of our in vivo and in vitro Gα12 manipulation experiments.
HIF-1α mediates and coordinates metabolic changes upon hypoxic responses, which would be required for maintenance of cellular energy balance, including lipid metabolism (52–54). Our findings shown here demonstrate that Gα12 promotes HIF-1α–dependent USP22 induction, maintaining SIRT1 levels. The data showing a bona fide increase in USP22 by enforced expression of Ad-Gα12QL in Gna12-KO primary hepatocytes further strengthens the concept that the ability of Gα12 to stabilize SIRT1 relies on HIF-1α. Our findings are consistent with the report that hypoxic stimuli increase SIRT1 in a HIF-1α–dependent manner (55). Likewise, transgenic mice with adipose tissue–selective expression of a dominant negative form of HIF-1α showed an impairment in energy expenditure with decreased thermogenesis (53). We also verified the role of RhoA/Rock in the regulation of HIF-1α by Gα12 (26–28). Overall, the outcomes of our study uncover the new Gα12 signaling cascade encompassing HIF-1α–driven USP22 expression that affects SIRT1 in response to altered metabolic environments.
Considering that a subset of GPCRs generally form oligomeric complexes with other GPCRs (56), it is quite challenging to define a single GPCR or its corresponding ligand or ligands responsible for numerous metabolic events. In the present study, however, we attempted to find possible GPCRs and/or ligands for our proposed mechanism, focusing on adenosine signaling as one of the candidates. In a recent study, adenosine signaling contributed to alcohol-induced fatty liver in mice (57), supportive of possible involvement of adenosine signaling in our proposed model. In contrast to our findings, it has been claimed that A2b receptor activation downregulated CPT1 and PPARα (57), which may be due to differences in experimental design (e.g., agonist treatment time: 30 minute to 12 hours vs. 24 hours). Thus, more detailed experiments may be necessary to define receptor activation and Gα12 coupling at the molecular level. Our results do not exclude the possibility of other G proteins (i.e., Gs or Gi) coupling because each adenosine receptor may also couple to the G proteins (37, 41). Nevertheless, our findings provide evidence that adenosine signaling affects the Gα12-mediated USP22-SIRT1 axis under different physiological conditions.
In summary, we discovered a function of Gα12 signaling in lipid metabolism. Since FA oxidation occurs mainly in mitochondria, the identified Gα12-signaling pathway may fill the missing link between cell-surface receptor activation and mitochondrial fuel oxidation. Moreover, our study identifies the regulatory role of Gα12 signaling in the SIRT1/PPARα pathway, delineating the molecular basis by which Gα12 regulates SIRT1. These findings suggest HIF-1α and USP22 as attractive targets for energy expenditure, which could be utilized to combat the obesity epidemic. Current and future investigation of the function and mechanism of this cascade may offer new insight into the understanding of energy metabolism and uncover targets for treating metabolic diseases.
Methods
Details of the materials and experimental protocols are provided in the Supplemental Methods.
Animal experiments.
All animals were maintained in a 12-hour light/12-hour dark cycle and fed ad libitum. Details of the generation of the Gna12-KO mice used in this study have been described previously (17). Male mice at 6 to 8 weeks of age, unless otherwise indicated, were used in this study. To minimize environmental differences, mice were housed for at least a week before each experiment. For the fasting/refeeding transition model, Gna12-KO mice and their age-matched WT littermates were fed ad libitum, fasted for 24 hours, and refed for 24 hours with free access to water. For a diet-induced obesity model, age-matched WT and Gna12-KO mice were subjected to ad libitum feeding of either ND or HFD with 60% kcal fat (D12492, Research Diets) for up to 16 weeks. After sacrifice of animals, tissues were dissected, snap-frozen, and processed for protein and RNA quantification (58).
Statistics.
Values are expressed as mean ± SEM. Statistical significance was tested by 2-tailed Student’s t test or 1-way ANOVA with Bonferroni’s or least significant difference (LSD) multiple comparison procedure where appropriate. Differences were considered significant at P < 0.05.
Accession number.
All original microarray data using liver tissues of each genotype were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE51694).
Study approval.
All animal studies were approved by the IRB and conducted under the guidelines of the IACUC of Seoul National University. Human NAFLD liver specimens were provided by the University of Kansas Liver Center Tissue Bank (Kansas City, Kansas, USA) between 2010 and 2011 (cohort no. 1) and Cedars-Sinai Medical Center in 2017 (cohort no. 2). All of the human specimens were procured with proper written, informed consent.
Author contributions
THK and YMY designed the studies, performed the experiments and analyzed data, and drafted the manuscript. CYH and JHK acquired samples and analyzed data. HO and SSK performed the metabolic cage and clamp studies, and CSC analyzed and interpreted data. BHY and YHC performed HPLC experiments for adenosine measurement. TSP performed lipidomic experiments. CHL and HK provided administrative and material support or did exploratory experiments. MN, ES, and YJYW collected, analyzed, and provided human samples and edited the manuscript. SGK designed and supervised the studies, analyzed and interpreted data, wrote the manuscript, and obtained funding.
Supplementary Material
Acknowledgments
We thank Melvin I. Simon (California Institute of Technology, Pasadena, California, USA) for the Gna12-KO mice and MEF cells, Patrick J. Casey (Duke University Medical Center, Durham, North Carolina, USA) for the adenovirus encoding mouse Gα12QL (Q229L), Richard D. Palmiter (University of Washington, Seattle, Washington, USA) for the mouse albumin enhancer/promoter (NB) construct, and Junichi Sadoshima (Rutgers New Jersey Medical School, Newark, New Jersey, USA) for the adenovirus encoding mouse SIRT1. We thank Yu-Jui Yvonne Wan, Ekihiro Seki, and Mazen Noureddin for providing human NAFLD liver samples from the University of Kansas Medical Center, Liver Tissue Bank (cohort no. 1 for YJYW) and Division of Digestive and Liver Diseases, Department of Medicine, Comprehensive Transplant Center, Cedars-Sinai Medical Center (cohort no. 2 for ES and MN). Min6 cells were kindly provided by Eun Young Park (Mokpo National University, Mokpo, South Korea). This research was supported mainly by a National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2018R1A2A1A05078694 to SGK). THK was supported partly by the Basic Science Research Program of the Ministry of Education (NRF-2018R1A6A3A11048112). YMY was supported partly by the Basic Science Research Program of the Ministry of Education (NRF-2014R1A6A3A01054056) and by the NIH/National Heart, Lung, and Blood Institute (T32HL134637). ES and MN were supported in part by the NIH (R01DK085252). HO and CSC were supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry for Health and Welfare of Korea (HI14C1135).
Version 1. 10/09/2018
In-Press Preview
Version 2. 11/12/2018
Electronic publication
Version 3. 12/03/2018
Print issue publication
Funding Statement
Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry for Health and Welfare of Korea
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(12):5587–5602.https://doi.org/10.1172/JCI97831.
Contributor Information
Tae Hyun Kim, Email: moneywater@snu.ac.kr.
Yoon Mee Yang, Email: janiceyang1223@gmail.com.
Chang Yeob Han, Email: hancy@snu.ac.kr.
Ja Hyun Koo, Email: jhkoo@snu.ac.kr.
Hyunhee Oh, Email: ohhee@gachon.ac.kr.
Su Sung Kim, Email: kss@ugenecell.com.
Byoung Hoon You, Email: dbqudgns87@naver.com.
Young Hee Choi, Email: choiyh@dongguk.edu.
Tae-Sik Park, Email: pts9918@gmail.com.
Chang Ho Lee, Email: jennysue@hanyang.ac.kr.
Hitoshi Kurose, Email: hikurose@phar.kyushu-u.ac.jp.
Mazen Noureddin, Email: mazen.noureddin@cshs.org.
Ekihiro Seki, Email: Ekihiro.Seki@cshs.org.
Yu-Jui Yvonne Wan, Email: yjywan@ucdavis.edu.
Cheol Soo Choi, Email: cschoi@gachon.ac.kr.
Sang Geon Kim, Email: sgk@snu.ac.kr.
References
- 1.Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 4.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114(2):147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worzfeld T, Wettschureck N, Offermanns S. G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends Pharmacol Sci. 2008;29(11):582–589. doi: 10.1016/j.tips.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Strathmann MP, Simon MI. G alpha 12 and G alpha 13 subunits define a fourth class of G protein alpha subunits. Proc Natl Acad Sci U S A. 1991;88(13):5582–5586. doi: 10.1073/pnas.88.13.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu N, Bradley L, Ambdukar I, Gutkind JS. A mutant alpha subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc Natl Acad Sci U S A. 1993;90(14):6741–6745. doi: 10.1073/pnas.90.14.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan AM, Fleming TP, McGovern ES, Chedid M, Miki T, Aaronson SA. Expression cDNA cloning of a transforming gene encoding the wild-type G alpha 12 gene product. Mol Cell Biol. 1993;13(2):762–768. doi: 10.1128/MCB.13.2.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly P, et al. The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2006;103(21):8173–8178. doi: 10.1073/pnas.0510254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang YM, et al. Gα12 gep oncogene deregulation of p53-responsive microRNAs promotes epithelial-mesenchymal transition of hepatocellular carcinoma. Oncogene. 2015;34(22):2910–2921. doi: 10.1038/onc.2014.218. [DOI] [PubMed] [Google Scholar]
- 11.Grzelinski M, et al. Critical role of G(alpha)12 and G(alpha)13 for human small cell lung cancer cell proliferation in vitro and tumor growth in vivo. Clin Cancer Res. 2010;16(5):1402–1415. doi: 10.1158/1078-0432.CCR-09-1873. [DOI] [PubMed] [Google Scholar]
- 12.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. G alpha12 is targeted to the mitochondria and affects mitochondrial morphology and motility. FASEB J. 2008;22(8):2821–2831. doi: 10.1096/fj.07-104224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature. 2006;444(7121):868–874. doi: 10.1038/nature05486. [DOI] [PubMed] [Google Scholar]
- 15.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 16.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu JL, Müller S, Mancino V, Offermanns S, Simon MI. Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci U S A. 2002;99(14):9352–9357. doi: 10.1073/pnas.102291599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res. 2014;329(1):85–93. doi: 10.1016/j.yexcr.2014.09.030. [DOI] [PubMed] [Google Scholar]
- 19.Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8(5):287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- 20.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A. 1999;96(13):7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 22.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20(5):1868–1876. doi: 10.1128/MCB.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009;9(2):123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashida S, et al. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol Cell Biochem. 2010;339(1-2):285–292. doi: 10.1007/s11010-010-0391-z. [DOI] [PubMed] [Google Scholar]
- 25.Lin Z, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell. 2012;46(4):484–494. doi: 10.1016/j.molcel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 26.Turcotte S, Desrosiers RR, Béliveau R. HIF-1alpha mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J Cell Sci. 2003;116(Pt 11):2247–2260. doi: 10.1242/jcs.00427. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi M, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha expression through RhoA activation in trophoblast cells. J Clin Endocrinol Metab. 2005;90(3):1712–1719. doi: 10.1210/jc.2004-1547. [DOI] [PubMed] [Google Scholar]
- 28.Ohta T, et al. Inhibition of the Rho/ROCK pathway enhances the efficacy of cisplatin through the blockage of hypoxia-inducible factor-1α in human ovarian cancer cells. Cancer Biol Ther. 2012;13(1):25–33. doi: 10.4161/cbt.13.1.18440. [DOI] [PubMed] [Google Scholar]
- 29.Ki SH, Choi MJ, Lee CH, Kim SG. Galpha12 specifically regulates COX-2 induction by sphingosine 1-phosphate. Role for JNK-dependent ubiquitination and degradation of IkappaBalpha. J Biol Chem. 2007;282(3):1938–1947. doi: 10.1074/jbc.M606080200. [DOI] [PubMed] [Google Scholar]
- 30.Vallerie SN, Hotamisligil GS. The role of JNK proteins in metabolism. Sci Transl Med. 2010;2(60):60rv5. doi: 10.1126/scitranslmed.3001007. [DOI] [PubMed] [Google Scholar]
- 31.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 32.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagao M, Kaziro Y, Itoh H. The Src family tyrosine kinase is involved in Rho-dependent activation of c-Jun N-terminal kinase by Galpha12. Oncogene. 1999;18(31):4425–4434. doi: 10.1038/sj.onc.1202832. [DOI] [PubMed] [Google Scholar]
- 34.Cho MK, et al. Role of Galpha12 and Galpha13 as novel switches for the activity of Nrf2, a key antioxidative transcription factor. Mol Cell Biol. 2007;27(17):6195–6208. doi: 10.1128/MCB.02065-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanuza-Masdeu J, Arévalo MI, Vila C, Barberà A, Gomis R, Caelles C. In vivo JNK activation in pancreatic β-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes. 2013;62(7):2308–2317. doi: 10.2337/db12-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tokarz VL, MacDonald PE, Klip A. The cell biology of systemic insulin function. J Cell Biol. 2018;217(7):2273–2289. doi: 10.1083/jcb.201802095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367(24):2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets--what are the challenges? Nat Rev Drug Discov. 2013;12(4):265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9(4):327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146(2):539–49.e7. doi: 10.1053/j.gastro.2013.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hampoelz B, Knoblich JA. Heterotrimeric G proteins: new tricks for an old dog. Cell. 2004;119(4):453–456. doi: 10.1016/j.cell.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Chisari M, Saini DK, Kalyanaraman V, Gautam N. Shuttling of G protein subunits between the plasma membrane and intracellular membranes. J Biol Chem. 2007;282(33):24092–24098. doi: 10.1074/jbc.M704246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Won HY, Min HJ, Lee WH, Kim SG, Hwang ES. Galpha12 is critical for TCR-induced IL-2 production and differentiation of T helper 2 and T helper 17 cells. Biochem Biophys Res Commun. 2010;394(3):811–816. doi: 10.1016/j.bbrc.2010.03.079. [DOI] [PubMed] [Google Scholar]
- 47.Kim KM, et al. Gα12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J Hepatol. 2018;68(3):493–504. doi: 10.1016/j.jhep.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121(11):4477–4490. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purushotham A, Xu Q, Li X. Systemic SIRT1 insufficiency results in disruption of energy homeostasis and steroid hormone metabolism upon high-fat-diet feeding. FASEB J. 2012;26(2):656–667. doi: 10.1096/fj.11-195172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schenk S, et al. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J Clin Invest. 2011;121(11):4281–4288. doi: 10.1172/JCI58554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem. 2011;286(25):22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishiyama Y, et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol. 2012;56(2):441–447. doi: 10.1016/j.jhep.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. J Biol Chem. 2010;285(43):32869–32877. doi: 10.1074/jbc.M110.135509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson GK, Tennant DA, McKeating JA. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J Hepatol. 2014;61(6):1397–1406. doi: 10.1016/j.jhep.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 55.Chen R, Dioum EM, Hogg RT, Gerard RD, Garcia JA. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem. 2011;286(16):13869–13878. doi: 10.1074/jbc.M110.175414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palczewski K. Oligomeric forms of G protein-coupled receptors (GPCRs) Trends Biochem Sci. 2010;35(11):595–600. doi: 10.1016/j.tibs.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng Z, et al. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest. 2009;119(3):582–594. doi: 10.1172/JCI37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim TH, Eom JS, Lee CG, Yang YM, Lee YS, Kim SG. An active metabolite of oltipraz (M2) increases mitochondrial fuel oxidation and inhibits lipogenesis in the liver by dually activating AMPK. Br J Pharmacol. 2013;168(7):1647–1661. doi: 10.1111/bph.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.