

Abstract
Breast cancer (BrCa) is the malignant tumor that most seriously threatens female health; however, the molecular mechanism underlying its progression remains unclear. Here, we found that conditional deletion of hypermethylated in cancer 1 (HIC1) in the mouse mammary gland might contribute to premalignant transformation in the early stage of tumor formation. Moreover, the chemokine (C-X-C motif) ligand 14 (CXCL14) secreted by HIC1-deleted BrCa cells bound to its cognate receptor GPR85 on mammary fibroblasts in the microenvironment and was responsible for activating these fibroblasts via the ERK1/2, Akt, and neddylation pathways, whereas the activated fibroblasts promoted BrCa progression via the induction of epithelial-mesenchymal transition (EMT) by the C-C chemokine ligand 17 (CCL17)/CC chemokine receptor 4 (CCR4) axis. Finally, we confirmed that the HIC1-CXCL14-CCL17 loop was associated with the malignant progression of BrCa. Therefore, the crosstalk between HIC1-deleted BrCa cells and mammary fibroblasts might play a critical role in BrCa development. Exploring the progression of BrCa from the perspective of microenvironment will be beneficial for identifying the potential prognostic markers of breast tumor and providing more effective treatment strategies.
Keywords: Cell Biology, Oncology
Keywords: Breast cancer, Chemokines

Introduction
Breast cancer (BrCa) is the most frequently diagnosed malignant tumor in women worldwide and represents the main cause of cancer-related death (1). Although systematic therapeutic approaches have decreased cancer-specific mortality, BrCa is still associated with high rates of cancer recurrence and metastasis (2). Therefore, exploring the mechanism underlying BrCa progression may be beneficial for diagnosis and treatment of the disease.
Recently, increasing evidence has shown that an appropriate tumor microenvironment (the soil) is necessary for the optimal growth of tumor cells (the seed) (3, 4). The tumor microenvironment includes various types of stromal cells that play a pivotal role in cancer progression (4). Among these cells, cancer-associated fibroblasts (CAFs) are the most abundant cell type involved in tumor development, orchestrating proliferation, invasion, and neoangiogenesis (5, 6). Notably, CAFs exhibit a mesenchymal-like phenotype and promote the metastasis of both premalignant and malignant mammary epithelial cells in BrCa (7). It is well accepted that CAFs are mainly derived from normal-associated fibroblasts (NAFs) and that they are induced by cancer-secreted cytokines, such as TGF-β and CXCL12/SDF-1 (8), or by loss of the expression of tumor-suppressor genes, such as PTEN, TP53, CDKN1A, and CAV1 (9–13). Reciprocally, the activated CAFs can cause BrCa epithelial cells to progress to more malignant stages (6, 14). Since it is accepted that the stroma is more genetically stable than cancerous epithelial cells, targeting the collateral interactions of CAFs may offer therapies that are less prone to the development of resistance. Therefore, focusing on the role and mechanism of CAFs in BrCa may provide a strategy for the treatment of BrCa patients.
Hypermethylated in cancer 1 (HIC1) is a tumor-suppressor gene located at 17p13.3. HIC1 resides completely within a CpG island that is frequently hypermethylated in human tumors, including breast, prostate, and lung cancer (15–17). HIC1 is located close to telomeric TP53, which is a sequence-specific transcriptional repressor belonging to the BTB/POZ and C2H2 zinc finger family (18). The N-terminal BTB/POZ domain of HIC1 is responsible for protein-protein interactions that are crucial for its biological function, and the C-terminal zinc finger domains are involved in sequence-specific binding to an HIC1-responsive element (HiRE) with a TGCC or GGCA core motif (19, 20). It has been reported that epigenetic silencing of HIC1 is one of the most common events in human cancer (15, 16, 21). Moreover, conventional knockout mice with homozygous deletion of HIC1 display embryonic lethality at midgestation (22), whereas heterozygous mutants develop a range of spontaneous tumors in an age-dependent manner (23). As a transcription factor, several downstream target genes of HIC1 have been identified; these include ACKR3, CXCR4, EFNA1, SNAI2, SIRT1, and IL6, which are associated with the modulation of angiogenesis, proliferation, and metastasis (16, 19, 21, 24–26). In previous work, we showed that HIC1 expression is silenced only in highly malignant triple-negative BrCa (TNBC) compared with other molecular subtypes of BrCa and that this silencing induces autocrine secretion of LCN2 that facilitates BrCa progression (21). The role and mechanism underlying the effect of HIC1 loss on modulation of the tumor microenvironment remain unsettled, but they are crucial to exploring the function of HIC1 in BrCa progression.
Results
HIC1 deletion induces hyperplasia and lactogenic defects in mammary gland in vivo.
Although our previous work indicated that HIC1 is silenced in some human cancers, including TNBC (16, 21, 26), its intrinsic role and the mechanism of its action in BrCa progression remain unknown. Here, to investigate the role of HIC1 in mammary gland malignant development in vivo, we generated Hic1–/– conditional knockout mice by crossing Hic1fl/fl mice with Wap-Cre mice in which Cre recombinase expression was driven by the mammary-specific whey acidic protein (Wap) promoter, which is activated at midpregnancy in luminal epithelial cells. Cre-positive Hic1fl/fl mice were used as the Hic1–/– group, and their Cre-negative Hic1fl/fl littermates, which were designated Hic1+/+, were used as controls (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI99974DS1). Western blot and quantitative reverse-transcriptase PCR (RT-qPCR) assays indicated that HIC1 deletion occurred only in mammary epithelium and not in liver, lung, or muscle (Supplemental Figure 1B). Notably, using whole-mount staining in 4-month-old, 8-month-old, and 12-month-old mice, we found striking hyperplastic abnormalities in the mammary ductal trees of Hic1–/– mice compared with their Hic1+/+ littermates (Figure 1A and Supplemental Figure 1C). Similarly, H&E-stained sections of the animals’ mammary glands showed that the epithelial layers in Hic1–/– mice were thicker than those in Hic1+/+ mice (Figure 1B). This effect was largely due to an increased population of epithelial cells. This was confirmed by immunofluorescence staining showing marked expression of the luminal marker keratin 8 (K8) (Figure 1C). In addition, greatly increased numbers of proliferative cells were observed in Hic1–/– mice compared with controls using Ki67 and cyclin D1 staining (Figure 1D). Moreover, deletion of HIC1 in MCF7 luminal BrCa cells increased their ability to form vasculogenic networks on Matrigel (Supplemental Figure 1G and Figure 2B). In contrast, restoration of HIC1 expression in MDA-MB-231 TNBC cells had the opposite effect (Supplemental Figure 1H). These findings indicate that HIC1 deletion may be associated with the premalignant development of mammary gland tissue.
Figure 1. HIC1 deletion induces hyperplasia of mammary gland in vivo.

(A) Representative whole-mount staining of the fourth inguinal mammary glands at the indicated ages (4 months and 8 months) were prepared from Hic1+/+ mice or Hic1–/– mice and stained with carmine aluminum (n = 6 for each group). M, months. (B) H&E staining of the mammary glands of 6-month-old mice. (C) Immunofluorescence staining of luminal epithelial marker (K8) and myoepithelial markers (α-SMA) in the mammary glands of 6-month-old, 8-month-old, and 12-month-old mice. (D) Immunohistochemical staining of Ki67 and cyclin D1 in mammary glands of 6-month-old mice. The dot plots show the mean value for each immunoreactivity score (IRS) with statistical evaluation. Data are shown as mean ± SEM. n = 6. *P < 0.05, 2-tailed Student’s t test. (E) Box plots of HIC1 mRNA levels in paired normal breast/BrCa tissues (left, paired t tests), non-TNBC/TNBC tissues (middle, 2-tailed Student’s t tests), and BrCa tissues at different stages (right, 1-way ANOVA followed by Bonferroni’s post hoc test). Data were obtained from the TCGA data set (TCGA_BRCA_exp_HiSeqV2-2015-02-24). *P < 0.05; **P < 0.01; ***P < 0.001. (F) Kaplan-Meier plots of the relapse-free survival of patients with BrCa in whole data sets stratified by HIC1 expression. Data were acquired from the Kaplan-Meier plotter database. P = 0.00027, log-rank test (28). Representative images in this figure were obtained from at least 3 animals of each genotype.
Figure 2. HIC1-deleted BrCa cells induce the activation of stromal fibroblasts in mammary gland.

(A) Representative 3 immunohistochemistry images for α-SMA in mammary glands of 6-month-old mice. Positive staining in the mammary gland stroma of Hic1–/– mice is indicated by red arrows (n = 3 for each group). (B) CRISPR-Cas9–mediated HIC1 deletion in MCF7 and T47D luminal BrCa cells. Cell lysates were analyzed by Western blot with antibodies against HIC1 and GAPDH. sg3 and sg4 represent 2 different interference sgRNA sequences. Ctrl, control. (C) Representative primary NAFs and CAFs isolated from human BrCa tissues. Western blot analysis of cell lysates was performed using antibodies against α-SMA, FAP, PDGFRα, and GAPDH. (D) Schematic showing primary NAFs cocultured with MCF7CtrlMCF7sgHIC1 or T47DCtrlT47DsgHIC1 luminal BrCa cells in a Transwell apparatus (0.4 μm pore size) for 4 days. (E) Western blot analysis of lysates of NAF6 cells that were cocultured with MCF7CtrlMCF7sgHIC1 or T47DCtrlT47DsgHIC1 luminal BrCa cells for 4 days. Antibodies against α-SMA, FAP, PDGFRα, and GAPDH were used. (F) Immunofluorescence staining for the detection of α-SMA expression in NAF6 cells that were cocultured with MCF7CtrlMCF7sgHIC1 or T47DCtrlT47DsgHIC1 luminal BrCa cells for 4 days.
Unexpectedly, we observed that pups born to Hic1–/– dams had lower weights than those reared by Hic1+/+ dams, although their 48-hour survival rates did not differ (Supplemental Figure 1D). This suggested that profound lactogenic defects might be present in Hic1–/– dams. Mammary gland whole mounts obtained from Hic1–/– dams at day 6 of lactation were smaller than those obtained from controls (Supplemental Figure 1C), indicating a deficiency in the amounts of secretory products. H&E staining of mammary glands showed the presence of lipid droplets in Hic1+/+ dams, whereas the Hic1–/– glands contained very few lipid droplets (Supplemental Figure 1E). Oil red O staining further indicated that the alveoli of Hic1–/– glands did not contain significant quantities of milk fat (Supplemental Figure 1F). These data suggest that HIC1 deletion also induces lactogenic defects in mammary glands in vivo.
Finally, based on the data in The Cancer Genome Atlas (TCGA) database, we observed that HIC1 mRNA levels were significantly lower in BrCa tissues than in normal tissues from the same patients in 113 cases (Figure 1E). Consistent with our previous results (21), HIC1 mRNA levels were lower in 123 TNBC cases than in 972 non-TNBC patients in the TCGA database (Figure 1E). Furthermore, the expression of HIC1 was also observed to be decreased in high-grade BrCa tissues compared with low-grade tissues both in the TCGA database and in an Oncomine data set (Zhao breast; https://www.oncomine.org/resource/login.html; ref. 27) (Figure 1E and Supplemental Figure 1I). Using the Kaplan-Meier method followed by the log-rank test, we further confirmed that HIC1 deletion correlates with lower relapse-free survival (Figure 1F) (28). These data suggest that HIC1 deletion is associated with poor prognosis of BrCa.
HIC1-deleted BrCa cells induce the activation of stromal fibroblasts in mammary gland.
It has been reported that HIC1 deletion may play a role in modulating the stromal microenvironment by causing stromal cells to secrete specific factors (16, 21). We explored whether HIC1-deleted mammary gland epithelial cells have the ability to affect stromal fibroblasts. Notably, Hic1–/– mice were found to have markedly activated fibroblasts in their mammary gland stroma compared with Hic1+/+ mice based on an evaluation of the expression of α-smooth muscle actin (α-SMA), which is frequently used as a classical marker of CAF in solid tumors (8, 29) (Figure 2A). To confirm this effect in vitro, HIC1-deleted MCF7 and T47D luminal BrCa cells (hereinafter referred to as MCF7CtrlMCF7sgHIC1 or T47DCtrlT47DsgHIC1) were obtained using the CRISPR-Cas9 system (Figure 2B), and primary mammary fibroblasts were freshly isolated from 4 human BrCa central and peripheral tumor areas (denoted as CAFs or NAFs, respectively) (Figure 2C). Figure 2C shows that the expression of α-SMA was higher in CAF2, -6, -8, and -10 than it was in the respective NAFs, albeit the expression of the other 2 CAF markers, fibroblast activation protein (FAP) and PDGFRα, did not follow the same trend. Next, a coculture system was used to simulate the in vivo situation. In this system, primary NAFs were placed in the lower chambers of a Transwell apparatus, and MCF7CtrlMCF7sgHIC1 or T47DCtrlT47DsgHIC1 cells were replated in the upper chambers (Figure 2D). After 4 days, we performed Western blot assays and immunofluorescence staining and found that, compared with that in the respective controls, expression of the CAF markers, especially α-SMA, was dramatically increased in NAF6 cells that were cocultured with MCF7sgHIC1T47DsgHIC1 cells; the phenotypes of the cells were highly similar to those of primary CAFs (Figure 2, E and F). Together, these data suggest that HIC1-deleted BrCa cells can directly induce the activation of stromal fibroblasts in mammary gland in vivo and in vitro.
CXCL14 is a direct target gene of HIC1.
Next, to identify potential downstream targets of HIC1, we analyzed the published data on the genome-wide transcriptome profiles of MDA-MB-231HIC1MDA-MB-231GFP and HBL-100shHIC1HBL-100shNC cells using Agilent Whole Human Genome Microarrays (21) (Supplemental Figure 2A). Among the differentially expressed genes encoding cytokines, 8 genes that may participate in the activation of mammary fibroblasts were identified (Figure 3A and Supplemental Figure 2B). RT-qPCR was performed to assay the expression of these 8 genes at the mRNA level in MCF7sgHIC1MCF7Ctrl, T47DsgHIC1T47DCtrl, and MDA-MB-231HIC1MDA-MB-231GFP cells. Chemokine (C-X-C motif) ligand 14 (CXCL14) was chosen as a potentially relevant cytokine based on results showing that altering HIC1 expression in these cells markedly modulated CXCL14 expression at both the mRNA and protein levels compared with the controls (Figure 3, A and B, and Supplemental Figure 2B). CXCL14 expression was also greatly increased in Hic1–/– mice at the mRNA and protein levels (Figure 3A and Supplemental Figure 2C). Similarly, the expression of Sirt1, a classical downstream gene of HIC1, was also observed to be enhanced in Hic1–/– mice (Figure 3A). These findings suggest that CXCL14 expression is modulated by HIC1.
Figure 3. CXCL14 is a direct target gene of HIC1.

(A) Left: relative RT-qPCR analysis of 8 differentially expressed genes (CCL2, CXCL14, CX3CL1, IL1B, IL24, IL8, TGFA, and VEGFA) after HIC1 deletion in MCF7 and T47D cells (n = 3). Right: relative RT-qPCR analysis of Hic1, Cxcl14, and Sirt1 mRNA levels in the mammary glands of Hic1–/– or Hic1+/+ mice (n = 5). (B) ELISA analysis of CXCL14 levels in the CM of MCF7sgHIC1MCF7Ctrl and T47DsgHIC1T47DCtrl cells. The supernatants were collected after culture of the cells for 24 hours or 48 hours (n = 4). (C) Schematic of the CXCL14 promoter region. The positions of selected consensus binding sites are indicated above the diagram; the lengths of the promoter constructs used in the reporter assays are shown below. (D) CXCL14 promoter activity after transfection of the full-length construct (–2000/+136) alone or together with HIC1 expression vectors. p–GL3-Basic, control for promoter constructs; pc3.1, control for the HIC1 expression vector. The results are expressed as the ratio of firefly luciferase to Renilla luciferase (n = 3). (E) CXCL14 promoter activity after cotransfection with 100 ng of the HIC1 expression vector and each of the promoter constructs. The –41/+136 construct had a significant repressive effect, despite its lower promoter activity (n = 3). (F) ChIP analysis of HIC1 at the CXCL14 promoter region in MCF7 and T47D cells (n = 3). Data are shown as mean ± SEM. n = 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, 2-tailed Student’s t tests (A, B, and E), 1-way ANOVA followed by Bonferroni’s post hoc test (D and F).
Given that HIC1 functions as a repressive transcription factor and directly binds to the promoter regions of target genes (20), we inferred that CXCL14 is a potential downstream target induced by HIC1. Indeed, as shown in Figure 3C, 11 putative HIC1-binding sites (TGCC/GGCA) were identified in the CXCL14 promoter. We constructed a series of CXCL14-truncated promoter/reporter fusion plasmids containing progressive deletions of the 5′ region of the gene from –2000 to +136 (Figure 3C). These constructs were then transfected together with the pcDNA3.1-His or the pcDNA3.1-HIC1 expression vector into 293T and MCF7 cells, and the promoter activities were measured using luciferase reporter assays. Figure 3D shows that, when transfected into 293T and MCF7 cells, the construct containing the full-length CXCL14 promoter resulted in higher activity than the basic construct. Moreover, transient transfection of the cells with HIC1 markedly inhibited CXCL14 promoter activity (Figure 3D) in a dose-dependent manner (Supplemental Figure 2D). The inhibitory effect of HIC1 on CXCL14 promoter activity in both cell lines was maintained in all the truncated constructs, including –2000/+136, –726/+136, –276/+136, and –41/+136 (Figure 3E). These results suggest that the regulatory region in the HIC1-mediated repression may be located within the –41 bp to +136 bp region of the CXCL14 promoter, which contains 2 putative HIC1-binding sites, M1 and M2 (Supplemental Figure 2E). We mutated both sites (GGGCA to AAATG) to abolish HIC1 binding (Supplemental Figure 2E). Supplemental Figure 2F shows that only the construct in which site M1 was mutated significantly decreased the repressive capacity of HIC1, suggesting that the putative M1 site in the CXCL14 promoter is essential for the HIC1-mediated inhibitory effect on CXCL14 expression. Finally, to further determine whether the CXCL14 promoter is indeed directly bound by HIC1, ChIP assays were performed in MCF7 and T47D cells using an antibody against HIC1. The pulled down DNA was amplified by ordinary PCR and RT-qPCR (Figure 3F) with primers that were designed based on the –41/+136 region of the CXCL14 promoter (Supplemental Figure 2E). As shown in Figure 3F, the CXCL14 promoter was markedly enriched in the HIC1-immunoprecipitated MCF7 and T47D chromatin, but absent from the chromatin that was immunoprecipitated by the control rabbit IgG. Taken together, these results demonstrate that HIC1 directly represses CXCL14 transcription.
CXCL14 derived from HIC1-deleted BrCa cells mediates the activation of mammary fibroblasts.
To explore whether CXCL14 activates mammary fibroblasts, various concentrations of rhCXCL14 were used to treat the primary mammary fibroblast NAF2 and NAF6 cells. Western blot assays and immunofluorescence staining showed that NAF2 and NAF6 cells were markedly activated after the treatment with rhCXCL14; the phenotypes of the treated cells were very similar to those of the primary corresponding CAF2 and CAF6 cells that were used as the positive controls (Figure 4, A and B). Similarly, immortalized NAF10 cells stably overexpressing CXCL14 were observed to be activated (Figure 4A). Moreover, CXCL14 treatment had an activating effect on NAF6 cells similar to that of CXCL12, but the effect was weaker than the effect observed after TGF-β treatment (Supplemental Figure 3A). Both CXCL12 and TGF-β are usually considered to be activators of mammary fibroblasts. In contrast, the addition of a neutralizing antibody to CXCL14 (αCXCL14) to the coculture system containing NAF6 cells and HIC1-deleted MCF7/T47D cells significantly suppressed the activation of NAF6 cells compared with the respective controls (Figure 4C). These results indicate that CXCL14 originating from HIC1-deleted BrCa cells plays a critical role in inducing the activation of mammary gland fibroblasts.
Figure 4. CXCL14 derived from HIC1-deleted BrCa cells mediates the activation of mammary fibroblasts.

(A) Left: NAF2 or NAF6 cells were treated with rhCXCL14 at various concentrations (0–200 ng/ml) for 4 days. The corresponding primary CAF2 or CAF6 cells were used as positive controls. Right: immortalized NAF10 cells stably overexpressed CXCL14. Cell lysates were analyzed by Western blot with antibodies against α-SMA, FAP, PDGFRα, and GAPDH. (B) Representative immunofluorescence staining detection of α-SMA, FAP, and PDGFRα expression in NAF6 or CAF6 cells treated similarly to the cells described in A. (C) NAF6 cells were cocultured with MCF7CtrlMCF7sgHIC1 or with T47DCtrlT47DsgHIC1 luminal BrCa cells for 4 days in the presence or absence of α-CXCL14 at 1 μg/ml or an isotype-matched IgG control. Cell lysates from NAF6 cells were analyzed by Western blot with antibodies against α-SMA, FAP, PDGFRα, and GAPDH. (D) Three MCF7 cell lines (Ctrl-NC, sgHIC1-NC, sgHIC1-shCXCL14) were injected bilaterally into the fourth mammary fat pads of female BALB/c nude mice (n = 10 per group). Tumor volumes were measured with calipers at the indicated time points. Data are shown as mean ± SD. ***P < 0.001, RM ANOVA followed by post hoc LSD test. (E) Photographs and weights of the tumors obtained from the animals described in D. Data are shown as mean ± SD. ***P < 0.001, 1-way ANOVA followed by Bonferroni’s post hoc test. (F) Representative immunohistochemical staining for Ki67 and stromal α-SMA in tumor tissues obtained from each experimental group. Dot plots show the mean value for the percentage of Ki67 or stromal α-SMA–positive cells with statistical evaluation (n = 5–6). Data are shown as mean ± SEM. n = 3 independent experiments. **P < 0.01; ***P < 0.001, 1-way ANOVA followed by Bonferroni’s post hoc test.
Next, to investigate the effect of the HIC1/CXCL14 axis on BrCa progression in vivo, 3 MCF7 cell lines (Ctrl-NC, sgHIC1-NC, and sgHIC1-shCXCL14) were injected bilaterally into the fourth mammary fat pads of female BALB/c nude mice (Supplemental Figure 3B). Figure 4, D and E, shows that both the volumes and the weights of the resulting tumors were greatly increased in the MCF7sgHIC1–NC group compared with the control MCF7Ctrl–NC group. However, knockdown of CXCL14 expression in MCF7sgHIC1 cells markedly rescued the HIC1 deletion-induced tumor burden compared with MCF7sgHIC1–NC cells. Moreover, IHC staining indicated that Ki67 and stromal α-SMA expression were greatly increased in MCF7sgHIC1–NC xenografts compared with the controls, whereas knockdown of CXCL14 expression in MCF7sgHIC1 cells markedly decreased the effects. These results were further confirmed by quantitative analysis (Figure 4F). Finally, using 2 Oncomine data sets, we found that the CXCL14 mRNA levels were higher in BrCa tissues (n = 93) than they were in normal breast tissues (n = 9) (Supplemental Figure 3C). Together, these data suggest that CXCL14 derived from HIC1-deleted BrCa cells mediates the activation of mammary fibroblasts and that this activation is responsible for BrCa progression.
GPR85 is a functional receptor for CXCL14 activities.
Based on our in vitro and in vivo findings, we next explored the mechanism through which CXCL14 mediates the activation of mammary fibroblasts. Supplemental Figure 4A shows that the ERK1/2 and Akt/p70 S6K pathways were significantly activated after rhCXCL14 treatment of NAF6 and NAF7 cells. CAF6 and CAF7 cells were used as positive controls. The ERK1/2 and Akt pathways were also activated in NAF6 cells when the cells were cocultured with HIC1-deleted T47D cells (Supplemental Figure 4B). In contrast, the addition of αCXCL14 to the coculture system abolished the effect (Supplemental Figure 4B), suggesting that the 2 pathways are potentially involved in the CXCL14-induced activation of mammary fibroblasts. To assess this possibility, 2 small-molecule inhibitors, U0126 (a MEK1/2 inhibitor) and LY294002 (a PI3K inhibitor), were used (Supplemental Figure 4C). Indeed, as shown in Supplemental Figure 4, D and E, and Supplemental Figure 5D, both U0126 and LY294002 obviously inhibited the expression of CAF markers, not only in rhCXCL14-treated NAF6/NAF8/NAF10 cells, but also in CAF10 cells, followed by inactivation of the ERK1/2 and Akt pathways.
Previous studies have noted that the neddylation pathway, a newly characterized posttranslational modification pathway, is activated in both lung and liver cancers (30, 31). We therefore explored whether this pathway is associated with the activation of mammary fibroblasts. Supplemental Figure 5A shows that the entire neddylation pathway was activated in CAF6/10 cells compared with the activity of the pathway in the respective NAFs. A slight activation of the neddylation pathway was also observed in NAF6 and NAF10 cells after treatment with rhCXCL14 for 4 days (Supplemental Figure 5B). Furthermore, when primary CAF10- or CXCL14-activated NAF10 cells were treated with the neddylation inhibitor MLN4924, expression of the CAF markers was markedly reduced, as determined by both Western blot and immunofluorescence assay (Supplemental Figure 5, C and D). These results suggest that the neddylation pathway is also involved in the activation of mammary fibroblasts. Taken together, the results presented here demonstrate that CXCL14 secreted by HIC1-deleted BrCa cells activates mammary fibroblasts through the ERK1/2, Akt, and neddylation pathways.
Given that the receptor of CXCL14 has not yet been identified, primary NAF6 cells were treated with biotin-CXCL14 to determine the potential receptor. Immunofluorescence assay showed that CXCL14 was localized on the cell membrane (Figure 5A), suggesting that a receptor on the fibroblast surface may mediate the biological effects of CXCL14. Next, HuProt human proteome microarrays containing 18,583 affinity-purified N-terminally GST-tagged proteins were used to identify CXCL14-binding proteins (Figure 5B). Notably, approximately 3.1% of the proteins on the microarrays, including 32 membrane proteins, were found to bind CXCL14 (Supplemental Figure 6, A–C). Ultimately, we focused on 2 known G protein–coupled receptors (GPCRs) and 5 transmembrane receptors (JMJD6, ROBO3, KLRC1, TNFRSF10C, F2R, SLAMF6, and GPR85) (Figure 5C and Supplemental Figure 6, D and E). Finally, as shown in Figure 5D, the orphan GPCR GPR85 (also called super conserved receptor expressed in brain 2 [SREB2]) was selected and subsequently confirmed to bind to CXCL14 in NAF6 cells using streptavidin-agarose pull-down assays. RNAi-mediated silencing of GPR85 in NAF6 cells and its overexpression in 293T cells was performed to assay whether GPR85 is the functional receptor for CXCL14 (Supplemental Figure 6, F and G). Being the accepted method for evaluating chemokine receptor activation (32), the calcium mobilization assay was used to measure changes in intracellular calcium ([Ca2+]i) upon CXCL14 stimulation. Figure 5E shows that NAF6 cells transfected with control siRNA (NC) responded to stimulation by both rhCXCL14 and rhCXCL12, whereas NAF6 cells transfected with GPR85-3 siRNA did not respond to rhCXCL14, but did respond to the positive control rhCXCL12. To evaluate the ability of CXCL14 to bind to GPR85, radioligand binding assays were next performed. Figure 5F shows that levels of specific binding for 125I-CXCL14 were markedly higher in 293T-GPR85 cells than in 293T-NC cells. Moreover,unlabeled CXCL14 was able to compete with the high level of the specific binding of labeled 125I-CXCL14 in GPR85 transfectants (IC50 = 0.436 nM, 0.0851–1.976 nM, 95% confidence interval; Figure 5G). Unlabeled CXCL12 and CXCL3, which have the highest sequence homology of CXCL14 among the CXC chemokine family, with approximately 30% amino acid identity and 55% similarity to CXCL14 when conservative changes are taken into account (33), were also able to compete with labeled 125I-CXCL14 for binding but with much less affinity than unlabeled CXCL14 (IC50 = 2.338 nM for CXCL12, and IC50 = 4.568 nM for CXCL3; Figure 5G). Next, Western blot analysis demonstrated that rhCXCL14 markedly activated the Akt and ERK1/2 pathways in NAF6-NC cells, but not in NAF6-siGPR85 cells, whereas activation of these signaling pathways was still observed in NAF6-siKLRC1 cells (Figure 5H and Supplemental Figure 6I). Notably, rhCXCL12 activated both the Akt and ERK1/2 pathways when GPR85 expression was reduced in NAF6 cells (Supplemental Figure 6H), suggesting that activation of the Akt and ERK1/2 pathways in NAFs by CXCL14 depends on GPR85. Next, we assayed the ability of CXCL14 to induce the activation of mammary fibroblasts after silencing of GPR85 expression. Figure 5I shows that inhibition of GPR85 expression in NAF6 cells followed by treatment with increasing concentrations of rhCXCL14 for 4 days attenuated the activation of expression of CAF markers compared with the effects observed in NAF6-NC cells. Similar effects were also noted in NAF6 cells cocultured with HIC1-deleted MCF7 cells; the BrCa cells had a stronger activating effect on NAF6-NC cells than on NAF6-siGPR85 cells (Figure 5J). Notably, knockdown of GPR85 expression in CAF6 cells inhibited CAF marker expression and Akt and ERK1/2 signaling (Supplemental Figure 6K). Besides, Western blot assays suggested that CXCL14-induced degradation of GPR85 occurs in NAF6 cells, whereas this effect was not observed after rhCXCL12 treatment (Supplemental Figure 6J). Taken together, these findings indicate that the effects of chemokine CXCL14 on the activation of mammary fibroblasts are dependent on GPR85, a functional receptor for CXCL14.
Figure 5. GPR85 is a functional receptor for CXCL14 activity.

(A) Confocal microscopy of NAF6 cells treated with 100 ng/ml biotin or biotin-CXCL14 at 4°C and stained with an antibody against Cy3-streptavidin. An isotype-matched IgG was used as a control. Cell nuclei were counterstained with DAPI. (B) Schematic of the procedure used to detect biotin-CXCL14–binding proteins using HuProt human proteome microarrays containing 18,583 affinity-purified N-terminally GST-tagged proteins. (C) Representative CXCL14-binding membrane proteins in the proteome microarrays. (D) Western blot validation using a streptavidin-agarose pull-down assay of proteome microarray determination that CXCL14 binds directly to GPR85. (E) Mobilization of [Ca2+]i in NAF6 cells that were transfected with control siRNA (NC) or GPR85-3 siRNA and then treated with 100 ng/ml HBSS, rhCXCL14, or rhCXCL12. The black arrows denote the times at which stimulation was initiated. (F) 125I-CXCL14 binding properties between 293T-NC and 293T-GPR85 cells. Data are shown as mean ± SD. n = 4 repetitions. **P < 0.01; ***P < 0.001, 2-tailed Student’s t test. (G) Binding assay with 10 nM 125I-CXCL14 in the presence or absence of increasing concentrations of unlabeled rhCXCL14, rhCXCL12, and rhCXCL3 for 293T cells that were transfected with GPR85. B, specific binding; T, total binding. (H) Knockdown of GPR85 expression by GPR85-3 siRNA in NAF6 cells in the presence or absence of 100 ng/ml rhCXCL14 for the indicated times (0, 30, and 60 minutes). Cell lysates were analyzed by Western blot with antibodies against p-Akt (Ser 473), Akt, p-ERK1/2, ERK1/2, and GAPDH. (I) Knockdown of GPR85 expression by GPR85-3 siRNA in NAF6 cells treated with rhCXCL14 at various concentrations (0–100 ng/ml) for 4 days. Cell lysates were analyzed by Western blot with antibodies against α-SMA, FAP, PDGFRα, and GAPDH. (J) NAF6 cells were transfected with control siRNA (NC) or GPR85-3 siRNA and then cocultured with MCF7Ctrl or MCF7sgHIC1 cells, respectively, for 4 days. NAF6 cell lysates were analyzed by Western blot with antibodies against α-SMA, FAP, PDGFRα, p-Akt (Ser 473), Akt, p-ERK1/2, ERK1/2, and GAPDH.
To investigate whether GPR85 on NAFs could affect the growth and metastasis of BrCa in vivo, MDA-231-LM2 cells mixed with NAF13-NC or NAF13-shGPR85-3 cells at a ratio of 3:1 were injected unilaterally into the fourth mammary fat pads of female BALB/c nude mice (Supplemental Figure 7A). Supplemental Figure 7, B and C, shows that both volumes and weights of the tumors were greatly decreased in the NAF13-shGPR85-3 group compared with the control NAF13-NC group. Moreover, bioluminescence imaging of the harvested lungs demonstrated that lung metastasis was also markedly suppressed in the NAF13-shGPR85-3 group compared with the control group (Supplemental Figure 7D), which was further confirmed using human cytokeratin staining of lung sections to assess human cancer cell metastasis (Supplemental Figure 7E). Collectively, these data suggest that GPR85 on NAFs significantly affects the growth and lung metastasis of TNBC xenografts. One possible explanation is that GPR85-deficient NAFs could not be activated by CXCL14 secreted from the TNBC cells. Therefore, the data suggest that the CXCL14/GPR85 axis is associated with growth and metastasis of TNBC.
Next, using IHC staining, we assayed the stromal phosphorylated ERK (p-ERK), p-Akt (Ser 473), and NEDD8 expression in orthotopic MCF7 xenografts, as described in Figure 4E, and MDA-231-LM2+NAF13 xenografts, as described in Supplemental Figure 7A. The staining reveals that the levels of stromal p-ERK, p-Akt (Ser 473), and NEDD8 were markedly increased in MCF7sgHIC1–NC xenografts compared with the respective controls, whereas knockdown of CXCL14 expression in MCF7sgHIC1 cells decreased these effects (Supplemental Figure 8A). Notably, these signaling pathways were similarly suppressed in the NAF13-shGPR85-3 group compared with the control NAF13-NC group (Supplemental Figure 8B); this was further confirmed by quantitative analysis (Supplemental Figure 8). In summary, the activation of stromal ERK, Akt, and neddylation pathways was suppressed in vivo either by inhibiting CXCL14 secretion in BrCa cells or by reducing GPR85 expression on NAF cells, indicating that the CXCL14/GPR85 axis is vital for activation of these signaling pathways.
CXCL14-activated fibroblasts induce migration of BrCa cells via CCL17.
It has been reported that CAFs induce the migration of cancer cells by secreting cytokine-like molecules (6, 14). Here, we found that CXCL14-activated NAF10 cells enhanced the migration of MDA-231-LM2 BrCa cells; this enhancement is similar to the function of primary CAF10 cells (Figure 6A). Next, we explored whether cytokines secreted by the CXCL14-activated fibroblasts are responsible for modulating the migration of BrCa cells. Cytokine arrays were used to detect the secreted soluble factors derived from conditioned medium (CM) of NAF10, CXCL14-activated NAF10, and CAF10 cells (Figure 6B). Figure 6B shows that the levels of C-C chemokine ligand 17 (CCL17), IL-5, and angiopoietin-2 were significantly increased in the CM of both CXCL14-activated NAF10 and CAF10 cells compared with the levels of these cytokines in the CM of NAF10 cells, suggesting that the cells may have similar phenotypes. Next, to assay the effects of these factors on BrCa cells, we used Boyden chambers in which MDA-231-LM2 cells were plated in the upper chambers and recombinant human CCL17, IL-5, or angiopoietin-2 was placed in the lower wells. The results showed that only CCL17 enhanced the migration of BrCa cells; the other 2 cytokines had no effect (Figure 6C). Consistent with the results obtained using cytokine arrays, ELISA showed that CCL17 was secreted at high levels into the CM of CXCL14-activated NAF10 and CAF10 cells (Supplemental Figure 9A). However, the increased secretion of CCL17 was not markedly observed in NAF13 cells using RNAi-mediated silencing of GPR85 compared with the control cells after rhCXCL14 treatment. (Supplemental Figure 9B). As shown in Figure 6D, the addition of αCCL17 to the coculture system dramatically suppressed the migration of BrCa cells compared with their respective controls. Furthermore, epithelial-mesenchymal transition (EMT) phenotypes were observed in MCF7 and MDA-231-LM2 cells after treatment with increasing concentrations of rhCCL17 for 4 days, as indicated by decreased E-cadherin expression, increased N-cadherin expression, and markedly increased migration and invasion abilities (Figure 6E and Supplemental Figure 9, C–E). Similarly, the migration abilities of MDA-MB-231 cells and breast epithelial cell MCF 10A cells were markedly enhanced by rhCCL17 treatment compared with the respective controls (Supplemental Figure 9F). These results confirm that CCL17 originating from CXCL14-activated fibroblasts that display phenotypes similar to CAFs can facilitate the migration of BrCa cells and is potentially associated with EMT.
Figure 6. CXCL14-activated fibroblasts induce migration of BrCa cells via CCL17.

(A) Upper panel: schematic showing the coculture of MDA-231-LM2 BrCa cells with primary NAF10, CXCL14-activated NAF10, or primary CAF10 cells in a Transwell apparatus (0.4 μm pore size) for 4 days. Lower panel: Boyden chamber assay of MDA-231-LM2 cells that were treated as above. (B) Upper panel: Human XL Cytokine Array Kits (R&D Systems) were used to measure the levels of 102 cytokines in the CM from diverse fibroblasts. Cytokines that were upregulated in the CM of CXCL14-activated NAF10 and CAF10 cells are indicated by colored boxes; they include CCL17 (red), IL-5 (green), and angiopoietin-2 (blue). Black frames indicate the positive controls, and the dashed boxes indicate the negative controls in each membrane. Lower panel: table showing the relative signal intensities of the 3 selected cytokines noted above. The signal intensities were quantified by densitometry using ImageJ software and normalized to the intensity of the internal positive controls. (C) Boyden chamber assay of MDA-231-LM2 cells plated with rhCCL17, rhIL-5, and rh angiopoietin-2 in the lower chambers at 100 ng/ml for 20 hours. (D) Boyden chamber assay of MDA-231-LM2 cells that were cocultured with primary NAF10, CXCL14-activated NAF10, or primary CAF10 cells in a Transwell apparatus for 4 days in the presence or absence of α-CCL17 (1 μg/ml) or an isotype-matched IgG control. (E) MCF7 or MDA-231-LM2 cells were treated with various concentrations (0–100 ng/ml) of rhCCL17 for 4 days, and lysates of the cells were analyzed by Western blot using antibodies against E-cadherin, N-cadherin, and GAPDH. (F) MCF7 or MDA-231-LM2 cells were treated with rhCCL17 at 100 ng/ml for the indicated times (0, 5, 15, 30, and 60 minutes). Cell lysates were analyzed by Western blot with antibodies against p-Akt (Ser 473), Akt, p-GSK-3β (Ser9), GSK-3β, and GAPDH. (G) Knockdown of CCR4 expression by siRNA-3 in MCF7 and MDA-231-LM2 cells in the presence or absence of rhCCL17 at 100 ng/ml for 4 days. Cell lysates were analyzed by Western blot with antibodies against E-cadherin, N-cadherin, p-Akt (Ser 473), Akt, and GAPDH. Data are shown as mean ± SEM. n = 3 independent experiments. ***P < 0.001, 1-way ANOVA followed by Bonferroni’s post hoc test.
It was recently shown that CC chemokine receptor 4 (CCR4), as the receptor for CCL17, can induce the migration of hepatocellular carcinoma cells via the Akt signaling pathway (34). Our mechanistic analyses revealed that CCL17 also activates the Akt/GSK-3β pathways through its receptor, CCR4, in both MCF7 and MDA-231-LM2 cells (Figure 6, F and G). Moreover, RNAi-mediated silencing of CCR4 in these cells markedly inhibited the development of the CCL17-induced EMT phenotype and the activation of Akt signaling (Supplemental Figure 9G and Figure 6G), suggesting that the CCL17/CCR4 axis enhances EMT in BrCa cells through activation of the Akt/GSK-3β pathways.
Finally, we observed that rhCCL17 greatly induced EMT of HIC1-deleted MCF7 BrCa cells, whereas it had little effect on the control cells, suggesting that HIC1 deletion in BrCa cells markedly increases their sensitivity to CCL17-induced EMT (Supplemental Figure 9H).
CCL17 secreted by CAFs promotes lung metastasis of BrCa xenografts.
Based on the above observations, we next assayed the effect of CCL17 secreted by CAFs on BrCa metastasis in vivo. MDA-231-LM2 cells mixed with primary NAF8 or CAF8 cells were injected bilaterally into the fourth mammary fat pads of female BALB/c nude mice. Two weeks later, when the xenografts became palpable, control IgG or αCCL17 was injected into the mice via the tail vein every 3 days. The treatment was maintained for 30 days, and the mice were then euthanized (Figure 7A and Supplemental Figure 10A). As shown in Figure 7B, bioluminescence imaging of the harvested lungs demonstrated that lung metastasis was markedly promoted in the CAF8 plus IgG group compared with the NAF8 plus IgG group. However, lung metastasis was greatly suppressed in the CAF8 plus αCCL17 group compared with the CAF8 plus IgG group (Figure 7B), and this was further confirmed using human cytokeratin staining of lung sections to assess human cancer cell metastasis (Figure 7C). Furthermore, the expression of N-cadherin and vimentin was markedly increased in the orthotopic xenografts in the CAF8 plus IgG group compared with those in the NAF8 plus IgG group; however, the expression of both proteins was greatly reduced in the CAF8 plus αCCL17 group (Figure 7C). Notably, Kaplan-Meier analysis also indicated that αCCL17 treatment greatly increased the survival of the mice xenografted with MDA-231-LM2 cells mixed with CAF8 cells compared with IgG treatment (Figure 7D). Collectively, these data suggest that CCL17 secreted by CAFs promotes the lung metastasis of BrCa xenografts.
Figure 7. CCL17 secreted by CAFs promotes lung metastasis of BrCa xenografts.

(A) Schematic showing the lung metastasis model of BrCa xenografts. MDA-231-LM2 BrCa cells mixed with primary NAF8 or CAF8 cells at a ratio of 3:1 were implanted bilaterally into the mammary fat pads of BALB/c nude mice (n = 12 per group). When the xenografts became palpable 2 weeks later, control IgG or αCCL17 (1 μg/mouse) was then injected via the tail vein every 3 days for 30 days; the mice were then euthanized. (B) Representative bioluminescence imaging of the harvested lungs (left) and quantification of their bioluminescent signals (right, n = 6). Data are shown as mean ± SD. *P < 0.05, 1-way ANOVA followed by Bonferroni’s post hoc test. (C) Representative immunohistochemical staining for human cytokeratin of each group’s lung tissues and for N-cadherin and vimentin in orthotopic xenografts. Dot plots show the mean values for the percentage of human cytokeratin-, N-cadherin–, or vimentin-positive cells with statistical evaluation (n = 5). Data are shown as mean ± SEM. n = 3 independent experiments. **P < 0.01; ***P < 0.001, 1-way ANOVA followed by Bonferroni’s post hoc test. (D) Kaplan-Meier survival curves of BALB/c nude mice that received the indicated treatments. *P < 0.05, log-rank test. (E) Kaplan-Meier plots of distant metastasis-free survival of systemically untreated patients (n = 543) stratified by CCL17 expression. Data were obtained from the Kaplan-Meier plotter database. P = 0.04, log-rank test (28).
Finally, we observed that the stromal CCL17 mRNA levels were higher in BrCa tissues (n = 53) than in normal breast tissues (n = 6) in an Oncomine data set (Finak breast; ref. 35) (Supplemental Figure 10B). Using the TCGA database, we also noted that total CCL17 mRNA levels were significantly higher in 133 BrCa tissues than in paired normal tissues (Supplemental Figure 10C). Furthermore, CCL17 expression was significantly increased in 123 high-metastatic TNBC tissues compared with low-metastatic luminal BrCa tissues (Supplemental Figure 10C). Using the Kaplan-Meier method followed by the log-rank test, we further confirmed that higher expression of CCL17 correlated with lower distant metastasis-free survival in 543 systemically untreated patients (Figure 7E), whereas this correlation was not observed in all cases or systemically treated patients (data not shown) (28). These data imply that CCL17 expression is associated with poor prognosis of BrCa.
The HIC1-CXCL14-CCL17 loop is associated with malignant progression in BrCa patients.
To determine whether our findings are clinically relevant, high-density tissue microarrays (TMA) containing 228 benign and malignant cases from BrCa patients were used to examine the expression of epithelial HIC1, stromal CXCL14, stromal CCL17, stromal GPR85, and epithelial CCR4 (Supplemental Figure 11A). As shown in Figure 8A and Supplemental Figure 11B, there were fewer cases with high expression of epithelial nuclear HIC1 among BrCa patients with malignancies than there were among patients with benign conditions. Notably, HIC1 expression was significantly associated with TNBC (P = 0.007 vs. benign), which has the worst clinical prognosis among all BrCa subtypes (Figure 8A and Supplemental Table 1). In particular, the expression of epithelial nuclear HIC1 correlated strongly with estrogen receptor (ER) and progesterone receptor (PR) expression (P < 0.001 and P = 0.001, respectively) (Supplemental Table 1). In contrast to HIC1, more cases with high expression of stromal CXCL14, stromal CCL17, stromal GPR85, and epithelial CCR4 were found among patients with malignant BrCa than in patients with benign diseases (Figure 8A; Supplemental Figure 11, B and C). Interestingly, the expression of stromal CXCL14 was also markedly correlated with TNBC subtype and ER/PR expression (P = 0.017 vs. benign, P = 0.001 and P < 0.001, respectively) (Figure 8A and Supplemental Figure 11B). Expression of stromal GPR85 was significantly lower in benign cases and luminal cases than in HER2+ TNBC patients (Supplemental Figure 11, B and C). Furthermore, the stromal CCL17 and epithelial CCR4 levels were higher in all 3 BrCa subtypes than in patients with benign conditions, but there was no significant difference among the 3 subtypes (Figure 8A; Supplemental Figure 11, B and C; Supplemental Table 1). Besides, the mRNA expression of stromal GPR85 and total CCR4 was observed to be upregulated in BrCa tissues compared with normal tissues in 2 Oncomine data sets and the TCGA database (Supplemental Figure 11, D and E). Thus, HIC1 and CXCL14/GPR85/CCL17/CCR4 expression are potential prognostic factors associated with poor prognosis of BrCa.
Figure 8. HIC1-CXCL14-CCL17 loop is associated with malignant progression in BrCa patients.

(A) Percentages of low and high expression of epithelial HIC1 (upper), stromal CXCL14 (middle), and stromal CCL17 (bottom) in benign breast tissue and various BrCa subtypes are shown as a pie chart. P vs. benign, χ2 test. (B) Representative IHC images showing the correlation between epithelial HIC1 expression and stromal CXCL14/CCL17 expression in 228 benign and malignant breast tumor samples. Broken lines indicate the margins of the tumor. (C) Schematic model showing how HIC1-mediated crosstalk between cancer cells and mammary fibroblasts promotes BrCa progression. Conditional deletion of HIC1 in the mouse mammary gland may contribute to premalignant transformation at the early stage of breast tumor formation. Moreover, chemokine CXCL14 secreted by HIC1-deleted BrCa cells binds to its cognate receptor GPR85 on mammary fibroblasts in the microenvironment, thereby activating the fibroblasts through the ERK1/2, Akt, and neddylation pathways. The activated fibroblasts in turn promote BrCa progression through induction of EMT by activation of the CCL17/CCR4 chemokine axis.
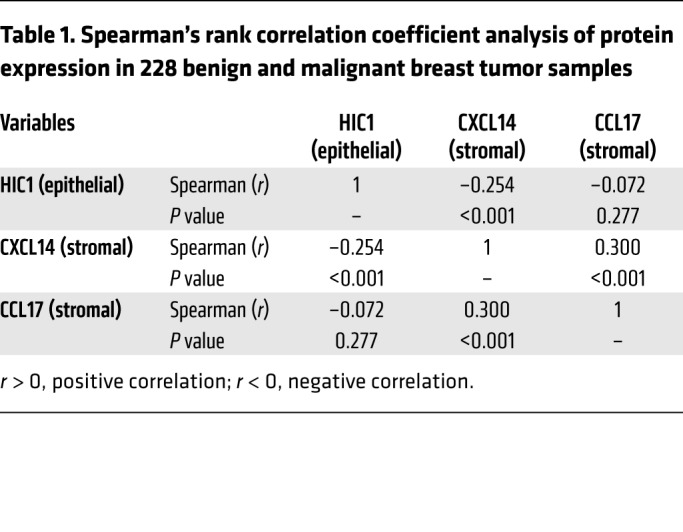
Finally, we found that the cases that had high epithelial nuclear HIC1 levels often displayed low stromal CXCL14 or CCL17 expression and vice versa (Figure 8B). Spearman’s rank correlation coefficient analysis was performed to determine whether the observed differences in expression of the 3 proteins had statistical significance (Table 1). Indeed, there was an inverse correlation between epithelial HIC1 and stromal CXCL14 levels and a positive correlation between stromal CXCL14 and CCL17 levels (Table 1). Moreover, CXCL14 expression tended to be positively associated with the expression of CCL17 in basal-like BrCa in the TCGA database (Supplemental Figure 11F). Collectively, especially with respect to the potential prognostic markers for the TNBC subtype, these data suggest that the HIC1-CXCL14-CCL17 loop is associated with malignant progression in BrCa patients.
Table 1. Spearman’s rank correlation coefficient analysis of protein expression in 228 benign and malignant breast tumor samples.
Discussion
The tumor-suppressor gene HIC1 has been widely documented as playing a vital role in the process of BrCa development (21, 25, 36, 37). However, the use of mice with a conditional knockout of HIC1 in the mammary gland to assay the specific function of the HIC1 gene in BrCa in vivo has not yet been reported. Here, we showed that conditional deletion of HIC1 in the mouse mammary gland may contribute to premalignant transformation at the early stage of tumor formation, inducing the formation of hyperplastic epithelium and increased expression of the proliferative markers Ki67 and cyclin D1 in mammary gland. Mechanistically, loss of HIC1 expression in lung cancer contributes to malignant transformation through the acquisition of chromosomal instability (38), which suggests that it may also correlate with mammary gland tumorigenesis. Furthermore, it has been reported that HIC1 acts as a nuclear modulator of the Wnt-signaling pathway by recruiting TCF4 and β-catenin to HIC1 bodies to exert its suppression functions (39). Indeed, we found that overexpression of HIC1 could repress stemness of BrCa cells in vitro (data not shown). In addition, lactogenic defects of mammary glands were also observed in Hic1–/– mice; this defect may be associated with Wnt signaling, which plays a significant role in the self-renewal of mammary stem cells and in mammary gland development (40–42).
It has been reported that BrCa cells and mammary fibroblasts cooperate and depend upon each other to promote the development of primary tumors to aggressive disease (43). In the work reported in this manuscript, we found that the chemokine CXCL14 secreted by HIC1-deleted BrCa cells induces a functional conversion of NAFs to CAFs; in turn, the activated CAFs could promote metastasis of BrCa cells through secretion of the chemokine CCL17 (Figure 8C). Moreover, using breast TMA, we confirmed that the HIC1-CXCL14-CCL17 loop is associated with malignant progression in BrCa patients.
Chemokine (C-X-C motif) ligand 14 (CXCL14), a small cytokine belonging to the CXC chemokine family, is also known as BRAK (for breast and kidney-expressed chemokine). CXCL14 is responsible for the broad chemotactic activity in activated NK cells, dendritic cells, monocytes, and macrophages (44–46). However, the effect of CXCL14 on cancer development remains controversial. Some reports have claimed antitumor and antiangiogenic effects of CXCL14 (47, 48), whereas others noted upregulated expression of CXCL14 in several cancer types, including BrCa and osteosarcoma (49, 50). A recent study also showed that CXCL14 expression in high stromal tissue, but not in epithelial tissue, is significantly associated with shorter survival in BrCa patients (51), raising the possibility that stromal CXCL14 may exert a procancer role in BrCa progression (52). Moreover, a general association between the activation of mesenchymal cells and CXCL14 has been reported in that CXCL14 is upregulated in activated hepatic stellate cells, which are the main source of extracellular matrix and myofibroblast production (53). Here, we found that CXCL14 has the role of activating mammary fibroblasts. It is well known that chemokines exert their effects by binding to specific transmembrane receptors, most of which are members of the 7-transmembrane GPCRs. Notably, the cognate receptor for chemokine CXCL14 has still not been definitively identified. In fact, induction of calcium influx by CXCL14 in prostaglandin E–treated (PGE-treated) monocytes has been observed, suggesting that a GPCR may be involved in CXCL14 signal transduction (44). For this purpose, using HuProt human proteome microarrays, we found that CXCL14 specifically binds to GPR85 on the cellular membrane of NAFs to exert its biological effects. GPR85, also known as SREB2, is a member of the highly conserved SREB receptor family. It is one of the orphan receptors that is the most evolutionarily conserved GPCR and shares 100% amino acid sequence identity with a number of species, including humans and mice (54). GPR85 has been reported to express virtually in all neurons, but most abundantly in hippocampal dentate gyrus and subventricular zone of the amygdala (55). Interestingly, CXCL14 is also considered to be a primordial or ancient CXC chemokine, based on sequence conservation among species and their homeostatic roles (56), that mediated functions within the developing CNS, including chemotaxis of neurons. Although the receptor for CXCL14 is as yet reported, as conserved ligands tend to bind to conserved receptors (57), the receptor for CXCL14 is probably also conserved. So far, there is no article reporting the function or expression of GPR85 in any other tissues except CNS. Here, expression of GPR85 protein in a variety of cells was observed, indicating that GPR85 levels in stromal cells (NAF/CAF13, Treg, and macrophage) were higher than in other cells (such as 293T, MCF7, Neuro-2A, and THP-1 cells, data not shown), which suggests that GPR85 may play a vital role in the microenvironment of BrCa. Furthermore, mechanistic analyses showed that the CXCL14/GPR85 chemokine axis is responsible for activating mammary fibroblasts through the ERK1/2, Akt, and neddylation pathways. Nevertheless, these pathways were found to be independent of each other in their activation of stromal fibroblasts (data not shown).
Finally, we showed that both CAFs and CXCXL14-activated NAFs can induce migration in vitro and metastasis in vivo of BrCa cells by secreting the potential chemokine CCL17. CCL17, originally known as thymus and activation-regulated chemokine (TARC), was shown to be expressed constitutively in thymus tissue and transiently in stimulated peripheral blood mononuclear cells (58). This chemokine is reported to specifically bind to and induce chemotaxis in T cells and to exert its effects by interacting with the chemokine receptor CCR4 (58, 59). CCR4 is a key receptor in the regulation of chemokine-dependent immune homeostasis and is selectively expressed by Tregs and Th2 cells. Recent studies have shown that colon and BrCa cells also express functional CCR4 receptors (60, 61) and that these receptors trigger remote induction of CCL17 in the lungs of mice and mediate CCR4-dependent pulmonary metastasis of BrCa cells (62). Among the stromal cells in the tumor microenvironment, tumor-associated macrophages have been reported to promote the process of EMT in hepatocellular carcinoma cells through the secretion of CCL17 (63). However, the role of the CCL17 secreted by CAFs in BrCa development is still unclear. Our data show that activated CAFs promote the migration of cancer cells in vitro and the metastasis of BrCa in vivo through the CCL17/CCR4 axis upon activation of the Akt/GSK-3β signaling pathway.
Notably, we found that HIC1-deleted BrCa cells display increased sensitivity to the induction of EMT by CCL17 compared with the respective control cells, suggesting that the crosstalk between HIC1-deleted BrCa cells and mammary fibroblasts is vital for BrCa development.
In conclusion, we find that the HIC1-CXCL14-CCL17-CCR4 loop is essential for BrCa progression. The activation of this loop in BrCa tissue may serve as a potential prognostic marker and may provide the basis for more effective treatment strategies.
Methods
Cell culture.
MDA-MB-231, MCF7, and T47D human BrCa cells, MCF 10A human breast epithelial cells, and 293T embryonic kidney cells were obtained from ATCC and grown according to standard protocols. MDA-231-LM2 human BrCa cells were provided and cultured as recommended by J. Massague (Memorial Sloan-Kettering Cancer Center, New York, New York, USA). Primary mammary fibroblasts were isolated from 13 cases of freshly removed BrCa patient tissue, and additional details are described in the Supplemental Methods. The cell lines were tested and authenticated by DNA typing in the Shanghai Jiao Tong University Analysis Core and were cultured in a 37°C water-saturated 5% CO2 atmosphere.
Mice.
Wap-Cre transgenic mice with a C57BL/6 background were initially generated by Shanghai Biomodel Organism Science & Technology Development Co. Hic1fl/fl transgenic mice with a C57BL/6 background were provided by Vladimir Korinek (Institute of Molecular Genetics, Academy of Sciences of the Czech Republic, Prague, Czech Republic) (64). All mice were kept under specific pathogen-–free (SPF) conditions. To ameliorate any suffering, mice were euthanized by CO2 inhalation.
Generation of conditional knockout mice.
A Cre-mediated specific HIC1 gene deletion in luminal mammary epithelial cells was achieved by crossing Hic1fl/fl mice with Wap-Cre mice to produce Hic1fl/+/Wap-Cre+ mice. Double heterozygous Hic1fl/+/Wap-Cre+ mice were then crossed with Hic1fl/fl mice to produce Hic1fl/fl/Wap-Cre+ mice. Next, Hic1fl/fl/Wap-Cre+ mice were bred with Hic1fl/fl mice to produce offspring that were used in our experiments. The Hic1fl/fl mice were used as the Hic1+/+ controls, and the Hic1fl/fl/Wap-Cre+ mice were used as the Hic1–/– group. To avoid problems in nursing pups that were caused by mothers with potentially defective mammary glands, only the male mice of the breeding pairs carried the Cre transgene. The genotypes of the offspring were determined by PCR amplification of DNA obtained from the tails of the animals. Primers for PCR-based genotyping are listed in the Supplemental Methods.
Luciferase reporter assays and ChIP.
The CXCL14 promoters and their truncated constructs were generated from genomic DNA of the human breast epithelial cell line MCF10A. All constructs and their mutants were inserted into the pGL3-basic reporter gene vector. ChIP was performed according to published protocols, with slight modifications (26). Additional details are provided in the Supplemental Methods.
Cytokine arrays.
Cytokine arrays (Proteome Profile Human XL Cytokine Array Kit, catalog c8052; R&D Systems) were used to survey the changes in 102 cytokines and chemokines in CM obtained from NAF10, CXCL14-activated NAF10, and CAF10 cells. NAF10 cells were treated with 100 ng/ml rhCXCL14 for 4 days, then seeded into 6-well plates containing unprocessed NAF10 and CAF10 cells. After 48 hours, the cell culture supernatants were collected and incubated overnight with the array. Next, the membranes were washed to remove unbound materials and incubated with a cocktail of biotinylated antibodies. Streptavidin-HRP and chemiluminescent detection reagents were then applied, and a signal was produced at each capture spot corresponding to the amount of protein bound. The membranes were exposed to x-ray film for 10 minutes, and profiles of the mean spot pixel densities were analyzed using Protein Array Analyzer for ImageJ (NIH).
Tumor xenografts.
To establish orthotopic BrCa xenografts, 5 × 106 MCF7 cells (MCF7NC–NC, MCF7sgHIC1–NC, MCF7sgHIC1–shCXCL14) were injected bilaterally into the fourth mammary fat pads of female BALB/c nude mice (n = 10 per group). To supplement estrogen for MCF7 growth, each mouse was implanted with a pellet containing 0.72 mg 17β-estradiol (catalog NE-121; Innovative Research) 3 days prior to MCF7 injection. Tumor growth was evaluated by monitoring tumor volume with calipers (length × width2 × 0.5) every 5 days. The animals were euthanized 6 weeks later, and the tumor xenografts were harvested for further investigation.
To establish orthotopic lung metastatic mouse models of MDA-231-LM2 BrCa cells, 1 × 106 MDA-231-LM2 cells mixed with primary NAF or CAF cells at a ratio of 3:1 were orthotopically implanted into the mammary fat pads of BALB/c nude mice. When the xenografts were palpable (approximately 0.5 cm in diameter), tail vein injection of control IgG or a neutralizing antibody against CCL17 (catalog MAB364; R&D Systems) (1 μg/mouse) was performed every 3 days for 4 weeks. The lungs of the animals bearing breast tumor xenografts that stably express luciferase were analyzed using the Xenogen IVIS Imaging System (PerkinElmer).
TMA.
High-density TMA of human BrCa clinical samples (catalog BRC2281) were obtained from a cohort of 228 patients and constructed by Superbiotek Inc. IHC staining was performed with specific antibodies against HIC1 (catalog bs15485R; Bioss), CXCL14 (catalog ab46010; Abcam), CCL17 (catalog ab195044; Abcam), GPR85 (catalog ab140783; Abcam), and CCR4 (catalog ab1669; Abcam). Additional details are provided in the Supplemental Methods.
Statistics.
All statistical analyses were performed using SPSS for Windows, version 13.0. Two-tailed Student’s t tests were used for comparisons between 2 groups, and 1-way ANOVA followed by Bonferroni’s post hoc test was used for multiple comparisons (3 or more groups). Kaplan-Meier curves for survival analyses were determined using the log-rank test. For multiple group comparisons and repeated measures of in vivo data, repeated-measures ANOVA (RM ANOVA), followed by a post hoc least significant difference (LSD) test, was performed. To analyze the relationship between protein levels and clinicopathological status in BrCa TMA, χ2 tests were applied. Spearman’s rank correlation coefficient analysis was performed to assess the relationship among epithelial HIC1, stromal CXCL14, and CCL17 in the BrCa TMA as well as the correlation of CXCL14 expression with CCL17 expression in the TCGA database. All experiments for cell cultures were performed independently at least 3 times and in triplicate each time. In all cases, P values of less than 0.05 were considered statistically significant.
Study approval.
All murine studies were in accordance with the animal welfare policy of the Shanghai Jiao Tong University School of Medicine and approved by the Shanghai Jiao Tong University School of Medicine IACUC (protocol no. A-2016-015). For all human studies, written, informed consent was received from participants prior to inclusion in the study where required. All samples were obtained in accordance with standard protocols of the Renji Hospital Ethics Committee of Shanghai Jiao Tong University School of Medicine.
Author contributions
JW, YW, ZH, JL, and XM conceived and designed the study. YW, XW, and LW acquired the data. YW and LW analyzed the data. YJ and JL provided resources. MH, Y Li, LH, Y Liang, TW, MY, GL, and GF supervised the study. YW and JW wrote and reviewed the manuscript.
Supplementary Material
Acknowledgments
The authors express their appreciation to Ceshi Chen and Binhua Zhou for comments. Research in the authors’ laboratory is supported by the National Natural Science Foundation of China (nos. 81272404, 81772806), the National Key Program (program 973) for Basic Research of China (2011CB510106), and the Program for Professor of Special Appointment (Eastern Scholar to JW) at Shanghai Institutions of Higher Learning.
Version 1. 09/11/2018
In-Press Preview
Version 2. 10/22/2018
Electronic publication
Version 3. 12/03/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2018;128(12):5235–5250.https://doi.org/10.1172/JCI99974.
Contributor Information
Yingying Wang, Email: 1181742801@qq.com.
Xiaoling Weng, Email: 3070701040@163.com.
Luoyang Wang, Email: wangnan235@163.com.
Mingang Hao, Email: rogerbao2001@163.com.
Yue Li, Email: 0903ly@163.com.
Lidan Hou, Email: houlidan0311@126.com.
Yu Liang, Email: 1131183@qq.com.
Tianqi Wu, Email: 395307534@qq.com.
Mengfei Yao, Email: yaomengfei@163.com.
Guowen Lin, Email: linguowen1024@126.com.
Yiwei Jiang, Email: angelajyw@126.com.
Guohui Fu, Email: fuguhu@263.net.
Zhaoyuan Hou, Email: houzy@sjtu.edu.cn.
Xiangjun Meng, Email: meng_xiangjun@yahoo.com.
Jinsong Lu, Email: lujjss@163.com.
Jianhua Wang, Email: jianhuaw2007@qq.com.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67(6):439–448. doi: 10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 3.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101. [PubMed] [Google Scholar]
- 4.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186–196. doi: 10.1016/j.addr.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 7.Dumont N, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia. 2013;15(3):249–262. doi: 10.1593/neo.121950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kojima Y, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trimboli AJ, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461(7267):1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Procopio MG, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17(9):1193–1204. doi: 10.1038/ncb3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123(6):1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 12.Trimis G, Chatzistamou I, Politi K, Kiaris H, Papavassiliou AG. Expression of p21waf1/Cip1 in stromal fibroblasts of primary breast tumors. Hum Mol Genet. 2008;17(22):3596–3600. doi: 10.1093/hmg/ddn252. [DOI] [PubMed] [Google Scholar]
- 13.Trimmer C, et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: a new genetically tractable model for human cancer associated fibroblasts. Cancer Biol Ther. 2011;11(4):383–394. doi: 10.4161/cbt.11.4.14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studebaker AW, et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008;68(21):9087–9095. doi: 10.1158/0008-5472.CAN-08-0400. [DOI] [PubMed] [Google Scholar]
- 15.Zheng J, et al. HIC1 modulates prostate cancer progression by epigenetic modification. Clin Cancer Res. 2013;19(6):1400–1410. doi: 10.1158/1078-0432.CCR-12-2888. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, et al. Hypermethylated in cancer 1(HIC1) suppresses non-small cell lung cancer progression by targeting interleukin-6/Stat3 pathway. Oncotarget. 2016;7(21):30350–30364. doi: 10.18632/oncotarget.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujii H, Biel MA, Zhou W, Weitzman SA, Baylin SB, Gabrielson E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene. 1998;16(16):2159–2164. doi: 10.1038/sj.onc.1201976. [DOI] [PubMed] [Google Scholar]
- 18.Wales MM, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1(6):570–577. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- 19.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123(3):437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Pinte S, Stankovic-Valentin N, Deltour S, Rood BR, Guérardel C, Leprince D. The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor: definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J Biol Chem. 2004;279(37):38313–38324. doi: 10.1074/jbc.M401610200. [DOI] [PubMed] [Google Scholar]
- 21.Cheng G, et al. HIC1 silencing in triple-negative breast cancer drives progression through misregulation of LCN2. Cancer Res. 2014;74(3):862–872. doi: 10.1158/0008-5472.CAN-13-2420. [DOI] [PubMed] [Google Scholar]
- 22.Carter MG, et al. Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum Mol Genet. 2000;9(3):413–419. doi: 10.1093/hmg/9.3.413. [DOI] [PubMed] [Google Scholar]
- 23.Chen WY, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33(2):197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- 24.Van Rechem C, et al. Scavenger chemokine (CXC motif) receptor 7 (CXCR7) is a direct target gene of HIC1 (hypermethylated in cancer 1) J Biol Chem. 2009;284(31):20927–20935. doi: 10.1074/jbc.M109.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W, et al. A potential tumor suppressor role for Hic1 in breast cancer through transcriptional repression of ephrin-A1. Oncogene. 2010;29(17):2467–2476. doi: 10.1038/onc.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao M, et al. HIC1 loss promotes prostate cancer metastasis by triggering epithelial-mesenchymal transition. J Pathol. 2017;242(4):409–420. doi: 10.1002/path.4913. [DOI] [PubMed] [Google Scholar]
- 27.Zhao H, et al. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol Biol Cell. 2004;15(6):2523–2536. doi: 10.1091/mbc.e03-11-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Györffy B, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 29.Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106(6):dju083. doi: 10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- 31.Xu J, et al. The neddylation-cullin 2-RBX1 E3 ligase axis targets tumor suppressor RhoB for degradation in liver cancer. Mol Cell Proteomics. 2015;14(3):499–509. doi: 10.1074/mcp.M114.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19(4):541–555. doi: 10.1016/j.ccr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hromas R, et al. Cloning of BRAK, a novel divergent CXC chemokine preferentially expressed in normal versus malignant cells. Biochem Biophys Res Commun. 1999;255(3):703–706. doi: 10.1006/bbrc.1999.0257. [DOI] [PubMed] [Google Scholar]
- 34.Cheng X, et al. Up-regulation of chemokine receptor CCR4 is associated with human hepatocellular carcinoma malignant behavior. Sci Rep. 2017;7(1):12362. doi: 10.1038/s41598-017-10267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, et al. HIC1 and miR-23~27~24 clusters form a double-negative feedback loop in breast cancer. Cell Death Differ. 2017;24(3):421–432. doi: 10.1038/cdd.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulay G, et al. Loss of hypermethylated in cancer 1 (HIC1) in breast cancer cells contributes to stress-induced migration and invasion through β-2 adrenergic receptor (ADRB2) misregulation. J Biol Chem. 2012;287(8):5379–5389. doi: 10.1074/jbc.M111.304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szczepny A, et al. The tumor suppressor Hic1 maintains chromosomal stability independent of Tp53. Oncogene. 2018;37(14):1939–1948. doi: 10.1038/s41388-017-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valenta T, Lukas J, Doubravska L, Fafilek B, Korinek V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 2006;25(11):2326–2337. doi: 10.1038/sj.emboj.7601147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boras-Granic K, Hamel PA. Wnt-signalling in the embryonic mammary gland. J Mammary Gland Biol Neoplasia. 2013;18(2):155–163. doi: 10.1007/s10911-013-9280-x. [DOI] [PubMed] [Google Scholar]
- 41.Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology. 2006;73(5):213–223. doi: 10.1159/000098207. [DOI] [PubMed] [Google Scholar]
- 42.Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010;6(6):568–577. doi: 10.1016/j.stem.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avgustinova A, et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat Commun. 2016;7:10305. doi: 10.1038/ncomms10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurth I, Willimann K, Schaerli P, Hunziker T, Clark-Lewis I, Moser B. Monocyte selectivity and tissue localization suggests a role for breast and kidney-expressed chemokine (BRAK) in macrophage development. J Exp Med. 2001;194(6):855–861. doi: 10.1084/jem.194.6.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Starnes T, et al. The chemokine CXCL14 (BRAK) stimulates activated NK cell migration: implications for the downregulation of CXCL14 in malignancy. Exp Hematol. 2006;34(8):1101–1105. doi: 10.1016/j.exphem.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 46.Shurin GV, et al. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174(9):5490–5498. doi: 10.4049/jimmunol.174.9.5490. [DOI] [PubMed] [Google Scholar]
- 47.Shellenberger TD, et al. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. 2004;64(22):8262–8270. doi: 10.1158/0008-5472.CAN-04-2056. [DOI] [PubMed] [Google Scholar]
- 48.Hata R, et al. Suppressed rate of carcinogenesis and decreases in tumour volume and lung metastasis in CXCL14/BRAK transgenic mice. Sci Rep. 2015;5:9083. doi: 10.1038/srep09083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allinen M, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 50.Lu J, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF-κB signaling. J Clin Invest. 2015;125(5):1839–1856. doi: 10.1172/JCI78437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sjöberg E, Augsten M, Bergh J, Jirström K, Östman A. Expression of the chemokine CXCL14 in the tumour stroma is an independent marker of survival in breast cancer. Br J Cancer. 2016;114(10):1117–1124. doi: 10.1038/bjc.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu XL, et al. Expression of CXCL14 and its anticancer role in breast cancer. Breast Cancer Res Treat. 2012;135(3):725–735. doi: 10.1007/s10549-012-2206-2. [DOI] [PubMed] [Google Scholar]
- 53.De Minicis S, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132(5):1937–1946. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 54.Matsumoto M, et al. The evolutionarily conserved G protein-coupled receptor SREB2/GPR85 influences brain size, behavior, and vulnerability to schizophrenia. Proc Natl Acad Sci U S A. 2008;105(16):6133–6138. doi: 10.1073/pnas.0710717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsumoto M, et al. A conserved mRNA expression profile of SREB2 (GPR85) in adult human, monkey, and rat forebrain. Brain Res Mol Brain Res. 2005;138(1):58–69. doi: 10.1016/j.molbrainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Huising MO, van der Meulen T, Flik G, Verburg-van Kemenade BM. Three novel carp CXC chemokines are expressed early in ontogeny and at nonimmune sites. Eur J Biochem. 2004;271(20):4094–4106. doi: 10.1111/j.1432-1033.2004.04347.x. [DOI] [PubMed] [Google Scholar]
- 57.Murphy PM. Molecular mimicry and the generation of host defense protein diversity. Cell. 1993;72(6):823–826. doi: 10.1016/0092-8674(93)90571-7. [DOI] [PubMed] [Google Scholar]
- 58.Imai T, Yoshida T, Baba M, Nishimura M, Kakizaki M, Yoshie O. Molecular cloning of a novel T cell-directed CC chemokine expressed in thymus by signal sequence trap using Epstein-Barr virus vector. J Biol Chem. 1996;271(35):21514–21521. doi: 10.1074/jbc.271.35.21514. [DOI] [PubMed] [Google Scholar]
- 59.Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997;272(23):15036–15042. doi: 10.1074/jbc.272.23.15036. [DOI] [PubMed] [Google Scholar]
- 60.Li JY, et al. The chemokine receptor CCR4 promotes tumor growth and lung metastasis in breast cancer. Breast Cancer Res Treat. 2012;131(3):837–848. doi: 10.1007/s10549-011-1502-6. [DOI] [PubMed] [Google Scholar]
- 61.Al-haidari AA, Syk I, Jirström K, Thorlacius H. CCR4 mediates CCL17 (TARC)-induced migration of human colon cancer cells via RhoA/Rho-kinase signaling. Int J Colorectal Dis. 2013;28(11):1479–1487. doi: 10.1007/s00384-013-1712-y. [DOI] [PubMed] [Google Scholar]
- 62.Olkhanud PB, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69(14):5996–6004. doi: 10.1158/0008-5472.CAN-08-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu F, Li X, Chen S, Zeng Q, Zhao Y, Luo F. Tumor-associated macrophage or chemokine ligand CCL17 positively regulates the tumorigenesis of hepatocellular carcinoma. Med Oncol. 2016;33(2):17. doi: 10.1007/s12032-016-0729-9. [DOI] [PubMed] [Google Scholar]
- 64.Pospichalova V, et al. Generation of two modified mouse alleles of the Hic1 tumor suppressor gene. Genesis. 2011;49(3):142–151. doi: 10.1002/dvg.20719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.