

Abstract
Methods for the asymmetric transfer hydrogenation (ATH) of ketones and imines are still being intensively studied and developed. Of foremost interest is the use of Noyori’s [RuCl(η6-arene)(N-TsDPEN)] complexes in the presence of a hydrogen donor (i-PrOH, formic acid). These complexes have found numerous practical applications and have been extensively modified. The resulting derivatives have been heterogenized, used in ATH in water or ionic liquids and even some attempts have been made to approach the properties of biocatalysts. Therefore, an appropriate modification of the catalyst that suits the specific requirements for the reaction conditions is very often readily available. The mechanism of the reaction has also been explored to a great extent. Model substrates, acetophenone (a ketone) and 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline (an imine), are both reduced by this Ru catalytic system with almost perfect selectivity. However, in each case the major product is a different enantiomer (S- for an alcohol, R- for an amine when the S,S-catalyst is used), which demanded an in-depth mechanistic investigation. Full-scale molecular modelling of this system enabled us to visualize the plausible 3D structures of the transition states, allowing the proposition of a viable explanation of previous experimental findings.
Keywords: asymmetric transfer hydrogenation, imines, ketones, chiral, molecular modelling
1. Introduction
It is a well-known fact that optically pure compounds play an inimitable role in various branches of the chemical industry, with pharmaceutical and fine chemical production processes being the most prominent ones [1,2]. Nowadays, enantioenriched chiral substances can be obtained in various ways, e.g., via chiral resolution of a racemic mixture, enzymatic catalysis, isolation of chiral compounds from natural resources, or catalytic asymmetric synthesis [3].
Chiral separation of enantiomers from a racemate prepared by standard organic techniques is still a widely-used method in asymmetric synthesis [4,5,6,7], mainly for economic reasons. However, chiral resolution brings unfavourable additional steps into the chain of synthetic reactions. Moreover, the other enantiomer is often of no value after the resolution.
Enzymatic catalysis stands in contrast to this classical method [8], exploiting the chirality imprinted in species of natural origin. Notwithstanding the superb selectivity achievable by means of enzymatic catalysis, the use of enzymes is often disadvantageous due to the strict conditions required to maintain their integrity and efficacy.
Catalytic asymmetric synthesis stands somewhere “in between” when compared to chiral resolution techniques and enzymatic reactions, particularly in terms of cost and efficacy. Without a doubt, it embodies one of the pillars of modern chemistry. Although it is endowed with an exquisite toolbox for the synthesis of optically pure substances [9,10,11], much still remains to be explored.
Among the most frequently used techniques for enantioselective synthesis is asymmetric hydrogenation, already a 40 year old method that exploits an astonishing variety of organometallic complex catalysts [12,13,14]. The asymmetric bias of the reaction is generally reached by employing chiral ligands, which provide the necessary chiral environment around the central metal atom. The first successful ligands capable of stereoselective reductions were DIOP [15,16] (a bisphosphine developed by Kagan and Dang in 1971) and CAMP [17] (a monophosphine introduced by Knowles and co-workers in 1972). Using rhodium-based precursors, these ligands formed in situ catalytic complexes able to hydrogenate unsaturated carboxylic acids, giving amino acids of good optical purity. It was indeed a huge breakthrough in chemistry, as Knowles justifiably pointed out [13]: “(...) this was the first time ever that anyone had obtained enzyme-like selectivity with a man-made catalyst! Never in our wildest imagination did we think a structure versus activity study would converge so quickly to a product with commercial potential. CAMP was our sixth candidate. As I look back from this perspective, I don’t think that even we were emotionally equipped to realize what we had done.”
During the 40 years of asymmetric hydrogenation development, a myriad of ligands and catalysts have emerged, giving >99% ees in reductions of C=C, C=O and C=N double bonds present in a variety of substrates. The majority of the ligands developed for asymmetric synthesis are those based on the phosphorus atom, which was thoroughly covered in a recent monograph [18]. In addition to these, hybrid P,N ligands [19,20] have also been developed. This review, however, focuses on a defined group of ligands containing nitrogen donor atoms.
To date the original Noyori-type η6-arene/N-arylsulfonyl-1,2-diphenylethylenediamine-ruthenium(II) catalysts have performed well in asymmetric transfer hydrogenations (ATH) of various groups of substrates [21,22,23,24,25,26,27,28] and have found numerous practical applications [29,30,31,32,33,34,35,36,37]. Naturally, a number of reviews have been published concerning both these remarkable catalytic hydrogenation systems [7,38,39,40,41,42,43] and their reaction mechanisms [44,45,46,47]. It should be pointed out that these complexes have also been used for asymmetric hydrogenations (AH) [48,49], which represents an extraordinary feature since both mechanisms (ATH and AH) are usually not achievable using a common catalyst. This review examines the ATH of imines and ketones using Noyori’s extraordinary η6-arene/N-arylsulfonyl-1,2-diphenylethylenediamine-ruthenium(II) catalysts and discusses some mechanistic considerations based on the latest findings. Hence, [RuCl(η6-arene)(amino alcohol)] species, which were investigated at the same time as the [RuCl(η6-arene)TsDPEN] complexes, will not be covered, although they have also been tested by Noyori for the reduction of aromatic ketones [50]. Another analogous group of ligands comprises tosylated diaminocyclohexane (TsCYDN) [51] and its derivatives. Although these ligands are very similar to the DPEN-type auxiliaries, they are beyond the scope of this review.
In the first instance, these catalytic complexes were used in homogeneously catalyzed ATH of ketones and imines in organic media [21]. Although these initial experiments proved the indisputable significance of the newly-discovered catalysts (see Section 2.1.), they suffered from difficult catalyst separation, which is a common issue in homogeneous catalysis. Consequently the chiral product, which was often synthesised for biological uses, contained expensive and environmentally unfriendly ruthenium compounds. Therefore, successive developments in this area have focused on greener applications of the catalysts in hand, which in particular meant their immobilization or the replacement of the solvent with water or ionic liquids. In Section 2.2. to Section 2.4., some examples are given on how this can be accomplished when unmodified catalysts are used in practice. However, the majority of cases have dealt with changes to the catalyst structure so as to improve its properties for a desired use. This large group of derivatives is covered in subchapter 3 which is further structured thematically. Hand in hand with these overall improvements came the possibility of catalyst recycling, which is of foremost practical importance.
The last subchapter of this review outlines the knowledge of the reaction mechanisms through which the ATH of ketones and imines operate under different circumstances. While the reaction mechanism is very well characterised in the case of ketones, a different concept had to be devised for the ATH of imines. The mechanistic pathways further change upon using different hydrogen donor molecules, or conducting the reactions in aqueous solutions.
2. Original η6-Arene/N-sulfonyldiamine-RuII Catalysts
2.1. The Pioneering Works on η6-Arene/N-sulfonyldiamine-RuII Catalysts
Beginning in 1995, Noyori’s η6-arene/N-arylsulfonyl-1,2-diphenylethylenediamine-ruthenium(II) catalysts 1(a-f) [52,53,54,55] (Figure 1) emerged. These complexes were able to accelerate the hydrogenations of a wide range of aldehydes, ketones and imines with great selectivity [7,21,56]. This subsection briefly treats the original RuII complexes in selected examples of their original applications.
Figure 1.

The basic set of η6-arene/N-sulfonyldiamine-RuII catalysts.
The RuII complexes are composed of two moieties: the η6-arene ligand and the chiral diamine auxiliary ligand. Benzene [53], p-cymene [53,55] and mesitylene [52,54,55] were first applied as the η6-arene ligands and shortly afterwards, hexamethylbenzene was tested as well [26]. The chiral diamine moiety was originally represented by N-tosyl-1,2-diphenylethylene-1,2-diamine (Ts-DPEN), N-(naphthalene-1-sulfonyl)-1,2-diphenylethylene-1,2-diamine (Nps-DPEN) and N-(2,4,6-trimethylphenylsulfonyl)-1,2-diphenylethylene-1,2-diamine (mesitylsulfonyl-DPEN).
The original ligand combinations are outlined in Table 1 along with their performance data. Catalyst 1a was found to be highly active in some cases, probably due to its η6-benzene ligand (entries 1 vs. 6). It was also described as efficacious in the ATH of 1-phenyl-3,4-dihydroisoquinoline derivatives [23], which proved unfeasible when using e.g., 1b. Perhaps the most frequently used catalyst is 1b, equipped with η6-p-cymene and TsDPEN, which has been shown to be able to efficiently hydrogenate imines and ketones with very high enantioselectivity (entries 2 and 3, refs. [53,57]). Catalyst 1c was mainly used in the ATH of ketones, furnishing even higher ees than 1b (entries 4 and 5, refs. [52,54]). Usage of catalyst 1d has been rather limited due to its lower activity. For instance, although it was demonstrated that 1d accelerated the hydrogenation of benzaldehyde-1-d (entry 6), this reaction catalyzed by 1a or 1b proceeded up to 20 times faster with a very similar ee [26]. Catalyst 1e has not been used very often in practice, owing to its very low solubility in most organic solvents [23]. However, similarly to 1a, it was reported to be able to catalyze the ATH of 1-phenyl-3,4-dihydroisoquinoline derivatives (entry 7, ref. [53]). Above all, catalyst 1f was tested in the ATH of 1-substituted 3,4-dihydroisoquinolines with very good enantioselectivity (entry 8, ref. [53]).
Table 1.
The basic set of η6-arene/N-sulfonyldiamine-RuII catalysts and their representative reactions.
| Entry | Catalyst | Substrate | Product | S/C [a]; Time; Temp.; Conversion (%) |
% ee (config) [b] |
Ref. [c] |
|---|---|---|---|---|---|---|
| 1 | (R,R)-1a | 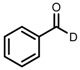 |
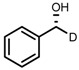 |
200; 0.5 h; 28 °C; 100 | 97 (R) | [26] |
| 2 | (S,S)-1b | 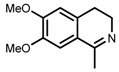 |
 |
200; 3 h; 28 °C; 99 | 95 (R) | [53] |
| 3 | (R,R)-1b [d] | 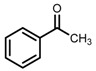 |
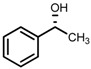 |
100; 20 h; 28 °C; > 99 | 97.7 (R) | [57] |
| 4 | (S,S)-1c | 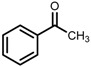 |
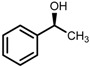 |
200; 20 h; 28 °C; 99 | 98 (S) | [54] |
| 5 | (S,S)-1c [d] | 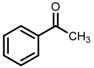 |
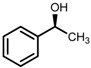 |
200; 15 h; r.t.; 99 | 97 (S) | [52] |
| 6 | (R,R)-1d | 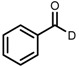 |
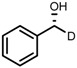 |
200; 7 h; 28 °C; 99 | 96 (R) | [26] |
| 7 | (S,S)-1e | 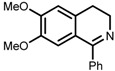 |
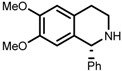 |
200; 8 h; 28 °C; 99 | 84 (R) | [53] |
| 8 | (R,R)-1f | 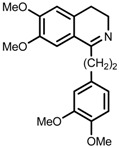 |
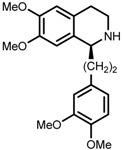 |
200; 12 h; 28 °C; 99 | 92 (S) | [53] |
[a] Molar ratio substrate/catalyst; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry; [d] Catalyst was formed in situ from the [RuCl2(η6-arene)]2 dimer and the corresponding N-arylsulfonylethylene-1,2-diamine.
Basically, there are two ways to use such a complex as a catalyst: generation in situ or use of a preformed complex. Preparation of the catalyst in situ involves reacting the [RuCl2(η6-arene)]2 dimer with the corresponding N-arylsulfonyl-diphenylethylenediamine immediately before the hydrogenation, without further purification. This approach was actually applied in the first published work [52], where [RuCl2(η6-mesitylene)]2 and (S,S)-TsDPEN (Ru:TsDPEN = 1:2 molar) were heated at 80 °C for 20 min and then used in the reduction of aromatic ketones. For example, hydrogenation of acetophenone in such a manner gave (S)-1-phenylethanol in 97% ee and 95% yield (Entry 5 in Table 1). Preparing the catalyst this way may substantially help in overcoming certain difficulties, such as: (1) the true catalytic complex is not commercially available (whereas the precursors are); (2) the complex isolation is difficult or infeasible; or (3) the complex is poorly soluble. Preparation of the complex also involves a recrystallization step, which may thus be avoided if problematic.
On the other hand, usage of a preformed complex can sometimes be more convenient in practice, as only one compound is used in the catalyzed reaction. Additionally, successful preparation of a crystalline Ru catalytic complex can allow its characterisation by single-crystal X-ray diffraction analysis, providing the molecular geometry, which can be used in molecular modelling amongst other applications. In the remainder of this section various applications of the type 1 catalysts are discussed.
2.2. ATH Catalyzed by 1 in Water
Reactions carried out in water are very desirable as they are “green chemistry compatible” – in fact, enzyme-catalyzed reactions in nature proceed exclusively in aqueous media. Generally, such reactions may either form homogeneous (i.e., both catalyst and substrate are soluble in water) or heterogeneous (one of the components being insoluble in water) systems [58]. The substrate and/or the catalyst often have low water solubility and, in contrast, the hydrogen donor can be hydrophilic. Numerous methods of overcoming these difficulties have been published.
In 2004, the ATH system under discussion was first reported to be practicable in water with surprisingly good results for the hydrogenation of aromatic ketones catalyzed by 1b. In this work [59], Wu, Xiao et al. showed that the reaction proceeded much faster in HCOONa/H2O than in the HCOOH/triethylamine azeotrope. While in the HCOONa/H2O system acetophenone was completely reduced within 2 h (entry 1 in Table 2), in the azeotrope it took a reaction time of 12 h to reach almost full conversion (entry 2). Their explanation of this observation was based on the assumption that 1b was more soluble in the substrate (organic phase) than in water. Following this, they revealed a strong pH dependence of the ATH of aromatic ketones in water using 1b and HCOOH/triethylamine [60], proposing two different catalytic cycles for the reaction. While under basic conditions they expected the standard metal-ligand bifunctional mechanism to take place [61], low pH values were connected with protonation of the amido nitrogen of the TsDPEN ligand, which caused its partial decoordination. This phenomenon explained the loss of activity of 1b under acidic conditions. Optimal pH values were described as between pH 5 and 8, which was effectively maintained by a suitable aqueous solution of HCOOH/triethylamine. Remarkably, this optimization of the reaction conditions allowed use of S/C ratios as high as 10,000.
Table 2.
ATH of acetophenone catalyzed by 1b in water [a].
| Entry | Catalyst config. | Solution | Time; Temp.; Conversion (%) | % ee (config) [b] | Ref. [c] |
|---|---|---|---|---|---|
| 1 | (R,R) | H2O/HCOONa | 2 h; 40 °C; >99 | 94 % (R) | [59] |
| 2 | (R,R) | HCOOH/Et3N | 12 h; 40°C; 98 | 97 % (R) | [59] |
| 3 | (R,R) | H2O/HCOONa/CTAB [d] | 4 h; 28 °C; >99 | 95 % (R) | [62] |
| 4 | (S,S) | H2O/HCOONa/PEG | 15 h; 40 °C; >99 | 96 % (S) | [63] |
[a] Molar ratio substrate/catalyst (S/C) = 100; The catalyst was formed in situ from the [RuCl2(η6-p-cymene)]2 dimer and TsDPEN; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry. [d] CTAB = cetyltrimethyl-ammonium bromide.
Catalyst 1b has also been integrated into micelle-like microreactors created from cetyltrimethylammonium bromide (CTAB) [62], which provided lipophilic reaction compartments where the ATH process could be accelerated. The model substrate, acetophenone, was reduced in up to 98% ee (Table 2, entry 3) by this catalytic system. Interestingly, the catalyst was reusable 6 times, where ee remained at ~95% and the catalyst activity decreased with each run. In addition to mere CTAB, the authors also tested combinations of surfactants, namely sodium dodecyl sulfate (SDS) with CTAB, where they found optimal reaction conditions with the ratio of SDS:CTAB being 2:1.
A remarkable work was published in 2007 [63], where a mixture of polyethylene glycol (PEG) and water was successfully applied in the ATH of aromatic ketones catalyzed by 1b, using HCOONa as the source of hydrogen (Table 2, entry 4). A mixture of PEG:H2O (9:1, v/v) was found to be the optimal reaction solvent, which allowed 14 repetitions of use without deterioration in enantioselectivity and with only slightly decreased reactivity. Using this method chiral 1-phenylethanol was obtained in 94-96% ee (S/C = 100, 40 °C, 15-20 h for consecutive runs 1-9, and 20-40 h for runs 10-15). After each run, the product was easily extracted with hexane and the polymeric phase washed with HCOOH to regenerate the sodium formate it contained. ICP analysis showed that >99.9% of 1b remained in the PEG phase.
Wang et al. [64] developed a reaction protocol for the ATH of ketones with 1b and HCOONa in a mixture of water/dichloromethane, which was rather problematic as the substrate and catalyst were more soluble in the organic solvent, whereas the HCOO– anion was present in the aqueous phase. The reaction was thus limited by the transport through the phase boundary. Their solution to this problem was to create an emulsion system by applying ultrasonic irradiation and a surfactant tetrabutylammonium iodide (TBAI), which caused a significant increase in the surface area between the organic and hydrophilic phases. Notably, this method facilitated the ATH of solid ketones, which is often slow or unfeasible when conducted in neat water due to their low water solubility. The high surface area created by emulsions provided proper contact of all the reaction components and the ATH was thus accelerated.
These examples demonstrate that the RuII catalysts 1 can also be effectively used in aqueous media. However, most ATHs in water using 1 have been described in conjunction with structural modifications of the catalyst, which is discussed further in section 3.1. and section 3.2.
2.3. Attempts to Immobilize Unmodified Complexes 1
Heterogeneous catalysts are heavily favoured in industry as they can be easily separated from the reaction mixture and the resulting product does not contain any residual catalyst, i.e., unwanted compound(s) of heavy metals, which are expensive and may be harmful to the environment. Unfortunately, the structure of catalyst 1 is not particularly suitable for heterogenization without any further modification. The only reactive groups of the species are Ru-Cl and NH2 of TsDPEN, which, however, directly participate in the ATH mechanism [61,65] and thus should not be occupied by the solid support. Therefore, examples of immobilization of the unmodified complexes 1 are very scarce.
In 2003, de Smet, Vankelecom, et al. [66] used selective polydimethylsiloxane (PDMS) membranes to separate catalyst 1b, aiming for a flow arrangement of ATH. After determination of the optimal conditions (mixture of methanol/isopropanol (30:70)), the membrane enabled the substrate and product to permeate and, simultaneously, was able to retain 1b completely. This way, catalyst phase and bulk phase were separated by the membrane. In the ATH of acetophenone, 4 consecutive runs at a constant ee (95%) were performed, however 5% leaching of Ru was observed. To the best of our knowledge, this interesting idea has not been developed since.
Šiklová and co-workers [67] attempted to immobilize 1b in the channels of MCM-41 molecular sieve functionalized by (3-aminopropyl)triethoxysilane (APTES). Their supported catalyst was used in the ATH of a cyclic imine (1-methyl-3,4-dihydroisoquinoline) to give 89% ee (Table 3, entry 1), which was the same value as they obtained for the homogeneous system. However, the reaction rate was lower in the case of the supported catalyst. This observation was ascribed to diffusion processes occurring within the channels of the solid material.
Table 3.
ATH of acetophenone catalyzed by immobilized 1b.
| Entry | Catalyst config. | Substrate | Solution | S/C [a]; Time; Temp.; Conversion (%) |
% ee (config) [b] |
Ref. [c] |
|---|---|---|---|---|---|---|
| 1 | (S,S) [d] | DHIQ [e] | HCOOH/Et3N; acetonitrile | 200; 7 h; r.t.; 99 | 89% (R) | [67] |
| 2 | (S,S) [f] | acetophenone | H2O/HCOONa; Et4N+Br | 100; 5 h; 30 °C; >99 | 93% (S) | [68] |
[a] Molar ratio substrate/catalyst; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry; [d] 1b was immobilized in functionalized MCM-41; [e] DHIQ = 1-methyl-3,4-dihydroisoquinoline; [f] 1b was immobilized in a nanocage of SBA-16.
A very interesting concept was developed by Li, Yang and co-workers [68], who encapsulated 1b in a mesoporous “cage”, i.e., in the pores of the SBA-16 material. This was done by narrowing the pore size of SBA-16 by silylation with diphenyldichlorosilane. This complex was able to perform the ATH of acetophenone with 93% ee (Table 3, entry 2) and was reported to be reusable at least 6 times, although with a gradual decrease in catalytic activity. The concept of a mesoporous nanocage represented by SBA-16 was also applied in different catalytic systems [69,70,71].
2.4. ATH Catalyzed by 1 in Ionic Liquids
Ionic liquids are a desirable medium for conducting catalyzed reactions as they are ranked amongst the green solvents and in addition, they can facilitate catalyst recycling. A few examples of their use in ATH with 1 have been published.
In 2005 [72], the ATH of acetophenone was examined using 1a in the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate, also known as [bmim][PF6]. The reaction, employing HCOOH/triethylamine, gave 1-phenylethanol in 93% ee (96% conv., 24 h). The recycling is discussed in section 3.4.
The performance of 1b in nine different ionic liquids was examined by Joerger et al. in 2006 [73]. Although the catalytic activity was found to be lower than under Noyori’s original conditions, high ees (up to 97%) were obtained for the ATH of acetophenone. The catalyst could be recycled no more than 4 times, despite employing various methods (bulb-to-bulb distillation of the product, extraction).
Huťka and Toma [74] optimized the reaction conditions for the ATH of a variety of ketones in several ionic liquids (ethylmethylimidazolium ethylsulfate, ECOENG500, [bmim][BF4]), where aryl alkyl ketones were reduced with the highest enantioselectivity. Their results further showed a very small dependence of ee upon temperature.
3. Modifications of the η6-Arene/N-sulfonyldiamine-RuII Complexes
Perhaps the most interesting feature of asymmetric catalysis is the modularity of catalytic complexes. On the basis of the first successful catalytic systems, numerous alternative species are derived which mimic or exceed the properties of the original complex [75]. This basic idea has inspired many researchers to explore possible modifications of known catalysts, which would lead to improved catalytic behaviour of these species. Herein we present an overview of direct modifications to the structure of catalyst 1.
The nature of the Noyori’s η6-arene/N-arylsulfonyl-1,2-diphenylethylenediamine-ruthenium(II) homogeneous catalyst 1 for the ATH of imines and ketones allows us to modify both moieties of the compound, i.e., the η6-arene ligand and the chiral diamine. The structure of the chelate diamine ligand (1) is responsible for the configuration of the major product enantiomer [61,76], while changing the ligand configuration readily changes the product configuration and (2) contributes to the asymmetric bias in enantioselective reactions [61]. However, the latter is mainly governed by the selection of the η6-arene ligand substituents, which affect the structure of transition states of the hydrogenation process and thus alter the reaction enantioselectivity [61,77]. Moreover, both the η6-arene ring and the N-sulfonyl fragment influence the catalyst activity [52,54,61,77].
Thus, successful ligand modification is not a trivial process owing to the complicated factors governing the catalyst activity and capability of asymmetric induction in hydrogenation reactions. In this text, the variations of 1 discussed are divided into several sections, according to the purpose of the modifications. Therefore, structural alterations leading to improvements e.g., in catalyst immobilization or ATH in water and ionic liquids are discussed. However, due to the large number of derivatives of 1, the scope of this review is limited and hence examples of straight-forward modifications of 1 (see e.g., [78,79,80,81,82,83]) will not be discussed in this work. Likewise, Wills’ well-known tethered complexes (see e.g., [84,85,86,87]) are not covered as they have been reviewed elsewhere [41,88].
3.1. Modifications of 1 Facilitating ATH in Aqueous Media
Small alterations of the diamine ligand in 1 led to the development of noteworthy catalytic systems enabling the implementation of ATH in water. This topic has also been reviewed recently [41,89,90]. In general, the derivatives generally bear a polar functional group which increases their water solubility. The structures are depicted in Figure 2.
Figure 2.

Modifications of TsDPEN used in ATH in water.
The first hydrophilic modifications of catalytic complex 1b appeared in 2001, when Bubert et al. [91] introduced a sulfonic acid group in the para- position of the N-arylsulfonyl fragment of TsDPEN. The novel ligand 2 furnished chiral 1-phenylethanol in 94% ee in the ATH of acetophenone using [RuCl2(η6-p-cymene)]2 (Table 4, entry 1). At the same time, the ligand was also used in rhodium- and iridium-based catalysts, showing both higher activity and selectivity in the ATH of acetophenone under identical conditions [92].
Table 4.
ATH of ketones and imines in water using modified diamine ligands [a].
| Entry | Ligand | Substrate | Solution | Time; Temp.; Conversion (%) |
% ee (config) [b] |
Ref. [c] |
|---|---|---|---|---|---|---|
| 1 | (S,S)-2 | acetophenone | H2O/i-PrOH/t-BuOK | 48 h; 22 °C; 96 | 94 (S) | [91] |
| 2 | (R,R)-3 | acetophenone | H2O/HCOONa; SDS [d] | 24 h; 40 °C; >99 | 95 (R) | [93] |
| 3 | (R,R)-3 | α-bromo-acetophenone | H2O/CH2Cl2/HCOONa; SDS | 24 h; 28 °C; 87 (isol. yield) | 94 | [93] |
| 4 | (R,R)-3 | DHIQ [e] | H2O/HCOONa; CTAB [f] | 10 h; 28 °C; 97 (isol. yield) | 95 (S) | [94] |
| 5 | (S,S)-4 | acetophenone | H2O/HCOONa | 0.5 h; 28 °C; 33 | 95 (S) | [95] |
| 6 | (R,R)-5c | acetophenone | H2O/HCOONa | 2 h; 40 °C; 100 | 94 (R) | [96] |
| 7 | (R,R)-5d | acetophenone | H2O/HCOONa | 2 h; 40 °C; 100 | 92 (R) | [96] |
[a] Molar ratio substrate/catalyst (S/C) = 100; The catalyst was formed in situ from the [RuCl2(η6-p-cymene)]2 dimer and the corresponding ligand; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry; [d] SDS = sodium dodecyl sulfate; [e] DHIQ = 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline; [f] CTAB = cetyltrimethylammonium bromide.
In 2003, Deng and co-workers disclosed a TsDPEN ligand modification bearing two ortho-sulfonate groups (3, Scheme 2) [93]. The ligand was easy to prepare and water-soluble. The first experiments on the ATH of ketones with 3 were carried out using the [RuCl2(η6-p-cymene)]2 ligand and sodium formate as a hydrogen source instead of HCOOH/triethylamine. In addition, the presence of sodium dodecyl sulfate (SDS) or 15-crown-5 was found to be necessary to increase the reaction rate.
Scheme 2.

Metal-ligand bifunctional mechanism: ATH of benzaldehyde using 30 and i-PrOH [61].
Under these conditions, a range of aromatic ketones were successfully reduced, including α-bromoacetophenone, which was complicated when the HCOOH/triethylamine azeotrope was used [97]. Chiral 1-phenylethanol was obtained in 95% ee (entry 2) and the ATH of α-bromoacetophenone in a H2O/CH2Cl2 mixture proceeded with 94% ee (entry 3).
The sulfonated ligand 3 was subsequently applied to the ATH of imines and iminiums under similar conditions [94]. However, when SDS was used as a surfactant both yield and enantioselectivity decreased. Therefore, other surfactants had to be tested and cetyltrimethylammonium bromide (CTAB) gave the best results (entry 4). A variety of cyclic imines (isoquinolines, β-carbolines, N-sulfonylimines) were successfully reduced this way, although in the case of some acyclic imines, this catalytic system has been found to be unsuitable. When the 3-based catalyst was tested for recycling, the enantioselectivity remained constant and yield slightly decreased with repeated catalyst use. The catalytic system developed was also utilized for the hydrogenation of tetra-alkyl-substituted iminium substrates, which were found to be more reactive than their tri-alkylated imine analogues.
In 2007 Li, Deng et al. introduced an o,o’-aminated TsDPEN ligand 4 for ATH in water conducted without the presence of surfactants [95]. Although the catalyst, formed in situ with [Ru(η6-p-cymene)Cl2]2, was not overly active in the ATH of acetophenone using HCOONa (entry 5), its [Cp*RhCl2]2 analogue furnished 97% conversion under identical conditions. Many other aromatic ketones were also reduced using this complex, mostly with excellent enantioselectivity (>95%).
Zhou and co-workers [96,98] synthesized water-soluble ligands 5a-5d, which, when combined with [Ru(η6-p-cymene)Cl2]2 and HCOONa, reduced aromatic ketones with great enantioselectivity (entries 6 and 7). Their water solubility was promoted by a quarternary ammonium group (imidazolium [98], or triethylammonium and pyridinium [96]) which was attached to the TsDPEN moiety. The ligands were tested for recycling (five or six consecutive runs) in the ATH of acetophenone, where their activity only slightly decreased and ee remained constant at 93% in the case of 5c (100% conv., S/C = 100, 2-3 h, 40 °C) [96]. The 5a/5b based catalysts showed great activity (quantitative conversions within 2 hours even after 6 runs), although erosion of ee was observed [98]. Ligands 5a and 5b were also used for ATH in ionic liquids [99] (see section 3.4.).
3.2. ATH Conducted on Immobilized Catalysts Derived from 1
A variety of approaches to the immobilization of 1 have emerged, offering solid catalysts applicable in heterogeneous ATHs of ketones and imines. This generally involved introduction of a functional group onto the aromatic rings of the diamine ligand, which provided the necessary linkage to the solid material.
Perhaps the first attempt at heterogenization of 1 was published in 1997 [100]. The TsDPEN ligand was replaced by N-[(4-vinylphenyl)sulfonyl]-1,2-diphenylethylene-1,2-diamine, which was subsequently copolymerized with styrene (1:10) into a linear polymer (6a), or with styrene and divinylbenzene (1:10:0.5) into a cross-linked polymer (6b). These two ligands were tested with [Ru(η6-benzene)Cl2]2, [Ru(η6-p-cymene)Cl2]2 and [Ir(COD)Cl2]2 precursors in the ATH of acetophenone using isopropanol as the hydrogen source. Although the iridium-based catalysts were both more active and selective (up to 94% ee at 96% yield after 48 h), their stability was disappointingly low. The ruthenium complexes were somewhat more stable (i.e., reusable up to 4 times) but they gave significantly lower ees and were less active (see Table 5, entry 1).
Table 5.
ATH of ketones and imines using immobilized derivatives of 1 [a].
| Entry | Ligand | Substrate | Solution | S/C [b]; Time; Temp.; Conversion (%) | % ee (config) [c] |
Ref. [d] |
|---|---|---|---|---|---|---|
| 1 | (S,S)-6b | acetophenone | i-PrOH/KOH | 20; 48 h; – ; 23 | 84 (S) | [100] |
| 2 | (S,S)-7a | acetophenone | HCOOH/Et3N/ CH2Cl2 |
100; 18 h; 30 °C; 71 | > 99 (S) | [101] |
| 3 | (S,S)-7b | acetophenone | HCOOH/Et3N | 100; 28 h; 30 °C; 95 | 96.7 (S) | [101] |
| 4 | (R,R)-8 | acetophenone | HCOOH/Et3N | 100; 20 h; 50 °C; 95 | 94 (R) | [102] |
| 5 | (R,R)-8 | acetophenone | H2O/HCOONa | 100; 1 h; 40 °C; 99 | 92 (R) | [103] |
| 6 | (R,R)-9a | acetophenone | HCOOH/Et3N | 100; 6 h; 40 °C; >99 | 97 (R) | [104] |
| 7 | (R,R)-9b | acetophenone | H2O/HCOONa; TBAB [e] | 100; 2 h; 40 °C; >99 | 96 (R) | [105] |
| 8 | (S,S)-10b | 2-cyano-acetophenone | HCOOH/Et3N | 100; 17 h; 30 °C; 98 (isol. yield) | 97 (S) | [107] |
| 9 | (S,S)-11 | DHIQ [f] | HCOOH/Et3N/CH2Cl2 | 100; 12 h; r.t.; 100 (isol. yield) | 91 (R) | [108] |
| 10 | (R,R)-12b | acetophenone | H2O/HCOONa | 100; 3 h; 40 °C; 100 | 98 (R) | [109] |
| 11 | (R,R)-13b | acetophenone | H2O/HCOONa | 100; 3 h; 40 °C; 100 | 98 (R) | [109] |
| 12 | (R,R)-13a | DHIQ [f] | H2O/HCOONa; CTAC [g] | 100; 24 h; r.t.; 69 | 94 (S) | [110] |
| 13 | (S,S)-14 | acetophenone | H2O/HCOONa | 100; 2 h; r.t.; >99 | 96 (S) | [111] |
| 14 | (S,S)-15 | DHIQ [f] | HCOOH/Et3N/CH2Cl2 | 100; 1.5 h; 40 °C; 99 | 94 (R) | [112] |
| 15 | (R,R)-16 | acetophenone | H2O/HCOONa | 100; 9 h; 40 °C; 100 | 97 (R) | [113] |
[a] The catalyst was formed in situ from the [RuCl2(η6-p-cymene)]2 dimer and the corresponding ligand; [b] Molar ratio substrate/catalyst; [c] See corresponding references for details on determination of ee and product configuration; [d] Reference containing data for a given entry. [e] TBAB = tetrabutylammonium bromide; [f] DHIQ = 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline; [g] CTAC = cetyltributyl-ammonium chloride.
In 1998 [101], Bayston et al. immobilized 1 onto aminomethylated polystyrene (PS), with (7b) or without (7a) polyethylene glycol (PEG) spacer units, creating orange/red polymeric beads. The PEG-containing ligand 7b was used with [Ru(η6-p-cymene)Cl2]2 in the ATH of acetophenone in a neat HCOOH/triethylamine mixture, and gave (S)-1-phenylethanol in 96.7% ee (Table 5, entry 3). Ligand 7a was more active when cosolvents were used (DMF, CH2Cl2), giving the product in amazing > 99% ee (entry 2). However, both catalysts were only reusable twice.
In 2004, many contributions in the field of heterogeneous ATH with modified 1 appeared. Li, Xiao and co-workers attempted to attach the TsDPEN ligand via its phenyl rings to a polyethylene glycol support, creating an immobilized ligand 8 (PTsDPEN) [102]. When hydrogenating acetophenone in neat HCOOH/triethylamine mixture using 8 combined with [Ru(η6-p-cymene)Cl2]2, 94% ee was obtained (entry 4). Efforts were made to reuse the catalyst by precipitation with diethyl ether, however, only three runs were practicable due to a rapid loss of catalytic activity. The use of 8 was also investigated for the ATH of ketones in water using HCOONa as a hydrogen donor, which caused a huge increase in the reaction rate [103]. In the ATH of acetophenone using 8 with [Ru(η6-p-cymene)Cl2]2, 1-phenylethanol (ee 92%) was obtained in full conversion within 1 hour (entry 5). Furthermore, the PTsDPEN-based catalyst was found to be much more stable in the aqueous phase, which enabled more efficient catalyst recycling by precipitation with diethyl ether. The first 12 consecutive runs were shown to be feasible at a constant ee without a significant loss in activity. Ligand 8 therefore seemed to be highly attractive.
Liu, Tu et al. chose the N-arylsulfonyl group of TsDPEN as a linker to a solid support, which was represented by amorphous silica gel (in 9a) and mesoporous silicas (MCM-41 in 9b and SBA-15 in 9c) [104,105]. These inorganic solids were selected for their high stability at high temperatures and in organic solvents. Surprisingly, cheap amorphous silica gel exceeded both MCM-41 and SBA-15 in performance. When conducting the ATH of acetophenone in neat HCOOH/triethylamine mixture with 9a and [Ru(η6-p-cymene)Cl2]2 [104], the product was obtained in full conversion after 6 hours (Table 5, entry 6). When catalyst recycling was attempted, no more than five runs were feasible, which was caused by severe leaching of the ruthenium. The best results were obtained when o- and m-fluorinated acetophenones were tested for recycling where 10 consecutive runs were successfully accomplished (ee 93-94%, activity decreasing). However, as the activity of this catalyst in the HCOOH/triethylamine azeotrope was only moderate, HCOONa was also used for the ATH in water [105], with tetrabutylammonium bromide (TBAB) applied as an additive, which was chosen after screening a number of surfactants and phase transfer catalysts. The reaction rate was markedly accelerated; optically enriched 1-phenylethanol was obtained quantitatively within 2 hours and in 96% ee (entry 7). In terms of reusability the acetophenone was recycled 6 times, and 11 runs were achieved in the case of 4’-bromoacetophenone. Further experiments were carried out on these complexes [106], giving results consistent with the initial data.
Two novel polystyrene-supported ligands 10a and 10b (Figure 3) were synthesized by attaching modified TsDPEN to aminomethylated polystyrene [107]. These were examined in the ATH step of the enantioselective synthesis of (S)-fluoxetine (entry 8). The final pharmaceutical product was obtained in 97% ee and contained less than 0.04 ppm of ruthenium.
Figure 3.

Immobilized TsDPEN-based ligands (I).
Another work dealing with the immobilization of 1b onto an inorganic support was published in 2007 by Huang and Ying [108]. They compared silica gel and siliceous mesocellular foam (MCF), with the latter giving the best results when used for immobilization and subsequently used in ATH. Ligand 11 immobilized on MCF, used in combination with [Ru(η6-p-cymene)Cl2]2 and HCOOH/triethylamine, catalyzed the ATH of 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline (entry 9) and several aromatic ketones with high yield and enantioselectivity. The ligand was reported to be reusable at least 6 times, although leaching of both Ru and ligand was observed. The group of Itsuno [109] modified the previously described polystyrene-supported ligand 6a [100] by introducing p-styrenesulfonate instead of styrene in the copolymerization (ligands 12a and 12b in Figure 4).
Figure 4.

Immobilized TsDPEN-based ligands (II).
Analogously, the cross-linked polymer (13a,13b) was obtained by copolymerization with p-styrenesulfonate and divinylbenzene. While the original ligands (6a,6b) were described as shrinking in water, these novel analogues were much more compatible with ATH in water thanks to the extra sulfonyl pendant groups. As cations, either Na+ (in 12a and 13a) or benzyltributylammonium (in 12b and 13b) were used, which allowed the modification of the lipophilic properties of the ligand. A very interesting observation was the enhanced enantioselectivity (98% ee) in the ATH of acetophenone using 12b and 13b with [Ru(η6-p-cymene)Cl2]2 and HCOONa (entries 10 and 11). The authors attributed this to the creation of microenvironments in the polymer structure causing stronger asymmetric bias. Reusability of the 13b-containing catalyst was also addressed, where 5 repeated uses were reported. Two years later [114], this work was extended by two more pendant groups, carboxylate and alkanesulfonate, both again with Na+ or benzyltributylammonium cations. Nevertheless, neither of these surpassed their first ligands containing the arenesulfonate group. In 2009 [110], they further examined this concept in the ATH of imines. The sulfonylated polymers complemented with Na+ or benzyltributylammonium were found suitable for the ATH of cyclic imines in water utilizing HCOONa (entry 12).
Inspired by previous works on PTsDPEN (8) [102,103] (vide supra), Chan et al. introduced a polyethylene glycol chain in the para-position of the N-arylsulfonyl fragment of TsDPEN, creating PEG-BsDPEN (14) [111]. They found that the enantioselectivity of the acetophenone hydrogenation was slightly dependent on the amount of HCOONa. The catalyst was fairly active and recyclable, which could be seen from the 8th run where 96% conversion and 95% ee were reached within 4 hours (the first run is listed as entry 13). This ligand was then successfully applied in the syntheses of (R)-salmeterol [115] and (R,R)-formoterol [116].
Li et al. [112] extended the possibilities of immobilization in siliceous mesocellular foam (MCF) by grafting it with magnetic nanoparticles, aiming for an easily-separable heterogeneous catalyst (ligand 15 in Scheme 4). The area of applicability of nanoparticles in catalysis has been reviewed very recently [117,118,119,120]. The activity of the 15-based catalyst was very good; for example, 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline was reduced with 94% ee within 1.5 h, when [Ru(η6-p-cymene)Cl2]2 and HCOOH/triethylamine in dichloromethane were used (entry 14). It was successfully recycled nine times, with a slight erosion of ee (94-90%) and a decrease in activity caused by mechanical stirring and mild Ru leaching (11 mol% after nine runs). Several aromatic ketones were tested as well with good results achieved.
Scheme 4.

Major differences in ATH of ketones and imines using the catalyst (S,S)-1b.
Another polyethylene glycol-supported ligand (16) was synthesized in 2010 [113]. In the ATH of acetophenone using [Ru(η6-p-cymene)Cl2]2 and HCOONa, 97% ee was observed (entry 15). However, the catalyst was not particularly suitable for recycling as only three consecutive runs could be performed due to a marked decrease in both activity and ee.
3.3. ATH Conducted on Dendrimeric Catalysts Derived from 1
Although the dendrimeric modifications of 1 partially overlap with the area of catalyst immobilization techniques treated in section 3.2., we set this group apart due to its exclusivity. The dendrimeric structures impart some unique properties to the catalysts, which are hardly achievable by other methods [121,122,123]. It is worth noting that the globular shape of the dendrimers allows for membrane filtration [124] or precipitation as a means of recycling the catalyst. Recent examples of dendrimeric catalysts derived from 1 are given above (Table 6).
Table 6.
ATH of acetophenone using dendritic catalysts [a].
| Entry | Ligand | Solution | Time; Temp.; Conversion (%) | % ee (config.) [b] | Ref. [c] | |
|---|---|---|---|---|---|---|
| 1 | (S,S)-17 [d] | HCOOH/Et3N | 20 h; 28 °C; >98 | 96.5 (S) | [125] | |
| 2 | (R,R)-18a | HCOOH/Et3N | 20 h; 28 °C; 99 | 97.6 (R) | [57] | |
| 3 | (R,R)-19 | HCOOH/Et3N | 20 h; 28 °C; 97 | 97.1 (R) | [57] | |
| 4 | (R,R)-20 [e] | HCOOH/Et3N | 20 h; 28 °C; 97 | 97.1 (R) | [128] | |
| 5 | (R,R)-21 [f] | HCOOH/Et3N/CH2Cl2 | 20 h; 28 °C; 97 | 96.1 (R) | [129] | |
| 6 | (S,S)-17 [e] | i-PrOH/i-PrOK | 48 h; 25 °C; 65 | 95 (S) | [130] | |
| 7 | (S,S)-22 | H2O/HCOONa; TBAI [g] | 4 h; 40 °C; > 99 | 97 (S) | [131] | |
[a] Molar ratio substrate/catalyst (S/C) = 100; The catalyst was formed in situ from the [RuCl2(η6-p-cymene)]2 dimer and the corresponding ligand; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry; [d] For all generations (n = 1-3), similar performances were observed; [e] Data for the third generation (n = 3); [f] Data for the second generation (n = 2); [g] TBAI = tetrabutylammonium iodide.
In 2001, Chen, Deng and co-workers [125] applied Fréchet’s polyether dendritic structures [126] to the TsDPEN ligand in the effort to find a recyclable catalyst. ATH of acetophenone using their dendritic ligands 17 (Figure 5) revealed they were recyclable up to five times, with constant enantioselectivity (ee > 96%) and decreasing catalyst activity.
Figure 5.

Dendritic TsDPEN-based ligands.
The same group also developed a set of ligands where the TsDPEN units were located at the periphery of the dendrimer [57], believing that the diamines would be more accessible by the substrate molecules than in the former case [125], which would accelerate the reaction. For the first generation, tetrakis(2-carboxyethoxymethyl) methane was chosen as the core unit bearing 4 TsDPEN units connected via amide linkages, with (18b) or without (18a) glycine residues. As the second generation, a more complex ligand 19 bearing 12 diamine units was synthesized on the basis of Newkome’s cascade polymers [127]. In the ATH of acetophenone, the performance of 18a, 18b and 19 was compared with their monomeric analogues to find that both activity and enantioselectivity were almost identical for dendrimeric and monomeric ligands. These ligands proved applicable in the stereoselective hydrogenations of a number of aromatic ketones and imines.
Finally, Deng’s group synthesized unique hybrid dendritic catalysts 20 [128] by coupling the Fréchet- and Newkome-type ligands mentioned above. Catalyst recycling by precipitation was in the scope of this study, while it was shown that polyether dendrons of the Fréchet type [125] (17) were more suitable for recycling (up to six runs) due to their higher stability. In contrast, the diamine units in hybrid dendrimers 20 were located too far from the polyether moiety and the catalysts based on such dendrimers were less stable. The hybrid dendrimers therefore help to reveal that proximity of the diamines and the polyether dendrons was important for the catalyst stability.
Alternative dendrimeric ligands 21 were described in 2005, linking the diamine to the dendrons via the phenyl rings of TsDPEN [129]. The dendrimers were synthesized up to the third generation, and generations 1 and 2 were slightly more active than their amino-functionalized relatives [125]. However, the activity of the third generation dropped significantly, probably due to the steric inaccessibility of the active site by the substrate, caused by the bulky dendrons. The second generation was also tested for recyclability, although substantial leaching of the catalyst was observed (about 10 mol% in each run, determined by ICP) and therefore only four consecutive runs could be performed with conversion dropping by 5% each time. An interesting feature of these ligands was the possibility of changing the N-arylsulfonyl moiety, leading to higher enantioselectivities in some hydrogenations.
The group of Rothenberg developed a multilayered cylinder for membrane-like separation of the catalyst [130]. The cylinder was made of macroporous α-alumina covered by a thin layer of nanoporous γ-alumina. Because the steric bulk of 1b had to be increased to prevent it penetrating the nanopores it was attached onto a third-generation dendrimer as reported by Deng (ligand 17) [125]. Their membrane cup was capable of entrapping more than 99.7% of the Ru compounds after 3 days of stirring. Although this ceramic cylinder was tested with 17 (entry 6), it has the potential to be applied in a variety of catalyzed reactions where the catalytic species has sufficiently high molecular weight.
A fluorinated dendritic TsDPEN ligand 22 (FTsDPEN), which surpassed all previous dendritic catalysts based on 1 in terms of reusability, was introduced very recently [131]. 22 was tested with [Ru(η6-p-cymene)Cl2]2 in 30 consecutive runs of ATH of acetophenone using aqueous HCOONa and TBAI (S/C = 100, 40 °C). During the first 16 runs, ee was maintained between 93-95% and reaction time was 4-8 hours. From run 16 to 26, 12-24 hours were needed to achieve full conversion, while ee was within the range of 88-93%. The final runs showed a significant decrease in the catalyst activity. The recyclability of 22 was therefore very promising in comparison with the preceding studies [63,128], none of which achieved so many effective repeated uses.
3.4. ATH Conducted in Ionic Liquids Using Modified Catalysts 1
This section covers the ligand modifications connected with usage in ionic liquids (Figure 6) and is closely linked with section 2.4. where unmodified complexes 1 are addressed.
Figure 6.

Ligands designed for ATH in ionic liquids.
Geldbach and Dyson [132] introduced an imidazolium group onto the η6-aromatic moiety of 1 creating its derivative 23, which allowed immobilization in the ionic liquid 1-butyl-2,3-dimethylimidazolium hexafluorophosphate. Since KOH with isopropanol as a hydrogen source caused a sharp decrease in the catalytic activity, the HCOOH/triethylamine azeotrope was selected as an alternative. In this arrangement, the ATH of acetophenone was examined in comparison with original 1b. The best recycling results were obtained with 1b placed in ionic liquid, which afforded 5 consecutive runs with negligible loss in activity and >99% ee (Table 7, entry 1). After each run, the product was extracted with hexane and the ionic liquid containing the catalyst was dried under reduced pressure.
Table 7.
ATH of acetophenone performed in ionic liquids.
| Entry | Ligand | Solution | S/C [a]; Time; Temp.; Conversion (%) |
% ee (config.) [b] | Ref. [c] |
|---|---|---|---|---|---|
| 1 | (R,R)-23 [d] | HCOOH/Et3N/[C4C1C1Im]PF6 [e] | 200; 24 h; 35 °C; > 99 | > 99 | [132] |
| 2 | (S,S)-24 [f] | HCOOH/Et3N/[bmim][PF6] [g] | 100; 24 h; r.t.; 98 | 92 (S) | [72] |
| 3 | (R,R)-5a [h] | HCOOH/Et3N/[bmim][PF6] [g] | 100; 8 h; 40 °C; 100 | 97 (R) | [99] |
[a] Molar ratio substrate/catalyst; [b] See corresponding references for details on determination of ee and product configuration; [c] Reference containing data for a given entry; [d] The catalyst was formed in situ from (R,R)-TsDPEN and the corresponding ligand; [e] 1-butyl-2,3-dimethylimidazolium hexafluoro-phosphate; [f] The catalyst was formed in situ from the [RuCl2(η6-benzene)]2 dimer and (S,S)-24; [g] [bmim][PF6] = 1-butyl-3-methylimidazolium hexafluorophosphate; [h] The catalyst was formed in situ from the [RuCl2(η6-p-cymene)]2 dimer and (R,R)-5a.
Ohta et al. [72] attached an imidazolium tag in the para- position of the TsDPEN ligand, creating 24. They found that both 1a (cf. section 2.4.) and the newly synthesized ligand, complexed to [Ru(η6-benzene)Cl2]2 in situ, were soluble in [bmim][PF6] under the reaction conditions and were suitable for recycling. The results obtained were very similar (5 consecutive runs, ~93% ee) but the imidazolium-containing ligand showed slightly better performance.
After successfully performing the ATH of aromatic ketones in water [98], Zhou et al. attempted to use their imidazolium-based ligands 5a and 5b (Scheme 2) also in ATH in the ionic liquid [bmim][PF6] [99]. They used the [Ru(η6-p-cymene)Cl2]2 precursor and HCOOH/triethylamine azeotrope for the ATH of acetophenone and obtained 1-phenylethanol in 97% ee (entry 3). Therefore, the reaction rate was about four times lower in comparison with the ATH in water [98]. Unfortunately, no more than 3 recycling runs were accomplishable due to a sharp decrease in catalytic activity.
3.5. Biomimetic Modifications of 1
Some very interesing works have dealt with modifications of 1 aiming to approach enzyme-like performance. Although artificial metalloenzymes have been receiving increasing attention lately [133,134,135], not many works have focused upon the area of Noyori-type ATH of ketones and imines. The group of Ward et al. have done the most in this field [136,137,138,139,140]. In 2005 [136], they successfully anchored a RuII complex [(η6-arene)RuCl(25)], bearing a biotinylated achiral ligand (25 in Figure 7), in a protein molecule of tetrameric streptavidin. The complex was tested in the ATH of acetophenone which revealed that the p-substituted ligand (p-25) was the best option. Interestingly, when η6-p-cymene was used in the complex, (R)-1-phenylethanol was produced, while with η6-benzene, the (S) enantiomer was observed. However, the ee values were modest and the catalytic activity was low. Therefore, they attempted to optimize the artificial enzyme by point mutations in streptavidin. It was shown that the mutations closest to the active site increased the catalytic activity, the most remote mutations enhanced the enantioselectivity, and double mutations combined the two. Eventually, after pH optimization with an appropriate buffer, (R)-1-phenylethanol was obtained in 85% ee and 90% conversion after 64 hours (S/C = 100, 55 °C). It needs to be emphasized that the induction of chirality was largely brought about by protein-substrate and protein-complex interactions.
Figure 7.

Ligands and complexes in biomimetic applications.
In 2006 [137], the Ward group continued the development of this intriguing hydrogenation system and tested 20 forms of streptavidin against 21 modifications of RuII complexes in the ATH of three different substrates at once. In the first instance, to accelerate the screening process, they screened only two forms of streptavidin with all 21 Ru catalysts. Based on the results, five catalysts were selected and further tested with 20 streptavidin isoforms. With this extensive pool of data, the authors confirmed that η6-p-cymene was pro-(R) and η6-benzene mainly pro-(S), although in the latter case it could be reversed to pro-(R) when certain mutations in streptavidin were applied. As a result, the most promising combinations of biotinylated complexes with particular streptavidin mutants were identified, and evaluated in the ATH of several ketone substrates. Furthermore, docking studies formed a significant part of this work, as they gave a valuable insight on the structure of the artificial metalloenzymes.
There has been a constant development in the area of Ward’s biotin-streptavidin species. Remarkably, one variant of their artificial hydrogenases, identified by employing a designed evolution protocol, was shown to reduce dialkyl ketones with up to 90% ee [138]. It has always been a great challenge to reduce such substrates with high enantioselectivity, since aromatic ketones facilitate the enantiodiscrimination by their disposition for the formation of a CH/π interaction with the η6-arene [77], which cannot be expected in the case of aliphatic ketones. Very recently, the concept was also applied to a cyclic imine [140], although with an iridium atom instead of ruthenium. Upon optimizing the reaction conditions, (R)-salsolidine was obtained in 96% ee and (S)-salsolidine could be synthesized in 78% ee.
A very interesting paper by Polborn and Severin [141] also belongs primarily in this section, in spite of the fact that it is based on immobilization of modified (η6-arene)RuCl(ethylenediamine) 26. This is because their work goes beyond ordinary immobilization and seeks to attain substrate specificity via molecular imprinting (see also [142]) by a product-like molecule. As the authors wished to selectively hydrogenate benzophenone on this novel catalyst, the diphenylphosphinato ligand was selected for the coordination to the Ru atom as a pseudosubstrate. This association complex was then copolymerized with ethylene glycol dimethacrylate (EGDMA). Finally, the diphenylphosphinato ligands were selectively removed and re-substituted by chloro ligands, which created a highly substrate-specific molecularly-imprinted cavity adjacent to the catalyst active site. The molecular imprinting led to significant acceleration of the ATH of benzophenone, which proceeded with notable substrate selectivity when more ketones were present in the mixture. The complex was also found to be regioselective to diarylketone groups in more complex substrates.
Weng, Tada and co-workers [143] used this concept as well, but they performed the immobilization of modified 1b (N-p-styrenesulfonyl-DPEN instead of TsDPEN, according to [100]) on functionalized solid SiO2. The molecular imprinting was carried out by the product molecule in question. Hence, after anchoring the catalyst, (R)-1-(o-fluorophenyl)ethanol (i.e., the product of ATH of o-fluoroacetophenone) was coordinated onto the complex as a template. This association complex, representing a monomeric unit, was subject to stacking of surface matrix overlayers. This was performed by four different methods, out of which photopolymerization was eventually utilized in the case of the catalytic species used for subsequent testing in ATH. The final step was the removal of the template from the immobilized complex, which formed the molecularly-imprinted cavity. The novel solid catalyst was thoroughly characterized by analytical methods and the heights of polymer matrices were determined, locating the optimal height for the hydrogenation process (2 nm). Eight different substrates were tested using this catalyst with HCOONa in water with SDS, and the results were promising. The reaction rate was indeed highest in the case of o-fluoroacetophenone (except for 2-acetylfuran which was apparently very compatible with the imprinted cavity) and the corresponding (R)-1-(o-fluorophenyl)ethanol was obtained with 91% ee, which was a considerably higher value than the previously reported one [106].
4. Mechanistic Considerations
For every chemical reaction it is highly desirable to have a deep understanding of its mechanism. In the case of asymmetric reactions, it is even more important as it provides the rationale for the enantiofacial discrimination which is not always a trivial issue. In this text, we focus on the reaction mechanism of asymmetric transfer hydrogenations on complexes 1.
4.1. ATH of Ketones Catalyzed by 1 in the Presence of Isopropanol and a Strong Base
In 2000 [144], Noyori et al. published a seminal work where the fundamental features of the ATH of ketones using 1, coined as metal-ligand (M-L) bifunctional catalysis, were proposed. The initial calculations upon this reversible system were performed using formaldehyde as a substrate, and simplified structures of the catalytic complexes, i.e., RuH(η6-benzene)(ethylenediamine) 27 or RuH(η6-benzene)(ethanolamine), were used (Scheme 1). According to these findings, the substrate does not coordinate directly to the Ru centre, but forms a C=O…H-N hydrogen-bonded intermediate 28 with the ruthenium complex, which evolves into a six-membered cyclic transition state (TS) 29. The hydrogen bond is supported by a so-called “NH effect”: the N-H bond in the chelating ligand is markedly more polar when in a complex with ruthenium. Thus, the six-membered TS results in a transfer of the proton from the NH group to the carbonyl oxygen, and a hydride transfer from Ru onto the C=O carbon atom. This novel mechanism was also compared with a β-elimination/insertion mechanism, which was shown to be energetically and sterically unfavoured.
Scheme 1.

The original metal-ligand bifunctional mechanistic concept [144].
The M-L bifunctional mechanism was further studied in the case of ATH of benzaldehyde using RuCl(η6-benzene)(ethanolamine) 30 and isopropanol as a hydrogen donor (Scheme 2) [61]. The Ru-hydride 32 conveys a proton and a hydride to the C=O bond of benzaldehyde, which affords the alcohol product and a 16e unsaturated Ru complex 31 [65]. Two TSs 33fav and 33dis are considered for the hydrogen transfer, one of which (33fav) has lower energy due to the CH/π interaction (vide infra). However, benzaldehyde is not a prochiral compound and thus both TSs yield benzyl alcohol. The 16e complex 31 is then regenerated to 32 through a hydrogen transfer from isopropanol, with acetone formed as a by-product. The Ru-hydride molecule 32 always arises from the 16e complex 31, which is initially formed from the Ru-Cl precursor 30 by elimination of HCl by a strong base like KOH or t-BuOK.
The other important mechanistic aspect is the enantioselectivity of ATH. In this hydrogenation system, several factors evoke the asymmetric bias: (1) Employing the chiral chelating ligands like TsDPEN makes Ru complexes 1 chiral at the Ru atom, which is an essential prerequisite for conducting asymmetric reactions with these complexes. The configuration of the chelate is responsible for the configuration of the major product enantiomer [61,76,144]. When an (S,S)-diamine is used, the configuration at Ru is R, and (S)-1-phenylethanol is afforded preferentially in the ATH of acetophenone. With an (R,R)-diamine, the configuration at Ru is S, and (R)-product is obtained accordingly; (2) The η6-arene ligand is capable of a weak, attractive CH/π interaction with the aromatic ring of the substrate molecule [77]. This, among others, explains why aromatic ketones and imines are reduced by these complexes much more easily than aliphatic substrates. The CH/π attraction is the reason why two TSs of different energies are formed, as shown in Scheme 9. The TS characterized by lower energy (denoted as favourable TS or favTS) is formed with higher probability and leads to the desired product enantiomer, whereas the less probable TS (called disfavourable TS or disTS) affords the unwanted product enantiomer. FavTS is stabilized by the CH/π interaction and for that reason, it is energetically more probable. The character of the CH/π interaction can be affected by the selection of the η6-arene ligand. When η6-benzene is used, C(sp2)H/π attraction takes place, which is different from the C(sp3)H/π interaction in the case of e.g., η6-hexamethylbenzene.
Therefore, the hydrogenation proceeds in the outer coordination sphere of the ruthenium atom, where the chelating ligand plays a pivotal role. According to Clapham’s classification of hydrogenations catalyzed by Ru-hydride complexes, this mechanism belongs to the TOL group [45].
4.2. ATH Catalyzed by 1 in the Presence of the HCOOH/Triethylamine Azeotrope
A great deal of ATHs using 1 are carried out in the presence of the HCOOH/triethylamine azeotrope. Utilizing formic acid entirely changes the reaction behaviour, rendering it almost irreversible because gaseous CO2 is formed as a by-product. This is in a marked contrast to the reductions using isopropanol as a hydrogen source, where the resulting acetone can react in the reverse direction and cause the overall reversibility. However, CO2 needs to be effectively removed by stirring or N2-bubbling in order to reach highest reaction rates and the desired irreversibility [145] as it is assumed that it can form a ruthenium-formate complex 35 by insertion into the Ru-H bond of 36 (Scheme 3) [146], which is not favoured in this context.
Scheme 3.

Structures observed in the reaction mixture when HCOOH/triethylamine is used in ATH.
In this case, the reaction mechanism has not yet been fully understood. In addition to the Ru-hydride 36 and the 16e complex 34, a Ru-formate species 35 is formed at the beginning of the reaction from HCOOH and 34 [146]. At low temperatures, only 35 was observed by NMR, while raising the temperature led to decarboxylation and formation of 36 [147]. Therefore, 36 is thought to arise from 35, which is formed first.
4.3. Mechanism of ATH Catalyzed by 1 in Water
As this review has extensively dealt with ATH of ketones and imines in aqueous media, some mechanistic considerations on these reactions are presented here. The group of Xiao recently published an in-depth study on the ATH of acetophenone conducted in water with 1b and HCOONa [148]. They arrived at a number of very interesting conclusions, promoting the role of H2O from a mere solvent to a highly important, actively involved species. It was found to facilitate the decarboxylation of 35 to 36, rendering the Ru-hydride 36 formation very fast with 35 not observable by NMR. The 16e complex 34 is supposedly stabilized by the water molecule via a transformation into Ru-aqua or Ru-hydroxyl species which were observed by NMR at −80 °C. Perhaps the most striking conclusion is the active participation of water in the TSs which was deduced from density functional theory (DFT) calculations. The water molecule is assumed to hydrogen-bond to the oxygen atom of the ketone C=O group, which lowers the energy of the TS by ~4 kcal mol–1, and thus facilitates hydrogen transfer. Furthermore, the catalytic cycles significantly differ under acidic, basic and neutral conditions, the latter of which are favoured. Acidic conditions severely decrease both the reaction rate and enantioselectivity as a result of the partial decoordination of the TsDPEN ligand through protonation of its amido nitrogen [60].
We believe that these selected mechanistic aspects demonstrate the importance of water in the mechanism of ATH conducted in aqueous solutions. The positive effects introduced by the H2O molecule to the reaction mechanism further support its use instead of traditional organic media.
4.4. ATH of Imines Catalyzed by 1 in the Presence of the HCOOH/Triethylamine Azeotrope
Initially, the stereoselective transfer hydrogenations of imines were carried out with HCOOH/triethylamine as isopropanol turned out to be completely ineffective [53]. Recently, it has been found that imines can only be reduced under acidic conditions [149], which conforms to the inapplicability of isopropanol as a hydrogen source. However, inherent imine protonation is not compatible with the original mechanism described in Section 4.1. In 2009, Wills and co-workers discussed all possible favTSs for the ATH of 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline catalyzed by 1 [82] which created a further discrepancy with the original mechanism with regard to the absolute configuration of the product of ATH (Scheme 4). If the standard six-membered favTS structure were applied with (S,S)-1b, the outcome [(S)-amine] would be a different enantiomer from the one observed experimentally, i.e., (R)-amine. This evidence suggests that a different pathway should be considered for the ATH of imines. This issue is further addressed in Section 4.5.
4.5. Molecular Modelling
Modelling the intermediates and TSs in silico grants us very specific information on their geometries. The M-L bifunctional mechanism was elucidated by performing high-precision ab initio calculations [61,77,144]. These computational methods are capable of providing 3D models which tell us much about the steric properties of a given compound. In addition, the energy values obtained can help us predict for example thermodynamic properties, reactivity, or even enantioselectivity [150].
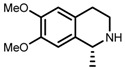
In our recent paper [151], we proposed the TS structures for the ATH of acetophenone (a ketone) and 1-methyl-3,4-dihydroisoquinoline (an imine) catalyzed by (S,S)-1b. This imine molecule was selected for its simplicity since all ruthenium complex structures were treated “as is” without any simplifications, which resulted in very high hardware demands. All calculated TSs conformed to the product configurations observed experimentally. (R)-1-methyl-1,2,3,4-tetrahydroisoquinoline and (S)-1-phenylethanol were thus obtained as the outcomes from favTSs.
All calculations were performed at the density functional theory (DFT) level, using the B3LYP [152,153] hybrid functional. For all atoms except ruthenium, the 6-31G(d,p) basis set was used; ruthenium was treated with the LANL2DZ basis set and effective core potential (ECP) [154]. The initial Ru-hydride model 36 was obtained by optimizing the X-ray coordinates of 1b published by Noyori in 1996 [53], where the chlorine atom was substituted by a hydrogen atom. As all other models were constructed and optimized “from scratch”, finding the TS structures was a rather difficult task. Optimizations of the TSs (Figure 8) were performed by the QST3 method, where three input geometries need to be specified: the reactant (37), the product (38) and an initial guess of the TS structure (39). Successful convergence of a TS optimization process is highly sensitive and the initial guesses need to be very precise. For that reason, the screening was accelerated by utilizing cut-off geometries of the Ru complex formed by replacing all three aromatic rings of TsDPEN with methyl groups. This operation significantly reduced the number of atoms present and the calculations were thus much faster, which allowed us to encounter the TS geometries more promptly. The optimized cut-off TSs (e.g., 40 in Figure 1) were then rebuilt into full-scale structures and used for the real TS optimizations.
Figure 8.

Cut-off input geometries for TS optimization. The C(sp3)H/π interaction is shown in green. The unnecessary hydrogen atoms have been omitted for clarity.
In this way we obtained the full-scale TS structures (41,42) depicted in Figure 9. The C(sp3)H/π (for 41) and C(sp2)H/π (42) attractions are depicted in green. It can be seen that the imine is N-protonated and only one hydrogen is transferred from Ru-H to the carbon atom of the C=N bond. The (S,S)-ligand leads to the (R) configuration of the amine product. Another important attraction, which has not been described before, was revealed between the =NH+- group of the substrate and the O=S fragment of TsDPEN ligand. It was found to significantly contribute to the stabilization of TSs.
Figure 9.

Optimized structures of favTSs utilizing C(sp3)H/π (41) or C(sp2)H/π (42) interactions (shown in green). The unnecessary hydrogen atoms have been omitted for clarity.
However, it was necessary to explore in detail which CH/π attractive forces (i.e., C(sp2)H/π or C(sp3)H/π) were exploited in this particular reaction. This was due to the use of η6-p-cymene in our calculations, which hypothetically allowed both options. The symmetry of the p-cymene ring allows four different sites which can be used in a CH/π attraction. Out of these, two were shown to be probable, one of them being C(sp3)H/π and the other C(sp2)H/π (in 41 or 42, shown in Figure 2).
To sum up, imines apparently cannot be reduced without the necessary N-protonation, which can occur in situ e.g., in the HCOOH/triethylamine H2-donor mixture. Upon hydrogenation by complexes 1, a different product configuration is observed while only ketones conform to the original M-L bifunctional mechanism. In the case of imines, different structures of the TSs have been suggested and they are in agreement both with the substrate protonation and the absolute configuration of the product.
5. Conclusions
The purpose of this review was to give an account on the group of Noyori’s renowned η6-arene/N-arylsulfonyl-1,2-diphenylethylenediamine-ruthenium(II) catalysts 1. The catalysts were discussed in the context of their immobilization, recyclability, use in aqueous media and other specific applications which very often bring about notable improvements. Although the original catalysts 1 could be used in some situations of this kind, most of the work dealt with slight modifications of the catalyst structure. Frequently, this led to enhancements in the catalytic activity, chemo-, regio- and enantio-selectivity, reusability, or extended the substrate scope.
It can be seen that this catalytic system for the ATH of ketones and imines is already very well established. The reaction mechanism has been largely characterized, which allows for precise optimization of reaction conditions for a given substrate. However, some aspects are still shrouded in secrecy, especially when considering the ATH of imines using the HCOOH/triethylamine azeotrope as a hydrogen donor.
Acknowledgements
This work has been financially supported by the Grant Agency of the Czech Republic (Grant GACR 104/09/1497), the Ministry of Education, Youth, and Sports of the Czech Republic (MEYS) (Grant CEZ: MSM 604 613 7301) and research intent no. AV0Z50200510. The access to the MetaCentrum computing facilities, provided under the programme “Projects of Large Infrastructure for Research, Development, and Innovations” LM2010005 funded by MEYS, is gratefully acknowledged.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References and Notes
- 1.Davies N.M., Teng X.W. Importance of chirality in drug therapy and pharmacy practice: Implications for psychiatry. Adv. Pharm. 2003;I:242–252. [Google Scholar]
- 2.Arnum P.V. Single-enantiomer drugs drive advances in asymmetric synthesis. Pharm. Technol. 2006;30:58–67. [Google Scholar]
- 3.Lin G.-Q., Li Y.-M., Chan A.S.C. Principles and Applications of Asymmetric Synthesis. John Wiley & Sons, Inc.; New York, NY, USA: 2001. [Google Scholar]
- 4.Liu W. Resolutions at Large Scale: Case Studies. In: Ager D., editor. Handbook of Chiral Chemicals. 2nd ed. CRC Taylor & Francis Group; Boca Raton, FL, USA: 2006. pp. 75–95. [Google Scholar]
- 5.Li Z.J., Grant D.J.W. Relationship between physical properties and crystal structures of chiral drugs. J. Pharm. Sci. 1997;86:1073–1078. doi: 10.1021/js970195h. [DOI] [PubMed] [Google Scholar]
- 6.Subramanian G. Chiral Separation Techniques: A Practical Approach. 3rd ed. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2006. [Google Scholar]
- 7.Gladiali S., Alberico E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006;35:226–236. doi: 10.1039/B513396C. [DOI] [PubMed] [Google Scholar]
- 8.Drauz K., Waldmann H. Enzyme Catalysis in Organic Synthesis. 2nd ed. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2002. [Google Scholar]
- 9.Noyori R. Asymmetric Catalysis In Organic Synthesis. John Wiley & Sons, Inc.; New York, NY, USA: 1994. [Google Scholar]
- 10.Blaser H.-U., Malan C., Pugin B., Spindler F., Steiner H., Studer M. Selective hydrogenation for fine chemicals: Recent trends and new developments. Adv. Synth. Catal. 2003;345:103–151. doi: 10.1002/adsc.200390000. [DOI] [Google Scholar]
- 11.Ojima I. Catalytic Asymmetric Synthesis. 3rd ed. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2010. [Google Scholar]
- 12.Noyori R. Asymmetric catalysis: Science and opportunities (Nobel Lecture) Angew. Chem. Int. Ed. 2002;41:2008–2022. doi: 10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Knowles W.S. Asymmetric hydrogenations (Nobel Lecture) Angew. Chem. Int. Ed. 2002;41:1998–2007. doi: 10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Knowles W.S., Noyori R. Pioneering perspectives on asymmetric hydrogenation. Acc. Chem. Res. 2007;40:1238–1239. doi: 10.1021/ar7000809. [DOI] [PubMed] [Google Scholar]
- 15.Dang T.P., Kagan H.B. The asymmetric synthesis of hydratopic acid and amino-acids by homogeneous catalytic hydrogenation. Chem. Commun. 1971:481. doi: 10.1039/c29710000481. [DOI] [Google Scholar]
- 16.Kagan H.B., Dang T.-P. Asymmetric catalytic reduction with transition metal complexes. I. A catalytic system of Rhodium(I) with (–)-2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane, a new chiral diphosphine. J. Am. Chem. Soc. 1972;94:6429–6433. doi: 10.1021/ja00773a028. [DOI] [Google Scholar]
- 17.Knowles W.S., Sabacky M.J., Vineyard B.D. Catalytic asymmetric hydrogenation. J. Chem. Soc. Chem. Commun. 1972:10–11. doi: 10.1039/c39720000010. [DOI] [PubMed] [Google Scholar]
- 18.Börner A. Phosphorus Ligands in Asymmetric Catalysis, Synthesis and Applications. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2008. [Google Scholar]
- 19.Tang W., Zhang X. New chiral phosphorous ligands for enantioselective hydrogenation. Chem. Rev. 2003;103:3029–3069. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- 20.Helmchen G., Pfaltz A. Phosphinooxazolines – A new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 21.Noyori R., Hashiguchi S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997;30:97–102. doi: 10.1021/ar9502341. [DOI] [Google Scholar]
- 22.Murata K., Okano K., Miyagi M., Iwane H., Noyori R., Ikariya T. A practical stereoselective synthesis of chiral hydrobenzoins via asymmetric transfer hydrogenation of benzils. Org. Lett. 1999;1:1119–1121. doi: 10.1021/ol990226a. [DOI] [Google Scholar]
- 23.Vedejs E., Trapencieris P., Suna E. Substituted isoquinolines by noyori transfer hydrogenation: Enantioselective synthesis of chiral diamines containing an aniline subunit. J. Org. Chem. 1999;64:6724–6729. doi: 10.1021/jo990594s. [DOI] [PubMed] [Google Scholar]
- 24.Koike T., Murata K., Ikariya T. Stereoselective synthesis of optically active a-hydroxy ketones and anti-1,2-diols via asymmetric transfer hydrogenation of unsymmetrically substituted 1,2-diketones. Org. Lett. 2000;2:3833–3836. doi: 10.1021/ol0002572. [DOI] [PubMed] [Google Scholar]
- 25.Okano K., Muratsugu S., Ikariya T. Stereoselective synthesis of optically active pyridyl alcohols via asymmetric transfer hydrogenation of pyridyl ketones. Tetrah. Lett. 2000;41:9277–9280. doi: 10.1016/S0040-4039(00)01695-6. [DOI] [Google Scholar]
- 26.Yamada I., Noyori R. Asymmetric transfer hydrogenation of benzaldehydes. Org. Lett. 2000;2:3425–3427. doi: 10.1021/ol0002119. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe M., Murata K., Ikariya T. Practical synthesis of optically active amino alcohols via asymmetric transfer hydrogenation of functionalized aromatic ketones. J. Org. Chem. 2002;67:1712–1715. doi: 10.1021/jo011076w. [DOI] [PubMed] [Google Scholar]
- 28.Xue D., Chen Y.-C., Cui X., Wang Q.-W., Zhu J., Deng J.-G. Transfer hydrogenation of activated C=C bonds catalyzed by ruthenium amido complexes: Reaction scope, limitation and enantioselectivity. J. Org. Chem. 2005;70:3584–3591. doi: 10.1021/jo0478205. [DOI] [PubMed] [Google Scholar]
- 29.Samano V., Ray J.A., Thompson J.B., Mook R.A., Jung D.K., Koble C.S., Martin M.T., Bigham E.C., Regitz C.S., Feldman P.L., et al. Synthesis of ultra-short-acting neuromuscular blocker GW 0430: A remarkably stereo- and regioselective synthesis of mixed tetrahydroisoquinolinium chlorofumarates. Org. Lett. 1999;1:1993–1996. doi: 10.1021/ol9911573. [DOI] [PubMed] [Google Scholar]
- 30.Meuzelaar G.J., van Vliet M.C.A., Maat L., Sheldon R.A. Improvements in the total synthesis of morphine. Eur. J. Org. Chem. 1999;1999:2315–2321. doi: 10.1002/(SICI)1099-0690(199909)1999:9<2315::AID-EJOC2315>3.0.CO;2-V. [DOI] [Google Scholar]
- 31.Miyagi M., Takehara J., Collet S., Okano K. Practical synthesis of (S)-1-(3-trifluoromethylphenyl)ethanol via Ruthenium(II)-catalyzed asymmetric transfer hydrogenation. Org. Proc. Res. Dev. 2000;4:346–348. doi: 10.1021/op000019m. [DOI] [Google Scholar]
- 32.Tietze L.F., Zhou Y., Töpken E. Synthesis of simple enantiopure tetrahydro-β-carbolines and tetrahydroisoquinolines. Eur. J. Org. Chem. 2000;2000:2247–2252. doi: 10.1002/1099-0690(200006)2000:12<2247::AID-EJOC2247>3.0.CO;2-4. [DOI] [Google Scholar]
- 33.Szawkalo J., Czarnocki Z. Enantioselective synthesis of some tetracyclic isoquinoline alkaloids by asymmetric transfer hydrogenation catalysed by a chiral ruthenium complex. Monatsh. Chem. 2005;136:1619–1627. doi: 10.1007/s00706-005-0341-8. [DOI] [Google Scholar]
- 34.Roszkowski P., Wojtasiewicz K., Leniewski A., Maurin J.K., Lis T., Czarnocki Z. Enantioselective synthesis of 1-substituted tetrahydro-b-carboline derivatives via the asymmetric transfer hydrogenation. J. Mol. Catal., A Chem. 2005;232:143–149. doi: 10.1016/j.molcata.2005.01.044. [DOI] [Google Scholar]
- 35.Roszkowski P., Maurin J.K., Czarnocki Z. Enantioselective synthesis of (R)-(–)-praziquantel (PZQ) Tetrahedron Asymmetry. 2006;17:1415–1419. doi: 10.1016/j.tetasy.2006.04.023. [DOI] [Google Scholar]
- 36.Szawkalo J., Czarnocki S.J., Zawadzka A., Wojtasiewicz K., Leniewski A., Maurin J.K., Czarnocki Z., Drabowicz J. Enantioselective synthesis of some tetrahydroisoquinoline and tetrahydro-b-carboline alkaloids. Tetrahedron Asymmetry. 2007;18:406–413. doi: 10.1016/j.tetasy.2007.01.014. [DOI] [Google Scholar]
- 37.Cheng J.-J., Yang Y.-S. Enantioselective total synthesis of (−)-(S)-stepholidine. J. Org. Chem. 2009;74:9225–9228. doi: 10.1021/jo9020826. [DOI] [PubMed] [Google Scholar]
- 38.Palmer M.J., Wills M. Asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry. 1999;10:2045–2061. doi: 10.1016/S0957-4166(99)00216-5. [DOI] [Google Scholar]
- 39.Wills M., Palmer M., Smith A., Kenny J., Walsgrove T. Recent developments in the area of asymmetric transfer hydrogenation. Molecules. 2000;5:4–18. doi: 10.3390/50100004. [DOI] [Google Scholar]
- 40.Everaere K., Mortreux A., Carpentier J.-F. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of carbonyl compounds with 2-propanol and ephedrine-type ligands. Adv. Synth. Catal. 2003;345:67–77. doi: 10.1002/adsc.200390030. [DOI] [Google Scholar]
- 41.Wang C., Wu X., Xiao J. Broader, greener, and more efficient: Recent advances in asymmetric transfer hydrogenation. Chem. Asian J. 2008;3:1750–1770. doi: 10.1002/asia.200800196. [DOI] [PubMed] [Google Scholar]
- 42.Nugent T.C., El-Shazly M. Chiral amine synthesis – recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010;352:753–819. doi: 10.1002/adsc.200900719. [DOI] [Google Scholar]
- 43.Fleury-Brégeot N., de la Fuente V., Castillón S., Claver C. Highlights of transition metal-catalyzed asymmetric hydrogenation of imines. ChemCatChem. 2010;2:1346–1371. doi: 10.1002/cctc.201000078. [DOI] [Google Scholar]
- 44.Bäckvall J.-E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002;652:105–111. doi: 10.1016/S0022-328X(02)01316-5. [DOI] [Google Scholar]
- 45.Clapham S.E., Hadzovic A., Morris R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004;248:2201–2237. doi: 10.1016/j.ccr.2004.04.007. [DOI] [Google Scholar]
- 46.Samec J.S.M., Backvall J.-E., Andersson P.G., Brandt P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006;35:237–248. doi: 10.1039/b515269k. [DOI] [PubMed] [Google Scholar]
- 47.Fabrello A., Bachelier A., Urrutigoïty M., Kalck P. Mechanistic analysis of the transition metal-catalyzed hydrogenation of imines and functionalized enamines. Coord. Chem. Rev. 2010;254:273–287. doi: 10.1016/j.ccr.2009.09.002. [DOI] [Google Scholar]
- 48.Sandoval C.A., Ohkuma T., Utsumi N., Tsutsumi K., Murata K., Noyori R. Mechanism of asymmetric hydrogenation of acetophenone catalyzed by chiral η6-Arene-N-tosylethylenediamine-ruthenium(II) complexes. Chem. Asian J. 2006;1:102–110. doi: 10.1002/asia.200600098. [DOI] [PubMed] [Google Scholar]
- 49.Sandoval C.A., Bie F., Matsuoka A., Yamaguchi Y., Naka H., Li Y., Kato K., Utsumi N., Tsutsumi K., Ohkuma T., et al. Chiral η6-Arene/N-tosylethylenediamine-ruthenium(II) complexes: Solution behavior and catalytic activity for asymmetric hydrogenation. Chem. Asian J. 2010;5:806–816. doi: 10.1002/asia.200900461. [DOI] [PubMed] [Google Scholar]
- 50.Takehara J., Hashiguchi S., Fujii A., Inoue S.-i., Ikariya T., Noyori R. Amino alcohol effects on the ruthenium(II)-catalysed asymmetric transfer hydrogenation of ketones in propan-2-ol. Chem. Commun. 1996:233–234. doi: 10.1039/cc9960000233. [DOI] [Google Scholar]
- 51.Püntener K., Schwink L., Knochel P. New efficient catalysts for enantioselective transfer hydrogenations. Tetrahedron Lett. 1996;37:8165–8168. doi: 10.1016/0040-4039(96)01853-9. [DOI] [Google Scholar]
- 52.Hashiguchi S., Fujii A., Takehara J., Ikariya T., Noyori R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral Ruthenium(II) complexes. J. Am. Chem. Soc. 1995;117:7562–7563. doi: 10.1021/ja00133a037. [DOI] [Google Scholar]
- 53.Uematsu N., Fujii A., Hashiguchi S., Ikariya T., Noyori R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996;118:4916–4917. doi: 10.1021/ja960364k. [DOI] [Google Scholar]
- 54.Fujii A., Hashiguchi S., Uematsu N., Ikariya T., Noyori R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture. J. Am. Chem. Soc. 1996;118:2521–2522. doi: 10.1021/ja954126l. [DOI] [Google Scholar]
- 55.Matsumura K., Hashiguchi S., Ikariya T., Noyori R. Asymmetric transfer hydrogenation of a,b-acetylenic ketones. J. Am. Chem. Soc. 1997;119:8738–8739. doi: 10.1021/ja971570a. [DOI] [Google Scholar]
- 56.Gladiali S., Alberico E. Transferhydrogenations. In: Beller M., Bolm C., editors. Transition Metals for Organic Synthesis. 2nd ed. Volume 2. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2004. pp. 145–166. [Google Scholar]
- 57.Chen Y.-C., Wu T.-F., Deng J.-G., Liu H., Cui X., Zhu J., Jiang Y.-Z., Choi M.C.K., Chan A.S.C. Multiple dendritic catalysts for asymmetric transfer hydrogenation. J. Org. Chem. 2002;67:5301–5306. doi: 10.1021/jo0257795. [DOI] [PubMed] [Google Scholar]
- 58.Dwars T., Oehme G. Complex-catalyzed hydrogenation reactions in aqueous media. Adv. Synth. Catal. 2002;344:239–260. doi: 10.1002/1615-4169(200206)344:3/4<239::AID-ADSC239>3.0.CO;2-O. [DOI] [Google Scholar]
- 59.Wu X., Li X., Hems W., King F., Xiao J. Accelerated asymmetric transfer hydrogenation of aromatic ketones in water. Org. Biomol. Chem. 2004;2:1818–1821. doi: 10.1039/b403627a. [DOI] [PubMed] [Google Scholar]
- 60.Wu X., Li X., King F., Xiao J. Insight into and practical application of pH-controlled asymmetric transfer hydrogenation of aromatic ketones in water. Angew. Chem. Int. Ed. 2005;44:3407–3411. doi: 10.1002/anie.200500023. [DOI] [PubMed] [Google Scholar]
- 61.Noyori R., Yamakawa M., Hashiguchi S. Metal-ligand bifunctional catalysis: A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001;66:7931–7944. doi: 10.1021/jo010721w. [DOI] [PubMed] [Google Scholar]
- 62.Wang F., Liu H., Cun L., Zhu J., Deng J., Jiang Y. Asymmetric transfer hydrogenation of ketones catalyzed by hydrophobic metal-amido complexes in aqueous micelles and vesicles. J. Org. Chem. 2005;70:9424–9429. doi: 10.1021/jo0514826. [DOI] [PubMed] [Google Scholar]
- 63.Zhou H.-F., Fan Q.-H., Huang Y.-Y., Wu L., He Y.-M., Tang W.-J., Gu L.-Q., Chan A.S.C. Mixture of poly(ethylene glycol) and water as environmentally friendly media for efficient enantioselective transfer hydrogenation and catalyst recycling. J. Mol. Catal., A Chem. 2007;275:47–53. doi: 10.1016/j.molcata.2007.05.029. [DOI] [Google Scholar]
- 64.Wang W., Li Z., Mu W., Su L., Wang Q. Highly efficient asymmetric transfer hydrogenation of ketones in emulsions. Catal. Commun. 2010;11:480–483. doi: 10.1016/j.catcom.2009.12.002. [DOI] [Google Scholar]
- 65.Haack K.-J., Hashiguchi S., Fujii A., Ikariya T., Noyori R. The catalyst precursor, catalyst, and intermediate in the Ru(II)-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. Engl. 1997;36:285–288. doi: 10.1002/anie.199702851. [DOI] [Google Scholar]
- 66.de Smet K., Pleysier A., Vankelecom I.F.J., Jacobs P.A. Recycling of homogeneous hydrogenation catalysts by dialysis coupled catalysis. Chem. Eur. J. 2003;9:334–338. doi: 10.1002/chem.200390028. [DOI] [PubMed] [Google Scholar]
- 67.Šiklová H., Leitmannová E., Kačer P., Červený L. Immobilization of Ru-TsDPEN catalyst on functionalized MCM-41. React. Kinet. Catal. Lett. 2007;92:129–136. doi: 10.1007/s11144-007-5140-2. [DOI] [Google Scholar]
- 68.Yang H., Li J., Yang J., Liu Z., Yang Q., Li C. Asymmetric reactions on chiral catalysts entrapped within a mesoporous cage. Chem. Commun. 2007:1086–1088. doi: 10.1039/b614635j. [DOI] [PubMed] [Google Scholar]
- 69.Yang H., Zhang L., Zhong L., Yang Q., Li C. Enhanced cooperative activation effect in the hydrolytic kinetic resolution of epoxides on [Co(salen)] catalysts confined in nanocages. Angew. Chem. Int. Ed. 2007;46:6861–6865. doi: 10.1002/anie.200701747. [DOI] [PubMed] [Google Scholar]
- 70.Yang H., Zhang L., Wang P., Yang Q., Li C. The enantioselective cyanosilylation of aldehydes on a chiral VO(Salen) complex encapsulated in SBA-16. Green Chem. 2009;11:257–264. doi: 10.1039/B815997A. [DOI] [Google Scholar]
- 71.Yang H., Zhang L., Su W., Yang Q., Li C. Asymmetric ring-opening of epoxides on chiral Co(Salen) catalyst synthesized in SBA-16 through the "ship in a bottle" strategy. J. Catal. 2007;248:204–212. doi: 10.1016/j.jcat.2007.03.006. [DOI] [Google Scholar]
- 72.Kawasaki I., Tsunoda K., Tsuji T., Yamaguchi T., Shibuta H., Uchida N., Yamashita M., Ohta S. A recyclable catalyst for asymmetric transfer hydrogenation with a formic acid-triethylamine mixture in ionic liquid. Chem. Commun. 2005:2134–2136. doi: 10.1039/B500320B. [DOI] [PubMed] [Google Scholar]
- 73.Joerger J.-M., Paris J.-M., Vaultier M. [Ru(arene)(diamine)] catalysts in ionic liquids: Recyclable catalytic systems for transfer hydrogenation. ARKIVOC. 2006:152–160. [Google Scholar]
- 74.Hut’ka M., Toma Š. Hydrogen transfer reduction of different ketones in ionic liquids. Chem. Mon. 2008;139:793–798. doi: 10.1007/s00706-008-0853-0. [DOI] [Google Scholar]
- 75.Václavík J., Kačer P., Červený L. Rational Design of Chiral Ruthenium Complexes for Asymmetric Hydrogenations. In: Poehler A.C., editor. Homogeneous Catalysts: Types, Reactions and Applications. Nova Science Publishers, Inc.; Hauppauge, NY, USA: 2011. in press. [Google Scholar]
- 76.Hayes A., Clarkson G., Wills M. The importance of 1,2-anti-disubstitution in monotosylated diamine ligands for ruthenium(II)-catalysed asymmetric transfer hydrogenation. Tetrahedron Asymmetry. 2004;15:2079–2084. doi: 10.1016/j.tetasy.2004.05.025. [DOI] [Google Scholar]
- 77.Yamakawa M., Yamada I., Noyori R. Angew. Chem. Int. Ed. 2001;40:2818–2821. doi: 10.1002/1521-3773(20010803)40:15<2818::AID-ANIE2818>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 78.Soleimannejad J., Sisson A., White C. Functionalized-arene ruthenium half-sandwich compounds as enantioselective hydrogen transfer catalysts. crystal structures of [RuCl{TsNCH(R)CH(R)NH2}(h6-C6H5OCH2CH2OH)] (R=H or Ph) Inorg. Chim. Acta. 2003;352:121–128. doi: 10.1016/S0020-1693(03)00151-8. [DOI] [Google Scholar]
- 79.Matsunaga H., Ishizuka T., Kunieda T. Highly efficient asymmetric transfer hydrogenation of ketones catalyzed by chiral ‘roofed’ cis-diamine-Ru(II) complex. Tetrahedron Lett. 2005;46:3645–3648. doi: 10.1016/j.tetlet.2005.03.165. [DOI] [Google Scholar]
- 80.Li X., Blacker J., Houson I., Wu X., Xiao J. An efficient Ir(III) catalyst for the asymmetric transfer hydrogenation of ketones in neat water. Synlett. 2006;2006:1155,1160. doi: 10.1002/chin.200638079. [DOI] [Google Scholar]
- 81.Liu J., Wu Y., Li X., Chan A.S.C. Structure effect of TsDPEN derivatives on enantioselectivity of asymmetric transfer hydrogenation. J. Organomet. Chem. 2008;693:2177–2180. doi: 10.1016/j.jorganchem.2008.03.018. [DOI] [Google Scholar]
- 82.Martins J.E.D., Clarkson G.J., Wills M. Ru(II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines. Organic Lett. 2009;11:847–850. doi: 10.1021/ol802801p. [DOI] [PubMed] [Google Scholar]
- 83.Martins J.E.D., Contreras Redondo M.A., Wills M. Applications of N’-alkylated derivatives of TsDPEN in the asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry. 2010;21:2258–2264. doi: 10.1016/j.tetasy.2010.07.013. [DOI] [Google Scholar]
- 84.Hannedouche J., Clarkson G.J., Wills M. A new class of “tethered” Ruthenium(II) catalyst for asymmetric transfer hydrogenation reactions. J. Am. Chem. Soc. 2004;126:986–987. doi: 10.1021/ja0392768. [DOI] [PubMed] [Google Scholar]
- 85.Cheung F.K., Hayes A.M., Hannedouche J., Yim A.S.Y., Wills M. “Tethered” Ru(II) catalysts for asymmetric transfer hydrogenation of ketones. J. Org. Chem. 2005;70:3188–3197. doi: 10.1021/jo050032a. [DOI] [PubMed] [Google Scholar]
- 86.Cheung F.K., Clarke A.J., Clarkson G.J., Fox D.J., Graham M.A., Lin C., Criville A.L., Wills M. Kinetic and structural studies on ‘tethered’ Ru(II) arene ketone reduction catalysts. Dalton Trans. 2010;39:1395–1402. doi: 10.1039/B915932K. [DOI] [PubMed] [Google Scholar]
- 87.Morris D.J., Hayes A.M., Wills M. The “reverse-tethered” Ruthenium (II) catalyst for asymmetric transfer hydrogenation: Further applications. J. Org. Chem. 2006;71:7035–7044. doi: 10.1021/jo061154l. [DOI] [PubMed] [Google Scholar]
- 88.Kačer P., Kuzma M., Leitmannová E., Červený L. Ruthenium complexes for asymmetric transfer hydrogenation. In: Chin H.F., editor. Organometallic Compounds: Preparation, Structure and Properties. Nova Science Publishers, Inc.; Hauppauge, NY, USA: 2009. pp. 361–387. [Google Scholar]
- 89.Wu X., Xiao J. Aqueous-phase asymmetric transfer hydrogenation of ketones – a greener approach to chiral alcohols. Chem. Commun. 2007:2449–2466. doi: 10.1039/b618340a. [DOI] [PubMed] [Google Scholar]
- 90.Wu X., Wang C., Xiao J. Asymmetric transfer hydrogenation in water with platinum group metal catalysts. Platinum Met. Rev. 2010;54:3–19. doi: 10.1595/147106709X481372. [DOI] [Google Scholar]
- 91.Bubert C., Blacker J., Brown S.M., Crosby J., Fitzjohn S., Muxworthy J.P., Thorpe T., Williams J.M.J. Synthesis of water-soluble aminosulfonamide ligands and their application in enantioselective transfer hydrogenation. Tetrahedron Lett. 2001;42:4037–4039. doi: 10.1016/S0040-4039(01)00623-2. [DOI] [Google Scholar]
- 92.Thorpe T., Blacker J., Brown S.M., Bubert C., Crosby J., Fitzjohn S., Muxworthy J.P., Williams J.M.J. Efficient rhodium and iridium-catalysed asymmetric transfer hydrogenation using water-soluble aminosulfonamide ligands. Tetrahedron Lett. 2001;42:4041–4043. doi: 10.1016/S0040-4039(01)00624-4. [DOI] [Google Scholar]
- 93.Ma Y., Liu H., Chen L., Cui X., Zhu J., Deng J. Asymmetric transfer hydrogenation of prochiral ketones in aqueous media with new water-soluble chiral vicinal diamine as ligand. Org. Lett. 2003;5:2103–2106. doi: 10.1021/ol0345125. [DOI] [PubMed] [Google Scholar]
- 94.Wu J., Wang F., Ma Y., Cui X., Cun L., Zhu J., Deng J., Yu B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by water-soluble catalyst in water. Chem. Commun. 2006:1766–1768. doi: 10.1039/b600496b. [DOI] [PubMed] [Google Scholar]
- 95.Li L., Wu J., Wang F., Liao J., Zhang H., Lian C., Zhu J., Deng J. Asymmetric transfer hydrogenation of ketones and imines with novel water-soluble chiral diamine as ligand in neat water. Green Chem. 2007;9:23–25. doi: 10.1039/B611809G. [DOI] [Google Scholar]
- 96.Zhou Z., Ma Q., Sun Y., Zhang A., Li L. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of aromatic ketones in water using novel water-soluble chiral monosulfonamide ligands. Heteroatom Chem. 2010;21:505–514. doi: 10.1002/hc.20641. [DOI] [Google Scholar]
- 97.Cross D.J., Kenny J.A., Houson I., Campbell L., Walsgrove T., Wills M. Tetrahedron Asymmetry. 2001;12:1801–1806. [Google Scholar]
- 98.Zhou Z., Sun Y. Water-soluble chiral aminosulfonamides as ligands for ruthenium(II)-catalyzed asymmetric transfer hydrogenation. Catal. Commun. 2009;10:1685–1688. doi: 10.1016/j.catcom.2009.05.013. [DOI] [Google Scholar]
- 99.Zhou Z., Sun Y., Zhang A. Asymmetric transfer hydrogenation of prochiral ketones catalyzed by aminosulfonamide-ruthenium complexes in ionic liquid. Cent. Eur. J. Chem. 2011;9:175–179. doi: 10.2478/s11532-010-0134-8. [DOI] [Google Scholar]
- 100.Ter Halle R., Schulz E., Lemaire M. Heterogeneous enantioselective catalytic reduction of ketones. Synlett. 1997:1257–1258. doi: 10.1055/s-1997-1018. [DOI] [Google Scholar]
- 101.Bayston D.J., Travers C.B., Polywka M.E.C. Synthesis and evaluation of a chiral heterogeneous transfer hydrogenation catalyst. Tetrahedron Asymmetry. 1998;9:2015–2018. doi: 10.1016/S0957-4166(98)00214-6. [DOI] [Google Scholar]
- 102.Li X., Chen W., Hems W., King F., Xiao J. Asymmetric transfer hydrogenation of ketones with a polymer-supported chiral diamine. Tetrahedron Lett. 2004;45:951–953. doi: 10.1016/j.tetlet.2003.11.104. [DOI] [Google Scholar]
- 103.Li X., Wu X., Chen W., Hancock F.E., King F., Xiao J. Asymmetric transfer hydrogenation in Wwater with a supported noyori-ikariya catalyst. Org. Lett. 2004;6:3321–3324. doi: 10.1021/ol0487175. [DOI] [PubMed] [Google Scholar]
- 104.Liu P.N., Gu P.M., Wang F., Tu Y.Q. Efficient heterogeneous asymmetric transfer hydrogenation of ketones using highly eecyclable and accessible silica-immobilized Ru-TsDPEN catalysts. Org. Lett. 2004;6:169–172. doi: 10.1021/ol036065z. [DOI] [PubMed] [Google Scholar]
- 105.Liu P.N., Deng J.G., Tu Y.Q., Wang S.H. Highly efficient and recyclable heterogeneous asymmetric transfer hydrogenation of ketones in water. Chem. Commun. 2004:2070–2071. doi: 10.1039/b408533g. [DOI] [PubMed] [Google Scholar]
- 106.Liu P.-N., Gu P.-M., Deng J.-G., Tu Y.-Q., Ma Y.-P. Efficient heterogeneous asymmetric transfer hydrogenation catalyzed by recyclable silica-supported Ruthenium complexes. Eur. J. Org. Chem. 2005;2005:3221–3227. doi: 10.1002/ejoc.200500074. [DOI] [Google Scholar]
- 107.Li Y., Li Z., Li F., Wang Q., Tao F. Preparation of polymer-supported Ru-TsDPEN catalysts and use for enantioselective synthesis of (S)-fluoxetine. Org. Biomol. Chem. 2005;3:2513–2518. doi: 10.1039/b505943g. [DOI] [PubMed] [Google Scholar]
- 108.Huang X., Ying J.Y. Asymmetric transfer hydrogenation over Ru-TsDPEN catalysts supported on siliceous mesocellular foam. Chem. Commun. 2007:1825–1827. doi: 10.1039/B615564B. [DOI] [PubMed] [Google Scholar]
- 109.Arakawa Y., Haraguchi N., Itsuno S. Design of novel polymer-supported chiral catalyst for asymmetric transfer hydrogenation in water. Tetrahedron Lett. 2006;47:3239–3243. doi: 10.1016/j.tetlet.2006.03.041. [DOI] [Google Scholar]
- 110.Haraguchi N., Tsuru K., Arakawa Y., Itsuno S. Asymmetric transfer hydrogenation of imines catalyzed by a polymer-immobilized chiral catalyst. Org. Biomol. Chem. 2009;7:69–75. doi: 10.1039/B815407B. [DOI] [PubMed] [Google Scholar]
- 111.Liu J., Zhou Y., Wu Y., Li X., Chan A.S.C. Asymmetric transfer hydrogenation of ketones with a polyethylene glycol bound Ru catalyst in water. Tetrahedron Asymmetry. 2008;19:832–837. doi: 10.1016/j.tetasy.2008.03.015. [DOI] [Google Scholar]
- 112.Li J., Zhang Y., Han D., Gao Q., Li C. Asymmetric transfer hydrogenation using recoverable ruthenium catalyst immobilized into magnetic mesoporous silica. J. Mol. Catal., A Chem. 2009;298:31–35. doi: 10.1016/j.molcata.2008.09.027. [DOI] [Google Scholar]
- 113.Zhou Z., Sun Y. Synthesis of polyethylene glycol supported chiral monosulfonamide and its application in asymmetric transfer hydrogenation of prochiral ketones. React. Kinet. Mech. Catal. 2010;99:391–396. doi: 10.1007/s11144-009-0137-7. [DOI] [Google Scholar]
- 114.Arakawa Y., Chiba A., Haraguchi N., Itsuno S. Asymmetric transfer hydrogenation of aromatic ketones in water using a polymer-supported chiral catalyst containing a hydrophilic pendant group. Adv. Synth. Catal. 2008;350:2295–2304. doi: 10.1002/adsc.200800362. [DOI] [Google Scholar]
- 115.Liu J., Zhou D., Jia X., Huang L., Li X., Chan A.S.C. A convenient synthesis of (R)-salmeterol via Rh-catalyzed asymmetric transfer hydrogenation. Tetrahedron Asymmetry. 2008;19:1824–1828. doi: 10.1016/j.tetasy.2008.07.021. [DOI] [Google Scholar]
- 116.Huang L., Liu J., Shan W., Liu B., Shi A., Li X. The asymmetric synthesis of (R,R)-formoterol via transfer hydrogenation with polyethylene glycol bound Rh catalyst in PEG2000 and water. Chirality. 2010;22:206–211. doi: 10.1002/chir.20728. [DOI] [PubMed] [Google Scholar]
- 117.Zhu Y., Stubbs L.P., Ho F., Liu R., Ship C.P., Maguire J.A., Hosmane N.S. Magnetic nanocomposites: A new perspective in catalysis. ChemCatChem. 2010;2:365–374. doi: 10.1002/cctc.200900314. [DOI] [Google Scholar]
- 118.Schätz A., Reiser O., Stark W.J. Nanoparticles as semi-heterogeneous catalyst supports. Chem. Eur. J. 2010;16:8950–8967. doi: 10.1002/chem.200903462. [DOI] [PubMed] [Google Scholar]
- 119.Ranganath K.V.S., Glorius F. Superparamagnetic nanoparticles for asymmetric catalysis-a perfect match. Catal. Sci. Technol. 2011;1:13–22. doi: 10.1039/c0cy00069h. [DOI] [Google Scholar]
- 120.Polshettiwar V., Luque R., Fihri A., Zhu H., Bouhrara M., Basset J.-M. Magnetically Recoverable Nanocatalysts. Chem. Rev. 2011 doi: 10.1021/cr100230z. in press. [DOI] [PubMed] [Google Scholar]
- 121.Kassube J., Gade L. Stereoselective Dendrimer Catalysis. In: Gade L., editor. Dendrimer Catalysis. Volume 20. Springer; Berlin/Heidelberg, Germany: 2006. pp. 61–96. [Google Scholar]
- 122.Méry D., Astruc D. Dendritic catalysis: Major concepts and recent progress. Coord. Chem. Rev. 2006;250:1965–1979. doi: 10.1016/j.ccr.2005.11.012. [DOI] [Google Scholar]
- 123.de Jesús E., Flores J.C. Dendrimers: Solutions for catalyst separation and recycling – a review. Ind. Eng. Chem. Res. 2008;47:7968–7981. doi: 10.1021/ie800381d. [DOI] [Google Scholar]
- 124.Dijkstra H.P., van Klink G.P.M., van Koten G. The use of ultra- and nanofiltration techniques in homogeneous catalyst recycling. Acc. Chem. Res. 2002;35:798–810. doi: 10.1021/ar0100778. [DOI] [PubMed] [Google Scholar]
- 125.Chen Y.-C., Wu T.-F., Deng J.-G., Liu H., Jiang Y.-Z., Choi M.C.K., Chan A.S.C. Dendritic catalysts for asymmetric transferhydrogenation. Chem. Commun. 2001:1488–1489. doi: 10.1039/b104160f. [DOI] [Google Scholar]
- 126.Hawker C.J., Frechet J.M.J. Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J. Am. Chem. Soc. 1990;112:7638–7647. doi: 10.1021/ja00177a027. [DOI] [Google Scholar]
- 127.Newkome G.R., Lin X. Symmetrical, four-directional, poly(ether-amide) cascade polymers. Macromolecules. 1991;24:1443–1444. doi: 10.1021/ma00006a042. [DOI] [Google Scholar]
- 128.Chen Y.-C., Wu T.-F., Jiang L., Deng J.-G., Liu H., Zhu J., Jiang Y.-Z. Synthesis of dendritic catalysts and application in asymmetric transfer hydrogenation. J. Org. Chem. 2005;70:1006–1010. doi: 10.1021/jo048317v. [DOI] [PubMed] [Google Scholar]
- 129.Liu W., Cui X., Cun L., Zhu J., Deng J. Tunable dendritic ligands of chiral 1,2-diamine and their application in asymmetric transfer hydrogenation. Tetrahedron Asymmetry. 2005;16:2525–2530. doi: 10.1016/j.tetasy.2005.07.008. [DOI] [Google Scholar]
- 130.Gaikwad A.V., Boffa V., Ten Elshof J.E., Rothenberg G. Cat-in-a-cup: Facile separation of large homogeneous catalysts. Angew. Chem. Int. Ed. 2008;47:5407–5410. doi: 10.1002/anie.200801116. [DOI] [PubMed] [Google Scholar]
- 131.Wang W., Wang Q. A fluorinated dendritic TsDPEN-Ru(II) catalyst for asymmetric transfer hydrogenation of prochiral ketones in aqueous media. Chem. Commun. 2010;46:4616–4618. doi: 10.1039/c002168g. [DOI] [PubMed] [Google Scholar]
- 132.Geldbach T.J., Dyson P.J. A versatile ruthenium precursor for biphasic catalysis and its application in ionic liquid biphasic transfer hydrogenation: Conventional vs. task-specific catalysts. J. Am. Chem. Soc. 2004;126:8114–8115. doi: 10.1021/ja048886k. [DOI] [PubMed] [Google Scholar]
- 133.Letondor C., Ward T.R. Artificial metalloenzymes for enantioselective catalysis: Recent advances. ChemBioChem. 2006;7:1845–1852. doi: 10.1002/cbic.200600264. [DOI] [PubMed] [Google Scholar]
- 134.Steinreiber J., Ward T.R. Artificial metalloenzymes as selective catalysts in aqueous media. Coord. Chem. Rev. 2008;252:751–766. doi: 10.1016/j.ccr.2007.09.016. [DOI] [Google Scholar]
- 135.Rosati F., Roelfes G. Artificial metalloenzymes. ChemCatChem. 2010;2:916–927. doi: 10.1002/cctc.201000011. [DOI] [Google Scholar]
- 136.Letondor C., Humbert N., Ward T.R. Artificial metalloenzymes based on biotin-avidin technology for the enantioselective reduction of ketones by transfer hydrogenation. Proc. Natl. Acad. Sci. USA. 2005;102:4683–4687. doi: 10.1073/pnas.0409684102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Letondor C., Pordea A., Humbert N., Ivanova A., Mazurek S., Novic M., Ward T.R. Artificial transfer hydrogenases based on the biotin-(strept)avidin technology: Fine tuning the selectivity by saturation mutagenesis of the host protein. J. Am. Chem. Soc. 2006;128:8320–8328. doi: 10.1021/ja061580o. [DOI] [PubMed] [Google Scholar]
- 138.Creus M., Pordea A., Rossel T., Sardo A., Letondor C., Ivanova A., LeTrong I., Stenkamp R.E., Ward T.R. X-Ray structure and designed evolution of an artificial transfer hydrogenase. Angew. Chem. Int. Ed. 2008;47:1400–1404. doi: 10.1002/anie.200704865. [DOI] [PubMed] [Google Scholar]
- 139.Ward T.R. Artificial metalloenzymes based on the biotin-avidin technology: Enantioselective catalysis and beyond. Acc. Chem. Res. 2010;44:47–57. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 140.Dürrenberger M., Heinisch T., Wilson Y.M., Rossel T., Nogueira E., Knörr L., Mutschler A., Kersten K., Zimbron M.J., Pierron J., et al. Artificial transfer hydrogenases for the enantioselective reduction of cyclic imines. Angew. Chem. Int. Ed. 2011;50:3026–3029. doi: 10.1002/anie.201007820. [DOI] [PubMed] [Google Scholar]
- 141.Polborn K., Severin K. Biomimetic catalysis with immobilised organometallic Ruthenium complexes: Substrate- and regioselective transfer hydrogenation of ketones. Chem. Eur. J. 2000;6:4604–4611. doi: 10.1002/1521-3765(20001215)6:24<4604::AID-CHEM4604>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 142.Locatelli F., Gamez P., Lemaire M. Molecular imprinting of polymerised catalytic complexes in asymmetric catalysis. J. Mol. Catal., A Chem. 1998;135:89–98. doi: 10.1016/S1381-1169(97)00288-4. [DOI] [Google Scholar]
- 143.Weng Z., Muratsugu S., Ishiguro N., Ohkoshi S.-i., Tada M. Preparation of surface molecularly imprinted Ru-complex catalysts for asymmetric transfer hydrogenation in water media. Dalton Trans. 2011;40:2338–2347. doi: 10.1039/C0DT00950D. [DOI] [PubMed] [Google Scholar]
- 144.Yamakawa M., Ito H., Noyori R. The metal-ligand bifunctional catalysis: A theoretical study on the Ruthenium(II)-catalyzed hydrogen transfer between alcohols and carbonyl compounds. J. Am. Chem. Soc. 2000;122:1466–1478. doi: 10.1021/ja991638h. [DOI] [Google Scholar]
- 145.Ikariya T., Blacker A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007;40:1300–1308. doi: 10.1021/ar700134q. [DOI] [PubMed] [Google Scholar]
- 146.Koike T., Ikariya T. Mechanistic aspects of formation of chiral Ruthenium hydride complexes from 16-electron ruthenium amide complexes and formic acid: Facile reversible decarboxylation and carboxylation. Adv. Synth. Catal. 2004;346:37–41. doi: 10.1002/adsc.200303152. [DOI] [Google Scholar]
- 147.Kuzma M., Pelantová H., Sedmera P., Kačer P., Šiklová H., Červený L. Study on Influence of Amines on Behavior of the Noyoris Catalysts Under Transfer Hydrogenation Conditions Studied by NMR. In: Somorjai G., editor. International Symposium on Relations between Homogeneous and Heterogeneous Catalysis XIII (ISHHC XIII) University of California; Berkeley, CA, USA: 2007. [Google Scholar]
- 148.Wu X., Liu J., di Tommaso D., Iggo J.A., Catlow C.R.A., Bacsa J., Xiao J. A multilateral mechanistic study into asymmetric transfer hydrogenation in water. Chem. Eur. J. 2008;14:7699–7715. doi: 10.1002/chem.200800559. [DOI] [PubMed] [Google Scholar]
- 149.Åberg J.B., Samec J.S.M., Backvall J.-E. Mechanistic investigation on the hydrogenation of imines by [p-(Me2CH)C6H4Me]RuH(NH2CHPhCHPhNSO2C6H4-p-CH). Experimental support for an ionic pathway. Chem. Commun. 2006:2771–2773. doi: 10.1039/B605838H. [DOI] [PubMed] [Google Scholar]
- 150.Balcells D., Maseras F. Computational approaches to asymmetric synthesis. New J. Chem. 2007;31:333–343. doi: 10.1039/b615528f. [DOI] [Google Scholar]
- 151.Václavík J., Kuzma M., Přech J., Kačer P. Asymmetric transfer hydrogenation of imines and ketones using chiral Ru(II)Cl(η6-p-cymene)[(S,S)-N-TsDPEN] as a catalyst: A computational study. Organometallics. 2011 submitted. [Google Scholar]
- 152.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 153.Becke A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 154.Hay P.J., Wadt W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985;82:270–283. doi: 10.1063/1.448799. [DOI] [Google Scholar]